Introduction
Chronic myeloid leukemia (CML) is a BCR-ABL
oncogene-driven malignant disease, characterized by markedly
elevated immature myeloid cells in bone marrow and peripheral
blood. CML cells progress slowly in the chronic phase (CP) for
about one to three years, and then proliferate more rapidly
stepping into the accelerated phase (AP) with an increase in blast
numbers. The accelerated phase lasts for only several months and
eventually converts to acute leukemia in the blast crisis phase
(BC) with more aggressive characteristics than de novo acute
leukemia. Most CML patients respond well to the tyrosine kinase
inhibitor (TKI) imatinib in the chronic phase, however, ~20–30%
patients develop resistance to imatinib (1–3). Some
of the patients are de novo resistant to imatinib, others
exhibit a good response in the beginning, however this response is
lost with the progression of this disease. Almost half of the
imatinib-resistant patients develop point mutations in the BCR/ABL
gene during the course of TKI treatment. Other drug resistance
mechanisms include BCR-ABL amplification, additional acquired gene
mutation and drug efflux (4,5).
Second and third generation tyrosine kinase inhibitors such as
dasatinib, ponatinib, are able to overcome imatinib resistance in
some patients. However, some mechanisms, for example, BCR/ABL point
mutation T315I-mediated resistance cannot be overcome by current
available clinical drugs thus highlighting the need for further
research on the mechanism of leukemogenesis of CML cells in order
to explore novel mechanism-based strategies with high efficacy and
low toxicity.
Epidermal growth factor receptor kinase substrate 8
(EPS8) is a cytoplasmic protein that acts as a substrate of
receptor and non-receptor tyrosine kinases such as EGFR, FGFR,
VEGFR and Src kinase. EPS8 functionally serves as an adaptor
protein associating with diverse partner proteins to form complexes
that regulate multiple signalling pathways. Physiologically, EPS8
forms a complex with Abi-1 and SOS-1 to regulate the Rac signalling
pathway promoting cytoskeletal remodelling. EPS8 also plays a role
in membrane flow, pseudopodium formation, morphogenesis of
microvilli, stereocilia function and length, cellular adhesion and
motility (6). Furthermore, EPS8 has
been identified as an oncogene, as it enables cellular
transformation in vitro and tumour formation in vivo
upon overexpression (7). EPS8 has
been documented to be highly expressed in a broad spectrum of solid
tumours, such as squamous carcinoma, cervical cancer, colon
carcinoma, and breast cancer (8–12).
However, only a few studies have addressed the role of EPS8 in
haematological malignancies. Microarray analysis by Kang et
al revealed that a high level of EPS8 predicted a poor
prognosis of infant acute lymphoblastic leukemia (ALL) patients
with MLL rearrangements (13). In
addition, we previously determined that increased expression of
EPS8 mRNA in bone marrow was related to a poor response to
chemotherapy and a poor prognosis in acute myeloid leukemia (AML)
and ALL patients (14,15). However, it remains unclear whether
EPS8 is implicated in CML and how EPS8 regulates the biological
functions of CML cells.
In the present study we performed q-RT-PCR to
demonstrate that CML patients expressed higher EPS8 mRNA than
healthy controls in bone marrow mononuclear cells. Then, we knocked
down the expression of EPS8 in the CML cell line K562 and observed
that attenuated EPS8 reduced proliferation, increased apoptosis,
arrested the cell cycle at the G1 phase and reduced adhesion and
migration. Notably, silencing EPS8 increased chemosensitivity both
in the imatinib sensitive cell line K562 and the resistant cell
line 32D-p210BCR/ABL-T315I. Mechanistically, knockdown
of EPS8 downregulated p-BCR/ABL and its downstream AKT/mTOR
signalling pathway. Notably, knockdown of EPS8 attenuated K562 cell
proliferation in BALB/c nude mice. Collectively, these data
revealed that EPS8 regulated the cell biology of CML. Targeting
EPS8 alone or combined with TKI may be promising therapeutic
strategies for refractory and relapsed CML patients.
Materials and methods
Cell lines and human samples
Bone marrow mononuclear cells were collected from
patients with CML at the Department of Hematology of Zhujiang
Hospital, Southern Medical University from 2013 to 2015. Some of
the RNA samples were purchased from KingMed Diagnostics (Guangzhou,
China). In total, 113 cases of CML (male n=60, female n=53)
including 50 cases of chronic phase (CP), 21 cases of accelerated
phase (AP) and 21 cases of blast crisis phase (BC) as well as 21
CML patients in complete remission (CR) and 21 normal control were
enrolled. In these cases 82 CML patients had clinical data of their
quantitative BCR-ABL-p210 level presented as the percentage of p210
to Abl as assessed by qRT-PCR and blast percentage in bone marrow.
All the patients had signed informed consents. The study was
approved by the Ethics Committee of Zhujiang Hospital, Southern
Medical University.
The human K562, KBM5, MEG01 cell lines were
purchased from Jennio Biotech Co. (Guangzhou, China) and maintained
in the laboratory of the Department of Hematology, Zhujiang
Hospital, Southern Medical University. The murine 32D-p210-T315I
and 32D-p210-WT myeloid precursor cell lines were kindly provided
as gifts by Professor Lin Qiu from the Chinese Academy of Medical
Sciences. The cells were cultured in RPMI-1640 containing 10% fetal
calf serum (FCS) at 37°C and 5% v/v CO2.
Establishment of the stably
transfected cell lines
A shRNA targeting EPS8 (CCCTATTGAATAAGGAC) or a
scrambled shRNA was inserted into the pLVX lentiviral vector
carrying the puromycin resistance gene to construct the transfer
vector pLVX-shRNA-EPS8-puro. Then, the pLVX-shRNA-EPS8-puro vector
and the control pLVX-GFP-puro vector were transfected into 293T
cells, along with the packaging vector to envelop the lentivirus.
The K562 cells were transfected with the lentivirus carrying the
pLVX-shRNA-EPS8-puro vector or control vector, followed by
puromycin selection for ~3 weeks to establish the stably
transfected K562-shRNA-EPS8 and K562-GFP cell lines. Similarly, two
efficient shRNA sequences targeting EPS8 and a scrambled shRNA were
synthesized to establish the stably transfected
32D-p210-T315I-shRNA1-EPS8, T315I-shRNA2-EPS8 and T315I-scramble
cell lines, respectively.
Quantitative real-time PCR
Total RNA was prepared using TRIzol Reagent
(Invitrogen, Carlsbad, CA, USA). RNase-free DNase I (Promega,
Madison, WI, USA) was used to remove the genomic DNA. Reverse
transcription was performed with the M-MLV Reverse transcriptase
cDNA synthesis kit (Promega). Quantitative real-time PCR was
performed with SYBR Green qPCR SuperMix (Invitrogen) on an ABI
PRISM 7500 Sequence Detection System according to the standard
SYBR-Green PCR protocol. 18S rRNA was used as the internal control.
Triplicate reactions were performed as follows: 40 cycles of a
two-step PCR (95°C for 15 sec, 60°C for 32 sec) after initial
denaturation (95°C for 2 min). The data are presented as the fold
change in expression, as determined with the 2−ΔΔCt
method (17). The primer sequences
were as follows: EPS8 forward, 5′-GATGGAGGAAGTGCAAGATG and reverse,
5′-GACTGTAACCACGTCTTCACA; 18S rRNA forward, 5′-CCTGGATACCGCAGCTAGGA
and reverse, 5′-GCGGCGCAATACGAATGCCCC.
Cell proliferation assay
Cells (1×104 cells/well) were plated in
96-well plates in quadruplicate and cultured in 10% FCS-containing
medium with or without the indicated concentrations of drugs. At 0,
20, 44 and 68 h, 10 µl of CCK-8 solution was added to the cells,
which were then incubated for an additional 4 h. Then, the
OD450 values were obtained using a microplate
reader.
