Introduction
Glioblastoma (GBM) is the most common
intra-parenchymal and lethal brain cancer while GBM patients are
left behind without any curable therapy to date (1,2). The
current standard therapy for GBM involves maximal surgical
resection followed by radiotherapy and tomozolomide (TMZ)
chemotherapy. However, this treatment strategy fails to eliminate a
subset of tumor cells that escape from therapeutic insults and
finally results in tumor recurrence, leading to reduced survival in
these patients (3,4). Among these treatments, radiation (IR)
plays a major role in the treatment of GBM patients. Factually, the
efficacy of this therapeutic modality is often limited by the
occurrence of radioresistance (5).
However, the molecular mechanisms responsible for the
radioresistance of human GBM are not yet clear.
Mitotic checkpoint serine/threonine kinase B (BUB1B)
is the mammalian homolog of yeast Mad3, but differs significantly
since BUB1B has a kinase domain that is absent in Mad3 (6). A reecent study showed that complete
deletion of BUB1B in the mouse germline results in early embryonic
death (7). Additionally,
BUB1B− (haplo-insufficient) mice display increased
megakaryopoiesis and increased chromosome instability, as well as
susceptibility to cancer (8,9).
Moreover, reduction in the BUB1B level or inhibition of BUB1B
kinase activity in human cancer cells results in massive chromosome
loss and apoptotic cell death (10). Another study recently identified
BUB1B as an essential element for the growth and survival of
rhabdomyosarcoma cells using a bar-coded, tetracycline-inducible
shRNA library screening and found that suppression of forkhead box
protein M1 (FOXM1) either by shRNAs or FOXM1 inhibitor siomycin A
resulted in reduction of BUB1B expression and decreased cell growth
(11). FOXM1 is a transcription
factor and is well known to play an essential role in the
regulation of a wide spectrum of biological processes including
cell proliferation, cell cycle progression, cell differentiation,
DNA damage repair, tissue homeostasis, angiogenesis and apoptosis
(12,13). Recent evidence shows that FOXM1
expression is elevated in human GBM and is essential for
maintaining neural, progenitor and GBM stem cells as a master
regulator of metastasis (14). In
addition, other studies indicate that radiation induces stimulation
of FOXM1 expression which is dependent on STAT3 phosphorylation
(15). Moreover, the FOXM1-RFC5
axis has been proven to mediate TMZ resistance, and thiostrepton
may serve as a potential therapeutic agent against TMZ resistance
in glioma cells (16). Based on
these findings, it has been proven that BUB1B plays an essential
role in tumor proliferation and is correlated with poor patient
prognosis in multiple types of cancer including breast, gastric,
colorectal and prostate cancer (17–20).
However, the functional role and mechanism of FOXM1/BUB1B signaling
in therapeutic resistance remain unclear.
In the present study, we identified that BUB1B
expression was enriched in GBM tumors and was functionally required
for tumor proliferation both in vitro and in vivo.
Clinically, BUB1B expression was associated with poor prognosis in
GBM patients and BUB1B-dependent radioresistance in GBM was
decreased by targeting BUB1B via shRNAs. Mechanistically, FOXM1
transcriptionally regulated BUB1B expression by binding to and then
activating the BUB1B promoter. Therapeutically, we found that FOXM1
inhibitor attenuated tumorigenesis and radioresistance of GBM both
in vitro and in vivo. Collectively, BUB1B promotes
tumor proliferation and induces radioresistance in GBM, implying
that BUB1B is a potential therapeutic target for GBM.
Materials and methods
Ethics
Protocols for the usage of experimental animals
(nude mice), human samples and cell lines in the present study were
approved by the Scientific Ethics Committee of Shaanxi Provincial
People's Hospital (Xi'an, China).
Reagents and antibodies
The following reagents and primary antibodies were
used in the present study: DMEM-F12, fetal bovine serum (FBS),
Accutase solution (Sigma-Aldrich, Billerica, MA, USA), alamarBlue
solution (all from Thermo Fisher Scientific, Waltham, MA, USA),
RIPA buffer, phosphatase inhibitor cocktail, protease inhibitor
cocktail (all from Sigma-Aldrich), Bradford protein assay (Bio-Rad
Laboratories, Hercules, CA, USA), albumin from bovine serum (BSA;
Sigma-Aldrich), PageRuler Plus prestained protein (Thermo Fisher
Scientific), iScript Reverse Transcription Supermix for qRT-PCR
(Bio-Rad Laboratories), BUB1B overexpression lentivirus
(pLenti-GIII-CMV-GFP-2A-Puro; BC018739), shBUB1B lentivirus
(piLenti-siRNA-GFP, iV002138), shFOXM1 lentivirus
(piLenti-siRNA-GFP, iV008091) [all from Applied Biological
Materials (abm) Inc., Richmond, BC, Canada], siomycin A
(ALX-380-243-MC05; Enzo Life Sciences, Farmingdale, NY, USA), Alexa
Fluor® 488 Annexin V/Dead Cell Apoptosis kit (Thermo
Fisher Scientific). Anti-BUB1B (rabbit; #4116; Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-FOXM1 (rabbit; ab184637;
Abcam, Cambridge, MA, USA), and β-actin (A5316; mouse;
Sigma-Aldrich) were used.
In vitro cell cultures
GBM cells U87, U251 and U138 and HEK-293T cells were
provided by Shaanxi Provincial People's Hospital. Cell lines were
cultured in DMEM/F12 medium containing 10% FBS supplement (vol %).
The culture medium was replaced every 5–10 days. Normal human
astrocytes were used as a control sample in the present study.
Radiation for in vitro GBM cells was performed using Thermo
Fisher t ICx radiation equipment according to the manufacturer's
protocol.
In vitro cell proliferation assay
GBM cells were transduced with the BUB1B or FOXM1
shRNA lentivirus for 3 days, and the cells were dissociated into a
single-cell suspension with Accutase. After the live cell number
was measured by the Trypan blue method with the Countess Automated
Cell Counter (Thermo Fisher Scientific), 200 µl of the cell
suspension containing 2,000 cells was added into 96-well plates.
The cell number was evaluated by alamarBlue according to the
manufacturer's protocol every 2 days after seeding.
Luciferase assays of the BUB1B
promoter
We amplified the −350/−216 bp region of the human
BUB1B promoter from RD genomic DNA by PCR using the primers
containing the restriction sites XhoI and HindIII,
respectively. These nucleotide sequences of primers were as
follows: sense primer, 5′-CTCGAGTAAGCCTGCTGCACTTCCAC-3′ and
antisense primer, 5′-AAGCTTCTCCTCCGTGCTCTCGCGTCT-3′ for the
−350/−216 bp fragment. These fragments were ligated into a
TOPO® TA vector (Thermo Fisher Scientific), and then
cloned into a pGL4.18 firefly luciferase reporter vector (Promega,
Madison, WI, USA) after digestion with XhoI and
HindIII. PCR-based site-directed mutagenesis was performed
to generate a single point mutation in FOXM1 binding sites of the
BUB1B promoter region using QuickChange II XL Site-Directed
Mutagenesis kit (Agilent Technologies, La Jolla, CA, USA).
