Introduction
Pancreatic cancer is a devastating disease. It is
attributed to obscure causes and generally displays a dismal
prognosis. Particularly, it remains intractable owing to the fact
that most types of pancreatic cancer are resistant to traditional
chemotherapies. Pancreatic ductal adenocarcinoma (PDAC) is by far
the most common tumor type of the exocrine pancreas, accounting for
85 to 90% of all pancreatic tumors (1). Although research on PDAC has been
carried out for half a century, its 5-year survival rate
(approximately 15-5%) remains the lowest as compared to other
digestive tract tumors (2). Only
20% of these patients have the chance to undergo curative
operation. More than 80% of the patients are diagnosed with PDAC at
later stages, for whom chemotherapy is the only choice (3). Gemcitabine is one of the few
FDA-approved agents used in the treatment of advanced pancreatic
cancer, and inhibits tumor growth by replacing cytidine during DNA
replication, thus blocking the biosynthesis of deoxyribonucleotide
by inactivating ribonucleotide reductase (4). Unfortunately, this therapy typically
prolongs the survival of fewer than 20% of the patients by only a
few months. Some of the patients become gemcitabine-resistant and
succumb to progressive disease after several courses, while the
vast majority of the patients have to bear the side effects with
little clinical remission after the treatment of gemcitabine
(5). Thus, evaluation of the
susceptibility of pancreatic cancer patients to gemcitabine before
clinical practice is crucial. Furthermore, unraveling of the
molecular mechanisms of gemcitabine resistance may improve the
efficacy of treatment.
Nude mice and commercial cell lines are used for the
research of the malignant biological characteristics of pancreatic
cancer, but immunodeficient nude mice have a poor track record for
predicting the response to therapy in clinical pancreatic cancer
patients. In addition, long culture duration makes commercial cells
prone to genetic drift (6,7). Compared to other tumor cell lines,
there are only 15 commercial PDAC cell lines. The classical cell
lines, such as Capan-1, Aspc-1, CFPAC, are derived from ascites,
lymph node metastases and other peritoneal metastases (8,9). As
far as we know, the primary tumor and their metastatic derivatives
usually undergo gene mutation, thus it is open to discussion
regarding whether the conclusion we have acquired from
metastasis-derived cells could be applied to primary tumors. Thus,
primary cells, which can imitate the epigenetic phenotypes, are
recommended to obtain a greater phenotypic heterogeneity as
compared to the disposable cell lines, and can upgrade the
pancreatic cancer cell line family. Few primary models are
available due to the limited amounts of clinical samples. On the
other hand, when rapid-growing fibroblasts overgrow or tumor cells
suffer bacterial contamination, the establishment of primary cells
fails. In this regard, patient-derived xenograft (PDX) models have
been utilized in order to overcome these difficulties. Moreover,
primary cells can be isolated from these xenograft-passaged tumors.
Both PDX models and primary cells assumedly play important roles in
the study of pancreatic cancer chemoresistance.
The mechanisms of the chemoresistance of pancreatic
cancer have been proposed as follows (10–12).
The microenvironment of PDAC has two marked features: lack of
vessels and rich in dense stroma. Lack of vessel leads to failure
of drug delivery. The stroma of PDAC is a dynamic environment
filled with various components, including pancreatic stellate
cells, matrix metalloproteinases, and growth factors. The
interaction between microenvironmental components and tumor cells
could impact the features of tumor cells themselves, such as the
chemoresistance. Previous studies on the internal biological
processes underlying drug resistance have recognized the role of
traditional drug-resistance related genes, such as MDR1, LRP and
RRM1, in PDAC chemoresistance. A recent study identified many new
protein (e.g., MCF2L, VMP1, and MAML3) that may lead to gemcitabine
resistance through the differenial expression spectrum between
gemcitabine-resistance strains and -sensitive strains (13). In the present study, our preliminary
screening results suggest that gemcitabine-resistant pancreatic
cancer cells exhibit a higher level of MCF2 transforming
sequence-like protein (MCF2L) mRNA, and therefore we chose MCF2 to
interrogate its role in the resistance of the primary cells to
gemcitabine. The goal of this study was to establish and validate
preclinical in vivo and in vitro models for screening
of the patients who are unable to respond to gemcitabine. In
addition, we attempted to identify the molecules involved in
gemcitabine resistance, which may serve as novel therapeutic
targets for PDAC.
Materials and methods
Establishment of pancreatic cancer PDX
(patient-derived xenograft) models
The human study protocol for the present study was
reviewed and approved by the Research Ethics Committee of Ruijin
Hospital, Shanghai Jiaotong University School of Medicine. All of
the eligible patients had been informed of the essentials of this
study, and written consent was obtained before enrollment of each
patient into this study. Surgically resected primary tumor tissues
from the patients with primary pancreatic cancer were harvested and
then separately placed in a sterile culture dish. After dissection
and removal of the necrotic areas, fatty tissues, blood clots and
connective tissues with forceps and scissors, each tumor specimen
was washed with 1% antibiotics-containing Dulbecco's phosphate
buffered saline (DPBS) twice and subsequently transferred into
another new dish where it was finely trimmed into a 20–30
mm3 fragment. Immediately following this process, the
tumor sample was implanted subcutaneously, by using an 18-gauge
trocar, in the fore and/or hind bilateral flanks of 6- to
8-week-old female BALB/c nude mice (Shanghai SLAC Laboratory Animal
Co., Ltd.). The general health of the mice was monitored daily and
growth of the tumor xenograft was monitored twice a week. Once the
first generation of xenografts (designed as P0) was established
(when the tumor size reached 500–800 mm3), serial
implantations in BALB/c nude mice were performed to expand the
xenograft tumors (i.e. P1, P2, P3, and beyond). Tumor size was
measured periodically using a digital caliper (Cal Pro, Sylvac,
Switzerland), and tumor volume was calculated as 0.5 × length ×
width2.
Establishment of pancreatic primary
cancer cells
Tumor samples were resected from the xenograft mouse
model and then placed in a sterile culture dish. After dissection
and removal of the necrotic areas, fatty tissues, blood clots and
connective tissues with forceps and scissors, the tumor specimens
were washed twice with DPBS that contains 100 U/ml penicillin and
0.1 mg/ml streptomycin. The samples were transferred into a new
dish where they were finely minced on ice with sterile scissors.
Approximately 15–20 ml of 1X Accumax solution was added to carry
out enzymatic dissociation of the cells from the primary tumor
tissues by vigorous pipetting. Following this process, the
resultant sample suspension was equally distributed between two
50-ml centrifuge tubes and incubated at 37°C for 1–2 h in a shaking
water bath. Then, the dissociated cells were re-suspended in 35–40
ml of 1X DPBS (for each 50 ml centrifuge tube) by pipetting. The
resultant suspension was subjected to filtration using 70-µm cell
strainers placed on two 50-ml centrifuge tubes, and the filtrates
were centrifuged at 1200 rpm for 5 min at room temperature. The
supernatants were aspirated and then washed with
antibiotic-containing DPBS twice. Finally, the primary cells were
harvested and cultured with serum-containing medium, which was
changed every second or third day. The cells were passaged using
trypsin/EDTA (Beyotime Biotechnology) when reaching sub-confluence.
Images of the cultures were captured with Olympus 1681 (Olympus,
Hamburg, Germany).
Immunofluorocytochemical
profiling
The expression of cytokeratin-8 (CK-8), epithelial
cell adhesion molecule (EpCAM) and pancreas/duodenum homeobox
protein-1 (PDX-1) in the isolated primary cells was evaluated by
immunofluorocytochemistry. For immunofluorescence labeling, one
drop of cell suspension at a low density was dripped on the cover
glass pretreated with poly-L-lysine, and then subjected to
incubation at 37°C for 2–4 h, which can make the cells fully
adherent. After washing with PBS, the cells were fixed with 4%
paraformaldehyde for 10 min at room temperature and then
permeabilized with 2 mg/l to 0.03 mg/l of Triton X-100 in PBS. The
nonspecific binding sites were blocked by incubating with 10 mg/l
bovine serum albumin (BSA) in PBS for 45 min at room temperature.
Slides were then incubated overnight at 4°C with the following
primary Abs: anti-CK-8 (1:500, Abcam), anti-PDX1 (1:500, Abcam),
anti-EpCAM (1:500, Santa Cruz Biotechnology), and the negative
control was incubated with 1X PBS. After incubation, the slides
were washed with PBS and further incubated with the donkey
anti-mouse, donkey anti-goat, donkey anti-rabbit secondary Ab
(dilution ratio 1:200, Life Technologies) for 2 h at room
temperature. After extensive washes, the slides were mounted in
glycerine (Beyotime) and observed using a confocal laser scanning
microscope (Radiance Plus; Bio-Rad Laboratories, Milan, Italy).