Apoptosis assay
Cells (5×105) were collected, and 1.24 µl
of Annexin V-APC was added to the cells. The cells were incubated
at room temperature for 15 min, centrifuged at 1,000 × g for 5 min,
and the supernatant was discarded. The cells were then resuspended
in 0.5 ml of cold 1X binding buffer. Ten microliters of 7-ADD or
propidium iodide (PI) was added on ice, and the cells were analyzed
by flow cytometry. The percentage of apoptotic cells (Annexin
V+) was quantified.
Cell cycle analysis
Cells (1×106) were collected and washed
twice with cold PBS. Then, cold 70% ethanol was added to the cells,
which were incubated overnight at 4°C. The following day, the cells
were washed once with PBS, and 500 µl of PBS containing 50 µl/ml
PI, 100 µg/ml RNase A, and 0.2% Triton X-100 were added, and the
cells were incubated for 30 min at 4°C in the dark. DNA content was
detected by flow cytometry.
Adhesion and migration assay
The 96-well plates were coated with fibronectin (30
mg/l) and dried overnight. PBS (20 µl) containing 3% BSA was added
to the 96-well plate at 37°C for 2 h, and then removed. Two hundred
microliters of cells (0.5×106 cells/ml) suspended in
RPMI-1640 and 10% FBS were added to the 96-well plates and
incubated for 1.5 h; the plate was then washed gently. The cells
that adhered to the bottom of the 96-well plate were observed under
a microscope, and the number of adherent cells was assessed using
the CCK-8 assay.
The cell migration assay was performed as previously
described. Briefly, the cells were suspended in serum-free
RPMI-1640 containing 0.1% BSA at a concentration of
1×106 cells/ml. A suspension of 0.1 ml of cells was
added to the upper chamber, and the cells were allowed to migrate
to the lower chamber, which contained RPMI-1640 and 10% FBS, for
6–8 h at 37°C in a 5% CO2 incubator. The cells that had
migrated to the lower chamber were observed under a microscope. The
number of cells in the lower chamber was assessed using the CCK-8
assay.
Chemotherapy drug sensitivity
assay
The cells (1×104 cells/100 µl/well) were
plated onto 96-well culture plates in quadruplicate in the presence
of different concentrations of either daunorubicin or imatinib for
the indicated time-points. Cell viability was assayed by adding
CCK-8 solution. The percentage of cell viability was evaluated by
assessing the absorbance at 450 nm and normalized to the
corresponding untreated control.
Western blotting
Total protein lysates were separated on sodium
dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto a polyvinylidene fluoride (PVDF) membranes. The
blots were subjected to a standard immunodetection procedure using
specific antibodies. The signals were enhanced with a
chemiluminescence substrate and detected using an image analyser.
The primary antibodies used were as follows: ERK (rabbit,
polyclonal, 1:1,000; cat. no. ab196883), p-ERK (rabbit, polyclonal,
1:1,000; cat. no. ab214362) (both from Abcam, Cambridge, UK); STAT5
(rabbit, monoclonal, 1:1,000; cat. no. 94205), p-STAT5 (rabbit,
polyclonal, 1:1,000; cat. no. 9351), Bcl-2 (rabbit, polyclonal,
1:1,000; cat. no. 2872) (all from CST, Danvers, MA, USA); Mcl-1
(rabbit, polyclonal, 1:1,000; cat. no. 16225-1-AP; Proteintech,
Rosemont, IL, USA); PTEN (rabbit, monoclonal, 1:1,000; cat. no.
ab32199), EPS8 (rabbit, polyclonal, 1:1,000; cat. no. ab96144)
(both from Abcam); phospho-BCR/ABL (rabbit, polyclonal, 1:500; cat
no. 3901), BCR/ABL (rabbit, polyclonal, 1:1,000; cat. no. 3902),
β-actin (mouse, monoclonal, 1:1,000; cat. no. 3700), mTOR (rabbit,
polyclonal, 1:1,000; cat. no. 2972), phospho-mTOR-s2448 (rabbit,
polyclonal, 1:1,000; cat. no. 2971), Akt (rabbit, polyclonal,
1:1,000; cat. no. 9272), phospho-Akt-t308 (rabbit, polyclonal,
1:1,000; cat. no. 5110), phospho-Akt-s473 (rabbit, monoclonal,
1:1,000; cat. no. 4060), GSK3β (rabbit, monoclonal, 1:1,000, cat.
no. 5676), phospho-GSK3β-s9 (rabbit, monoclonal, 1:1,000; cat. no.
9322) (all from CST); 4EBP1 (mouse, polyclonal, 1:1,000; cat. no.
60246-1-Ig; Proteintech); phospho-4EBP1-T37/46 (rabbit, polyclonal,
1:1,000; cat. no. 9459; CST); and GAPDH (mouse, monoclonal,
1:1,000; cat. no. KC-5G4; KangChen Bio-tech).
Xenograft model
BALB/c nu/nu mice were purchased from Southern
Medical University. Nude mice, 6–8 weeks, were implanted
subcutaneously with 1×107 K562-scramble or
K562-shRNA-EPS8 cells, irrespectively. Tumor volume was assessed
every 3 days. Mice were sacrificed on day 24 after inoculation and
the subcutaneous tumors were weighed. All the procedures were
performed according to an approved Southern Medical University,
Animal Care and Use Committee protocol.
Statistical analyses
The data are reported as the mean ± standard
deviations (SDs). One-way ANOVA followed by the
Student-Newman-Keuls' pairwise multiple comparison test were used
to compare the EPS8 mRNA expression among CML patients at different
phases with healthy donors, as well as differences obtained in the
proliferation, apoptosis, cell cycle, adhesion and migration
assays. Spearman's rank correlation coefficient was used to analyze
the correlation between EPS8 mRNA expression and clinical
characteristics. A two-tailed independent Student's t-test was used
to compare the means in the chemotherapy drug sensitivity assay.
Differences were considered significant when P<0.05.