Sequences were verified by DNA sequencing. HEK293T or U87 cells
were transfected using Lipofectamine™ 2000 with 1 µg of empty
vector, or BUB1B promoter luciferase reporter and cultured for 2
weeks. Luciferase assays were performed using Bright-Glo reagent
(Promega) on the Victor3 counter (PerkinElmer, Walthan, MA,
USA).
RNA isolation and quantitative
real-time PCR
RNA was isolated using RNeasy Mini kit (Qiagen,
Valencia, CA, USA) according to the manufacturer's instructions.
RNA concentration was determined using NanoDrop 2000c (Thermo
Fisher Scientific). cDNA was synthesized using iScript Reverse
Transcription Supermix for qRT-PCR according to the manufacturer's
protocol. The reverse-transcribed cDNA was analyzed by quantitative
RT-PCR (qRT-PCR), GAPDH or 18S was used as an internal control.
Each qRT-PCR included a 10 µl reaction mixture/well that included
2.5 µl cDNA, 0.5 µl forward primer (0.5 µM), 0.5 µl reverse primer
(0.5 µM), 1.5 µl of DNase/RNase-free distilled water, and 5 µl
SYBR-Green reagent (Qiagen). The following cycles were performed
during DNA amplification: 94°C for 2 min, 40 cycles of 94°C (30
sec), 60°C (30 sec), and 72°C (40 sec). The primer sequences were
as follows: FOXM1 forward, TCTCCTCTTTCCCTGGTCCT and FOXM1 reverse,
ATAGCAAGCGAGTCCGCATT; BUB1B forward, CCAGGCTTTCTGGTGCTTAG and BUB1B
reverse, GTGCTTCCCAGTTTCACTCC; GAPDH forward, CGGAG
TCAACGGATTTGGTCGTAT and GAPDH reverse, AGCCT TCTCCATGGTGGTGAAGAC;
18S forward, GGCCCTGT AATTGGAATGAGTC and 18S reverse, CCAAGATCCAACT
ACGAGCTT.
Western blotting
The cell lysates were prepared in RIPA buffer
containing 1% protease and 1% phosphatase inhibitor cocktail on
ice. Protein concentrations were determined using the Bradford
method. Equal amounts of protein lysates (10 µg/lane) were
fractionated on NuPAGE Novex 4–12% Bis-Tris Protein gel and
transferred to a polyvinylidene difluoride (PVDF) membrane (both
from Thermo Fisher Scientific). Subsequently, the membranes were
blocked with 5% skimmed milk for 1 h and then treated with the
relevant antibody at 4°C overnight. Protein expression was
visualized with ECL Western Blotting System (GE Healthcare Life
Sciences, Pittsburgh, PA, USA). β-actin served as a loading
control.
Flow cytometric analysis
Cells were harvested after the incubation period and
washed in cold phosphate-buffered saline (PBS) for 3 times. The
washed cells were re-centrifuged (from step 2), the supernatant was
discarded and 5×105 cells were suspended in 100 µl 1X
Annexin-binding buffer. Alexa Fluor® 488 Annexin V (5
µl) was added and 1 µl 100 µg/ml propidium iodide (PI) working
solution was prepared according to the protocol. The cells were
incubated at room temperature for 15 min, and then 400 µl 1X
Annexin-binding buffer was added, mixed gently and the samples were
kept on ice. The stained cell population was analyzed by measuring
the fluorescence emission at 530 and 575 nm with 488 nm
excitation.
In vivo intracranial xenograft tumor
models
Six-week-old nude mice (female) were purchased from
Xi'an Jiaotong University and were used for GBM intracranial
implantation. All animal experiments were carried out at Shaanxi
Provincial People's Hospital. The GBM suspension (1×105
cells in 5 µl of PBS) transduced with the non-target or shBUB1B
lentivirus was injected into the brains of nude mice after
anesthesia. At least 5 mice were used for each group. Drug
treatment was carried out through tail vein injection, and started
from 5 days after tumor cells were implanted. Mice were monitored
once a day for symptoms related to tumor growth including an arched
back, unsteady gait, paralysis of legs and body weight loss. Mice
were euthanized by an overdose of anesthesia of ketamine and
xylazine after a total body weight loss of 40% or severe symptoms
were observed.
Statistical analysis
All data are presented as mean ± SD. The number of
replicates for each experiment is stated in the figure legends.
Statistical differences between 2 groups were evaluated by
two-tailed t-test. Comparison among multiple groups was performed
by one-way analysis of variance (one-way ANOVA) followed by
Dunnett's post test. The statistical significance of the
Kaplan-Meier survival plot was determined by log-rank analysis.
Flow cytometric results were analyzed with χ2 analysis.
Statistical analysis was performed by Prism 6 (GraphPad Prism 5.0;
GraphPad Software, Inc., La Jolla, CA, USA), unless mentioned
otherwise in the figure legends. P<0.05 was considered to
indicate a statistically significant result.