Exome library preparation and
sequencing
The genomic DNA of primary cells was sequenced by
next generation sequencing (NGS) to uncover the exome mutations of
KRAS and TP53. A total of 150 ng to 1 µg of DNA extracted from
primary cells by MALBAC (multiple annealing and looping-based
amplification cycles) was sheared into fragments around 175 bp
using the Covaris system (Covaris). The sheared DNA was purified
with Agencourt AMPure XP SPRI beads (Beckman Coulter). The DNA was
blunted with 5′-phosphorylated ends using the NEB Quick Blunting
kit and ligated to truncated PE P7 adaptors and barcoded P5
adaptors using NEBNext Quick Ligation Module. After cleanup with
Agencourt AMPure XP SPRI beads and nick fill-in with Bst
polymerase large fragment (New England Biolabs), the DNA fragments
with adaptors were enriched by PCR. A total amount of 500 ng of DNA
pooled from four barcoded libraries was used for hybridization and
posthybridization amplification following the manufacturer's
protocol (SureSelectXT Target Enrichment System for Illumina
Paired-End Sequencing Library, version 1.3.1, February 2012,
pp37-60; Agilent Technologies). The posthybridization amplification
product was quality checked and sequenced with Illumina HiSEq.
2000/2500 2X 100-bp paired-end (PE) reads.
CCK-8 assay
Cells (5×104) were seeded in 200 µl
aliquots into 96-well plates. At the indicated time points, 20 µl
CCK-8 solution (Dojindo, Tokyo, Japan) was added to the cells, and
the plates were incubated at 37°C in 5% CO2 humidified
chamber for an additional 2 h. The absorbance at 450 nm was
measured to determine the number of viable cells in each well. All
experiments were performed in quintuplicate.
In vitro migration assay
To quantify the in vitro motility of the 7
cell isolates, 8 µm Transwell cell culture chambers (Corning
Costar, Milan, Italy) were used. For migration assay, the 7 cell
isolates were starved in medium containing 1% FBS (fetal bovine
serum) for 24 h before the assay. A cell suspension (200 µl;
2×105 cells/ml) in RPMI-1640 with 1% FBS was added to
the upper chambers, whereas 600 µl of RPMI-1640 with 10% FBS was
placed in the lower wells. Thereafter, the cells were incubated at
37°C in 5% CO2 humidified chamber for 24 and 48 h. The
number of cells that migrated through the filters and seeded to the
lower chamber was photographed at each interval. Migration
experiments were conducted in quintuplicate.
Endothelial tube formation assay
Briefly, each well of 96-well culture plates was
coated with 100 µl Matrigel (BD Biosciences, Franklin Lakes, NJ,
USA), which was left to solidify at 37°C for 2 h. Human umbilical
vein endothelial cells (HUVECs) were re-suspended in the
supernatants obtained from each primary cell. Cell resuspension
solution (200 µl) containing 5×104 HUVECs were then
added to the solidified Matrigel. After 3 h of incubation at 37°C
with 5% CO2, the tubule numbers were assessed under a
light microscope.
Western blot analysis of EMT
markers
For western blot analysis, cells at confluence were
washed twice with PBS and harvested by trypsinization. Cells were
lysed in RIPA supplemented with cocktail and PMSF (Solarbio), and
the protein concentration was determined using a Bradford assay
(Bio-Rad Laboratories). Total protein extract (50 µg) of each
sample was separated by 10% sodium dodecyl sulfate polyacrylamide
gel electrophoresis and electroblotted on polyvinylidene fluoride
membranes (Merk Millipore). The transferred membranes were probed
with specific primary Ab at 1:1000 dilutions: anti-E-cadherin,
anti-N-cadherin, anti-vimentin, anti-catenin, anti-claudin.
Anti-GAPDH at a dilution of 1:1000 (Cell Signaling Technology) was
used as an internal control. After 3 washes, anti-mouse and
anti-rabbit horseradish peroxidase-conjugated secondary Ab (1:5000,
Cell Signaling Technology) were added to the membranes. Bands were
visualized by incubating the membranes with enhanced
chemiluminescence reagent (Thermo Fisher Scientific) and exposing
the membranes to X-ray film.
Real-time qPCR analysis
Cells, harvested by trypsinization, were subjected
to RNA extraction using the RNeasy kit (Qiagen, Milan, Italy). Two
microliters of total RNA (1 µg) was used for cDNA synthesis by
using reverse transcription (RT) reaction (ReverTra Ace-α-™,
Toyobo, Osaka, Japan). cDNAs was subjected to real-time
quantitative PCR analysis by using specific sets of primers
designed for the MCF2L gene (5′-AAGCCCGGTTATCACCTTCC-3′ and
5′-GGAGGTCCATTTGTCCCGT-3′). GAPDH gene (5′-GGAGCGAGATCCCTCCAAAAT-3′
and 5′-GGCTGTTGTCATACTTCTCATGG-3′) was used as an internal
control.
In vivo efficacy study
We chose the most gemcitabine sensitive cell line
PC-07-0049 and the most resistant cell line PC-07-0037 to conduct
animal tests. Gemcitabine was purchased from Melonepharma Co., Ltd.
(Dalian, China) and was formulated in distilled water for the in
vivo study. Tumor tissues were cut into small fragments of ~30
mm3 under sterile conditions. BALB/c nude mice were
implanted subcutaneously with a tumor fragment by using a trocar.
When the average tumor size reached 150–200 mm3, tumor
size-matched mice were randomly assigned to two groups with 5 mice
in each group. The tumor-bearing mice were administered gemcitabine
at 30 mg/kg (i.p., thrice per week), or vehicle (250 µl, i.p.,
thrice per week) for two weeks. Tumor volumes and body weights were
measured using calipers thrice a week.
Statistical analysis
The difference in tumor volumes between treatment
groups was analyzed for significance using one-way ANOVA followed
by Dunnett's test. The difference in expression profile of the
MCF2L mRNA between different PDX model-derived cells was analyzed
for significance using the Student's t-test. The statistical
analyses were performed using the IBM SPSS Statistics 19.0
software. P<0.05 was considered to indicate a statistically
significant difference.
Results
Patient information, PDX modeling, and
primary pancreatic cell isolation
According to the method mentioned above, we
harvested 28 tumor specimens, which were derived from patients
clinically diagnosed with primary pancreatic cancer but without
known or suspected peritoneal metastases, to establish the PDX
(patient-derived xenograft) models. It should be specified that the
patients who were selected for this study had neither been
subjected to adjuvant chemotherapy/radiotherapy nor undergone
radical pancreaticoduodenectomy. As anticipated, we acquired 28 PDX
models after the initial inoculation; the P0 tumor formation rate
in this regard was 100%. However, merely 18 of the 28 tumor
specimens had the capacity to expand the xenograft numbers in the
PDX mice over 10 successive generations; the remainder failed to
re-establish tumor xenografts after the second passage. As to these
expandable PDX models, the average time it took to form xenografts
with a size of 500 mm3 was 36±7.8 days. As indicated in
Table I, postoperative pathological
examination of the donors showed that 15 patients had pancreatic
ductal adenocarcinoma (of which 8 were identified in the head of
the pancreas and 7 were found in its tail). The other 3 patients
were confirmed to suffer from pancreatic duct mucus adenoma,
duodenal papillary adenocarcinoma, and pancreatic adenosquamous
carcinoma, respectively. The clinical information (i.e.
demographic) of the 18 patients is shown in Table II. In addition, we further
performed isolation of the primary cells from 7 of the 18
xenografts, which were able to be expanded for more than 10
generations in the PDX mice. Of the 7 primary cell isolates,
PC-07-0038 became inert after the 19th passage, which suggested the
occurrence of cell growth arrest. Accordingly, 6 constantly growing
primary cell isolates, namely PC-07-0001, PC-07-0015, PC-07-0034,
PC-07-0037, PC-07-0045, and PC-07-0049, were selected for the later
drug sensitivity assessment.
 | Table I.Pathologic examination of the
patients whose tumor specimens enabled the establishment of PDX
models and the time required for P0 tumor formation. |
Table I.
Pathologic examination of the
patients whose tumor specimens enabled the establishment of PDX
models and the time required for P0 tumor formation.
| PDX model | Pathological
type | Time spent on P0
tumor formationa
(days) |
|---|
| PC-07-0001 | PDAC arising from
the head of the pancreas | 48±7 |
| PC-07-0015 | PDAC arising from
the head of the pancreas | 27±9 |
| PC-07-0019 | Duodenal papillary
adenocarcinoma | 20±3 |
| PC-07-0020 | PDAC arising from
the head of the pancreas | 104±14 |
| PC-07-0021 | PDAC arising from
the head of the pancreas | 62±9 |
| PC-07-0022 | PDAC arising from
the head of the pancreas | 34±4 |
| PC-07-0023 | PDAC arising from
the body and tail of the pancreas | 27±4 |
| PC-07-0034 | PDAC arising from
the body and tail of the pancreas | 36±4 |
| PC-07-0035 | PDAC arising from
the head of the pancreas | 12±2 |
| PC-07-0037 | PDAC arising from
the body and tail of the pancreas | 35±3 |
| PC-07-0038 | PDAC arising from
the head of the pancreas | 28±6 |
| PC-07-0040 | PDAC arising from
the head of the pancreas | 34±10 |
| PC-07-0041 | PDAC arising from
the head of the pancreas | 33±7 |
| PC-07-0043 | Pancreatic
intraductal papillary mucinous carcinoma | 27±12 |
| PC-07-0045 | PDAC arising from
the body and tail of the pancreas | 40±10 |
| PC-07-0048 | Pancreatic
adenosquamous carcinoma | 35±11 |
| PC-07-0049 | PDAC arising from
the head of the pancreas | 29±4 |
| PC-07-0053 | PDAC arising from
the head of the pancreas | 23±3 |
| Table II.Demographic characteristics of the
patients included in the study. |
Table II.