Results
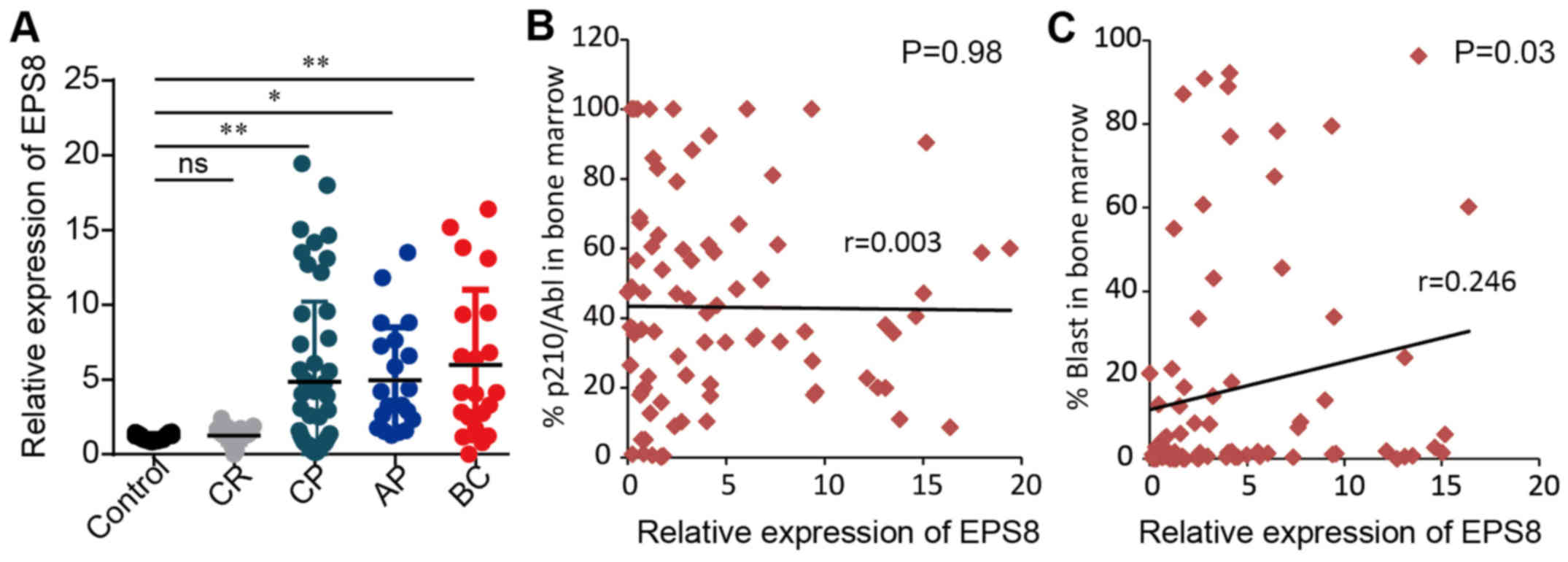
CML patients express a high level of
EPS8 mRNA
A published microarray data of 91 cases of CML
patients reported by Radich et al (16) revealed an increase in EPS8 mRNA
expression of CML patients compared to healthy controls, although
the diffence was not statistically significant. Furthermore, there
was a significant increase in EPS8 expression in the blast crisis
phase of CML patients compared to healthy controls. To validate the
result of these microarray data, we assessed EPS8 mRNA expression
in bone marrow mononuclear cells from 113 cases of CML in CP (50
cases), AP (21 cases), BC (21 cases), CR (21 cases), and 21 healthy
controls using quantitative real-time PCR. CML patients in CP, AP
and BC exhibited significantly higher expression of EPS8 than those
in complete remission and healthy donors (P<0.05). Moreover,
there was a tendency for patients in BC to have increased EPS8
expression compared to patients in CP and AP (P>0.05) (Fig. 1A). We further performed correlation
analysis between EPS8 expression and clinical features. There was
no significant difference in EPS8 expression between the sex
(P=0.74), age (P=0.85) and BCR-ABL level (P=0.98) of CML patients,
however there was a correlation to blast percentage in bone marrow
(P=0.03) (Fig. 1B and C; Table I). Collectively, these data revealed
that EPS8 may play a role in CML leukemsogenesis.
 | Table I.Correlation analysis between EPS8
expression and the clinical features of CML patients. |
Table I.
Correlation analysis between EPS8
expression and the clinical features of CML patients.
| Clinical
features | Eps8 expression
(mean rank) | P-value |
|---|
| Male (n=60) | 49.83 | 0.74 |
| Female (n=53) | 48.99 |
|
|
| Correlation
coefficient (Spearmans r) |
|
| Age (years) | 0.019 | 0.85 |
| p210 | 0.003 | 0.98 |
| Blast
percentage | 0.246 | 0.03 |
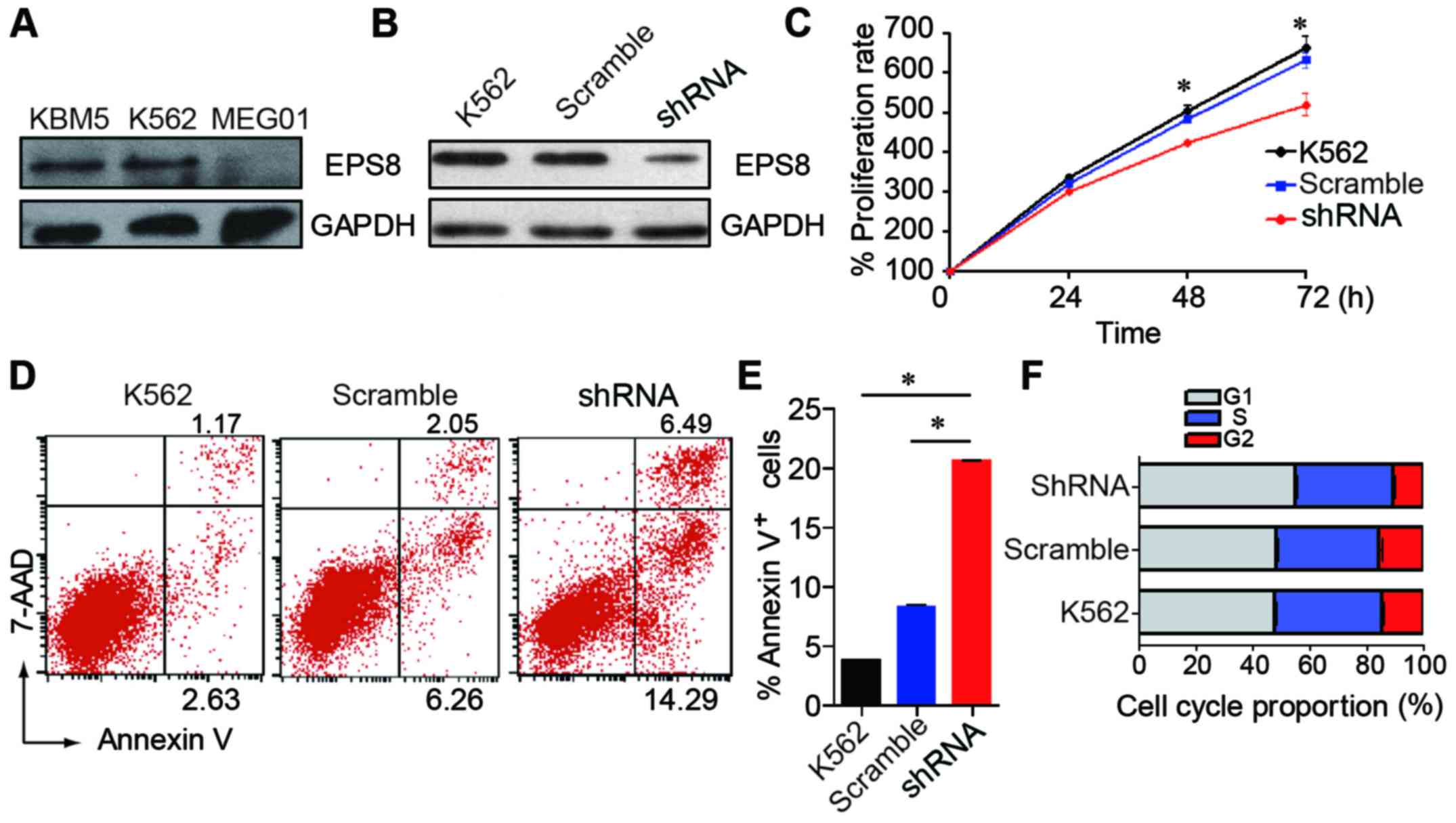
Knockdown of EPS8 inhibits CML cell
proliferation, induces apoptosis and arrests the cell cycle in the
G1 phase
To evaluate the effect of EPS8 on the biological
function of CML cells, we knocked down the expression of EPS8 in
CML cell line K562. EPS8 expression was detected in three human CML
cell lines. The K562 and KBM5 cells expressed a higher level of
EPS8, while the MEG01 cells exhibited a lower expression (Fig. 2A). Then, the K562 cells were
transfected with a lentivirus carrying an EPS8-targeted shRNA
vector to knockdown EPS8. As a result, EPS8 expression was
attenuated in the K562-shRNA-EPS8 cells that were stably transduced
with the lentivirus (Fig. 2B).
To investigate the role of EPS8 in CML cells, we
assessed the proliferation, apoptosis and cell cycle after EPS8
silencing. We first examined the proliferation of K562-shRNA-EPS8
cells and determined that knockdown of EPS8 reduced the
proliferation of the K562 cells (Fig.
