Introduction
Retinoblastoma (RB) is the most common intraocular
malignant neoplasm of infancy and childhood. Although enucleation
is an effective therapy for children with RB, one has to consider
the resulting visual impairment and cosmetic deformity. External
beam radiation therapy (EBRT) was proven to be the first effective
eye salvage therapy for advanced RB, but it increased the risk of
secondary tumors in children with germline RB1 mutations
[for review see (1)]. In the 1990s,
systemic chemotherapy with focal therapy (laser and cryotherapy)
was the standard treatment for intraocular RB (1). Intra-arterial chemotherapy (IAC),
delivered via the internal carotid artery, was first used to
enhance the effectiveness of EBRT, but is now a standard primary
treatment in ocular centers to avoid enucleation or systemic
treatment and a second-line therapy after failure of intravenous
chemoreduction (1–3). To date, ophthalmic artery chemosurgery
and intravitreous chemotherapy have completely replaced EBRT,
reduced the use of systemic chemotherapy and diminished
enucleations (2,3).
DNA topoisomerase (topo) enzymes regulate DNA
metabolism and affect replication, transcription, recombination,
chromatin assembly, DNA repair and ultimately cell division.
Important chemotherapeutic agents target these enzymes. Inhibitors
of topo II enzymes, such as etoposide, stabilize DNA-topo II
complexes by blocking DNA relegation. Trapping the enzyme in a
complex with cleaved DNA causes direct double-strand DNA damage
that then leads to p53 stabilization, finally causing apoptosis
(4,5).
The DNA-damaging agent cisplatin is likewise used
extensively as a chemotherapeutic drug. Since 1994 chemotherapy
with cisplatin and vincristine combined with focal therapy has been
successfully used for RB treatment. Cisplatin acts as an alkylating
or chelating agent, capable of forming adducts with macromolecules
such as cellular DNA. This results in DNA cross-links and induces
cell cycle arrest (6). The
inability to repair the DNA damage ultimately mediates the
cytotoxicity of this anticancer agent.
Another commonly used drug regiment includes a
combination of vincristine, etoposide and carboplatin (VEC) for
intravenous administration (7).
However, management of RB is limited not only by drug
dosage-related side-effects, but also by drug resistance to
chemotherapy. Resistance to chemotherapy leading to poor outcome
and survival remains a challenge for developing strategies for
therapeutic interventions in all types of cancer and in
vitro chemoresistant cell line models are an indispensable
resource towards delineating the development of novel drugs.
In the present study, we set out to characterize
three etoposide- and three cisplatin-resistant RB cell lines with
regard to morphological and functional changes compared to their
respective parental, chemosensitive counterparts.
Materials and methods
Cell culture
The human RB cell line RB-355, established and first
described by Griegel et al (1990) (8), and formerly donated by K. Heise, was
kindly provided by Dr H. Stephan. The RB cell lines Y-79 (9) and WERI-Rb1 (10), originally purchased from the Leibniz
Institute DSMZ (German Collection of Microorganisms and Cell
Cultures), were likewise kindly provided by Dr H. Stephan. All RB
cell lines were last tested and authenticated in September 2015.
Mutation analyses were conducted using an MLPA kit (SALSA MLPA kit
P047 RB1; MRC-Holland, Amsterdam, The Netherlands) and reactions
were performed according to the manufacturer's instructions.
Additional sequencing of the RB1 gene was performed for all
RB cell lines. However, most recent STR analyses (March 2017)
confirmed the authenticity of the cell lines.
The cell lines were cultivated as suspension
cultures in Dulbecco's modified Eagle's medium (DMEM) with 15%
fetal bovine serum (FBS) (both from PAN-Biotech GmbH, Aidenbach,
Germany), 100 U penicillin/ml and 100 µg streptomycin/ml, 4 mM
L-glutamine (both from Gibco, Karlsruhe, Germany), 50 µM
β-mercaptoethanol (Carl Roth, Karlsruhe, Germany) and 10 µg
insulin/ml (PAN-Biotech) at 37°C, 10% CO2 and 95%
humidity. No approval from an Ethics Committee was required for
work with the human cell lines.
Chemoresistant RB cell lines
All chemoresistant RB cell lines characterized were
generously provided by Dr H. Stephan. To generate these cell lines,
established Y-79, WERI-Rb1 and RB-355 cells (see above) were
continuously treated with consecutively increasing concentrations
of etoposide or cisplatin (both from Teva, Berlin, Germany) until
the chemoresistant sublines exhibited a at least 10-fold higher
IC50 value in WST-1 viability assays than the respective
parental controls (11). The
chemoresistant cell lines were subsequently cultivated as described
above for RB cell lines with additional treatment of the
appropriate cytostatic drug twice a week (every 3–4 days). For
details on final concentrations of the drugs used, see Table I.
 | Table I.Concentrations of the
chemotherapeutic agents used to treat the RB cell lines. |
Table I.
Concentrations of the
chemotherapeutic agents used to treat the RB cell lines.
| Cytostatic
drug | Cell line | Final concentration
(µmol/ml) |
|---|
| Cisplatin | WERI-Rb1 | 8 |
| (1 mg/ml) | Y-79 | 5 |
|
| RB-355 | 6 |
| Etoposide | WERI-Rb1 | 5 |
| (20 mg/ml) | Y-79 | 3 |
|
| RB-355 | 1 |
Cell proliferation and apoptosis
detection
Cell proliferation was determined by
5-bromo-2′-deoxyuridine (BrdU; Sigma, Steinheim, Germany)
incorporation. For BrdU immunocytochemistry 10 µM BrdU was added to
the cells 4 h prior to paraformaldehyde (PFA; Sigma) fixation.
Cells were incubated with a rat anti-BrdU antibody (1:1,000;
ab6326; Abcam, Cambridge, UK) and proliferating cells were
visualized using a goat anti-rat antibody labelled with Alexa
Flour® 488 (1:1,000; A11006; Life Technologies,
Darmstadt, Germany). In order to determine changes in apoptosis
levels, cells were stained with 4′,6-diamidino-2-phenylindole
(DAPI; Sigma) or Click-iT® Plus TUNEL assay for in
situ apoptosis detection (cat. #C10617; Thermo Fisher
Scientific, Darmstadt, Germany), following the manufacturer's
protocol and pycnotic nuclei were manually counted as previously
described by our group (12).
Growth kinetic
To determine growth kinetics, 3×105 RB
cells were seeded in 500 µl DMEM with supplements in a 24-well
plate and vital cells were counted manually using the trypan blue
exclusion method. Cells were seeded in triplicates and counted at
several time points (24, 48, 72 and 96 h). In the case of the
extremely slow-growing Y-79 cisplatin-resistant RB cell line, we
recorded long-term growth curves over a period of 336 h with longer
counting intervals and plotted the values logarithmically to
visualize differences between resistant and control cells.
Colony formation assay
Soft agarose assays were performed as previously
described in detail (13). The
colony formation efficiency (%)/visual field was determined by
counting the colonies and single cells in 5 visual fields
(magnification, ×10)/cell line in triplicates and images were
captured using a Leica DMIL or a Nikon Eclipse TS2 microscope
equipped with a digital camera and ProgRes Capture Basic 1.2.01 or
IC Capture 2.4 (The Imaging Source) software.
Chicken chorioallantoic (CAM)
assays
In order to test for changes in the migration and
tumor formation capacity following etoposide and cisplatin
resistance, RB cells were grafted onto the chick chorioallantoic
membrane (CAM) as described in a recent publication by our group
(14). Mainly following the
metastasis model protocol published by Zijlstra et al (2002)
(15), and visualized by Palmer
et al (2011) (16), 50 µl
cell suspension [1×106 chemoresistant or control cells
in phosphate-buffered saline (PBS)] was grafted onto the CAM area.
For each RB cell line, 30 eggs were grafted (15 with parental and
15 with chemoresistant RB cells) in at least 3 independent
experiments. All in vivo chick CAM experiments were
conducted according to the relevant national guidelines of the
responsible authority, the State Office for Nature, Environment and
Consumer Protection (LANUV) as well as to the Directive 2010/63/EU
of the European Parliament and of the council of September 22, 2010
on the protection of animals used for scientific purposes, which
does not comprise any restrictions for the use of non-mammalian
embryos. However, the Institutional Animal Care and Use Committee
(IACUC) of the Medical Faculty of the University Hospital Essen
approved the CAM assays and no ethical approval was required as
according to the German Animal Experiment and Welfare Guidelines,
ethical approval is only essential when animals are intended to
live beyond hatching.