Results
BUB1B expression is highly enriched in
GBM cells
In the present study, we first sought to
characterize the role of BUB1B in GBM tumors. To this end, we
analyzed the microarray data from a microarray database published
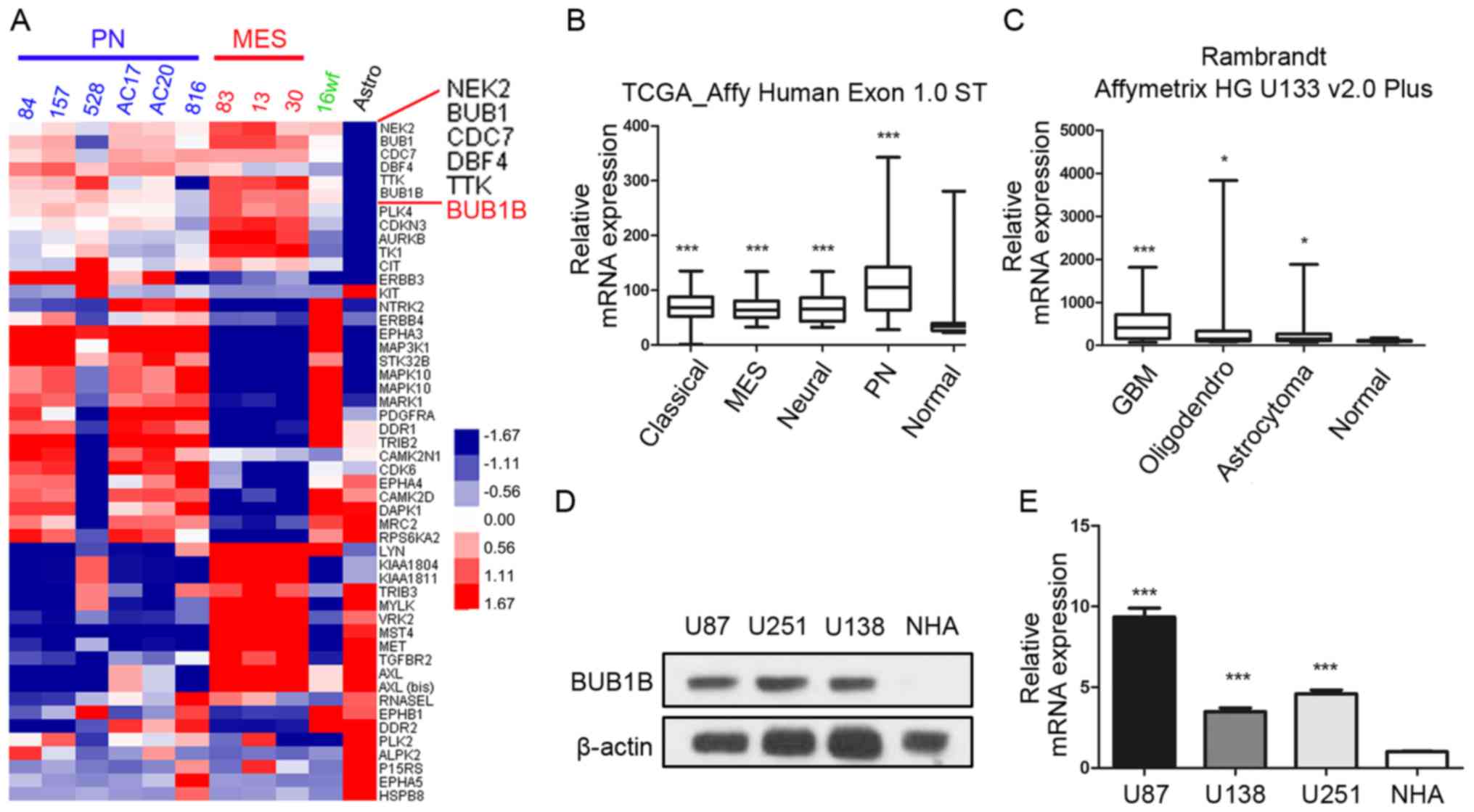
in 2013 (21) and found that BUB1B
was one of the most upregulated kinase-encoding genes in GBM
samples compared to normal human astrocyte (NHA) cells (Fig. 1A). Additionally, we found that BUB1B
expression was significantly enriched in all the 4 subgroups of GBM
according to The Cancer Genome Atlas (TCGA) database [classical,
mesenchymal (MEN), neural and proneural (PN)] compared with
non-tumor tissue (Fig. 1B).
Furthermore, the data from the Rembrandt database also demonstrated
that BUB1B mRNA expression in GBM was significantly higher than
that in non-tumor and other types of glioma (Fig. 1C). To confirm this, western blotting
using 3 GBM cell lines (U87, U251 and U138) and NHAs was performed.
The results exhibited higher expression of BUB1B protein in the GBM
cells (Fig. 1D). Similar to the
protein expression, qRT-PCR data demonstrated the same result
(Fig. 1E). Taken together, these
data indicated that BUB1B is preferentially expressed in GBM.
BUB1B is functionally required for GBM
proliferation both in vitro and in vivo
To examine the biological role of BUB1B in GBM, we
selected one in vitro GBM cell line (U87) which had a
relatively higher BUB1B expression and transduced the cells with
either a lentiviral shRNA clone for BUB1B (shBUB1B) or a
non-targeting shRNA (shNT). The efficiency for lentiviral infection
was confirmed by GFP fluorescence (Fig.
2A). qRT-PCR and western blot analysis indicated that BUB1B
expression was significantly reduced in the shBUB1B U87 cells
(Fig. 2B and C). Furthermore, in
vitro cell growth kinetics of the shBUB1B-transfected U87 cells
was proportionally diminished to the reduction levels of BUB1B
(Fig. 2D). Next, we investigated
the effect of BUB1B knockdown on in vivo tumor formation. To
this end, we used U87 cell-derived mouse intracranial tumor models.
A longer survival was observed in the mouse xenografted shBUB1B
group, highlighting potent antitumorigenic effects of BUB1B
knockdown (Fig. 2E). We then
harvested U87-implanted mouse brains and H&E or IHC staining
for BUB1B was performed. The results indicated that the mice
xenografted with shNT-transfected U87 cells rapidly formed lethal
hypervascular GBM-like tumors while BUB1B expression was decreased
in the shBUB1B-transfected U87 cell xenografted mouse brains
(Fig. 2F). Similar results were
obtained when using U251 GBM cells (Fig. 2G-I). Moreover, the survival analysis
from the Rembrandt database indicated that overall survival was
significantly longer in the BUB1BLow expression group
than that noted in the BUB1BHigh expression group
(Fig. 2J). Collectively, these
findings indicate that BUB1B is required for GBM proliferation both
in vitro and in vivo.