2C). Then, we assessed cell apoptosis and the results revealed
that the K562-shRNA-EPS8 cells exhibited enhanced apoptosis
compared with their parental cells (Fig. 2D and E). Then, the cell cycle was
evaluated and it was determined that more K562-shRNA-EPS8 cells
were present in the G1 phase, and fewer cells were present in the S
phase compared with the corresponding control cells (Fig. 2F). These data revealed that EPS8
regulated the proliferation, apoptosis and cell cycle in CML
cells.
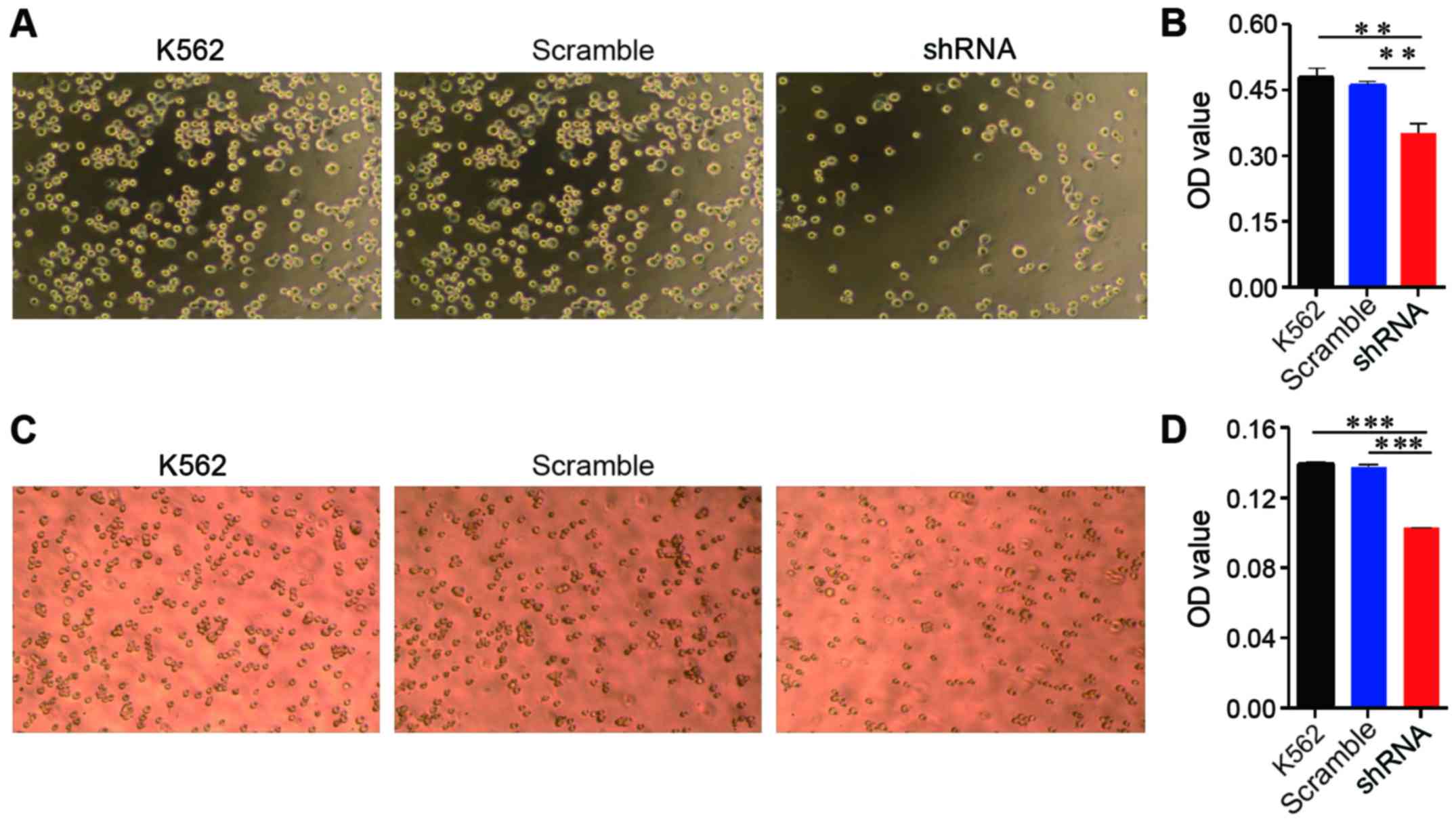
Attenuated EPS8 expression leads to
impaired adhesion and migration
EPS8 plays an important role in cytoskeletal
remodelling and cellular motility; therefore, we evaluated the
effect of EPS8 on adhesion and migration of K562 cells. The K562
cells and their derived cells were plated on fibronectin-coated
96-well plates for 1.5 h and then washed. Less K562-shRNA-EPS8
cells remained on the fibronectin-coated plates compared with the
control cells (Fig. 3A and B).
Cellular migration was assessed using the Transwell assay, in which
the cells were placed into the upper chamber, which contained
serum-free medium, and the lower chamber contained medium with 10%
FBS. Fewer K562-shRNA-EPS8 cells were observed in the lower chamber
compared with the K562-scramble cells demonstrating that the
attenuation of EPS8 expression impaired the migration of the K562
cells (Fig. 3C and D).
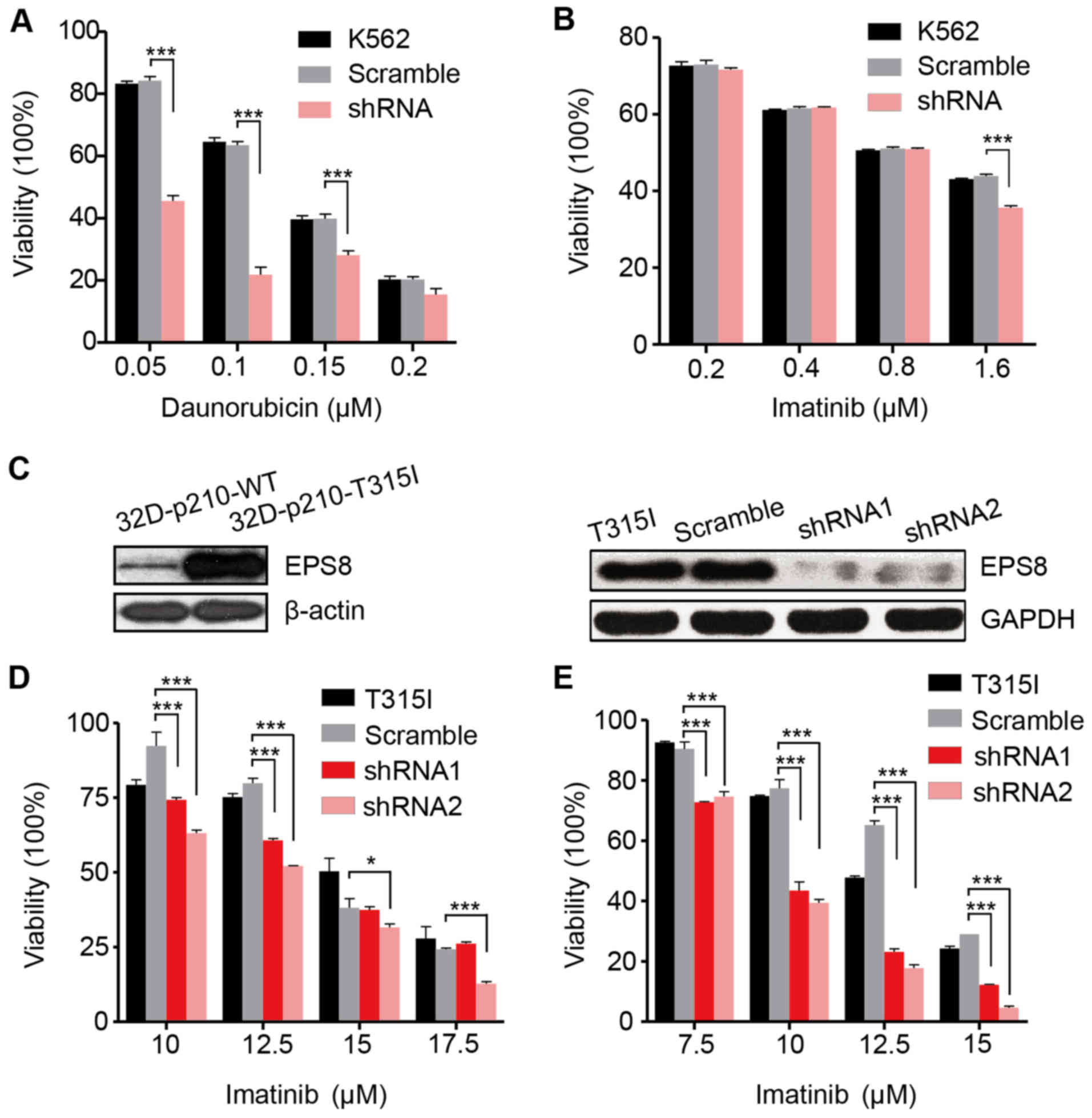
EPS8 is implicated in the
chemosensitivity of CML cells
We then investigated the effect of EPS8 on the
chemosensitivity of the K562 cells. Daunorubicin is a widespread
used common chemotherapy drug to treat myeloid leukemia. In the
present we determined that the viability of the K562-shRNA-EPS8
cells was significantly reduced in the presence of indicated
concentrations of daunorubicin indicating that EPS8 knockdown
increases their chemosensitivity (Fig.