Harvesting of tissue
The duration of the chick CAM assay is limited to a
7–9 day window prior to hatching. Seven days after grafting
(E10-17) chick embryos were anesthetized by cooling on ice and
sacrificed by decapitation. CAM tumors were excised, measured,
photographed and fixed for 1 h at 4°C in 4% PFA in 0.1 M phosphate
buffer (pH 7.4). For cryo-embedding, the tumor tissue was incubated
for 30 min in PBS (pH 7.3) containing 15% sucrose, followed by a
30-min incubation in PBS containing 30% sucrose and finally
embedded in OCT compound (Tissue-Tek; Germany), and sectioned at 10
µm using a cryostat. Images and measurements of tumors forming on
the upper CAM were captured with a Nikon stereo dissecting
microscope SMZ 1000 equipped with a Nikon digital camera and Nikon
EclipseNet software. Exemplarily, images were captured with a
3D-Profilomter VR-3200 microscope (Keyence, Neu-Isenburg, Germany)
to visualize changes in the 3D volume of the tumors.
Immunocytochemistry
The localization of the tumors forming in the upper
CAM after grafting was visualized on hematoxylin and eosin
(H&E)-stained cryosections. The human origin of the tumors
forming in the chicken CAM after grafting human RB cells was
verified using a mouse anti-human nuclear antibody (MAB 128; Merck
Millipore, Darmstadt, Germany) at a dilution of 1:100 in PBS
containing 0.1% Triton, 4% bovine serum albumin (BSA) and 1% normal
goat serum (NGS) overnight at 4°C. The reaction was visualized
using a goat anti-mouse antibody labelled with Alexa
Flour® 488 (A11001; Molecular Probes, Camarillo, CA,
USA), diluted 1:1,000 in PBS with 1% BSA for 2 h at room
temperature.
For β-tubulin stainings, 1×105 cells were
stained on coverslips as previously described (17). In brief, cells were fixed with 4%
PFA in PBS for 1 h following 3 washes with PBS. Afterwards, cells
were incubated with ice-cold 100% methanol for 5 min on ice, washed
3 times with PBS and incubated for 1 h in blocking solution [PBS,
0.3% Triton, 4% BSA, 5% NGS (Dako, Hamburg, Germany)]. Anti
β-tubulin primary antibody (T-4026; Sigma-Aldrich, Munich, Germany)
was diluted 1:200 in PBS containing 0.1% Triton, 4% BSA and 1% NGS
and cells were incubated overnight at 4°C. Following 3 washes with
PBS, goat anti-mouse secondary Alexa 488-coupled antibody
(Molecular Probes, Germany) was used at 1:1,000 dilutions in PBS/1%
BSA (Roth). Finally, cells were counterstained with DAPI to
visualize the nucleus. As controls, in all cases PBS was
substituted for the primary antisera in order to test for
non-specific labeling. No specific cellular staining was observed
when the primary antiserum was omitted.
Measurements of cell and nuclear
size
For measurements of cell and nuclear sizes, images
from β-tubulin-stained cells on coverslips were acquired using a
Nikon Eclipse E600 microscope equipped with a digital camera. For
each cell line, 5 equally distributed, x-shape rendered visual
fields/coverslip were captured, and the cytoplasmic and nuclear
outline of 7 cells/visual field were measured using the Nikon
Eclipse net measurement software.
Statistical analysis
All assays were performed at least in triplicate.
Statistical analyses were conducted using GraphPad Prism 6. Data
represent means ± SEM of 2 to 5 independent experiments from
independent RB cell cultures. Results were analyzed by a Students
t-test or one-way ANOVA and Newman-Keuls post hoc test and
considered significantly different at *P<0.05, **P<0.01 or
***P<0.001. Statistics on the growth curves was performed using
a free web interface http://bioinf.wehi.edu.au/software/compareCurves/,
which uses the compareGrowthCurves-function from a statistical
modeling package called ‘statmod’, available from the R Project for
Statistical Computing: http://wwww.r-project.org, previously described
elsewhere (18).
Results
Etoposide and cisplatin resistance
changes RB cell line morphology
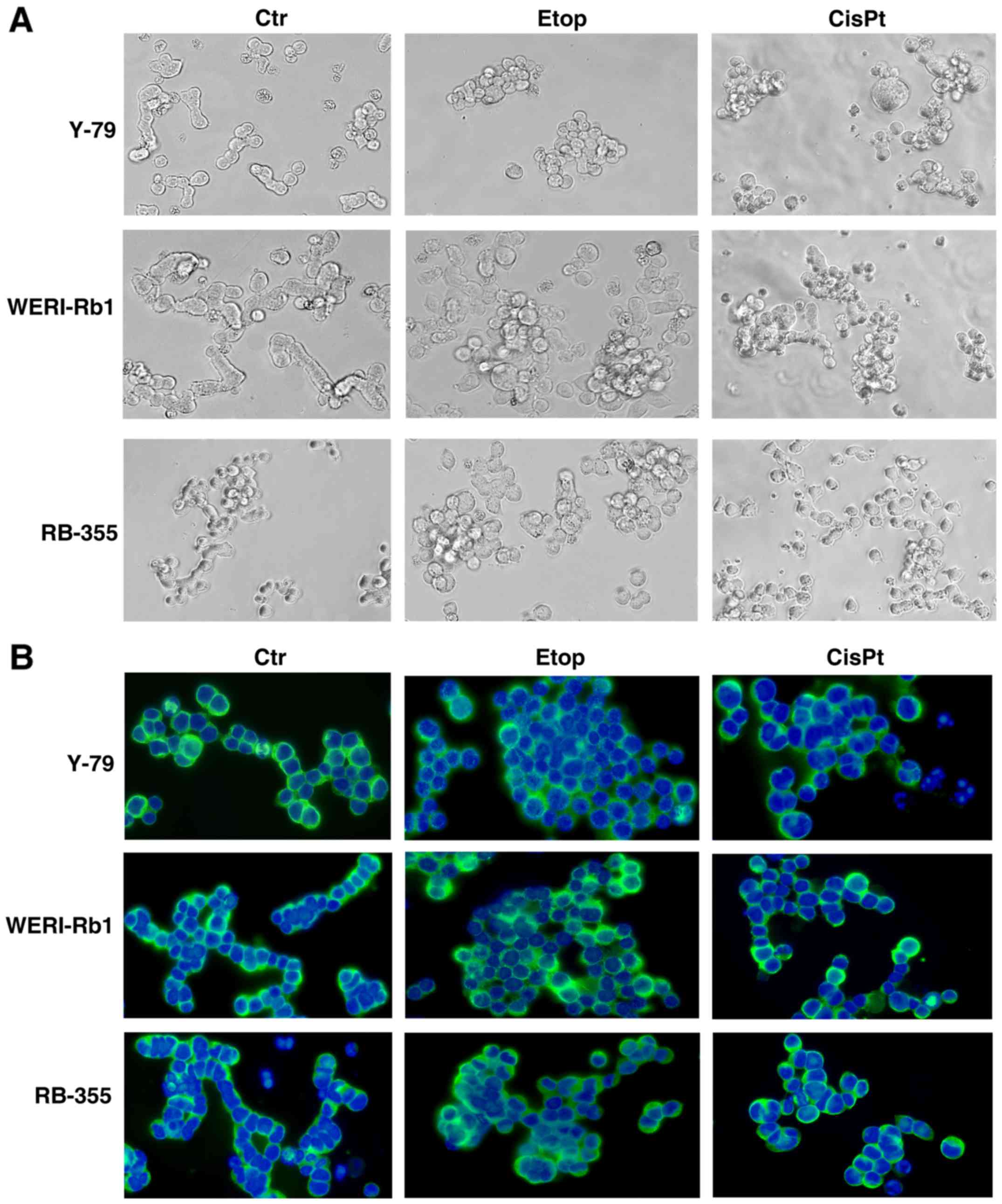
In order to check for morphological changes
following etoposide and cisplatin resistance, we compared the
appearance of parental and chemoresistant Y-79, WERI-Rb1 and RB-355
cells a) in cell culture (Fig. 1A)
and b) after seeding on poly-D-lysine-coated coverslips and
immunocytochemical staining with β-tubulin and DAPI counterstaining
(Fig. 1B). While all parental
controls and particularly WERI-Rb1 cells frequently form chain-like
structures when seeded on coverslips, all chemoresistant RB cell
lines tended to form clusters, which were more pronounced in
etoposide-resistant cell lines than in cisplatin-resistant cells.
However, the etoposide- and cisplatin-resistant cell lines seemed
to become partially adherent and sometimes neurite-like processes
could be observed.