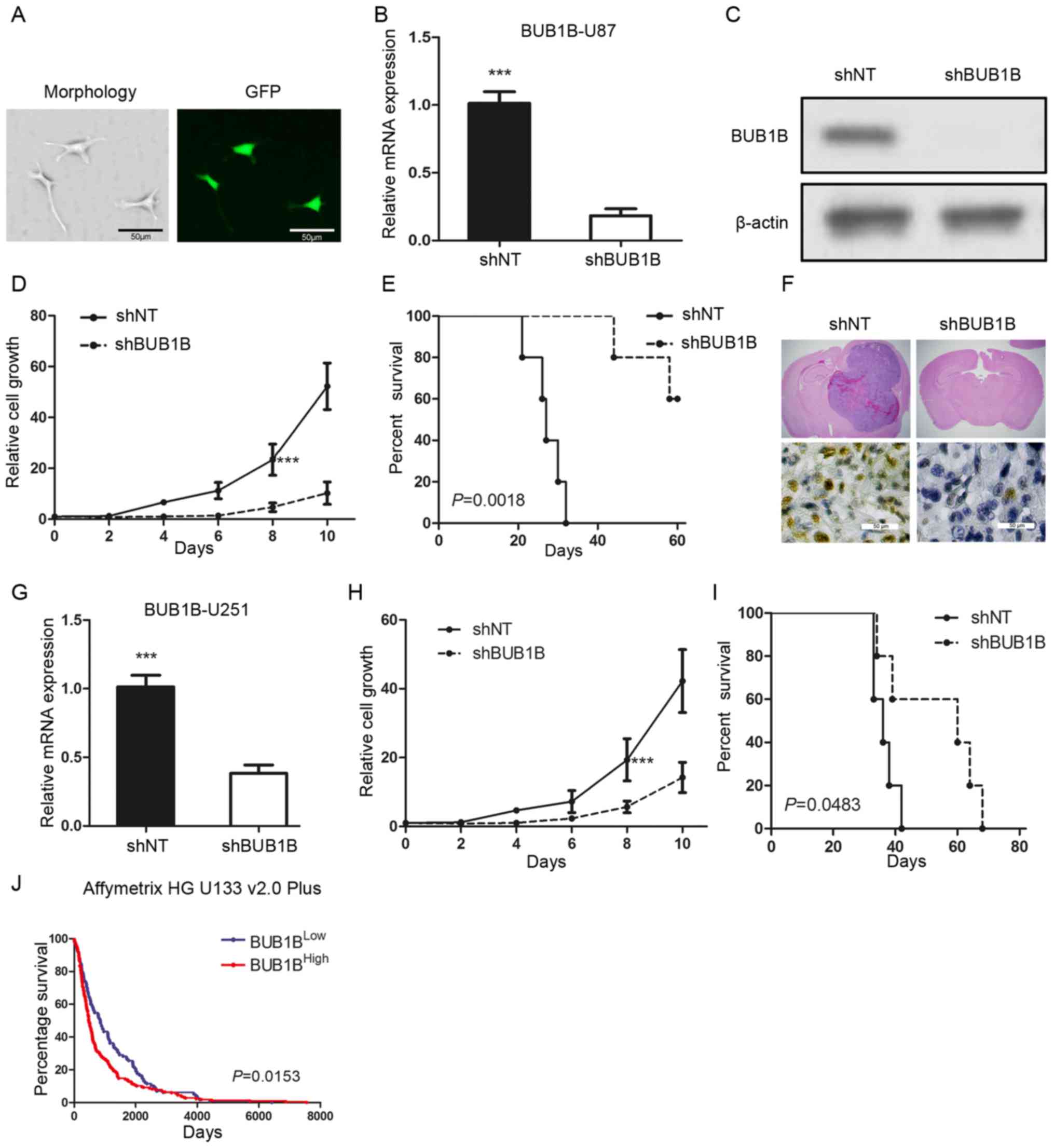 | Figure 2.BUB1B is functionally required for
GBM proliferation both in vitro and in vivo. (A)
Fluorescence image shows that the pGFP-shBUB1B lentivirus was
successfully transfected into U87 cells. (B) qRT-PCR of U87 cells
transduced with shNT or shBUB1B (n=3, ***P<0.001, with t-test).
(C) Western blotting of U87 cells transduced with shNT or shBUB1B.
β-actin served as a control. (D) In vitro cell growth assay
showed that shBUB1B reduced cell proliferation of U87 cells (n=6,
***P<0.001, with one-way ANOVA). (E) Kaplan-Meier analysis of
nude mice harboring intracranial tumors derived from U87 cells
transduced with shNT or shBUB1B (n=5, P=0.0018, with log-rank
test). (F) Representative images of H&E (upper panel) or BUB1B
(lower panel) stained mouse brain section after the intracranial
transplantation of U87 cells transduced with shNT or shBUB1B. (G)
qRT-PCR of U251 cells transduced with shNT or shBUB1B (n=3,
***P<0.001, with t-test). (H) In vitro cell growth assay
showed that hBUB1B reduced cell proliferation of U251 cells (n=6,
***P<0.001, with one-way ANOVA). (I) Kaplan-Meier analysis of
nude mice harboring intracranial tumors derived from U251 cells
transduced with shNT or shBUB1B (n=5, P=0.0483, with log-rank
test). (J) Analysis of the Rembrandt data revealed an inverse
correlation between BUB1B expression and post-surgical survival of
GBM patients (P=0.0153, with log-rank test). |
FOXM1-binding region is critical for
the activation of the BUB1B promoter
To further assess the regulatory mechanism of BUB1B
in GBM, we performed a luciferase reporter assay with constructs
driven by a human BUB1B promoter (Fig.
3A). qRT-PCR and western blot analyses indicated that BUB1B
expression was significantly reduced after FOXM1 inhibition via
shRNA (Fig. 3B and C). Thus, we
performed luciferase assay to measure the BUB1B promoter activity
with reduced expression of FOXM1 in HEK293T or U87 cells. The
region (−350/−216 bp) of the human BUB1B promoter was constructed
and cloned into a luciferase reporter vector based on a previous
study (11). Then, empty vector
(EV) or reporter for BUB1B promoter (pGL-BUB1B) was transduced into
HEK293T or U87 cells followed by shNT or shFOXM1 transfection. As
expected, shRNA-mediated knockdown of FOXM1 resulted in a marked
decrease in transcriptional activity of the FOXM1-binding region in