4A). However, as an imatinib-sensitive cell line, the
K562-shRNA-EPS8 cells did not exhibit further increased sensitivity
to imatinib except in the high drug concentration (Fig. 4B).
To investigate whether knockdown of EPS8 can
overcome imatinib resistance, we employed a murine
imatinib-resistant cell line 32D-p210T325I-BCR/ABL
(32D-p210-T315I), which was generated by transfecting myeloid
precursor cells with the vector carrying prototype
imatinib-resistant BCR/ABL point mutation T315I in CML patients.
Compared to the imatinib sensitive cell line
32D-P210BCR/ABL (32D-p210-WT) which expressed native
BCR/ABL protein p210, 32D-p210-T315I cells expressed a higher level
of EPS8 protein (Fig. 4C). To
silence EPS8 expression, LV-EPS8-shRNA1 and LV-EPS8-shRNA2 vectors
were transfected into 32D-p210-T315I cells, respectively. EPS8
expression was considerably decreased compared with the controls
(Fig. 4C). The results revealed
that knockdown of EPS8 reduced the cell viability in a dose- and
time-dependent manner. Following 24 and 48 h of incubation, cell
viability in the EPS8-shRNA1 or EPS8-shRNA2 groups was
significantly lower than the scramble group. Therefore,
downregulation of EPS8 increased the sensitivity of 32D-p210-T315I
cells to imatinib (Fig. 4D and
E).
In accordance with these results, we determined that
knockdown of EPS8 notably increased apoptosis in imatinib-treated
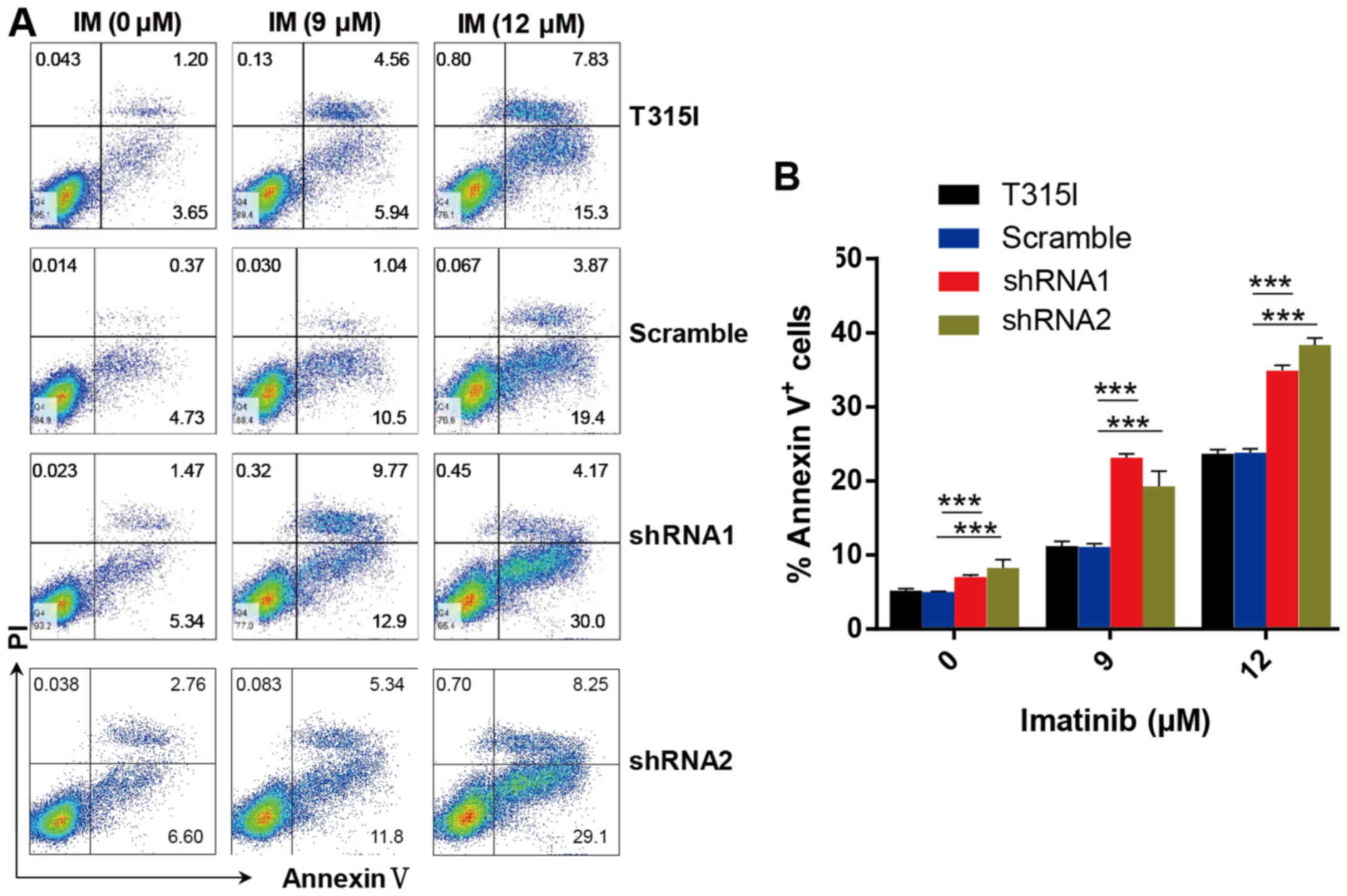
32D-p210-T315I cells in a dose-dependent manner. Apoptosis was
markedly increased in EPS8-shRNA cells compared with the control
groups after imatinib treatment for 24 h (Fig. 5A and B). These results indicated
that EPS8 protected imatinib-resisitant CML cells against
drug-induced apoptosis.
