Introduction
Hepatocellular carcinoma (HCC) ranks as the third
leading cause of cancer mortality worldwide and the 5-year survival
rate of HCC patients is less than 17% (1). HCC afflicts nearly 466,100 individuals
and causes approximately 422,100 deaths every year in China. Due to
the lack of early symptoms and reliable diagnostic markers for
early detection, 60% of patients with HCC are diagnosed at a
locally advanced or metastatic stage (1,2). The
exact molecular mechanisms which are responsible for this dismal
clinical course remain largely unknown. Consequently, it is
essential to further elucidate the molecular mechanisms determining
HCC metastasis and recurrence.
Chemokines have been shown to play important roles
in many aspects of tumor cell biology, including regulation of
tumor cell growth, metastasis and host immune response (3). Chemokine (C-C motif) ligand 2 (CCL2),
also known as monocyte chemoattractant protein-1 (MCP-1), is a
major chemokine that recruits monocytes and macrophages to the
sites of inflammation. Recent studies have revealed that a high
level of tumor CCL2 expression is associated with unfavorable
patient prognosis in various types of cancer (4–6). CCL2
preferentially binds to the C-C chemokine receptor type 2 (CCR2),
which is expressed in various tissues including thymus, lung,
liver, kidney, pancreas and ovary (7). CCL2 has been reported to play an
important role in epithelial-mesenchymal transition (EMT) and
metastasis (8). Targeting
tumor-infiltrating macrophages via the CCL2/CCR2 axis was examined
as a therapeutic strategy against HCC (9). Shih et al (10) demonstrated that binding of CCL2 to
the corresponding CCR2 receptor induced the expression of
microRNA-21 and subsequently led to the activation of Rac1 and MMP9
and promoted the migration, invasion and EMT of HCC cells. However,
the specific mechanisms and the downstream targets mediated in the
CCL2/CCR2-induced invasion of HCC cells remain unclear and need
further investigation.
Several studies have demonstrated that sonic
Hedgehog (Hh) is overexpressed and plays important roles in many
types of cancer, including pancreatic cancer (11,12),
breast cancer (13) and HCC
(14). The Hh signaling pathway,
initiated through the binding of secreted Hh ligands to a
12-pass-transmembrane receptor called Patched 1 (Ptch1), results in
smoothened (SMO) dissociation. The activated SMO then initiates an
intracellular signaling cascade that eventually leads to the
activation of the Gli-1 transcription factor and then to the
upregulation of the downstream target genes (15). The expression of SMO and Gli-1 is
presumed to be a marker of the Hh pathway activation (16). Previous studies have demonstrated
that significant hyperactivation of the Hh signaling has been
observed in liver injury and cirrhosis which often lead to the
development of HCC lesions (17).
The activation of the Hh pathway in HCC could induce EMT and thus
promote HCC cell invasion and metastasis by upregulating vimentin
expression and downregulating E-cadherin expression (18).
In the present study, we focused on the role of the
CCL2/CCR2 axis in HCC and the possible mechanisms of the CCL2/CCR2
axis in HCC cell invasion. We found that the activation of CCR2 by
its ligand CCL2 increased the expression of SMO and Gli-1,
resulting in Hh pathway activation, EMT and HCC cell invasion.
Materials and methods
Cell culture and reagents
The human HCC cell line MHCC97H was kindly provided
by the Stem Cell Bank of the Chinese Academy of Sciences. The
MHCC97H cells were cultured in Dulbecco's modified Eagle's medium
(DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal
bovine serum (FBS), 100 U/ml penicillin and 100 µg/ml streptomycin
in a humidified atmosphere of 5% CO2 at 37°C.
Recombinant human CCL2 was purchased from PeproTech (Rocky Hill,
NJ, USA). Cyclopamine was purchased from Selleck Chemicals
(Houston, TX, USA). The MHCC97H cells in log phase growth were
cultured in 6-well plates in media containing only 1% FBS for 24 h
before the drug treatment. Subsequently, the drugs were
administered at indicated concentrations in medium containing 1%
FBS and the plates were incubated for another 48 h before a
Matrigel invasion assay was performed. The antibodies were
purchased from different resources as follows: CCR2-specific rabbit
polyclonal antibody (Proteintech, Chicago, IL, USA), Ptch-specific
rabbit polyclonal antibody (Proteintech), sonic Hh homolog
(SHH)-specific rabbit polyclonal antibody (Proteintech),
SMO-specific rabbit polyclonal antibody (Bioworld Technology,
Minneapolis, MN, USA), Gli-1-specific mouse monoclonal antibody
(Santa Cruz Biotechnology, Dallas, TX, USA), E-cadherin-specific
rabbit monoclonal antibody (Cell Signaling Technology, Danvers, MA,
USA), Snail-specific rabbit monoclonal antibody (Cell Signaling
Technology), vimentin-specific rabbit monoclonal antibody (Cell
Signaling Technology) and β-actin specific mouse monoclonal
antibody (Santa Cruz Biotechnology). Horseradish
peroxidase-conjugated goat anti-rabbit IgG and goat anti-mouse IgG
(Santa Cruz Biotechnology) were used as the secondary antibody.