Demographic characteristics of the
patients included in the study.
| PDX model | Sex | Age |
Diabetesa | CA199b |
|---|
| PC-07-0001 | F | 55 | N | >>1000 |
| PC-07-0015 | M | 78 | Y | 25.5 |
| PC-07-0019 | M | 76 | Y | 109 |
| PC-07-0020 | M | 65 | Y | >>1000 |
| PC-07-0021 | F | 78 | N | 2.7 |
| PC-07-0022 | F | 71 | Y | 5.6 |
| PC-07-0023 | M | 70 | Y | >>1000 |
| PC-07-0034 | M | 62 | N | 790 |
| PC-07-0035 | M | 67 | Y | 399 |
| PC-07-0037 | F | 59 | Y | >>1000 |
| PC-07-0038 | F | 82 | Y | >>1000 |
| PC-07-0040 | F | 63 | N | >>1000 |
| PC-07-0041 | M | 69 | N | 360 |
| PC-07-0043 | F | 74 | N | 21 |
| PC-07-0045 | M | 65 | Y | >>1000 |
| PC-07-0048 | M | 69 | N | 2.6 |
| PC-07-0049 | F | 47 | N | 39 |
| PC-07-0053 | F | 70 | N | >>1000 |
Size, shape, arrangement and growth of
the cultured primary cells
All of the isolated cells were shown to have the
capability of completely attaching to the bottom of the culture
dish. Cell culture medium testing showed that RPMI-1640
supplemented with 10% FBS provided the optimum growth conditions
for the primary cell isolates FC-07-0001, FC-07-0015, FC-07-0034,
and FC-07-0049. By contrast, the isolates FC-07-0037, FC-07-0038
and FC-07-0045 displayed best growth rates when cultivated in
DMEM/F12 supplemented with 2, 5, and 10% FBS, respectively. The
average volume of FC-07-0001, FC-7-0038, and FC-07-0049 was
comparatively larger than that of FC-07-0015, FC-07-0034,
FC-07-0037, and FC-07-0045 (data not shown). The morphologic
features of the isolated cells are specified in Table III. As for the cell expandability,
the primary cells isolated from the PDX models PC-07-0015,
PC-07-0034, PC-07-0037, PC-07-0045, and PC-07-0049 could still
maintain an active state (i.e. being capable of growing well) when
they were multiplied over 20 passages in culture. As indicated
above, cells isolated from the PDX model PC-07-0038 were shown to
go into an inert state when they had undergone 19 passages.
| Table III.Morphologic features of the cells
isolated from xenografts of the PDX models. |
Table III.
Morphologic features of the cells
isolated from xenografts of the PDX models.
| Primary cells from
the PDX model | Shape | Arrangement | Other notes |
|---|
| PC-07-0001 | Long quadrilateral
or triangular | Being scattered
disorderly |
|
| PC-07-0015 | Ovoid or round | Being fused into
irregular sheets |
|
| PC-07-0034 | Ovoid or round | Without clear
boundary; being fused into a monolayer |
|
| PC-07-0037 | A variety of
shapes | Being fused into
round sheets | Small nuclei |
| PC-07-0038 | Circular or
polygonal | Being scattered
disorderly | Large nuclei; many
granules in the cytoplasm |
| PC-07-0045 | Round | Being fused into
round or irregular ball; well-defined contour | Large nuclei; some
are multinucleated |
| PC-07-0049 | Polygonal | Being scattered
disorderly | Large volume; small
nuclei |
All the primary cell isolates express
CK-8, EpCAM and PDX-1
The expression of CK-8, EpCAM, and PDX-1 in the
primary cells isolated from the 7 PDX models was evaluated by
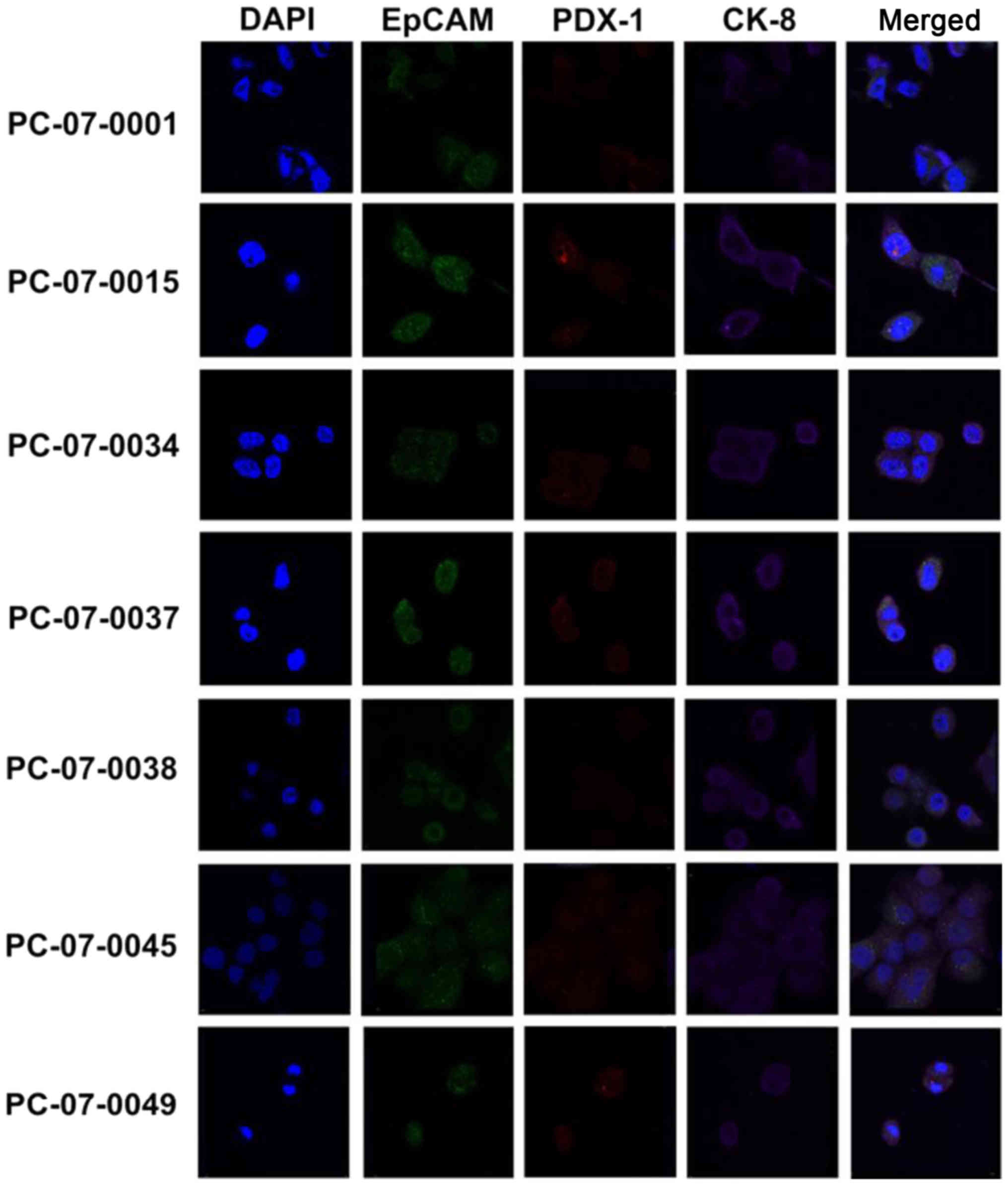
immunofluorocytochemical assay. As demonstrated in Fig. 1, all 7 primary cell isolates were
shown to express CK-8, EpCAM and PDX-1. Notably, the merged images
revealed that CK-8 (purple fluorescence), EpCAM (green
fluorescence), and PDX-1 (red fluorescence) were located in the
cytosol, plasma membrane, and nucleus/cytosol, respectively.