EPS8 regulates apoptosis and
proliferation signalling pathways in CML cells
To determine the molecular mechanism through which
EPS8 regulates CML cells, key molecules in main signalling pathways
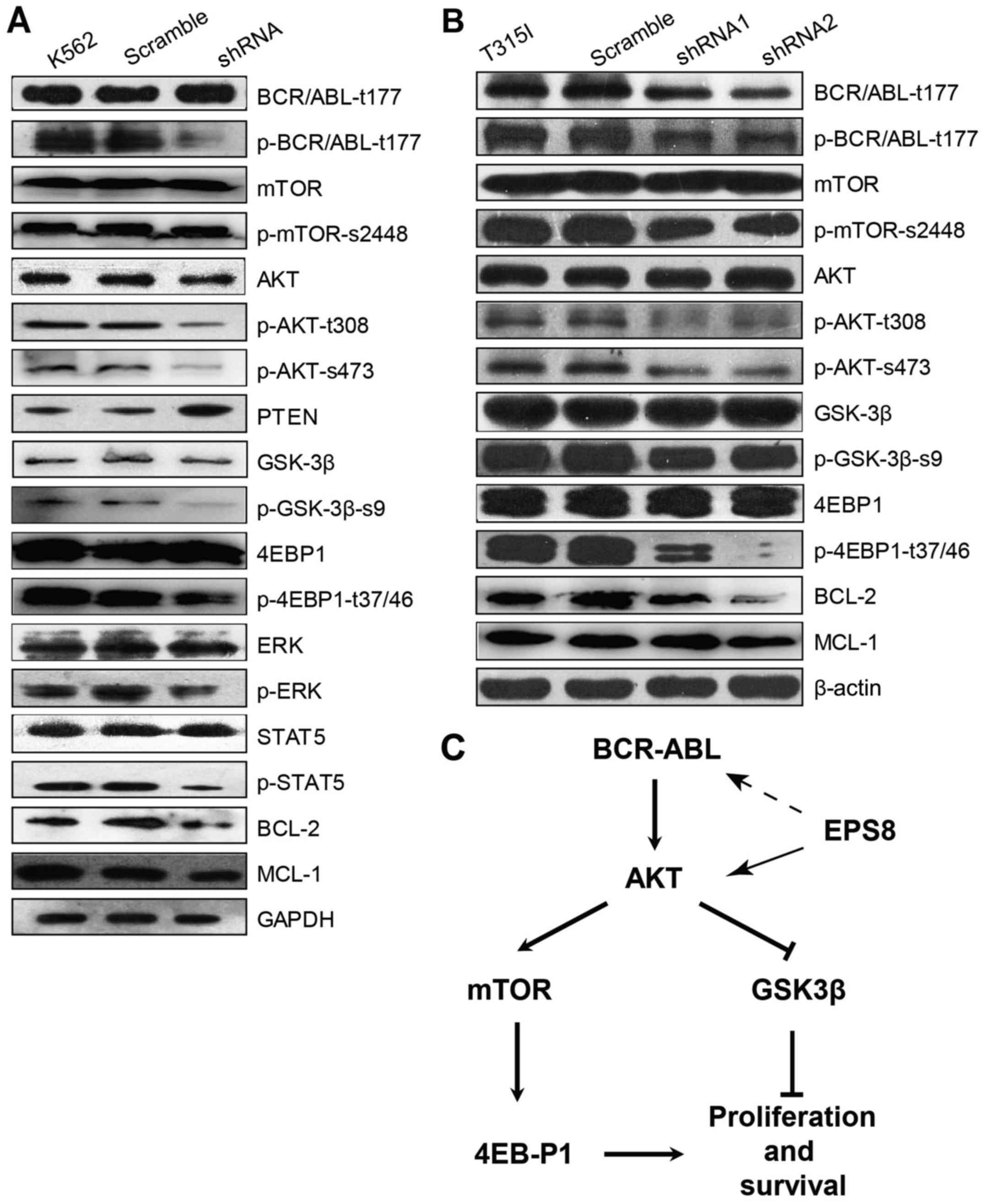
involved in leukemogenesis were assessed. Notably, phosphorylated
(p)-BCR-ABL was significantly decreased in EPS8-knockdown K562
cells compared with the control group, suggesting that EPS8
regulated BCR-ABL. Furthermore, our results illustrated that p-AKT
and phosphorylation of its downstream proteins mTOR, eukaryotic
translation initiation factor 4E binding protein 1 (EIF4EBP1) and
glycogen synthase kinase 3β (GSK3β), as well as, p-STAT5 and p-ERK
levels were reduced in K562-shRNA-EPS8 cells compared with the
control cells. Since Mcl-1 and Bcl-2 function as anti-apoptotic
markers in various cancers, we explored whether their expression
was affected by EPS8. Indeed, we found that the expression of Mcl-1
and Bcl-2 was suppressed by EPS8 silencing. These data revealed
that EPS8 inhibition-induced apoptosis was related with the
reduction of anti-apoptotic factors and inhibition of proliferation
and anti-apoptotic pathway activity (Fig. 6A).
Given that EPS8 regulated the anti-apoptotic
signalling and increased imatinib-induced apoptosis in
imatinib-resistant 32D-p210-T315I cells, we next investigated the
effect of EPS8 on BCR-ABL expression and its downstream PI3K/Akt
signalling pathway in 32D-p210-T315I cells and their derived cells.
In accordance with K562-shRNA-EPS8 cells, 32D-p210-T315I-shRNA-EPS8
cells exhibited decreased total BCR-ABL and p-BCR-ABL proteins.
Furthermore, EPS8 silencing led to a decrease of p-Akt (T308,
S476), p-4EBP1 (T37/46), p-mTOR (S2448) and p-GSK3-β (Fig. 6B). Collectively, these data revealed
that EPS8 knockdown interfered with the BCR-ABL/PI3K/AKT/mTOR
pathway (Fig. 6C).
Knockdown of EPS8 attenuated
proliferation of K562 cells in vivo
Finally, we determined the efficacy of the
inhibition of EPS8 on the proliferation of BCR-ABL positive cells
in vivo. Since no EPS8 inhibitor was available, we utilized
the shRNA to knockdown EPS8 in K562 cells in a xenograft model.