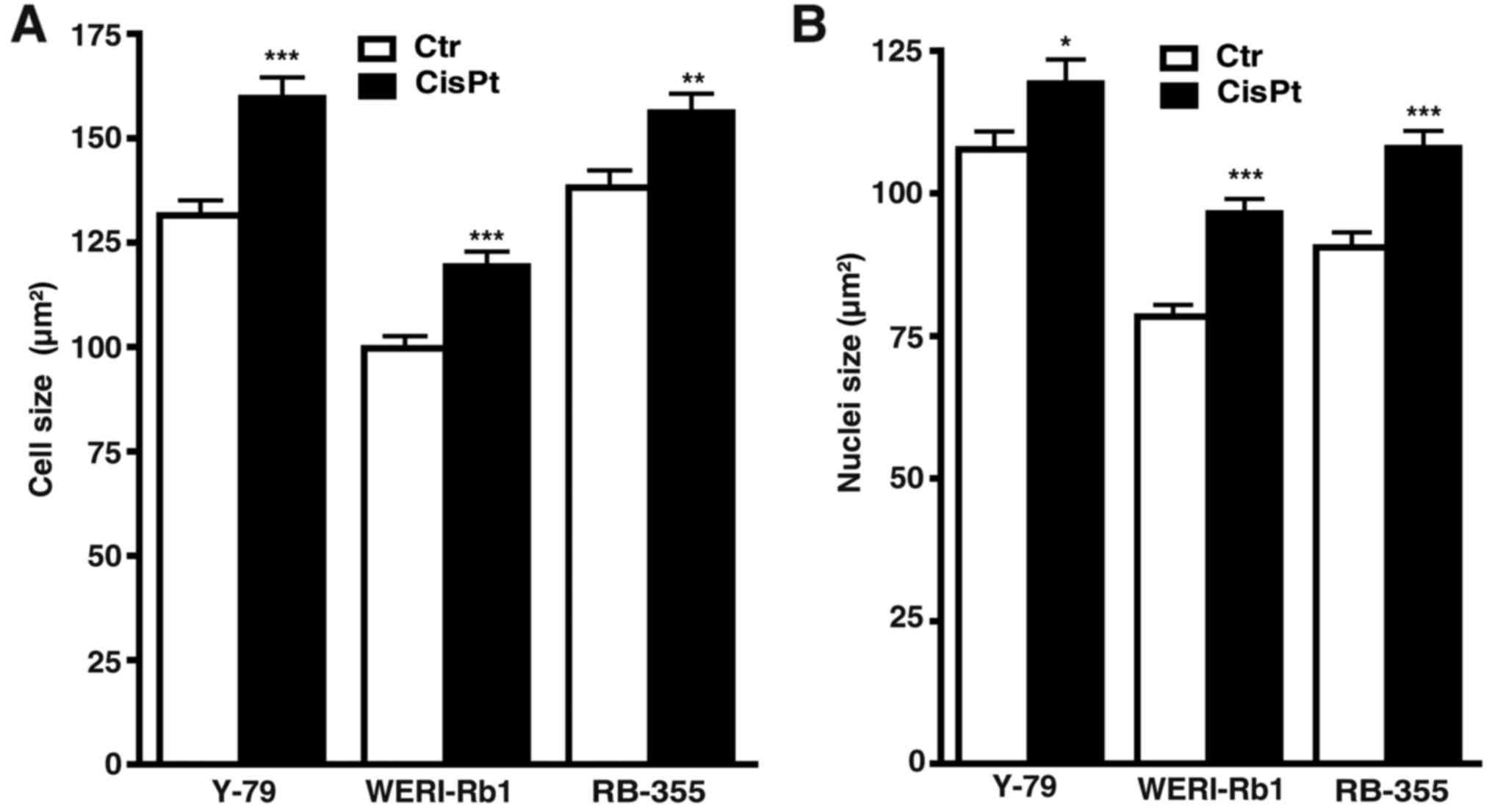
Measurements of cell and nuclear size revealed that
compared to their parental counterparts, cisplatin-resistant cells
were significantly bigger and had larger nuclei (Fig. 2A and B), whereas etoposide-resistant
cells did no exhibit significant changes in either size (data not
shown).
Etoposide resistance significantly
increases the growth and proliferation of RB cell lines
The etoposide-resistant cell lines Y-79 and
WERI-Rb1, established from two well-established RB cell lines,
originally derived from unilateral RB tumors (10), exhibited significantly higher growth
rates compared to the parental etoposide-sensitive control cells
(Fig. 3A and B). In semi-adherent
RB-355 RB cells, likewise derived from an unilateral RB tumor, but
exhibiting different growth kinetics (19), cell growth was not significantly
affected by etoposide resistance (Fig.
3C).
As revealed by WST-1 assays, etoposide resistance
resulted in a significant increase in Y-79 cell viability. WERI-Rb1
etoposide-resistant cells displayed a slightly, but not
significantly increased viability, whereas etoposide resistance did
not significantly alter the viability of RB-355 cells compared to
the parental controls (Fig.
3D).
Cell proliferation was significantly increased in
all three etoposide-resistant RB cell lines analyzed as reflected
by significantly higher numbers of BrdU-positive cells (Fig. 3E).
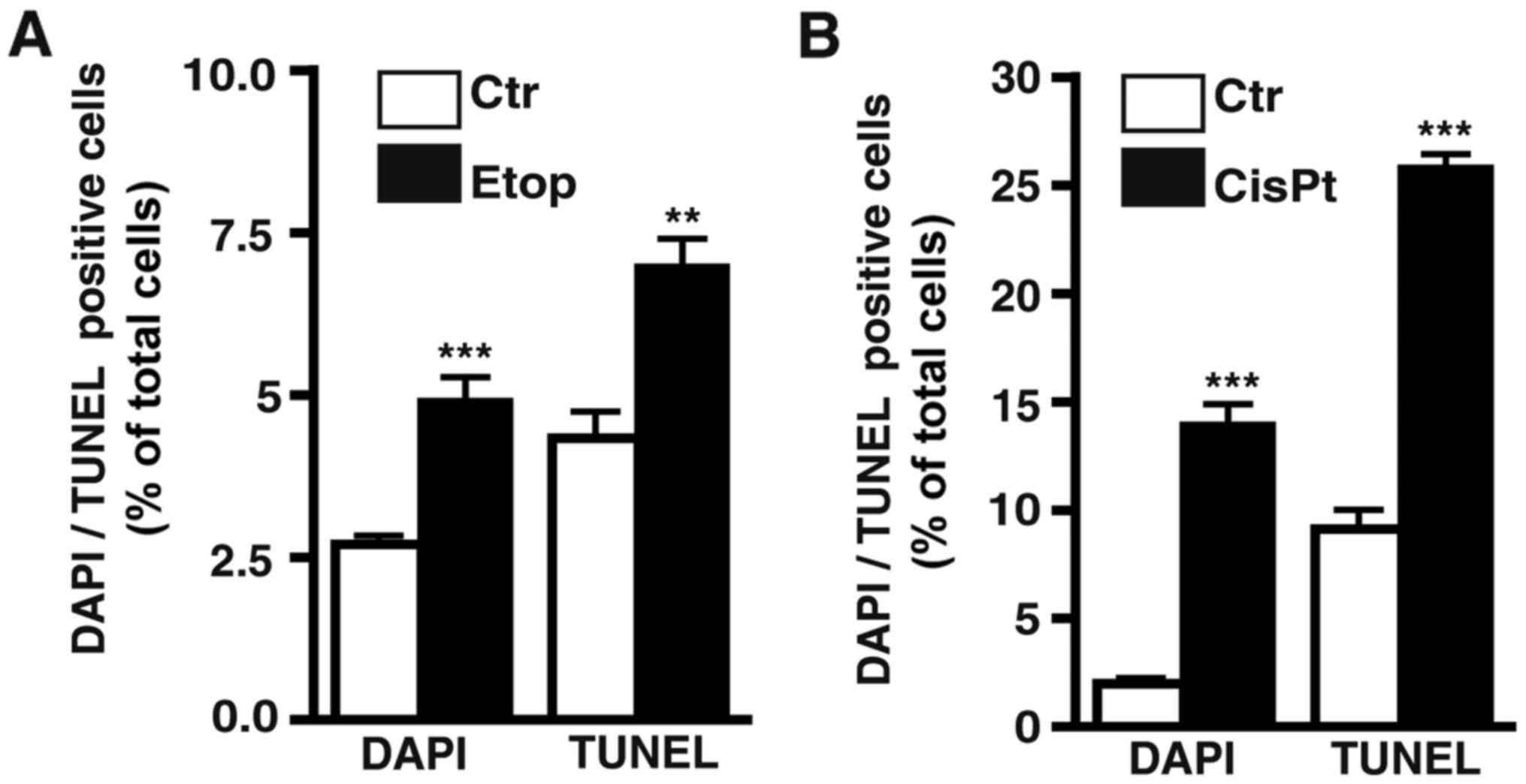
Etoposide treatment still induces apoptosis in
etoposide-resistant RB cell lines. As revealed by DAPI cell counts
(Fig. 3F) and confirmed by TUNEL
assays (Fig. 4A) continuous
treatment with etoposide still significantly induced apoptosis in
the WERI-RB1 etoposide-resistant RB cells, whereby the increase in
cell death levels in the Y-79 and RB-355 etoposide-resistant cell
lines was not significant. The remaining pro-apoptotic effect of
etoposide seemed to counterbalance its pro-proliferative effect
after induction of resistance (Fig.