both HEK293T and U87 cells (Fig. 3D and
E). Moreover, we transiently transfected either EV or BUB1B
promoter wild-type (WT) or BUB1B promoter mutation type (mutation)
into HEK293T cells, and luciferase assay showed a marked decrease
in transcriptional activity of the mutated BUB1B promoter in the
HEK293T cells (Fig. 3F).
Altogether, these data suggest that the FOXM1-binding region was
critical for BUB1B promoter activation in GBM cells.
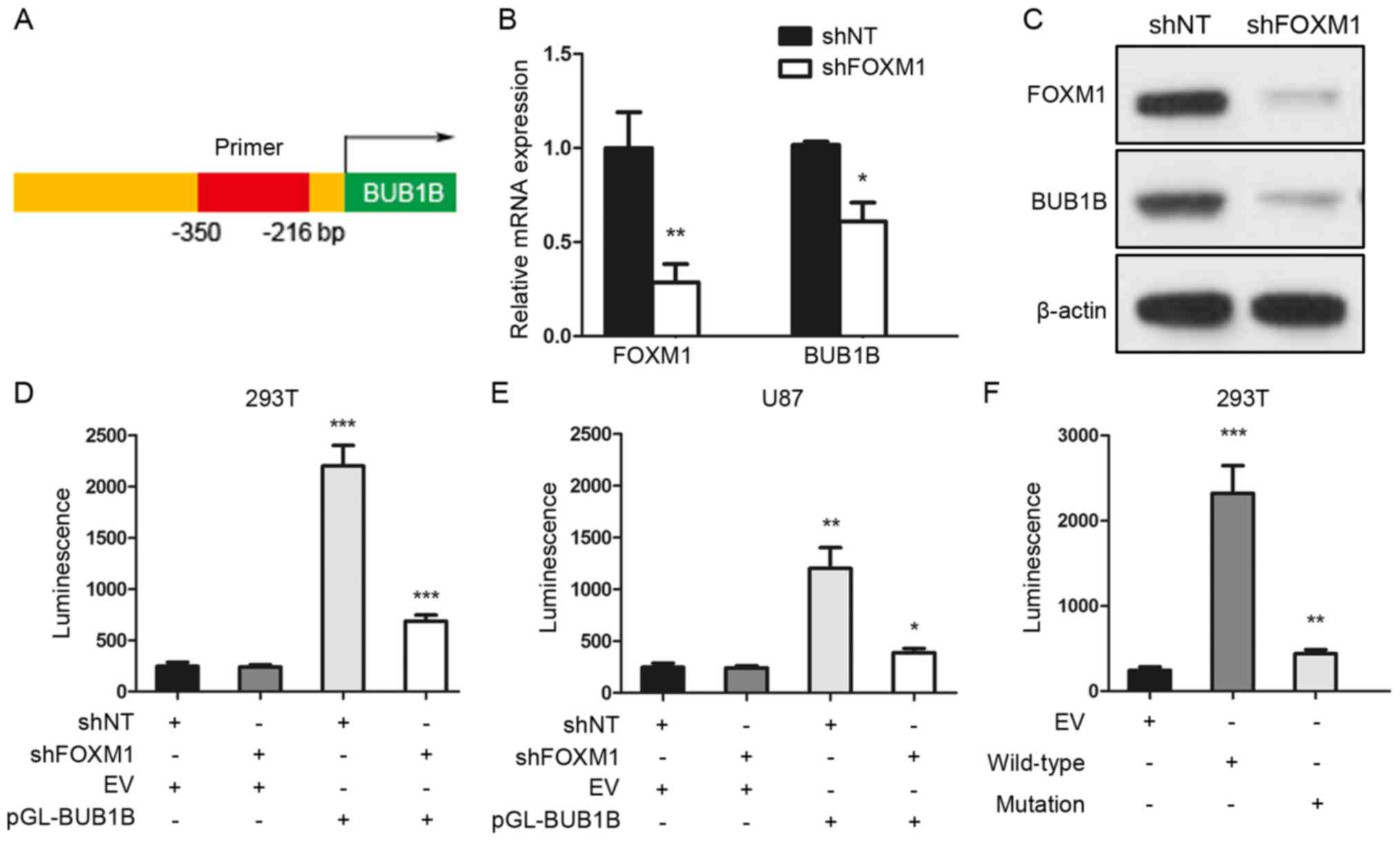 | Figure 3.FOXM1-binding region is critical for
the activation of the BUB1B promoter. (A) Schematic drawing of the
promoter region of human BUB1B. (B) qRT-PCR for FOXM1 and BUB1B
expression in U87 cells transduced with shNT or shFOXM1 (n=3,
*P<0.05, **P<0.01, with t-test). (C) Western blotting of U87
cells transduced with shNT or shFOXM1. β-actin served as a control.
(D and E) Luciferase activity assay showed that shRNA-mediated
knockdown of FOXM1 resulted in a marked decrease in transcription
activity of the FOXM1-binding region in both (D) HEK293T and (E)
U87 cells (n=3, *P<0.05, **P<0.01, ***P<0.001, with
one-way ANOVA followed by Dunnett's post test). (F) Luciferase
activity assay showed a decrease in transcription activity of
mutated BUB1B promoter in the HEK293T cells (n=3, **P<0.01,
***P<0.001, with one-way ANOVA followed by Dunnett's post
test). |
BUB1B-mediated radioresistance is
essential for GBM recurrence
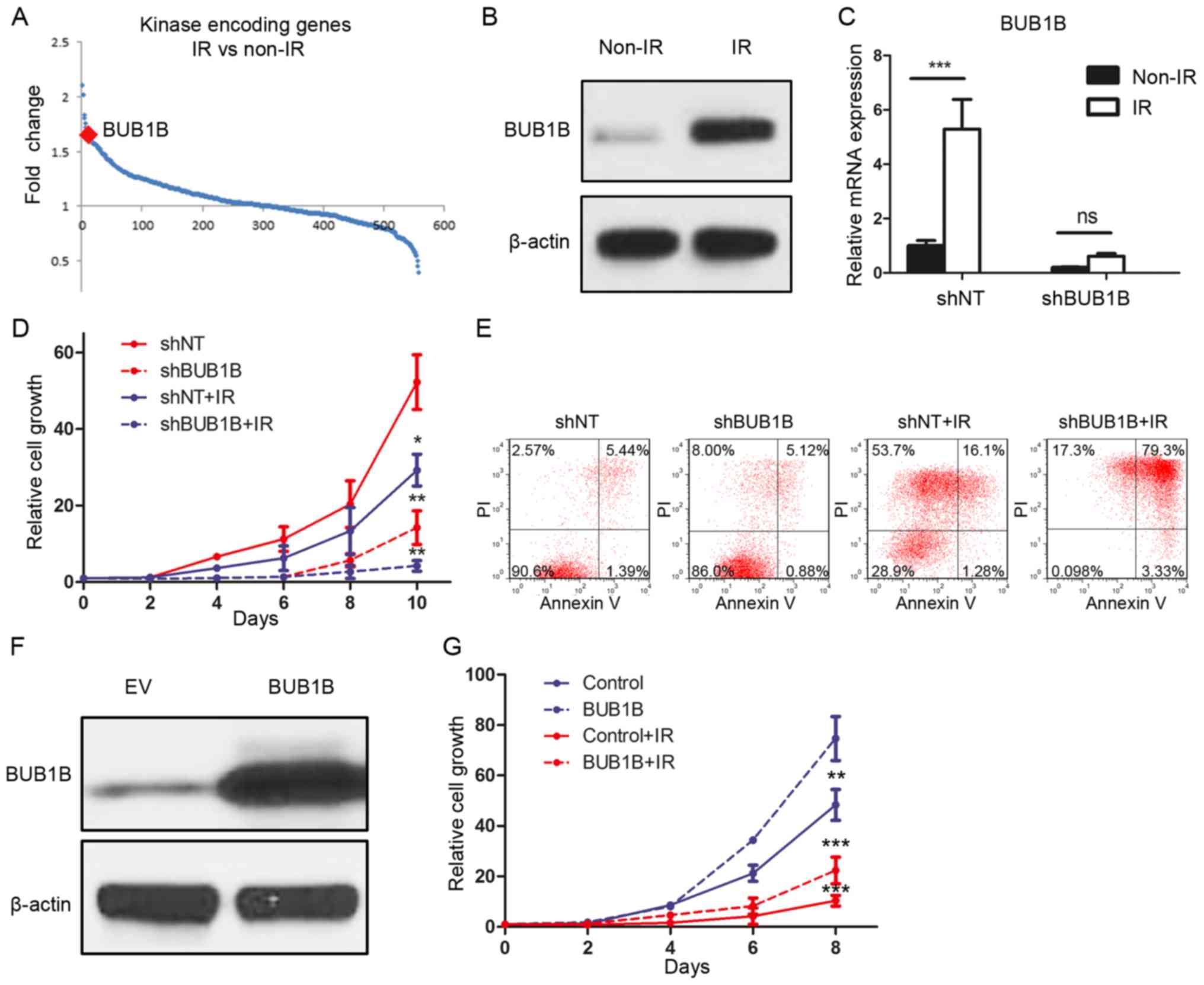
To identify the functional role of BUB1B in the
radioresistance of GBM, DNA microarray analysis focusing on 668
kinase-encoding genes in GBM cell lines was performed. The results
demonstrated that BUB1B was one of the top genes which was
upregulated after radiation of 12 Gy (Fig. 4A). To confirm this, we treated U87
cells with radiotherapy of 12 Gy or without and purified protein at
72 h after treatment. Western blot analysis indicated that the
BUB1B expression level was significantly elevated after radiation
(Fig. 4B). To investigate whether
BUB1B is necessary for GBM cells to maintain radioresistance, we
then combined BUB1B knockdown along with radiotherapy. qRT-PCR
results showed that the post-radiation elevation of BUB1B was
reduced by shBUB1B (Fig. 4C). In
vitro cell proliferation was markedly decreased by shBUB1B when
combined with radiation (Fig. 4D).