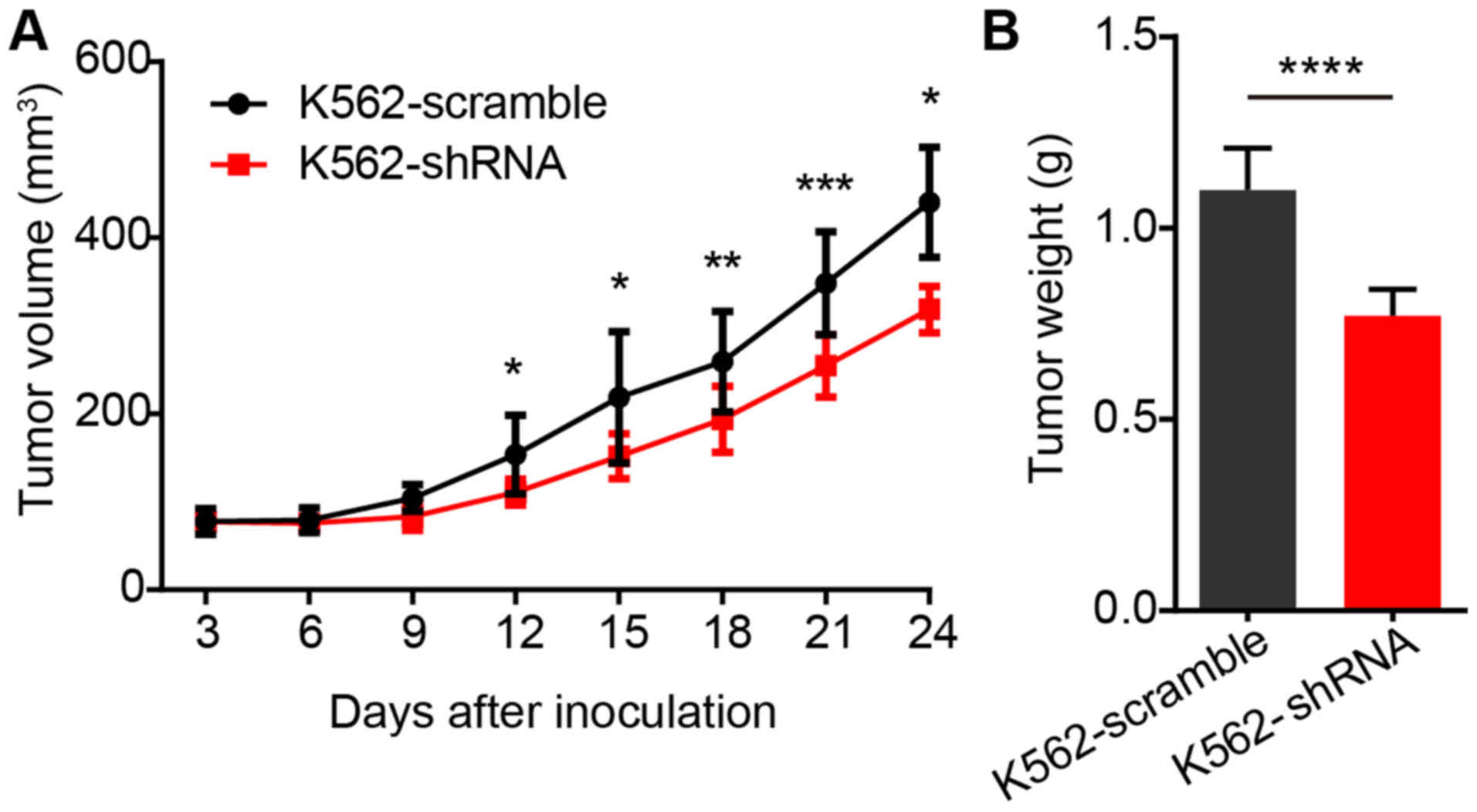
BALB/c nude mice were implanted subcutaneously with
1×107 K562-scramble or K562-shRNA-EPS8 cells, and the
tumor volume was observed every 3 days. The results revealed that
the mice bearing K562-shRNA-EPS8 cells had a significantly smaller
tumor volume compared to those bearing the K562-scramble cells
(Fig. 7A). Consistently, tumor
weight in mice harboring K562-shRNA-EPS8 cells at 24 days after
inoculation was significantly lower than that in the control mice
(Fig. 7B).
Discussion
EPS8 is a cytoplasmic non-receptor kinase and is
well recognized as an oncogene in a variety of solid tumours
(6). However, its role in
haematological malignant diseases is less documented. A gene
expression profiling study of 97 cases of infant ALL identified
EPS8, along with six other genes as prognostic factors. The
high-EPS8 cohort had an event-free survival of 20%, while the
low-EPS8 cohort had a rate of 65% (13). Our previous study revealed similar
results. We found that EPS8 expression in AML patients was elevated
compared with the control group. Moreover, EPS8 expression was
negatively related to the response of AML patients to chemotherapy
(15). Furthermore, we addressed
the expression of the EPS8 mRNA in 107 cases of ALL patients, and
there was a correlation between the expression of EPS8 and MDR1, as
well as EPS8 and WT1. A high EPS8/MDR1/WT1 expression profile was
associated with a higher risk of relapse. In addition, patients
with high expression of EPS8/MDR1/WT1 had a shorter event-free
survival (14). Notably, consistent
with the reported CML microarray data, we revealed in our present
study that our cohort CML patients had a higher expression of EPS8
compared to the controls, indicating that EPS8 plays a role in CML
disease. In addition, our data demonstrated that EPS8 mRNA
expression was not correlated with BCR-ABL, suggesting that EPS8
may be a BCR-ABL independent factor for CML patients. However, our
study did not contain follow-up clinical information, therefore
future investigation to address the relationship between EPS8
expression and drug resistance or survival in CML patients is
warranted.
To determine the role in CML cells, we knocked down
the expression of EPS8 in CML cell line K562 and determined that
silencing of EPS8 led to reduced proliferation, increased apoptosis
and cell cycle arrest in the G1 phase which was consistent with
other studies on EPS8. Xu et al reported that the stable
overexpression of EPS8 accelerated proliferation and reduced the
serum withdrawal-induced apoptosis of pituitary tumour cells
(19). Wang et al found that
EPS8 expression was increased in oral squamous carcinoma compared
with normal keratinocytes. The NH4 cell line exhibited mildly
elevated EPS8 expression and reduced invasion, while the NH12 cell
line, which expressed a high level of EPS8, was extremely
aggressive. EPS8 overexpression in NH4 cells led to proliferation
that was similar to that of NH12 cells (20). Similar results were observed in
other cancers. The results in the present study indicated that EPS8
was involved in CML as well.
One of the characteristics of leukemia cells is
invasion to other organs. Cytoskeletal proteins have an important
role in AML cell migration (21).
EPS8 is an important regulator of the cytoskeleton and is reported
to be related to the migration and invasion of many solid tumours
by regulating MMP9, E-cadherin and N-cadherin (7). EPS8 was demonstrated to upregulate the
expression of CXCL5 and CXCL12 via the transcription factor FOXM1
to enhance migration (22). Yap
et al demonstrated that EPS8 promoted oral squamous cancer
cell migration and invasion by activating Rac in an
integrin-dependent manner (23).
EPS8, Abi-1 and SOS-1 form a tricomplex and induce Rac-specific
guanine nucleotide exchange factor (GEF) activity, which has been
revealed to play a critical role in ovarian cancer metastasis
(9). In the present study the
ability of adhesion and migration was attenuated in K562-shRNA-EPS8
cells compared with the control, which was consistent with previous
research indicating that EPS8 may contribute to migration from bone
marrow and infiltration to other organs.
Gorsic et al revealed that EPS8 knockdown
increased the chemosensitivity of lymphoblastic cell lines, small
cell lung cancer cell lines and bladder cancer cell lines to
cisplatin (24). Chen et al
reported that EPS8 reduced the chemosensitivity of cervical cancer
cells to cisplatin and paclitaxel (25). Notably, our results demonstrated
that EPS8 regulated the sensitivity of CML cells, including
imatinib-sensitive and imatinib-resistant cells, to chemotherapy
and tyrosine kinase inhibitors.
To explore the molecular mechanism involved in the
EPS8-mediated effect on the response of CML cells to imatinib, we
detected BCR-ABL and its downstream proteins in imatinib-sensitive
cell line K562 and imatinib-resistant cell line 32D-p210-T315I and
their derivatives. EPS8 is a critical signaling molecule and
integrates multiple pathways. EPS8 has been reported to regulate
PI3K/AKT/mTOR (19,20) and MEK/ERK (8) signalling which control cell
proliferation, survival, differentiation and apoptosis. Consistent
with the evidence that EPS8 knockdown inactived Akt and downstream
4EBP1 phosphorylation in colon cancer cells (26), our study provided an extension of
this notion, revealing for the first time that the phosphorylation
of Akt (S308, S476), GSK3-β (S9), and 4EBP1 (T37/46) were decreased
in CML cells after EPS8 silencing. Notably, we determined that the
active form of the oncoprotein BCR-ABL was decreased after EPS8
knockdown, highlighting the critical role of EPS8 in BCR-ABL
positive cells. The exact underlying mechanism revealing how EPS8
regulates BCR-ABL remains unclear. Although EPS8 is an adaptor
protein, it may be less likely that EPS8 directly combines with
BCR-ABL to form a complex since recently Reckel et al
performed a quantitative comparative proteomic study in p190 or
p210 transfected BaF3 cells and EPS8 was not identified as an
interactor of BCR-ABL (27).
Despite the fact that BCR-ABL is an oncoprotein which is capable of
autophosphorylation, proliferation and survival signalling can also
regulate its phosphorylation. It is more possible that knockdown of
EPS8 leads to attenuation of proliferation and survival signalling,
which subsequently decreases the phosphorylation of BCR-ABL. A
future study is warranted to test this hypothesis.