Isolated primary cells display high
frequency of KRAS and TP53 mutations
By analyzing the whole-exome sequencing data of the
7 primary cells, we found that all of them contained several
mutations in the KRAS oncogene and TP53 tumor-uppressor gene. The
mutation frequency of KRAS was 100% (7/7), while that of TP53 was
71.4% (5/7). In all 7 primary isolates, KRAS was mutated at codon
12. PC-07-0015, PC-07-0034, PC-07-0045 and PC-07-0049 had a G to R
transition (GGT-GRT); PC-07-0001 showed a G to A transversion
(GGT-GAT); and PC-07-0037 and PC-07-0038 had a G to D transition
(GGT-GDT). TP53 inactivating mutations were found in 5 out of 7
cell isolates (Table IV).
| Table IV.Mutation loci of the key oncogene
KRAS and tumor-suppressor gene TP53 in the isolated primary
cells. |
Table IV.
Mutation loci of the key oncogene
KRAS and tumor-suppressor gene TP53 in the isolated primary
cells.
| Primary cells | TP53 | KRAS |
|---|
| PC-07-0001 | R248Q, R116Q,
R209Q | G12A |
| PC-07-0015 | C238R, C199R, C79R,
C106R | G12R |
| PC-07-0034 |
| G12R |
| PC-07-0037 | H179R, H140R, H20R,
H47R | G12D |
| PC-07-0038 | R175H, R136H, R16H,
R43H | G12D |
| PC-07-0045 | R175H, R136H, R16H,
R43H | G12R |
| PC-07-0049 |
| G12R |
Proliferation, motility and
angiopoietic ability of isolated primary cells
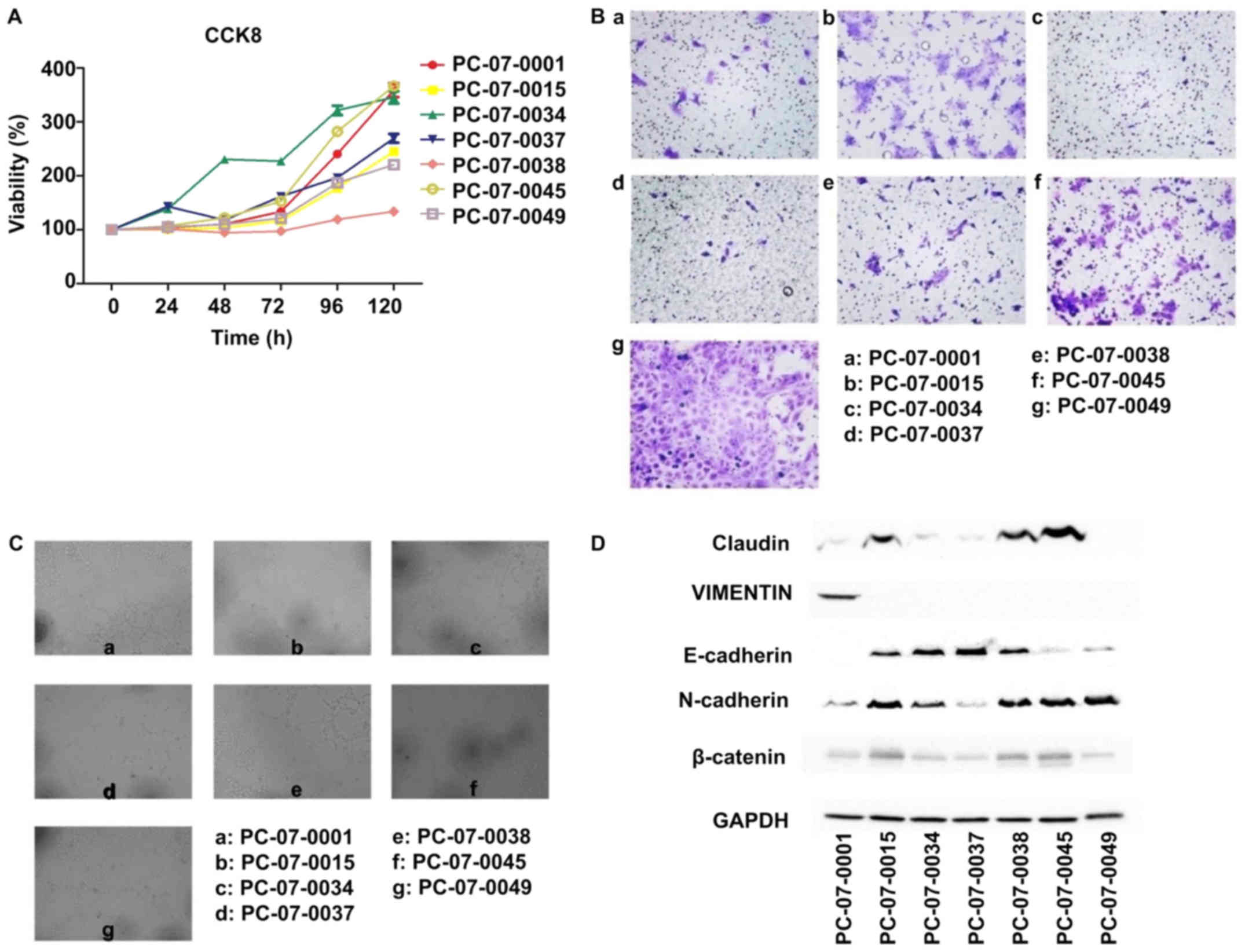
The results of the CCK-8 assay showed that the
relative proliferation rates of the 7 cells were different.
PC-07-0034 demonstrated the fastest growth rate, with a doubling
time of approximately 36 h, whereas PC-073-0038 exhibited the
slowest growth rate, with a doubling time of 145 h (Fig. 2A). The Transwell assay showed that
the number of PC-07-0049 cells passing through the basement
membrane was the highest, which indicated that the motility of
PC-07-0049 was the most advanced in all 7 cells, followed by
PC-07-0045 and PC-07-0015. In contrast, there were few PC-07-0034
and PC-07-0037 cells passing through the membrane indicating that
the migration rates of PC-07-0034 and PC-07-0037 were the slowest
(Fig. 2B). In the endothelial tube
formation assay, following treatment with the culture media of
PC-07-0001, PC-07-0038 and PC-07-0049, the HUVECs were capable of
forming integrated tubes; on the contrary, the culture media of
PC-07-0037 and PC-07-0045 were unable to induce endothelial tube
formation. Imperfect tubes were found in HUVECs treated with
culture media of PC-07-0015 and PC-07-0034 (Fig. 2C).
Expression of EMT markers in the
isolated primary cells
Results of the western blot analysis demonstrated
that PC-07-0045 and PC-07-0049 expressed the lowest level of
E-cadherin and the highest level of N-cadherin (Fig. 2D). However, the results of
PC-07-0037 were reversed. Claudin and β-catenin were most highly
expressed in PC-07-0015, PC-07-0038 and PC-07-0045. The expression
of vimentin was solely found in PC-07-0001.