3E) resulting in an absent overall effect of etoposide on cell
viability in the WERI-Rb1 and RB-355 etoposide-resistant cells
(Fig. 3D). By contrast, in Y-79
etoposide-resistant cells, displaying significantly increased cell
viabilities (Fig. 3D), the strong
pro-proliferative effect of etoposide-resistance appeared to
predominate (Fig. 3E).
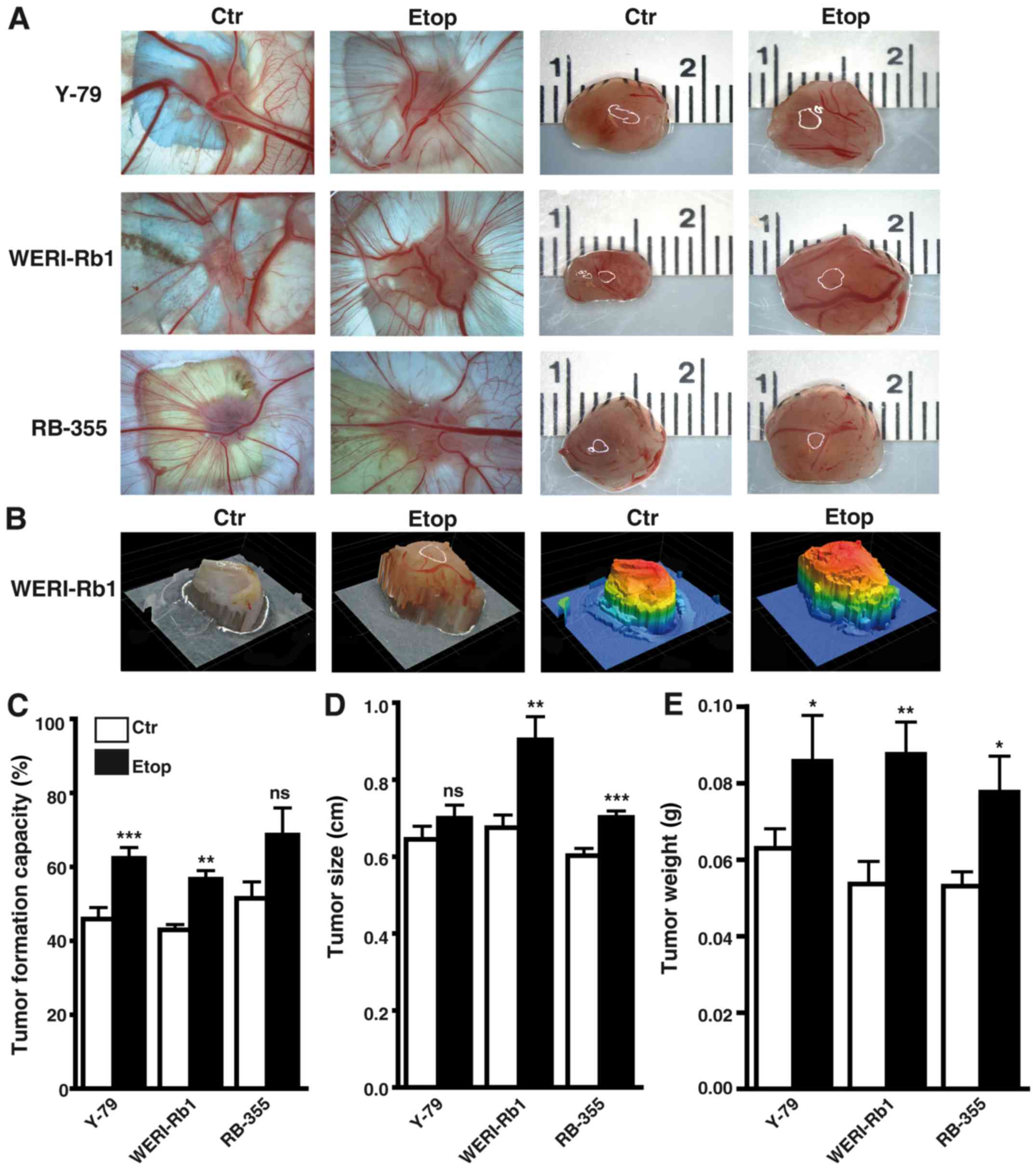
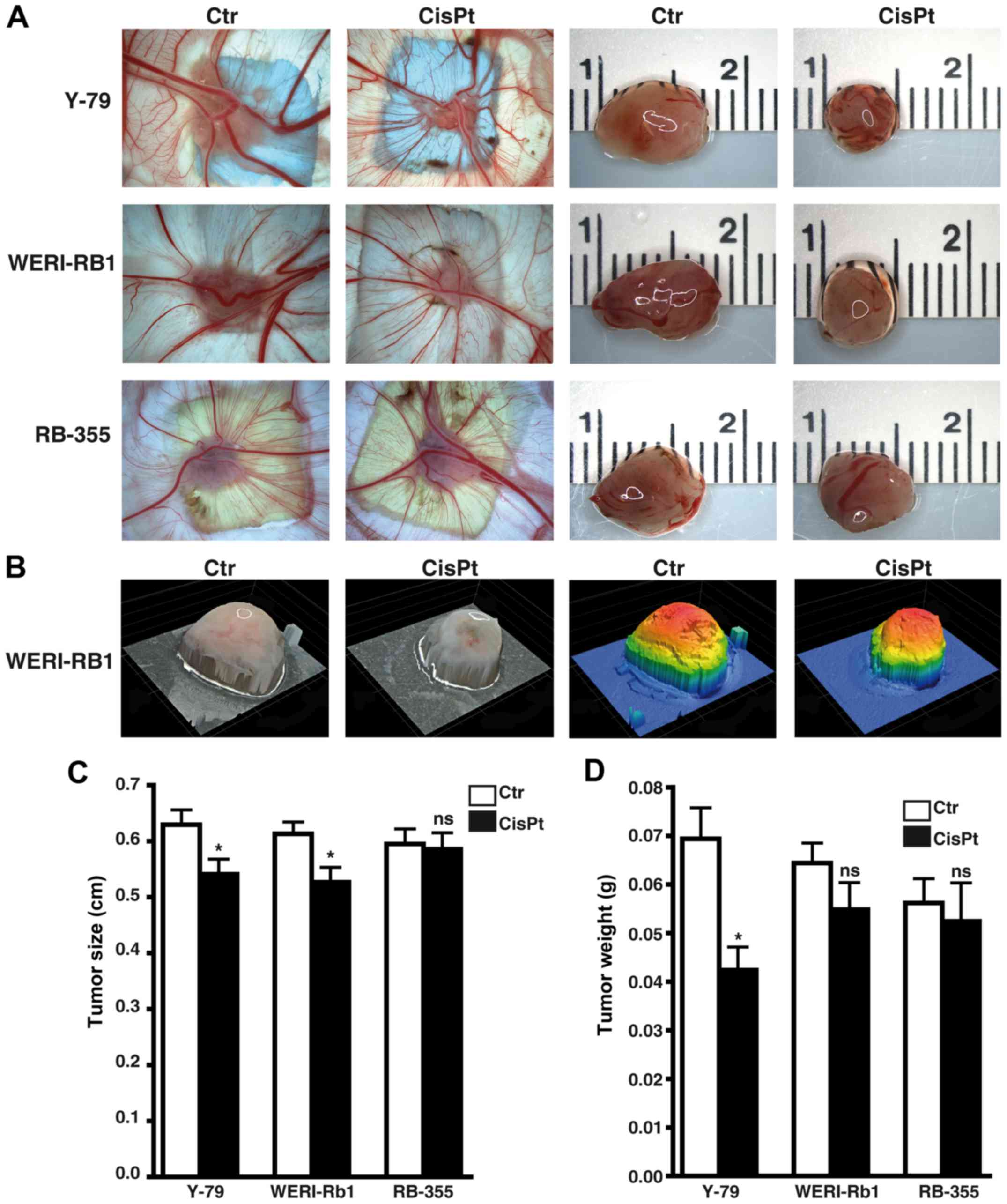
Etoposide resistance significantly
increases the incidence, weight, and size of tumors developing from
RB cell lines
Photo-documentation (Fig. 5A and B), counts (Fig. 5C) and measurements of the tumors
(Fig. 5D and E) developing from
Y-79, WERI-Rb1 and RBL-355 cells grafted on the upper CAM revealed
that the tumor formation capacity of Y-79 and WERI-Rb1
etoposide-resistant cell lines was significantly increased compared
to their parental counterparts (Fig.
5C). Tumor formation capacity of semi-adherent RB-355
etoposide-resistant cells likewise increased, but did not reach
significance. However, the etoposide-resistant RB cell lines
developed larger tumors (Fig. 5D)
and tumors of significantly higher weight (Fig. 5E) when compared with the
chemosensitive control cells.