In conclusion, our data revealed that EPS8 regulated
multiple biological functions such as proliferation, apoptosis, the
cell cycle, drug sensitivity of CML cells possibly by mediating the
regulation of the BCR-ABL/AKT/mTOR signalling pathway. Strategies
that target EPS8 in CML patients may help to overcome resistance to
tyrosine kinase inhibitors. The EPS8 inhibitor alone or combined
with a tyrosine kinase inhibitor may be promising customized
strategies to treat refractory and resistant CML patients.
Acknowledgements
The present study was supported by the Major Program
for Health Medical Collaborative Innovation of Guangzhou (grant no.
201704020216), the Startup Project for Clinical Trials of Southern
Medical University (grant no. LC2016ZD027), the Special Project for
the Development of Science and Technology of Guangdong Province
(grant no. 2016A020213005), the Competitive Allocation Project of
the Comprehensive Strategic Cooperation of the Chinese Academy of
Sciences of Guangdong Province (grant no. 2013B091500072), the
National Science Foundation of China (no. 81372249 and 81300431)
and the Youth Scientific Fund of Southern Medical University (no.
PY2014N072).
References
|
1
|
Huang R, Kang Q, Liu H and Li Y: New
insights into the molecular resistance mechanisms of chronic
myeloid leukemia. Curr Cancer Drug Targets. 16:323–345. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Druker BJ, Guilhot F, O'Brien SG, Gathmann
I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM,
Stone RM, et al IRIS Investigators, : Five-year follow-up of
patients receiving imatinib for chronic myeloid leukemia. N Engl J
Med. 355:2408–2417. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hochhaus A, O'Brien SG, Guilhot F, Druker
BJ, Branford S, Foroni L, Goldman JM, Müller MC, Radich JP, Rudoltz
M, et al IRIS Investigators, : Six-year follow-up of patients
receiving imatinib for the first-line treatment of chronic myeloid
leukemia. Leukemia. 23:1054–1061. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gorre ME, Mohammed M, Ellwood K, Hsu N,
Paquette R, Rao PN and Sawyers CL: Clinical resistance to STI-571
cancer therapy caused by BCR-ABL gene mutation or amplification.
Science. 293:876–880. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thomas J, Wang L, Clark RE and Pirmohamed
M: Active transport of imatinib into and out of cells: Implications
for drug resistance. Blood. 104:3739–3745. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li YH, Xue TY, He YZ and Du JW: Novel
oncoprotein EPS8: A new target for anticancer therapy. Future
Oncol. 9:1587–1594. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maa MC, Hsieh CY and Leu TH:
Overexpression of p97Eps8 leads to cellular transformation:
Implication of pleckstrin homology domain in p97Eps8-mediated ERK
activation. Oncogene. 20:106–112. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen C, Liang Z, Huang W, Li X, Zhou F, Hu
X, Han M, Ding X and Xiang S: Eps8 regulates cellular proliferation
and migration of breast cancer. Int J Oncol. 46:205–214. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen H, Wu X, Pan ZK and Huang S:
Integrity of SOS1/EPS8/ABI1 tri-complex determines ovarian cancer
metastasis. Cancer Res. 70:9979–9990. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ding X, Zhou F, Wang F, Yang Z, Zhou C,
Zhou J, Zhang B, Yang J, Wang G, Wei Z, et al: Eps8 promotes
cellular growth of human malignant gliomas. Oncol Rep. 29:697–703.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schoenherr C, Serrels B, Proby C,
Cunningham DL, Findlay JE, Baillie GS, Heath JK and Frame MC: Eps8
controls Src- and FAK-dependent phenotypes in squamous carcinoma
cells. J Cell Sci. 127:5303–5316. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun J, Jin LY, Wang HY, Zhou JQ, Li YJ, He
YZ and Li YH: Correlation of Eps8 with proliferation, metastasis
and prognosis of malignant tumors. Zhongguo Shi Yan Xue Ye Xue Za
Zhi. 21:493–497. 2013.(In Chinese). PubMed/NCBI
|
|
13
|
Kang H, Wilson CS, Harvey RC, Chen IM,
Murphy MH, Atlas SR, Bedrick EJ, Devidas M, Carroll AJ, Robinson
BW, et al: Gene expression profiles predictive of outcome and age
in infant acute lymphoblastic leukemia: A Children's Oncology Group
study. Blood. 119:1872–1881. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
He YZ, Liang Z, Wu MR, Wen Q, Deng L, Song
CY, Wu BY, Tu SF, Huang R and Li YH: Overexpression of EPS8 is
associated with poor prognosis in patients with acute lymphoblastic
leukemia. Leuk Res. 39:575–581. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang L, Cai SH, Xiong WY, He YJ, Deng L
and Li YH: Real-time quantitative polymerase chain reaction assay
for detecting the eps8 gene in acute myeloid leukemia. Clin Lab.
59:1261–1269. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Radich JP, Dai H, Mao M, Oehler V,
Schelter J, Druker B, Sawyers C, Shah N, Stock W, Willman CL, et
al: Gene expression changes associated with progression and
response in chronic myeloid leukemia. Proc Natl Acad Sci USA.
103:pp. 2794–2799. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cross DA, Alessi DR, Cohen P, Andjelkovich
M and Hemmings BA: Inhibition of glycogen synthase kinase-3 by
insulin mediated by protein kinase B. Nature. 378:785–789. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu M, Shorts-Cary L, Knox AJ,
Kleinsmidt-DeMasters B, Lillehei K and Wierman ME: Epidermal growth
factor receptor pathway substrate 8 is overexpressed in human
pituitary tumors: Role in proliferation and survival.
Endocrinology. 150:2064–2071. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang H, Patel V, Miyazaki H, Gutkind JS
and Yeudall WA: Role for EPS8 in squamous carcinogenesis.
Carcinogenesis. 30:165–174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Capala ME, Vellenga E and Schuringa JJ:
ELMO1 is upregulated in AML CD34+ stem/progenitor cells, mediates
chemotaxis and predicts poor prognosis in normal karyotype AML.
PLoS One. 9:e1115682014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang H, Teh MT, Ji Y, Patel V,
Firouzabadian S, Patel AA, Gutkind JS and Yeudall WA: EPS8
upregulates FOXM1 expression, enhancing cell growth and motility.
Carcinogenesis. 31:1132–1141. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yap LF, Jenei V, Robinson CM, Moutasim K,
Benn TM, Threadgold SP, Lopes V, Wei W, Thomas GJ and Paterson IC:
Upregulation of Eps8 in oral squamous cell carcinoma promotes cell
migration and invasion through integrin-dependent Rac1 activation.
Oncogene. 28:2524–2534. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gorsic LK, Stark AL, Wheeler HE, Wong SS,
Im HK and Dolan ME: EPS8 inhibition increases cisplatin sensitivity
in lung cancer cells. PLoS One. 8:e822202013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen YJ, Shen MR, Chen YJ, Maa MC and Leu
TH: Eps8 decreases chemosensitivity and affects survival of
cervical cancer patients. Mol Cancer Ther. 7:1376–1385. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maa MC, Lee JC, Chen YJ, Chen YJ, Lee YC,
Wang ST, Huang CC, Chow NH and Leu TH: Eps8 facilitates cellular
growth and motility of colon cancer cells by increasing the
expression and activity of focal adhesion kinase. J Biol Chem.
282:19399–19409. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Reckel S, Hamelin R, Georgeon S, Armand F,
Jolliet Q, Chiappe D, Moniatte M and Hantschel O: Differential
signaling networks of Bcr-Abl p210 and p190 kinases in leukemia
cells defined by functional proteomics. Leukemia. 31:1502–1512.
2017. View Article : Google Scholar : PubMed/NCBI
|