Viability of the isolated primary
cells in response to gemcitabine
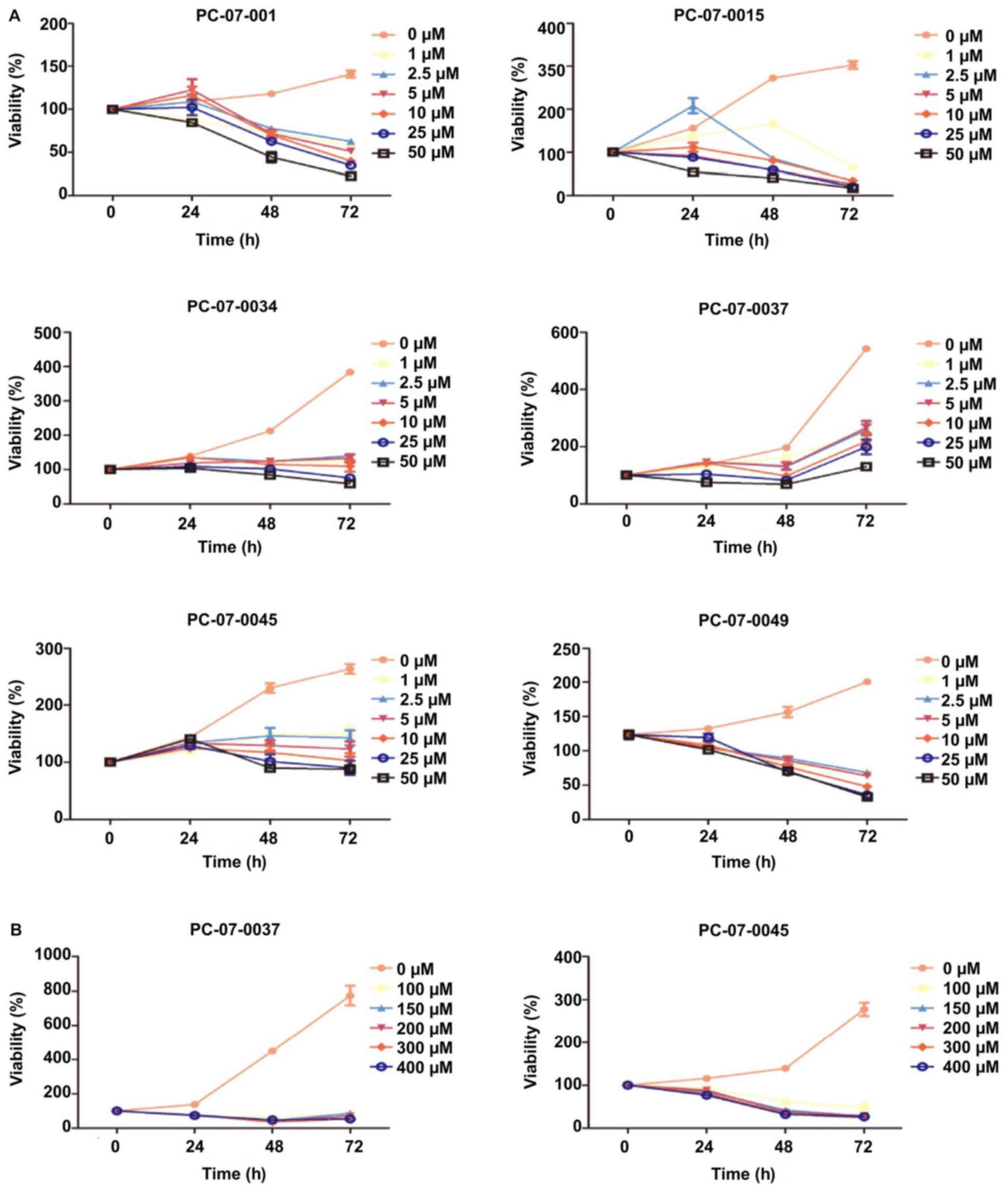
The dose response of each primary cell to
gemcitabine was evaluated by the CCK-8 assay. The 7 isolated
primary cells were treated with the following concentrations of
gemcitabine: 0, 2.5, 5, 10, 20, and 50 µM. The IC50
values of the cells derived from PC-07-0001, PC-07-0015 and
PC-07-0049 were 4.332, 1.534 and 9.697 µM, respectively. As for the
cells from PC-07-0034, PC-07-0037 and PC-07-0045, the
IC50 values were 55.18, 409.6 and 62.87 µM respectively.
Nevertheless, PC-07-0037 and PC-07-0045 cells subjected to
treatment with 50 µM gemcitabine were still alive even at 72 h
post-treatment, suggesting they were less sensitive to gemcitabine
(Fig. 3A). When subjected to higher
doses (100, 150, 200, 300 and 400 µM) of gemcitabine, their
vulnerabilities significantly increased (Fig. 3B). As a consequence, in view of
their vulnerabilities to lower and higher doses of gemcitabine,
these cells could be divided into the sensitive groups (PC-07-0001,
PC-07-0015, and PC-07-0049) and the resistant groups (PC-07-0034,
PC-07-0037, and PC-07-0045). The IC50 values of the two
groups were significantly different (P<0.05).
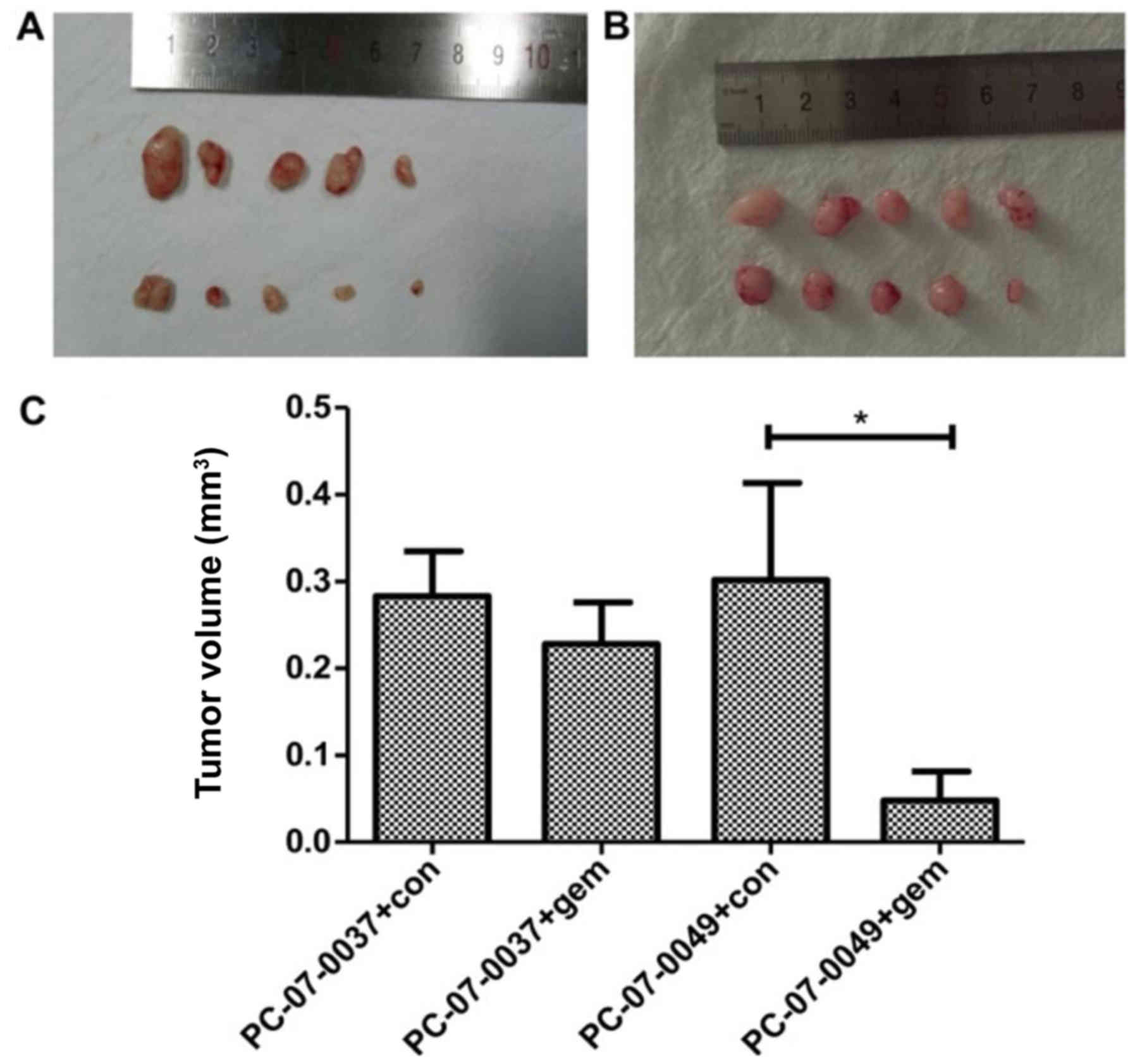
The PC-07-0049 and PC-07-0037 PDX mice
respond differently to gemcitabine
At 2-weeks post-treatment, the volume of the tumor
in the PC-07-0049 PDX mice (1,579.87±274.8 mm3) was
significantly smaller than that of the control group
(3,318.63±549.47 mm3) (P<0.05), which indicates that
gemcitabine significantly inhibited the growth of inoculated
sensitive pancreatic cancer cells in mice (Fig. 4A and C). However, as for the
resistant xenograft (i.e., PC-07-0037 implantation), there was no
significant difference between the gemcitabine treatment and
control groups (Fig. 4B and C).