Moreover, we performed flow cytometry for apoptosis with Annexin V
(AV) antibody and PI using shBUB1B-transduced U87 cells with (12
Gy) or without radiation. The results indicated that the
percentages of cells undergoing early
(AV+/PI−) and late
(AV+/PI+) apoptosis were both markedly
increased after radiation when combined with BUB1B knockdown in
comparison to radiation alone (Fig.
4E). Finally, BUB1B or EV was overexpressed in U138 cells
(Fig. 4F). In vitro cell
proliferation assay showed that both cell growth and
radioresistance of U138 cells were enhanced by exogenous BUB1B
(Fig. 4G). Taken together,
BUB1B-dependent radioresistance was essential for GBM tumorigenesis
and recurrence after therapeutic treatment.
FOXM1 inhibitor reduces tumorigenicity
and radioresistance of GBM
Based on the inhibitory effects of BUB1B silencing
on GBM cell proliferation and tumorigenicity, we sought to identify
a potential target for the clinical treatment of GBM. To this end,
FOXM1 inhibitor siomycin A was used to clarify the effects of BUB1B
inhibition on GBM (Fig. 5A). To
characterize the efficacy of this inhibitor, we investigated the
IC50 value of siomycin A in different GBM cell lines. As
expected, in vitro sensitivity of these GBM cells was
correlated with the BUB1B expression level, and normal astrocytes
showed more resistance to the FOXM1 inhibitor (Fig. 5B). qRT-PCR and western blotting
indicated that siomycin A treatment significantly reduced the
expression of BUB1B in the U87 cells (Fig. 5C and D). Furthermore, in
vitro cell proliferation was significantly decreased by
siomycin A when combined with radiation compared with radiotherapy
alone (Fig. 5E). More importantly,
systemic treatment of the U87-derived mouse model with siomycin A
significantly attenuated tumor growth, thereby extending the
survival of tumor-bearing mice compared to the vehicle-treated
counterparts (Fig. 5F and G). Flow
cytometric analysis indicated that the percentage of cells
undergoing apoptosis was markedly increased after radiation when
combined with siomycin A treatment in comparison to that noted
following radiation alone (Fig.
5H). To address whether siomycin A combined with radiation
enhances cell apoptosis due to synergism or addictive effects,
isobologram analysis was performed. The results indicated that
siomycin A (20 µM) increased the radiosensitivity of the U87 cells
via a synergistic effect (Fig. 5I).
Additionally, luciferase assay showed that the BUB1B promoter
activity was decreased by siomycin A treatment in the U87 cells
(Fig. 5J). Finally, in vitro
cell proliferation assay showed that both siomycin A-mediated
reduction in the cell growth of U138 cells could be partially, but
not completely, rescued by exogenous BUB1B (Fig. 5K). In conclusion, siomycin A reduced
the tumorigenicity and radioresistance of GBM via inhibition of the
FOXM1/BUB1B signaling pathway.
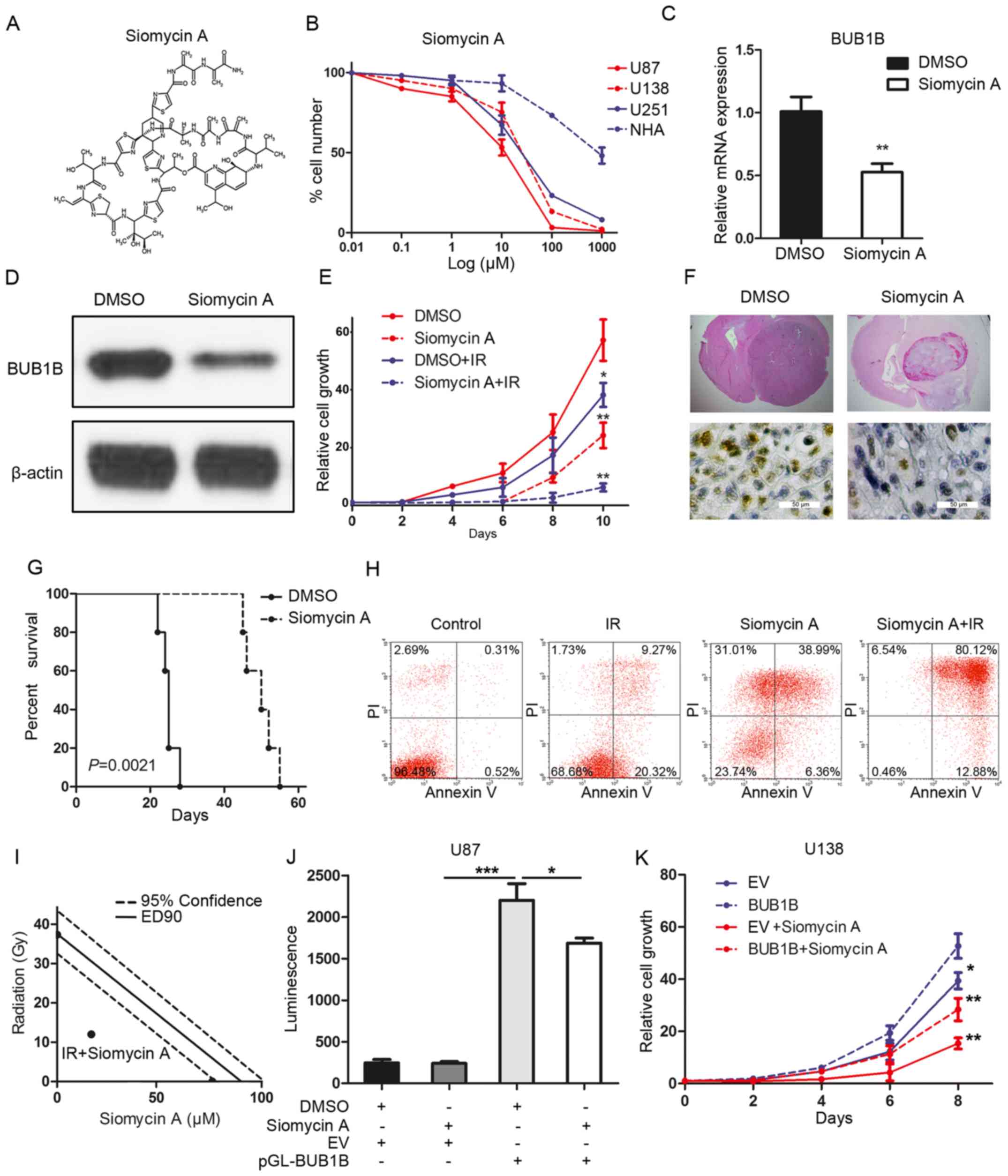 | Figure 5.Siomycin A reduces tumorigenicity and
radioresistance of GBM. (A) Chemical structure of siomycin A. (B)
In vitro cell viability assay for siomycin A with 3 GBM cell
lines (U138, U251 and U87) compared with normal astrocytes (NHA)
(n=6, P<0.05, with one-way ANOVA). (C) qRT-PCR for BUB1B
expression in U87 cells treated with or without siomycin A in U87
cells (n=3, **P<0.01, with t-test). (D) Western blotting for
BUB1B expression in U87 cells treated with or without siomycin A.