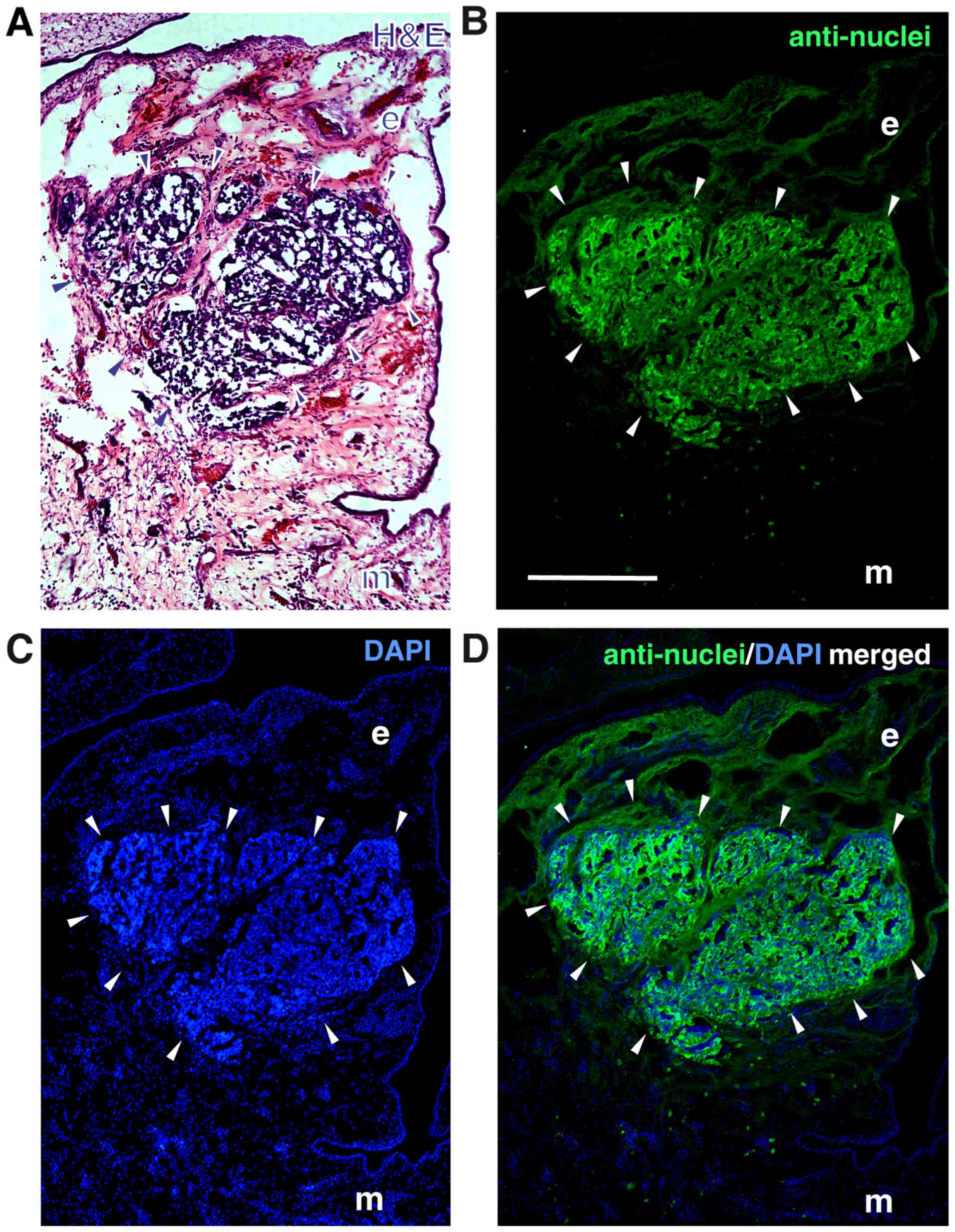
The localization of the tumors developing in the
upper chicken CAM at the border between CAM ectoderm and mesoderm 7
days after grafting human RB cells was visualized by H&E
staining of tumor cryosections (Fig.
6A). The human nature of the tumors was verified
immunocytochemically, using an anti-human nuclear antibody
(Fig. 6B-D).
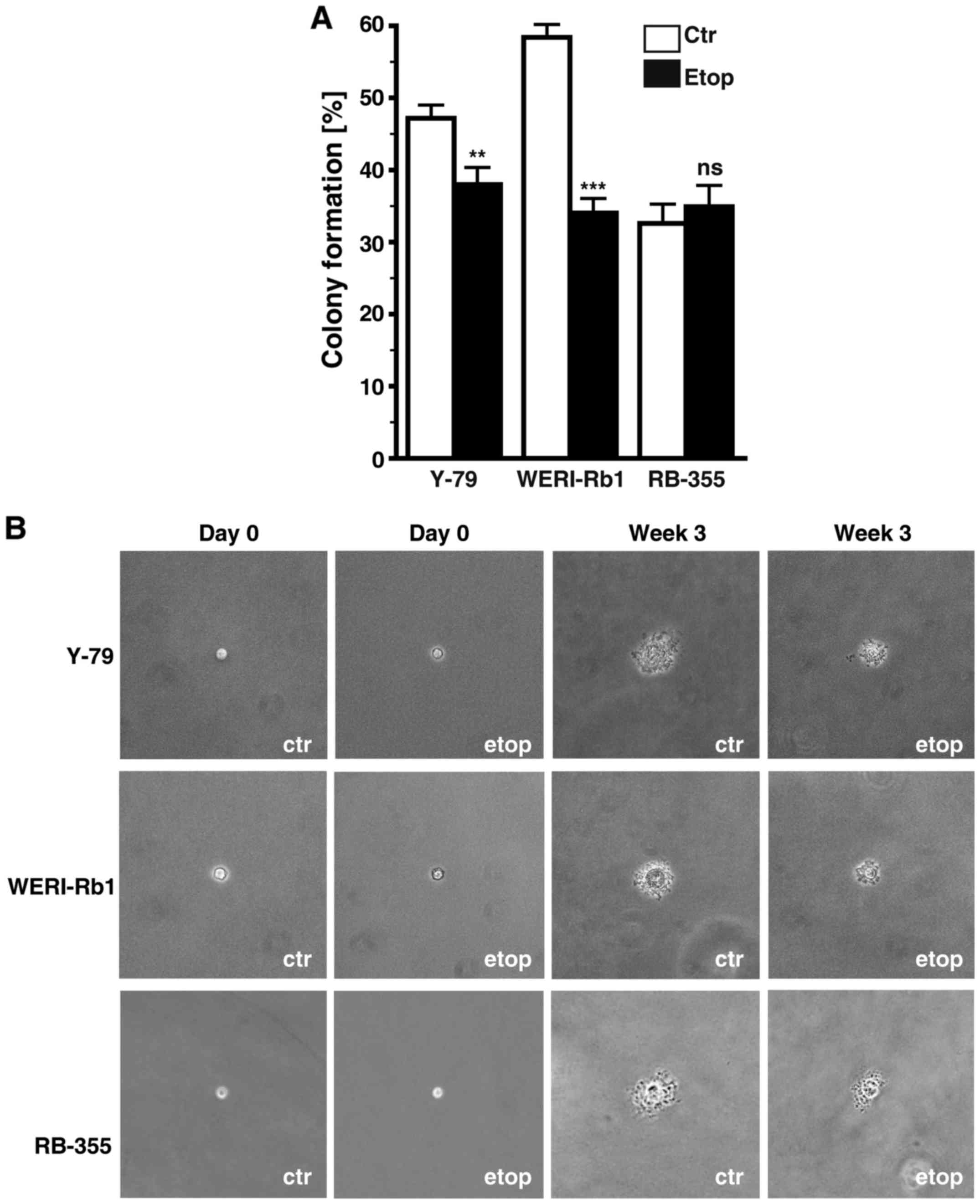
Etoposide resistance significantly
affects anchorage independent growth of RB cells
Etoposide resistance changed the anchorage
independent growth of Y-79 and WERI-Rb1 cells as reflected by a
significant decrease in their colony formation capacity in soft
agarose (Fig. 7A). However, all
three etoposide-resistant cell lines analyzed formed considerably
smaller colonies when compared with the chemosensitive control
cells (Fig. 7B).
Cisplatin-resistant RB cell lines
display decreased growth and proliferation rates
All cisplatin-resistant cell lines analyzed
displayed significantly lower growth kinetics compared to the
control cells (Fig. 8A-C). Among
the three cisplatin-resistant RB cell lines investigated, Y-79
cells were the slowest in growth and thus, we had to record
long-term growth curves over a period of 336 h (instead of 96 h)
and plot the values logarithmically to visualize differences
between resistant and control cells (Fig. 8A). As revealed by WST-1 assays
cisplatin resistance likewise resulted in a significant reduction
in cell viability of all three cisplatin-resistant cell lines
(Fig. 8D). Cell proliferation was
significantly decreased in the Y-79 and WERI-Rb1
cisplatin-resistant RB cell lines as reflected by significantly
lower numbers of BrdU-positive cells (Fig. 8E), but remained unchanged in the
RB-355 cisplatin-resistant cells (Fig.