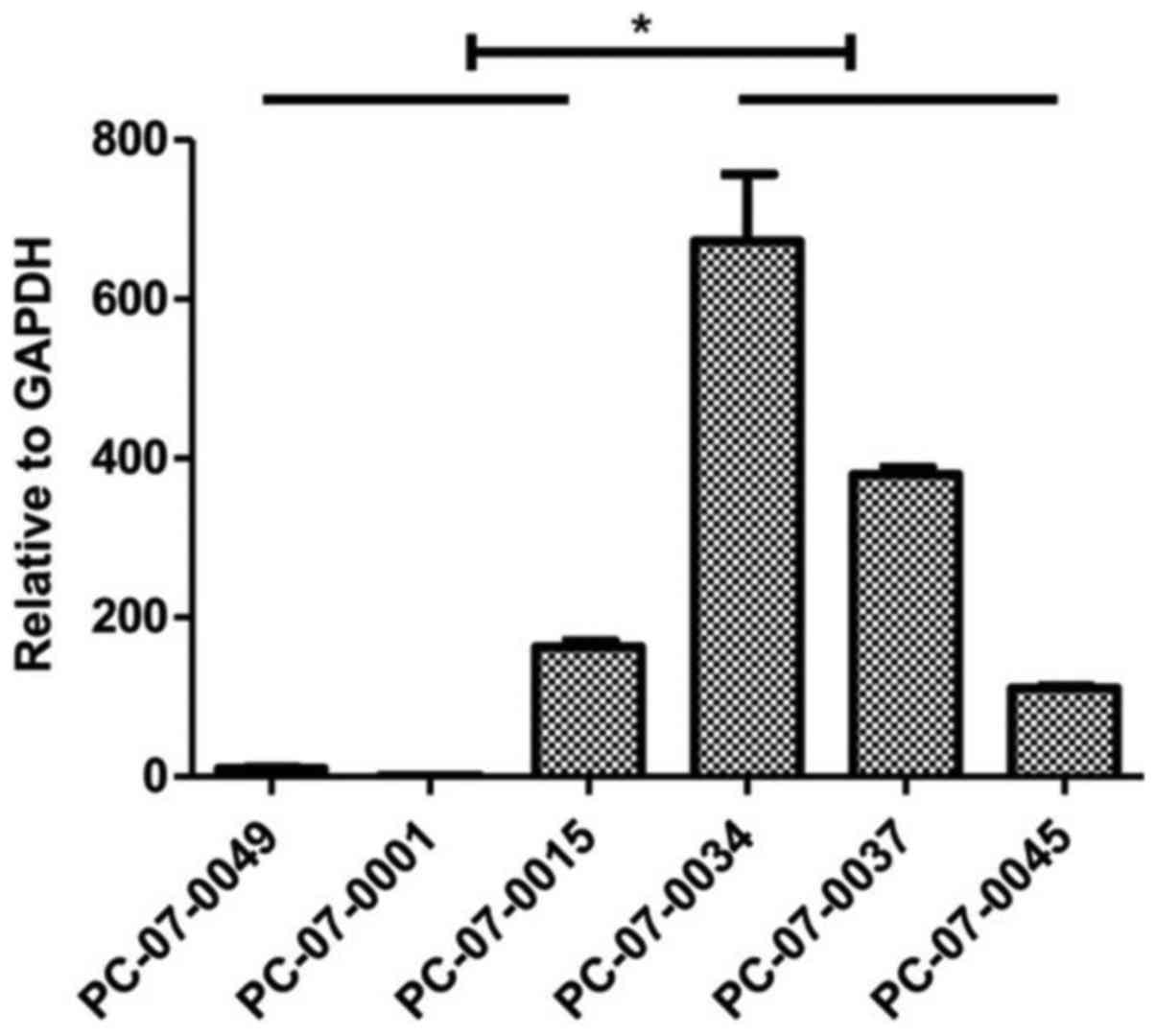
mRNA of MCF2L is highly expressed in the
chemoresistant groups. The results of the real-time qPCR assay
indicated that expression levels of MCF2 transforming sequence-like
protein (MCF2L) mRNA were significantly higher in the resistant
cells, as compared to that noted in the sensitive cells (P<0.05)
(Fig. 5).
Discussion
To achieve better outcomes of gemcitabine treatment,
it is important to evaluate the therapeutic effects of gemcitabine
on pancreatic cancer patients before clinical practice. In general,
research associated with drug sensitivity testing is inseparable
from the research tools in vitro. Immortalized cells provide
a substitute for a limited number of tissue specimens, and
therefore becomes an important part of the in vitro
experimental platform (14).
However, there are only 15 pancreatic cancer cell lines commonly
used for testing (15). Many
classical cell lines are not derived from primary tumors, but from
metastatic tissues instead. Whether the experimental conclusions
from metastasis-derived cell lines can be used to explain the
mechanisms involved in primary tumor formation is still
questionable, as accumulation of gene mutations occurs while
primary tumors become metastatic. Furthermore, the commercial cells
are prone to genetic drift after long-term culture and may not
represent patient characteristics (16–18),
which indicates that their derived xenograft models could not
accurately predict drug response during clinical therapy. While the
PDX (patient-derived xenograft) models are more advantageous for
testing drug susceptibility, the primary cells isolated from the
PDX models are therefore capable of evaluating drug responses in
vitro. Primary cells have been successfully established from
pancreatic ductal adenocarcinoma, neuroendocrine tumor, biliary
papilloma and normal tissue by means of direct adherent culture
methods or trypsin enzyme digesting method (19–22).
To validate whether the cells we harvested through
the above-mentioned are exactly tumor cells of human pancreatic
ductal adenocarcinoma, we assessed the expression of pancreatic
ductal-specific marker PDX-1, pancreatic tumor marker CK8 and
epithelial-specific marker EpCAM. After the body becomes mature,
PDX-1 specifically expresses in the pancreas as a transcription
factor and is considered to be a master gene that controls
pancreatic endocrine cell development (23). It contributes to the development of
the pancreas and participates in the differentiation of pancreas
islet cells. PDX-1 is expressed in pancreatic ductal epithelial
cells, β cells that secrete insulin, and delta cells that release
somatostatin (24). A series of
cytoskeleton markers, for example, cytokeratin, could be used to
identify whether the human pancreatic cancer cells were of ductal
epithelial origin (25).
Cytokeratin, belonging to the cytoskeleton protein family, is one
of the intermediate neurofilament protein expressed by epithelial
cells. It plays an important role in maintaining epithelial cell
morphology. CK-8 is an alkaline cell keratin and is mainly
expressed in the cytoplasm of epithelial malignant cells. As a
characteristic marker of epithelial cells, CK-8 could become a new
target for cancer treatment. High expression of CK-8 protein could
alter the epithelial phenotype and promote malignant transformation
of cells (26,27). Epithelial cell adhesion molecule
(EpCAM) is a calcium-independent epithelial cell adhesion molecule,
and it is also a novel tumor marker identified in recent years.
EpCAM is widely expressed in lung cancer, colorectal cancer,
pancreatic cancer and thyroid cancer, but is seldom expressed in
normal epithelium. In this regard, we considered it a cancer cell
marker of isolated primary cells (28,29).
In our study, we found that all of the 7 cells expressed CK-8,
EpCAM and PDX-1. CK-8 was shown to be located in the cytoplasm,
EPCAM in the plasma membrane, and PDX-1 in the nucleus and
cytoplasm. Thus, we confirmed that all the isolated cells were
pancreatic ductal epithelial tumor cells.
We found that morphologic and phenotypic features of
the primary cells varied between different PDX models, which may be
attributed to the genetic and epigenetic diversities between
individual patients. Such diversities conduce to the difference in
their clinical manifestations and differential drug sensitivity. We
analyzed the proliferation rate, migration capacity, ability of
blood vessel formation, and genomic characteristics of the primary
cells derived from the different PDX models. Similar to the
development of all epithelial tumors, our study also indicated that
activation of oncogenes and inactivation of tumor-suppressor genes,
due to genetic mutations, account for the occurrence of pancreatic
cancer (30,31). By analyzing the exome sequencing
data of the 7 primary cells, we found that all the primary cells
contained several mutations in the well-known oncogene KRAS and
tumor-suppressor gene TP53, and their mutation frequencies are
consistent with published sequencing data (32,33).
KRAS mutation was found in all 7 cell isolates and were all mutated
at codon 12. Of the 7 cells, 4 (PC-07-0015, PC-07-0034, PC-07-0045
and PC-07-0049) had a G to R transition (GGT-GRT). PC-07-0001
showed a G to A transversion (GGT-GAT), whereas PC-07-0037 and
PC-07-0038 had a G to D transition (GGT-GDT). TP53 inactivating
mutations were found in 5 out of 7 cell lines, which was also
consistent with a previous report described as follows. Dong et
al reported that most P53 mutations are located in one of 4
missense mutation hot spots, including a.a.129-146, a.a.171-179,
a.a.234-260 and a.a.270-287 (34,35).
In our study, all the cells had 3–4 mutation loci, 8
of which were located at the common loci reported in the previous
study. The CCK-8 assay indicated that the proliferation rate of the
7 cells were different. PC-07-0034 grew the fastest, with a
doubling time of around 36 h. By contrast, the doubling time of
PC-07-0038 was 145 h. The proliferation rates of the remaining
cells just fell in between. The digestive passage proportion of
cells also reflects its growth rate. PC-07-0034 should be passaged
into 3 or 4 culture dishes. Cell confluence (more than 90%) will be
achieved in 24 h if they are passaged into two dishes. We also
found several differences in the migration ability of these
isolated cells. Transwell assay showed that the number of
PC-07-0049 cells migrating across the basement membrane was the
largest, which indicated that the motility of PC-07-0049 was the
highest among all 7 cells. In contrast, there were few PC-07-0037
cells that accessed across the membrane, indicating that the
migration capacity of PC-07-0037 was weak. An important mechanism
of tumor invasion is the occurrence of epithelial-mesenchymal
transition (EMT). Our western blot data demonstrated that
PC-07-0045 and PC-07-0049 expressed the highest levels of
E-cadherin and the lowest level of N-cadherin. However, PC-07-0037
displayed opposite results. The differential motility of the 3
cells may be explained by levels of EMT, which are affected by the
expression of the two epithelial cadherins.