(E) In vitro cell growth assay showed that siomycin A
decreased cell proliferation of U87 cells when combined with
radiation (n=6, *P<0.05, **P<0.01, with one-way ANOVA). (F)
Representative images of H&E (upper panel) or BUB1B (lower
panel) stained mouse brain section after the intracranial
transplantation of U87 cells and then followed by continuous 7-day
siomycin A treatment or placebo by tail vein injection. (G)
Kaplan-Meier analysis was performed for the comparison of survival
in U87 cell-implanted mice treated with or without siomycin A (n=5,
P=0.0021, with log-rank test). (H) Flow cytometric analysis of
apoptosis using U87 cells pre-treated with siomycin A and then
treated with or without radiation (12 Gy) (P<0.05, with
χ2 analysis). (I) Isobologram analysis results for
siomycin A (20 µM) combined with radiation in U87 cells. (J)
Luciferase activity assay showed that siomycin A treatment resulted
in a decrease in transcription activity of FOXM1-binding region in
U87 cells (n=3, *P<0.05, ***P<0.001, with one-way ANOVA
followed by Dunnett's post test). (K) In vitro cell growth
assay for U138 cells pre-transduced with empty vector or exogenous
BUB1B overexpression vector and then treated with or without
siomycinA (n=6, *P<0.05, **P<0.01, with one-way ANOVA). |
Discussion
Accumulating data indicate that BUB1B is a critical
protein involved in the growth and survival of multiple types of
cancer including breast, gastric, colorectal and prostate cancer
(17–20). Our findings revealed that knockdown
of BUB1B led to the loss of GBM in vitro cell growth and
in vivo tumorigenicity. Furthermore, we identified the
forkhead box protein M1 (FOXM1)/BUB1B pathway as a pivotal
regulator of radioresistance in GBM besides its main function in
cell mitosis. In addition, deletion of this axis induced cell
apoptosis when combined with radiotherapy. These findings showed us
that targeting of BUB1B could become an efficient supporting
therapy for GBM.
FOXM1 is a typical proliferation-associated
transcription factor. However, the transcriptional downstream
targets of FOXM1 in tumor recurrence and therapy resistance remain
to be determined (22). FOXM1 was
reported as an upregulator of enhancer of zeste homolog 2 (EZH2)
and is functionally required for glioma stem cells to maintain
stemness and therapy resistance via forming a complex with MELK
(23). Wan et al (11) demonstrated that BUB1B is a direct
transcriptional target of FOXM1 and FOXM1 expression drives a BUB1B
promoter construct containing a consensus FOXM1 binding site
(−350/−216 bp region). Similarly, in the present study, using a
luciferase reporter assay we found that FOXM1 regulates the
activity of the BUB1B promoter in GBM and this cell signaling
pathway is essential for tumor proliferation and tumorigenicity in
GBM cells.
Recent evidence indicates that the FOXM1-Sox2
signaling axis promotes clonogenic growth and radioresistance of
GBM, suggesting that FOXM1 targeting combined with radiation may be
a potentially effective therapeutic approach for GBM (24). Moreover, FOXM1 also contributes to
the maintanence of stemness and radioresistance in GBM by
upregulation of the DNA damage repair signaling or MELK-mediated
oncogenic regulation of EZH2 (23,25).
Nonetheless, whether there are other mechanisms involved in
FOXM1-dependent tumorigenesis and therapy resistance in GBM
warrants further investigation. Herein, we found that blocking the
FOXM1/BUB1B pathway using a selective FOXM1 inhibitor siomycin A
reduced tumor growth and enhanced the radiosensitivity of GBM
cells, indicating that FOXM1 promotes radioresistance of GBM cells,
at least partially, dependent on its transcriptional regulation of
BUB1B. However, whether or not BUB1B promotes tumor growth and
radioresistance in GBM through its kinase activity warrents further
investigation. Further study is needed to design and synthesize a
specific small-molecule inhibitor directly targeting BUB1B to show
more efficiency for suppressing tumor growth and
radioresistance.
Endoreduplication is the replication of the genome
during the cell cycle without the subsequent completion of mitosis
and/or cytokinesis (26). A wide
variety of agents including microtubule inhibitors, topoisomerase
II inhibitors and DNA damaging agents have been reported as able to
induce endoreduplication (27).
Endoreduplication can be associated with cell differentiation but
also frequently occurs in malignant cells and may play a role in
maintaining cell fate (28,29). Numerous cell cycle-related proteins
including p21, aurora B, cyclin-dependent kinase 1 (CDK1), cyclin
B1 and B2, have been identified to be involved in endoreduplication
(11,30). A recent study evaluated the effects
of FOXM1 knockdown on breast cancer cells and showed both
endoreduplication and or mitotic catastrophe (31). Another study showed that BUB1B
knockdown induces endoreduplication and mitotic catastrophe in
rhabdomyosarcoma (11). However,
regardless of these findings, it is still unclear whether the
function of BUB1B in GBM depends on endoreduplication, or not.
Thus, further study should be performed to answer these
questions.
Altogether, in the present study, we identified that
BUB1B expression was enriched in GBM tumors and was functionally
required for tumor proliferation both in vitro and in
vivo. Clinically, BUB1B expression was associated with poor
prognosis in GBM patients and BUB1B-dependent radioresistance in
GBM could be decreased by targeting BUB1B via shRNAs.
Mechanistically, FOXM1 transcriptionally regulated BUB1B expression
by binding to and then activating the BUB1B promoter.
Therapeutically, we found that FOXM1 inhibitor attenuated
tumorigenesis and radioresistance of GBM both in vitro and
in vivo. Altogether, BUB1B promotes tumor proliferation and
induces radioresistance in GBM, indicating that BUB1B is a
potential therapeutic target for GBM.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Shaanxi Province, China (2012K17-01-03).
References
|
1
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Meyer MA: Malignant gliomas in adults. N
Engl J Med. 359:18502008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim SH, Ezhilarasan R, Phillips E,
Gallego-Perez D, Sparks A, Taylor D, Ladner K, Furuta T, Sabit H,
Chhipa R, et al: Serine/threonine kinase MLK4 determines
mesenchymal identity in glioma stem cells in an NF-κB-dependent
manner. Cancer Cell. 29:201–213. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheng P, Wang J, Waghmare I, Sartini S,
Coviello V, Zhang Z, Kim SH, Mohyeldin A, Pavlyukov MS, Minata M,
et al: FOXD1-ALDH1A3 signaling is a determinant for the
self-renewal and tumorigenicity of mesenchymal glioma stem cells.