8E).
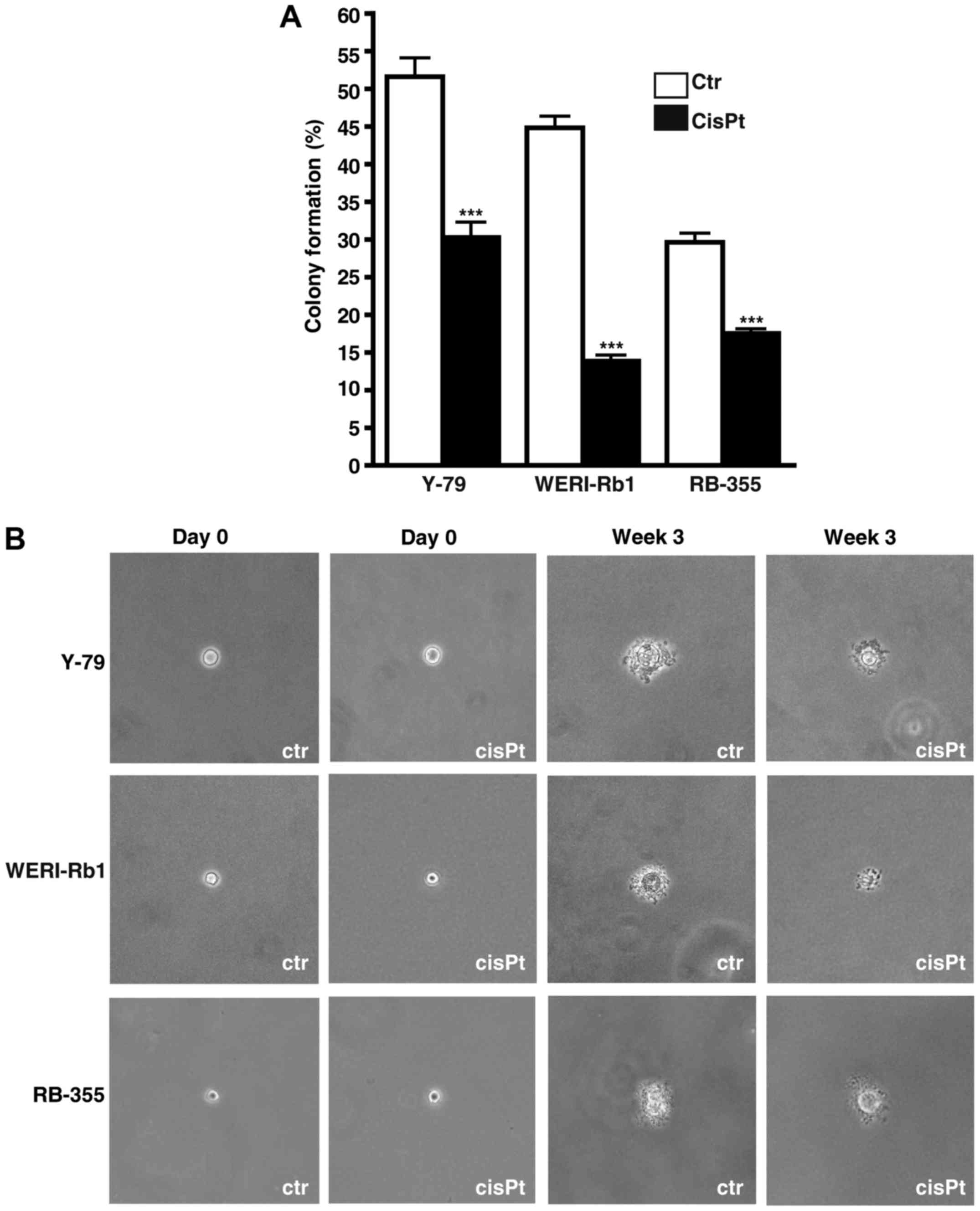
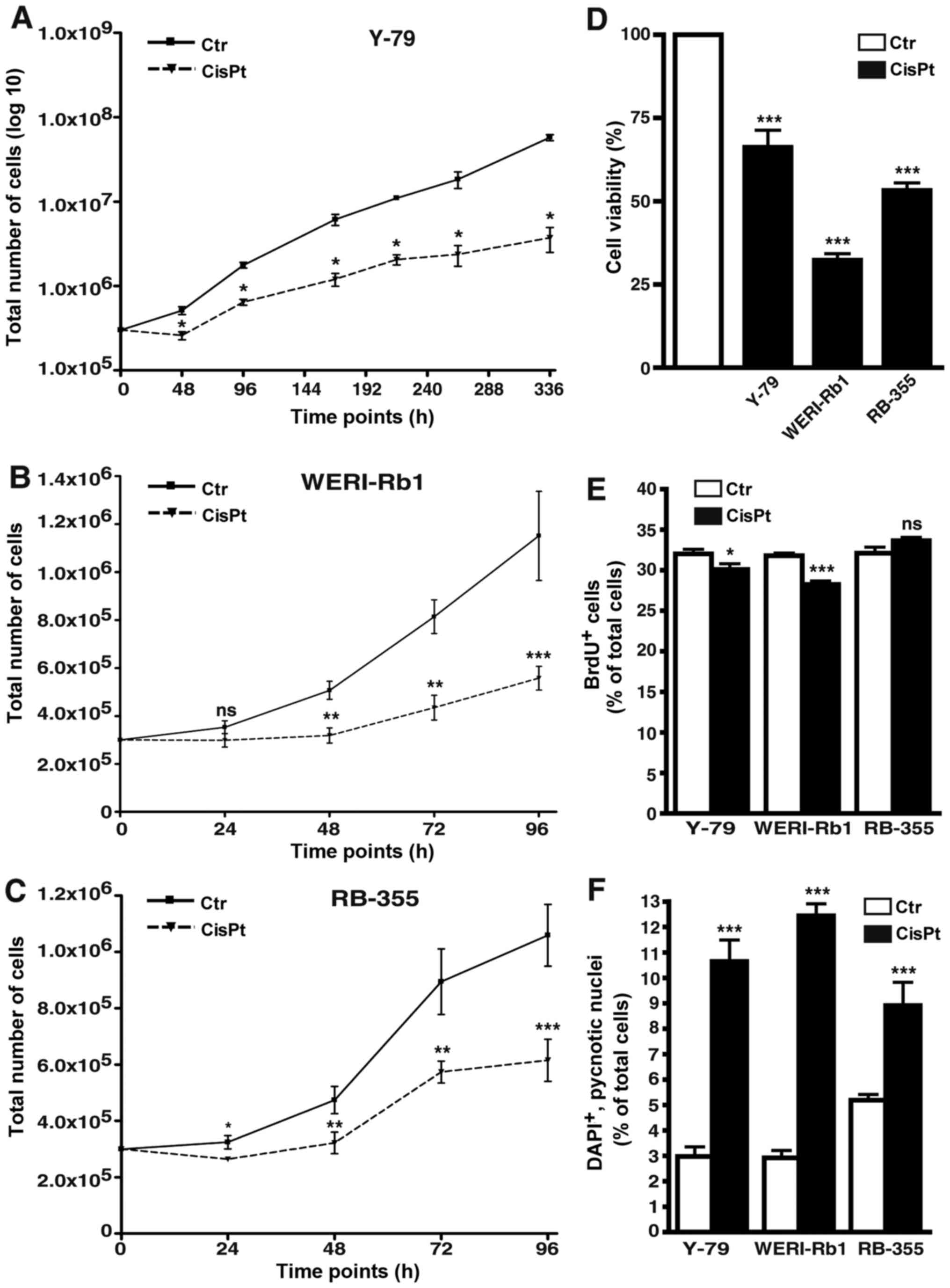 | Figure 8.Effects of cisplatin resistance on RB
cell growth, viability, proliferation and apoptosis. (A-C) Growth
curves, (D) WST-1 assays, (E) BrdU counts and (F) DAPI cell counts
of cisplatin-resistant (CisPt) Y-79 (A), WERI-Rb1 (B) and RBL-355
cells (C) vs. control cells (Ctr) revealed that cisplatin
resistance led to a significant decrease in growth kinetics (A-C),
cell viability (D) and proliferation (E). Cisplatin treatment,
however, still significantly increased apoptosis levels in all
cisplatin-resistant cell lines (F). Values are means of at least 3
independent experiments ± SEM; *P<0.05, **P<0.01,
***P<0.001 compared to the control group calculated by Student's
t-test. |
Cisplatin treatment increases
apoptosis in resistant RB cell lines
As revealed by DAPI cell counts (Fig. 8F) and confirmed by TUNEL assays
(Fig. 4B) cisplatin treatment still
significantly increased the apoptosis levels in the
cisplatin-resistant RB cell lines.