Vimentin is the most important intermediate filament
in ectomesenchymal cells, and it is absent in normal epithelial
cells. Its expression has been noted to be upregulated during tumor
transformation (36,37). The expression of vimentin was only
found in PC-07-0001, indicating that the motility of PC-07-0001 may
be associated with vimentin. β-catenin and E-cadherin have been
shown to be colocalized in intercellular adhesion connection
(38,39). The expression level of β-catenin is
associated with the malignant degree of prostate cancer, lung
cancer, and colorectal cancer (40). Claudin is a vital protein in cell
membrane tight junction, whose abnormal expression leads to
structural damage and functional impairment of epithelial cells
(41). The expression of claudin is
closely related to the occurrence and development of a wide variety
of tumors (42–44). Claudin and β-catenin were highly
expressed in PC-07-0015, PC-07-0038 and PC-07-0045. The motility of
the 3 cells may be related to the expression of β-catenin and
claudin. Similar to most malignant tumors, the basis of tumor
growth and metastasis is the formation of tumor blood vessels,
which is also designated as ‘angiogenesis’ (45,46).
In the endothelial tube formation assay, following treatment with
the culture media of PC-07-000, PC-07-0038 and PC-07-0049, the
HUVECs were capable of forming integrated tubes. On the contrary,
the culture media of PC-07-0037 and PC-07-0045 were unable to
induce endothelial tube formation.
Chemotherapeutic drugs may have serious side
effects, such as gastrointestinal reactions, liver and kidney
damage, and bone marrow suppression, even though they are able to
kill tumors. Thus, if we can evaluate the sensitivity of these
drugs before clinical trial, we can administer them to the
drug-sensitive population for effective treatment, while avoiding
or minimizing the side effects of the drugs. Gemcitabine is the
first line drug for advanced pancreatic cancer, which was certified
by the U.S. Food and Drug Administration (FDA) in 1997. However,
the clinical response rate of gemcitabine is still lower than 10%.
Over 90% of the patients are resistant to gemcitabine. Long-term
administration of gemcitabine has been shown to lead to acquired
drug resistance and side-effect in sensitive patients. In
traditional chemotherapeutic regimens, identification of the drug
sensitivity of patients before taking medication was unknown.
Patients diagnosed with pancreatic cancer were all recommended to
receive gemcitabine treatment. Many patients did not have a
satisfactory outcome, yet suffered the side effects of gemcitabine
instead. Thus, the sensitivity of patients to gemcitabine should be
evaluated before clinical application. Only the drug sensitive
population should be administered gemcitabine. In our experiment,
we found that the sensitivity of the cells to gemcitabine was
inconsistent. When PC-07-0049 was treated with 10 µM gemcitabine
for 72 h, cell proliferation was significantly inhibited. Moreover,
cell debris and cell necrosis increased. However, when PC-07-0037
was treated with 10 µM gemcitabine for 72 h, no obvious cell
proliferation inhibition was observed, and cell morphology appeared
normal. As for the in vivo tests, when the PDX models
subcutaneously inoculated with the 2 cells were treated with the
same concentration of gemcitabine, the volume of the tumor
xenografts in the drug-resistant PC-07-0037 PDX model administered
gemcitabine did not differ from that of the untreated control. In
contrast, the tumor volume of the drug-sensitive PC-07-0049 PDX
model (3,318.63±549.47 mm3) was obviously reduced as
compared to that of the untreated control (1,579.87±274.8
mm3). Therefore, we concluded that we can selectively
and specifically apply therapeutic agents to patients, according to
the results of the sensitivity test performed in the PDX models.
This will strategically not only increase therapeutic efficacy of
chemotherapy but also reduce its side effects.
It has become an urgent issue that drug resistance
greatly reduces therapeutic effects. Recent studies have identified
various proteins that may lead to gemcitabine resistance by means
of proteomics, RNA-seq and whole-genome siRNA library to measure
the expression spectrum of gemcitabine resistance between resistant
and sensitive strains. We tested and verified the mRNA expression
levels of these proteins in the primary cells and found that,
compared to the gemcitabine-ensitive cells, the
gemcitabine-resistant cells had a high level of MCF2L expression.
MCF2L, also named DBS/DBLs Big Sister, belongs to the DBL family.
MC2L is one of the guanine nucleotide exchange factor and
potentially links pathways that signal through RAC1, RHOA and CDC42
(47). It can catalyze guanine
nucleotide exchange on RHOA and CDC42 and interacts specifically
with the GTP-bound form of RAC1, suggesting that it functions as an
effector of RAC1. There are two types of endogenous DBS subtypes:
the molecular weight of the first type is 130 kDa (DBS-130), which
is located in the Golgi apparatus; the other one (DBS-80) is
located in the endoplasmic reticulum. A previous study has
demonstrated that the DBS-130 inhibitors can reduce the motility of
MDA-MB-231 cells in Transwell and scratch assays (48,49).
DBS becomes activated and highly tumorigenic via truncation of the
N-terminus, and the activation of DBS can promote cell
proliferation during the development of hemocytes (50). However, the role of MCF2L/DBS in the
drug resistance of tumor cells has not yet been studied. Our study
found that the gemcitabine-resistant cells had a high level of
MCF2L expression, as compared to the gemcitabine-sensitive cells.
Consequently, we conclude that MCF2L may play an important role in
gemcitabine resistance, which still needs to be further
confirmed.
Limitations of the study. Pancreatic cancer is
deadly and often does not cause any signs or symptoms at the early
stage. The patients are usually diagnosed at the late stage when
they feel something wrong with their bodies (e.g. indigestion,
heartburn, unexplained weight loss, and abdominal pain). Currently,
there is no standard diagnostic tool or established early detection
method for pancreatic cancer. The delay in diagnosis leads to loss
of therapeutic golden chance. Gemcitabine is a recommended
chemotherapeutic agent used in the treatment of pancreatic cancer.
Unfortunately, this treatment is very challenging due to the
occurrence of chemoresistance. In this regard, many patients do not
show a satisfactory outcome, but suffer the side effects of
gemcitabine instead. The purpose of this study was to establish a
PDX model for pre-clinical assessment of chemoresistance in
pancreatic cancer rather than for development of a clinical
diagnostic strategy. We expect that future improvement of the
technique for establishing the PDX model can make this pre-clinical
platform more useful for exploring the mechanisms of
chemoresistance. Regarding the pitfalls of this study, we recognize
that inadequate acquisition of the patient follow-up information
weakened the value and clinical significance of this study;
understanding of the correlation between patient responsiveness and
therapeutic efficacy may help unravel the underlying mechanisms of
gemcitabine resistance. Therefore, our future goal is to pursue
further studies on the downstream genes associated with gemcitabine
resistance.
In conclusion, the in vivo PDX models and
in vitro primary cell models derived from the clinical
samples provide a powerful support for the susceptibility testing
of gemcitabine and drug development. MCF2L can be considered to
play an important role in gemcitabine resistance.
References
|
1
|
Weissmueller S, Manchado E, Saborowski M,
Morris JP IV, Wagenblast E, Davis CA, Moon SH, Pfister NT,
Tschaharganeh DF, Kitzing T, et al: Mutant p53 drives pancreatic
cancer metastasis through cell-autonomous PDGF receptor β
signaling. Cell. 157:382–394. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yachida S, Jones S, Bozic I, Antal T,
Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al:
Distant metastasis occurs late during the genetic evolution of
pancreatic cancer. Nature. 467:1114–1117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Minami K, Shinsato Y, Yamamoto M,
Takahashi H, Zhang S, Nishizawa Y, Tabata S, Ikeda R, Kawahara K,
Tsujikawa K, et al: Ribonucleotide reductase is an effective target
to overcome gemcitabine resistance in gemcitabine-resistant
pancreatic cancer cells with dual resistant factors. J Pharmacol
Sci. 127:319–325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu CP, Xue XJ, Liang N, Xu DG, Liu FJ, Yu
XS and Zhang JD: Effect of chemoradiotherapy and neoadjuvant
chemoradiotherapy in resectable pancreatic cancer: A systematic
review and meta-analysis. J Cancer Res Clin Oncol. 140:549–559.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wennerström AB, Lothe IM, Sandhu V, Kure
EH, Myklebost O and Munthe E: Generation and characterisation of
novel pancreatic adenocarcinoma xenograft models and corresponding
primary cell lines. PLoS One. 9:e1038732014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kato M, Shimada Y, Tanaka H, Hosotani R,
Ohshio G, Ishizaki K and Imamura M: Characterization of six cell
lines established from human pancreatic adenocarcinomas. Cancer.
85:832–840. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Smith HS: In vitro properties of
epithelial cell lines established from human carcinomas and
nonmalignant tissue. J Natl Cancer Inst. 62:225–230.
1979.PubMed/NCBI
|
|
9
|
Chen WH, Horoszewicz JS, Leong SS, Shimano
T, Penetrante R, Sanders WH, Berjian R, Douglass HO, Martin EW and
Chu TM: Human pancreatic adenocarcinoma: In vitro and in vivo
morphology of a new tumor line established from ascites. In Vitro.