Cancer Res. 76:7219–7230. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Noda SE, El-Jawahri A, Patel D,
Lautenschlaeger T, Siedow M and Chakravarti A: Molecular advances
of brain tumors in radiation oncology. Semin Radiat Oncol.
19:171–178. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Taylor SS, Ha E and McKeon F: The human
homologue of Bub3 is required for kinetochore localization of Bub1
and a Mad3/Bub1-related protein kinase. J Cell Biol. 142:1–11.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baker DJ, Jeganathan KB, Cameron JD,
Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC,
Roche P, et al: BubR1 insufficiency causes early onset of
aging-associated phenotypes and infertility in mice. Nat Genet.
36:744–749. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie
S, Mahmood R, Yang YM, Xu M and Rao CV: Slippage of mitotic arrest
and enhanced tumor development in mice with BubR1
haploinsufficiency. Cancer Res. 64:440–445. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Q, Liu T, Fang Y, Xie S, Huang X,
Mahmood R, Ramaswamy G, Sakamoto KM, Darzynkiewicz Z, Xu M, et al:
BUBR1 deficiency results in abnormal megakaryopoiesis.
Blood. 103:1278–1285. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kops GJ, Foltz DR and Cleveland DW:
Lethality to human cancer cells through massive chromosome loss by
inhibition of the mitotic checkpoint. Proc Natl Acad Sci USA.
101:8699–8704. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wan X, Yeung C, Kim SY, Dolan JG, Ngo VN,
Burkett S, Khan J, Staudt LM and Helman LJ: Identification of
FoxM1/Bub1b signaling pathway as a required component for growth
and survival of rhabdomyosarcoma. Cancer Res. 72:5889–5899. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Raychaudhuri P and Park HJ: FoxM1: A
master regulator of tumor metastasis. Cancer Res. 71:4329–4333.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Monteiro LJ, Khongkow P, Kongsema M,
Morris JR, Man C, Weekes D, Koo CY, Gomes AR, Pinto PH, Varghese V,
et al: The Forkhead Box M1 protein regulates BRIP1 expression and
DNA damage repair in epirubicin treatment. Oncogene. 32:4634–4645.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu M, Dai B, Kang SH, Ban K, Huang FJ,
Lang FF, Aldape KD, Xie TX, Pelloski CE, Xie K, et al: FoxM1B is
overexpressed in human glioblastomas and critically regulates the
tumorigenicity of glioma cells. Cancer Res. 66:3593–3602. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maachani UB, Shankavaram U, Kramp T,
Tofilon PJ, Camphausen K and Tandle AT: FOXM1 and STAT3 interaction
confers radioresistance in glioblastoma cells. Oncotarget.
7:77365–77377. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peng WX, Han X, Zhang CL, Ge L, Du FY, Jin
J and Gong AH: FoxM1-mediated RFC5 expression promotes temozolomide
resistance. Cell Biol Toxicol. Feb 9–2017.(Epub ahead of print).
doi: 10.1007/s10565-017-9381-1. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hudler P, Britovsek NK, Grazio SF and
Komel R: Association between polymorphisms in segregation genes
BUB1B and TTK and gastric cancer risk. Radiol Oncol. 50:297–307.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mansouri N, Movafagh A, Sayad A, Heidary
Pour A, Taheri M, Soleimani S, Mirzaei HR, Shargh Alizadeh S,
Azargashb E, Bazmi H, et al: Targeting of BUB1b gene expression in
sentinel lymph node biopsies of invasive breast cancer in iranian
female patients. Asian Pac J Cancer Prev. 17(S3): 317–321. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hahn MM, Vreede L, Bemelmans SA, van der
Looij E, van Kessel AG, Schackert HK, Ligtenberg MJ, Hoogerbrugge
N, Kuiper RP and de Voer RM: Prevalence of germline mutations in
the spindle assembly checkpoint gene BUB1B in individuals with
early-onset colorectal cancer. Genes Chromosomes Cancer.
55:855–863. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fu X, Chen G, Cai ZD, Wang C, Liu ZZ, Lin
ZY, Wu YD, Liang YX, Han ZD, Liu JC, et al: Overexpression of BUB1B
contributes to progression of prostate cancer and predicts poor
outcome in patients with prostate cancer. Onco Targets Ther.
9:2211–2220. 2016.PubMed/NCBI
|
|
21
|
Mao P, Joshi K, Li J, Kim SH, Li P,
Santana-Santos L, Luthra S, Chandran UR, Benos PV, Smith L, et al:
Mesenchymal glioma stem cells are maintained by activated
glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc
Natl Acad Sci USA. 110:8644–8649. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Z, Zhang G and Kong C: FOXM1
participates in PLK1-regulated cell cycle progression in renal cell
cancer cells. Oncol Lett. 11:2685–2691. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim SH, Joshi K, Ezhilarasan R, Myers TR,
Siu J, Gu C, Nakano-Okuno M, Taylor D, Minata M, Sulman EP, et al:
EZH2 protects glioma stem cells from radiation-induced cell death
in a MELK/FOXM1-dependent manner. Stem Cell Reports. 4:226–238.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee Y, Kim KH, Kim DG, Cho HJ, Kim Y,
Rheey J, Shin K, Seo YJ, Choi YS, Lee JI, et al: FoxM1 promotes
stemness and radioresistance of glioblastoma by regulating the
master stem cell regulator Sox2. PLoS One. 10:e01377032015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ganguly R, Mohyeldin A, Thiel J, Kornblum
HI, Beullens M and Nakano I: MELK-a conserved kinase: Functions,
signaling, cancer, and controversy. Clin Transl Med. 4:112015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Edgar BA and Orr-Weaver TL:
Endoreplication cell cycles: More for less. Cell. 105:297–306.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cortés F, Mateos S, Pastor N and Domínguez
I: Toward a comprehensive model for induced endoreduplication. Life
Sci. 76:121–135. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee HO, Davidson JM and Duronio RJ:
Endoreplication: Polyploidy with purpose. Genes Dev. 23:2461–2477.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kaźmierczak A: Endoreplication in
Anemia phyllitidis coincides with the development of
gametophytes and male sex. Physiol Plant. 138:321–328. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim JA, Lee J, Margolis RL and Fotedar R:
SP600125 suppresses Cdk1 and induces endoreplication directly from
G2 phase, independent of JNK inhibition. Oncogene. 29:1702–1716.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wonsey DR and Follettie MT: Loss of the
forkhead transcription factor FoxM1 causes centrosome amplification
and mitotic catastrophe. Cancer Res. 65:5181–5189. 2005. View Article : Google Scholar : PubMed/NCBI
|