Cisplatin significantly influences
weight and size of tumors developing from RB cell lines
Photo-documentation (Fig. 9A and B) and measurements revealed
that tumors developing from Y-79 and WERI-Rb1 cisplatin-resistant
RB cells were significantly smaller (Fig. 9C). Y-79 cisplatin-resistant cells
likewise exhibited significantly lower weights than tumors
developing from chemosensitive control cells (Fig. 9D), whereby the same tendency did not
reach significance in the WERI-Rb1 cisplatin-resistant cells.
Compared to the respective parental controls, tumors forming from
RB-355 cisplatin-resistant cells neither decreased nor increased in
size and weight (Fig. 9C and D).
Cisplatin resistance did not significantly influence the tumor
formation capacity of the three cell lines investigated (data not
shown).
Cisplatin resistance significantly
effects anchorage independent growth of RB cells
Soft agarose assays revealed that cisplatin
resistance significant decreased the colony formation capacity of
all cisplatin-resistant RB cell lines analyzed (Fig. 10A). However, all
cisplatin-resistant cell lines formed considerably smaller colonies
compared to the control cells (Fig.
10B).
Discussion
Drug resistance and relapse are the major issues
associated with chemotherapy, which is regarded as the mainstay of
globe preserving treatment in retinoblastoma (RB). In the present
study presented, we provide a morphological and functional
characterization of three etoposide- and three cisplatin-resistant
RB cell lines.
Regarding morphological changes, we observed a
significant increase in cell and nuclear size in the
cisplatin-resistant RB cells, but no significant changes in the
etoposide-resistant cells. In this context, Żuryń et al
(2016) reported that incubation of HL-60 cells with etoposide
resulted in an enlargement of the cells and irregularities in shape
(20).
We showed that compared to the cells of origin,
etoposide-resistant RB cell lines were highly proliferative,
displayed a significantly increased tumor formation capacity and
formed larger tumors. Thus, these cells obviously become more
aggressive than their parental counterparts. Surprisingly,
etoposide treatment still induced apoptosis in the
etoposide-resistant RB cell lines. In line with our finding, it has
been shown that etoposide-resistant melanoma cells likewise display
reduced but still detectable apoptotic activities, but activation
of the mitochondrial pro-apoptotic pathway was no longer detectable
after exposure to etoposide (21).
In contrast to etoposide resistance, we found
cisplatin resistance to significantly lower growth kinetics and to
induce tumors with equal or diminished weights. Thus, compared to
the cells of origin, cisplatin-resistant RB cells do not display
increased tumorigenicity and aggressiveness.
Soft agarose assays of the present study presented
revealed that etoposide- and cisplatin-resistant RB cell lines
exhibited significantly reduced colony formation capacities and
formed considerably smaller colonies compared to their parental
counterparts. As RB cells normally grow as aggregates (14), the disruption of the united cell
structure by single cell seeding in soft agarose seems to be
responsible for the failure of chemoresistant RBs to form colonies.
Conversely, growth in aggregates is potentially favorable for the
development of RB cell chemoresistance. In this context, it has
already been shown that cells grown in contact with each other,
e.g. as tumor spheroids in culture, are more resistant to
alkylating agents and cisplatin than the same cells after
disaggregation (22). Moreover,
cell growth in aggregates determines chemoresistance of follicular
lymphoma cells as these cells display resistance to drugs used in
lymphoma therapy when cultured in 3 dimensions rather than in
suspension (23). Along this line,
a number of studies showed that the chemosensitivity of cancer
cells is affected by the extent of cell adhesion and expression of
intercellular adhesion molecules [reviewed in ref. (24)]. The neuronal L1 cell adhesion
molecule (L1CAM) has attracted attention as it is expressed in a
variety of tumors and high expression was associated with poor
prognosis (25). It has been shown
that L1CAM expression confers increased cell growth and
tumorigenesis of colorectal cancer cells (26). Further along this line, a
combination of anti-L1CAM antibody and cisplatin was found to
improve the therapeutic response in cholangiocarcinoma by enhancing
tumor growth inhibition compared to treatment with the drug alone
(27). Recently, Jo et al
showed that L1CAM increased the adhesion-mediated proliferation and
resistance of Y-79 and SNUOT-Rb1 RB cells to carboplatin,
vincristine and etoposide (28).
Among other mechanisms, ATP-binding cassette (ABC)
transporters were found to contribute to the process of drug
resistance in cancer (29,30) as overexpression of these drug
transporters in tumor cells reduces the intracellular drug level by
increasing its efflux (31).
Kachalaki et al found that ABCB1 [multi-drug resistance
(MDR) P-glycoprotein/MDR1] may play a role in acute myeloid
leukemia cell resistance to etoposide (32). However, the overexpression of ABCB1
reduced the sensitivities of ovarian cancer lines to cisplatin
(33). In RB the expression of
P-glycoprotein has likewise been linked to chemotherapy resistance
(34–36). Recently, it has been shown that
silencing of the ATP-binding cassette subfamily G member 2 (ABCG2)
inhibits multidrug resistance of WERI-Rb1 RB cancer stem cells,
including their resistance to etoposide and cisplatin treatment
(37).
Hypoxic tumor microenvironment is another factor
that determines the therapeutic response in many tumors. It has
been shown that hypoxia attenuates etoposide mediated G2/M arrest
and apoptosis induction and thus promotes etoposide resistance in
neuroblastoma cells (38). Sudhakar
et al found no expression of hypoxia-inducible factor-1α
(HIF-1α) in the normal adult retina, but observed positive HIF-1α
immunoreactivity in 83% RB tumors analyzed (39). Along this line, brain-derived
neurotrophic factor (BDNF) protects RB cells from
chemotherapy-induced apoptosis and thereby contribute to their
chemoresistance via induction of HIF-1α expression (40).
It has been reported that clusterin, a
cytoprotective chaperone protein known to protect various retinal
cells, is overexpressed in several types of malignant tumors. Song
et al found clusterin to be expressed in human RB and to
exert anti-apoptotic effects on cisplatin-induced apoptosis and
prevent cell death. Therefore, the authors hypothesized that
clusterin can contribute to cisplatin resistance of RB (41).
Finally, microRNAs (miRNAs) have been identified to
directly or indirectly influence the development of cancer drug
resistance [for review see (42,43)].
In this regard, Jia et al recently showed that after
overexpression of miR-3163 etoposide and cisplatin resistance of RB
cancer stem cells significantly decline (37).
After a first, more descriptive functional
characterization in the present study, ongoing experiments may
address the question of which mechanisms underlie the development
of etoposide and cisplatin resistance in RB cell lines and how
these can be circumvented. Currently, one can state that
etoposide-resistant RB cells display therapeutically undesirable
features such as fast growth, high proliferation rate and a
significantly increased tumor formation capacity compared to the
tumor cells of origin. Thereby, etoposide resistance seems to
aggravate the course of the disease and potentially worsens patient
prognosis. By contrast, compared to their parental counterparts,
cisplatin-resistant RB cells continue to exhibit higher apoptosis
rates, decreased growth kinetics and equal or diminished tumor
weights. It takes an extended treatment period to induce cisplatin
resistance and cisplatin-resistant RB cells do not display more
aggressive features than the cells of origin. Thus, finally
cisplatin resistance has less severe consequences for RB
patients.
References
|
1
|
Yousef YA, Soliman SE, Astudillo PPP,
Durairaj P, Dimaras H, Chan HSL, Héon E, Gallie BL and Shaikh F:
Intra-arterial chemotherapy for retinoblastoma: a systematic
review. JAMA Ophthalmol. Mar 17–2016.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shields CL, Lally SE, Leahey AM, Jabbour
PM, Caywood EH, Schwendeman R and Shields JA: Targeted
retinoblastoma management: When to use intravenous, intra-arterial,
periocular, and intravitreal chemotherapy. Curr Opin Ophthalmol.