18:24–34. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Koay EJ, Truty MJ, Cristini V, Thomas RM,
Chen R, Chatterjee D, Kang Y, Bhosale PR, Tamm EP, Crane CH, et al:
Transport properties of pancreatic cancer describe gemcitabine
delivery and response. J Clin Invest. 124:1525–1536. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Olive KP, Jacobetz MA, Davidson CJ,
Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA,
Caldwell ME, Allard D, et al: Inhibition of Hedgehog signaling
enhances delivery of chemotherapy in a mouse model of pancreatic
cancer. Science. 324:1457–1461. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moir JA, Mann J and White SA: The role of
pancreatic stellate cells in pancreatic cancer. Surg Oncol.
24:232–238. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zagorac S: Identification and functional
characterization of epigenetic determinants of pancreatic CSCs.
2015.PhD dissertation, la Universidad Autónoma de Madrid,
11–16–2015. http://hdl.handle.net/10486/669541
|
|
14
|
Ku JL, Yoon KA, Kim WH, Jang Y, Suh KS,
Kim SW, Park YH and Park JG: Establishment and characterization of
four human pancreatic carcinoma cell lines. Genetic alterations in
the TGFBR2 gene but not in the MADH4 gene. Cell Tissue Res.
308:205–214. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rückert F, Aust D, Böhme I, Werner K,
Brandt A, Diamandis EP, Krautz C, Hering S, Saeger HD, Grützmann R,
et al: Five primary human pancreatic adenocarcinoma cell lines
established by the outgrowth method. J Surg Res. 172:29–39. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Briske-Anderson MJ, Finley JW and Newman
SM: The influence of culture time and passage number on the
morphological and physiological development of Caco-2 cells. Proc
Soc Exp Biol Med. 214:248–257. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang-Liu CM and Woloschak GE: Effect of
passage number on cellular response to DNA-damaging agents: Cell
survival and gene expression. Cancer Lett. 113:77–86. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Esquenet M, Swinnen JV, Heyns W and
Verhoeven G: LNCaP prostatic adenocarcinoma cells derived from low
and high passage numbers display divergent responses not only to
androgens but also to retinoids. J Steroid Biochem Mol Biol.
62:391–399. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mohamed A, Blanchard MP, Albertelli M,
Barbieri F, Brue T, Niccoli P, Delpero JR, Monges G, Garcia S,
Ferone D, et al: Pasireotide and octreotide antiproliferative
effects and sst2 trafficking in human pancreatic neuroendocrine
tumor cultures. Endocr Relat Cancer. 21:691–704. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Murakami S, Ajiki T, Hori Y, Okazaki T,
Fukumoto T and Ku Y: Establishment of a novel cell line from
intraductal papillary neoplasm of the bile duct. Anticancer Res.
34:2203–2209. 2014.PubMed/NCBI
|
|
21
|
Kong D, Nishino N, Shibusawa M and Kusano
M: Establishment and characterization of human pancreatic
adenocarcinoma cell line in tissue culture and the nude mouse.
Tissue Cell. 39:217–223. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lawson T, Ouellette M, Kolar C and
Hollingsworth M: Culture and immortalization of pancreatic ductal
epithelial cells. Methods Mol Med. 103:113–122. 2005.PubMed/NCBI
|
|
23
|
Park JY, Hong SM, Klimstra DS, Goggins MG,
Maitra A and Hruban RH: Pdx1 expression in pancreatic precursor
lesions and neoplasms. Appl Immunohistochem Mol Morphol.
19:444–449. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fendrich V and Lauth M: The role of
pancreatic and duodenal homeobox 1 as a therapeutic target in
pancreatic cancer. Expert Opin Ther Targets. 18:1277–1283. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moll R, Franke WW, Schiller DL, Geiger B
and Krepler R: The catalog of human cytokeratins: Patterns of
expression in normal epithelia, tumors and cultured cells. Cell.
31:11–24. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Merjava S, Brejchova K, Vernon A, Daniels
JT and Jirsova K: Cytokeratin 8 is expressed in human
corneoconjunctival epithelium, particularly in limbal epithelial
cells. Invest Ophthalmol Vis Sci. 52:787–794. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu F, Chen Z, Wang J, Shao X, Cui Z, Yang
C, Zhu Z and Xiong D: Overexpression of cell surface cytokeratin 8
in multidrug-resistant MCF-7/MX cells enhances cell adhesion to the
extracellular matrix. Neoplasia. 10:1275–1284. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Munz M, Baeuerle PA and Gires O: The
emerging role of EpCAM in cancer and stem cell signaling. Cancer
Res. 69:5627–5629. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Went PT, Lugli A, Meier S, Bundi M,
Mirlacher M, Sauter G and Dirnhofer S: Frequent EpCam protein
expression in human carcinomas. Hum Pathol. 35:122–128. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jones S, Zhang X, Parsons DW, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et
al: Core signaling pathways in human pancreatic cancers revealed by
global genomic analyses. Science. 321:1801–1806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Deer EL, González-Hernández J, Coursen JD,
Shea JE, Ngatia J, Scaife CL, Firpo MA and Mulvihill SJ: Phenotype
and genotype of pancreatic cancer cell lines. Pancreas. 39:425–435.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Almoguera C, Shibata D, Forrester K,
Martin J, Arnheim N and Perucho M: Most human carcinomas of the
exocrine pancreas contain mutant c-K-ras genes. Cell. 53:549–554.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yin X, Su J, Zhou X, Guo J, Wei W and Wang
Z: K-ras-driven engineered mouse models for pancreatic cancer.
Discov Med. 19:15–21. 2015.PubMed/NCBI
|
|
34
|
Moore PS, Sipos B, Orlandini S, Sorio C,
Real FX, Lemoine NR, Gress T, Bassi C, Klöppel G, Kalthoff H, et
al: Genetic profile of 22 pancreatic carcinoma cell lines. Analysis
of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 439:798–802.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dong M, Nio Y, Yamasawa K, Toga T, Yue L
and Harada T: p53 alteration is not an independent prognostic
indicator, but affects the efficacy of adjuvant chemotherapy in
human pancreatic cancer. J Surg Oncol. 82:111–120. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dave JM and Bayless KJ: Vimentin as an
integral regulator of cell adhesion and endothelial sprouting.
Microcirculation. 21:333–344. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hong SM, Li A, Olino K, Wolfgang CL,
Herman JM, Schulick RD, Iacobuzio-Donahue C, Hruban RH and Goggins
M: Loss of E-cadherin expression and outcome among patients with
resectable pancreatic adenocarcinomas. Mod Pathol. 24:1237–1247.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang J, Dokurno P, Tonks NK and Barford D:
Crystal structure of the M-fragment of α-catenin: Implications for
modulation of cell adhesion. EMBO J. 20:3645–3656. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Al-Rawi R: β-Catenin as a biomarker in
diagnosis of tumors with special emphasis on colorectal carcinoma.
Biom J. 3:12017.
|
|
41
|
Overgaard CE, Mitchell LA and Koval M:
Roles for claudins in alveolar epithelial barrier function. Ann NY
Acad Sci. 1257:167–174. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chiurillo MA: Role of the Wnt/β-catenin
pathway in gastric cancer: An in-depth literature review. World J
Exp Med. 5:84–102. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Van Itallie CM and Anderson JM: Claudin
interactions in and out of the tight junction. Tissue Barriers.
1:e252472013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jamieson C, Sharma M and Henderson BR:
Targeting the β-catenin nuclear transport pathway in cancer. Semin
Cancer Biol. 27:20–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Folkman J: Fighting cancer by attacking
its blood supply. Sci Am. 275:150–154. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xu Z, Pothula SP, Wilson JS and Apte MV:
Pancreatic cancer and its stroma: A conspiracy theory. World J
Gastroenterol. 20:11216–11229. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Snyder JT, Rossman KL, Baumeister MA,
Pruitt WM, Siderovski DP, Der CJ, Lemmon MA and Sondek J:
Quantitative analysis of the effect of phosphoinositide
interactions on the function of Dbl family proteins. J Biol Chem.
276:45868–45875. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fitzpatrick ER, Hu T, Ciccarelli BT and
Whitehead IP: Regulation of vesicle transport and cell motility by
Golgi-localized Dbs. Small GTPases. 5:1–12. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu Z, Adams HC III and Whitehead IP: The
rho-specific guanine nucleotide exchange factor Dbs regulates
breast cancer cell migration. J Biol Chem. 284:15771–15780. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Klinger MB, Guilbault B and Kay RJ: The
RhoA- and CDC42-specific exchange factor Dbs promotes expansion of
immature thymocytes and deletion of double-positive and
single-positive thymocytes. Eur J Immunol. 34:806–816. 2004.
View Article : Google Scholar : PubMed/NCBI
|