25:374–385. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abramson DH, Shields CL, Munier FL and
Chantada GL: Treatment of retinoblastoma in 2015: Agreement and
disagreement. JAMA Ophthalmol. 133:1341–1347. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Beck WT, Mo YY and Bhat UG: Cytotoxic
signalling by inhibitors of DNA topoisomerase II. Biochem Soc
Trans. 29:702–703. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Clifford B, Beljin M, Stark GR and Taylor
WR: G2 arrest in response to topoisomerase II inhibitors: The role
of p53. Cancer Res. 63:4074–4081. 2003.PubMed/NCBI
|
|
6
|
Yuan L, Yu WM and Qu CK: DNA
damage-induced G2/M checkpoint in SV40 large T antigen-immortalized
embryonic fibroblast cells requires SHP-2 tyrosine phosphatase. J
Biol Chem. 278:42812–42820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Villegas VM, Hess DJ, Wildner A, Gold AS
and Murray TG: Retinoblastoma. Curr Opin Ophthalmol. 24:581–588.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Griegel S, Hong C, Frötschl R, Hülser DF,
Greger V, Horsthemke B and Rajewsky MF: Newly established human
retinoblastoma cell lines exhibit an ‘immortalized’ but not an
invasive phenotype in vitro. Int J Cancer. 46:125–132. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reid TW, Albert DM, Rabson AS, Russell P,
Craft J, Chu EW, Tralka TS and Wilcox JL: Characteristics of an
established cell line of retinoblastoma. J Natl Cancer Inst.
53:347–360. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McFall RC, Sery TW and Makadon M:
Characterization of a new continuous cell line derived from a human
retinoblastoma. Cancer Res. 37:1003–1010. 1977.PubMed/NCBI
|
|
11
|
Stephan H, Boeloeni R, Eggert A, Bornfeld
N and Schueler A: Photodynamic therapy in retinoblastoma: Effects
of verteporfin on retinoblastoma cell lines. Invest Ophthalmol Vis
Sci. 49:3158–3163. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Haubold M, Weise A, Stephan H and Dünker
N: Bone morphogenetic protein 4 (BMP4) signaling in retinoblastoma
cells. Int J Biol Sci. 6:700–715. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Philippeit C, Busch M and Dünker N:
Epigenetic control of trefoil factor family (TFF) peptide
expression in human retinoblastoma cell lines. Cell Physiol
Biochem. 34:1001–1014. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Busch M, Philippeit C, Weise A and Dünker
N: Re-characterization of established human retinoblastoma cell
lines. Histochem Cell Biol. 143:325–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zijlstra A, Mellor R, Panzarella G, Aimes
RT, Hooper JD, Marchenko ND and Quigley JP: A quantitative analysis
of rate-limiting steps in the metastatic cascade using
human-specific real-time polymerase chain reaction. Cancer Res.
62:7083–7092. 2002.PubMed/NCBI
|
|
16
|
Palmer TD, Lewis J and Zijlstra A:
Quantitative analysis of cancer metastasis using an avian embryo
model. J Vis Exp. 51:28152011.
|
|
17
|
Weise A and Dünker N: High trefoil factor
1 (TFF1) expression in human retinoblastoma cells correlates with
low growth kinetics, increased cyclin-dependent kinase (CDK)
inhibitor levels and a selective down-regulation of CDK6. Histochem
Cell Biol. 139:323–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Elso CM, Roberts LJ, Smyth GK, Thomson RJ,
Baldwin TM, Foote SJ and Handman E: Leishmaniasis host response
loci (lmr1-3) modify disease severity through a Th1/Th2-independent
pathway. Genes Immun. 5:93–100. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Busch M and Dünker N: Trefoil factor
family peptides - friends or foes? Biomol Concepts. 6:343–359.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Żuryń A, Krajewski A, Szulc D, Litwiniec A
and Grzanka A: Activity of cyclin B1 in HL-60 cells treated with
etoposide. Acta Histochem. 118:537–543. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Helmbach H, Kern MA, Rossmann E, Renz K,
Kissel C, Gschwendt B and Schadendorf D: Drug resistance towards
etoposide and cisplatin in human melanoma cells is associated with
drug-dependent apoptosis deficiency. J Invest Dermatol.
118:923–932. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kerbel RS, St Croix B, Florenes VA and Rak
J: Induction and reversal of cell adhesion-dependent multicellular
drug resistance in solid breast tumors. Hum Cell. 9:257–264.
1996.PubMed/NCBI
|
|
23
|
Gravelle P, Jean C, Familiades J, Decaup
E, Blanc A, Bezombes-Cagnac C, Laurent C, Savina A, Fournié JJ and
Laurent G: Cell growth in aggregates determines gene expression,
proliferation, survival, chemoresistance, and sensitivity to immune
effectors in follicular lymphoma. Am J Pathol. 184:282–295. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
St Croix B and Kerbel RS: Cell adhesion
and drug resistance in cancer. Curr Opin Oncol. 9:549–556. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fogel M, Gutwein P, Mechtersheimer S,
Riedle S, Stoeck A, Smirnov A, Edler L, Ben-Arie A, Huszar M and
Altevogt P: L1 expression as a predictor of progression and
survival in patients with uterine and ovarian carcinomas. Lancet.
362:869–875. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gavert N, Conacci-Sorrell M, Gast D,
Schneider A, Altevogt P, Brabletz T and Ben-Ze'ev A: L1, a novel
target of beta-catenin signaling, transforms cells and is expressed
at the invasive front of colon cancers. J Cell Biol. 168:633–642.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cho S, Lee TS, Song IH, Kim AR, Lee YJ,
Kim H, Hwang H, Jeong MS, Kang SG and Hong HJ: Combination of
anti-L1 cell adhesion molecule antibody and gemcitabine or
cisplatin improves the therapeutic response of intrahepatic
cholangiocarcinoma. PLoS One. 12:e01700782017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jo DH, Lee K and Kim JH, Jun HO, Kim Y,
Cho YL, Yu YS, Min JK and Kim JH: L1 increases adhesion-mediated
proliferation and chemoresistance of retinoblastoma. Oncotarget.
8:15441–15452. 2017.PubMed/NCBI
|
|
29
|
Szakács G, Paterson JK, Ludwig JA,
Booth-Genthe C and Gottesman MM: Targeting multidrug resistance in
cancer. Nat Rev Drug Discov. 5:219–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Longley DB and Johnston PG: Molecular
mechanisms of drug resistance. J Pathol. 205:275–292. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu CP, Calcagno AM and Ambudkar SV:
Reversal of ABC drug transporter-mediated multidrug resistance in
cancer cells: Evaluation of current strategies. Curr Mol Pharmacol.
1:93–105. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kachalaki S, Baradaran B, Majidi J,
Yousefi M, Shanehbandi D, Mohammadinejad S and Mansoori B: Reversal
of chemoresistance with small interference RNA (siRNA) in etoposide
resistant acute myeloid leukemia cells (HL-60). Biomed
Pharmacother. 75:100–104. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu DD, Li XS, Meng XN, Yan J and Zong ZH:
MicroRNA-873 mediates multidrug resistance in ovarian cancer cells
by targeting ABCB1. Tumour Biol. 37:10499–10506. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chan HS, Thorner PS, Haddad G and Gallie
BL: Multidrug-resistant phenotype in retinoblastoma correlates with
P-glycoprotein expression. Ophthalmology. 98:1425–1431. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chan HS, Lu Y, Grogan TM, Haddad G,
Hipfner DR, Cole SP, Deeley RG, Ling V and Gallie BL: Multidrug
resistance protein (MRP) expression in retinoblastoma correlates
with the rare failure of chemotherapy despite cyclosporine for
reversal of P-glycoprotein. Cancer Res. 57:2325–2330.
1997.PubMed/NCBI
|
|
36
|
Filho JPS, Correa ZM, Odashiro AN,
Coutinho AB, Martins MC, Erwenne CM and Burnier MN Jr:
Histopathological features and P-glycoprotein expression in
retinoblastoma. Invest Ophthalmol Vis Sci. 46:3478–3483. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jia M, Wei Z, Liu P and Zhao X: Silencing
of ABCG2 by microRNA-3163 inhibits multidrug resistance in
retinoblastoma cancer stem cells. J Korean Med Sci. 31:836–842.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang D, Zhu Q, Zhang X, Zhang L, He Q and
Yang B: Hypoxia promotes etoposide (VP-16) resistance in
neuroblastoma CHP126 cells. Pharmazie. 65:51–56. 2010.PubMed/NCBI
|
|
39
|
Sudhakar J, Venkatesan N, Lakshmanan S,
Khetan V, Krishnakumar S and Biswas J: Hypoxic tumor
microenvironment in advanced retinoblastoma. Pediatr Blood Cancer.
60:1598–1601. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gao Y, Jing M, Ge R and Lang L: Induction
of hypoxia-inducible factor-1α by BDNF protects retinoblastoma
cells against chemotherapy-induced apoptosis. Mol Cell Biochem.
414:77–84. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Song HB, Jun HO and Kim JH, Yu YS, Kim KW,
Min BH and Kim JH: Anti-apoptotic effect of clusterin on
cisplatin-induced cell death of retinoblastoma cells. Oncol Rep.
30:2713–2718. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Magee P, Shi L and Garofalo M: Role of
microRNAs in chemoresistance. Ann Transl Med. 3:3322015.PubMed/NCBI
|
|
43
|
Ayers D and Vandesompele J: Influence of
microRNAs and long non-coding RNAs in cancer chemoresistance.
Genes. 8:pii: E952017. View Article : Google Scholar
|