RNAi transfections
Loss-of-function analysis was performed using siRNAs
targeting Gli-1 and CCR2 which were purchased from GenePharm
(Shanghai, China): siRNA against Gli-1
(5′-GGCUCAGCUUGUGUGUAAUTT-3′, 5′-AUUACACACAAGCUGAGCCTT-3′), siRNA
against CCR2 (′-5CTGTCCAC-3′, ′-5CCCAAAGACCCACTCAT-3′) and a
negative control siRNA (′-5UUCUCCGAA-3′, ′-5ACGUGACACGUUCGGAGA-3′).
The cells were seeded in 6-well plates and were transfected with
100 nM siRNA using Lipofectamine 2000 (Invitrogen; Life
Technologies, Carlsbad, CA, USA) according to the manufacturer's
instructions. The knockdown of each target gene was confirmed by
western blot analysis. The cells were used for subsequent
experiments 48 h after transfection.
Quantitative real-time polymerase
chain reaction (qRT-PCR) analysis
Total RNA was extracted from the HCC cells using the
Fastgen1000 RNA isolation system (Fastgen, Shanghai, China)
according to the manufacturer's protocol. Total RNA was
reverse-transcribed into cDNA using a PrimeScript RT reagent kit
(Takara Bio, Dalian, China). Real-time PCR was used to
quantitatively examine the expression of E-cadherin, vimentin, SMO
and Gli-1 at the mRNA level. The PCR primer sequences were as
follows: for Snail forward, ′-5GGCTCCTT-3′ and reverse,
′-5CTGGAGATCCTTG-3′; for E-cadherin forward, ′-5ATTCTGATTCT-3′ and
reverse, ′-5AGTCCTGGTCCTCTTC-3′; for vimentin forward,
′-5CGGGAGAAATTGCAG-3′ and reverse, ′-5AAGGTCAAGACGTGCCA-3′; for SMO
forward, ′-5ACGAGGACGTGGAGGG-3′ and reverse,
5′-CGCACGGTATCGGTAGTTCT-3′; for Gli-1 forward,
5′-GGGATGATCCCACATCCTCAGTC-3′ and reverse,
5′-CTGGAGCAGCCCCCCCAGT-3′; for β-actin forward,
5′-AGCGAGTATCCCCCAAAGTT-3′ and reverse, 5′-GGGCACGAAGGCTCATCATT-3′.
The expression of each target gene was determined using β-actin as
the normalization control. The PCR reactions consisted of 30 sec at
95°C, followed by 40 cycles at 95°C for 5 sec, 30 sec at 60°C and
30 sec at 72°C. After each qRT-PCR, a dissociation curve analysis
was conducted. Relative gene expression was calculated using the
2−ΔΔCt method (19).
Western blot analysis
Total protein was extracted by RIPA lysis buffer
(Beyotime Institute of Biotechnology, Guangzhou, China) and the
concentration of protein was determined using the BCA protein assay
kit (Pierce, Rockford, IL, USA) according to the manufacturer's
instructions. The proteins were subjected to SDS-PAGE using a 10%
polyacrylamide gel with a 5% stacking gel, and then the protein was
transferred to polyvinylidene difluoride (PVDF) membranes. The
membranes were initially blocked with 5% non-fat dry milk in
Tris-buffered saline-Tween (TBS-T) for 2 h and then probed with
antibodies against E-cadherin (1:1,000 dilution), Snail (1:1,000
dilution), vimentin (1:1,000 dilution), CCR2 (1:500 dilution), SMO
(1:500 dilution), Gli-1 (1:1,000 dilution) or β-actin (1:1,000
dilution, loading control). After co-incubation with the primary
antibodies at 4°C overnight, the membranes were blotted with the
secondary antibody (1:10,000 dilution) for 2 h at 37°C.
Chemiluminescence detection of bound antibodies was performed using
an enhanced chemiluminescence ECL Plus system and a Molecular
Imager ChemiDoc XRS system (Bio-Rad Laboratories, Hercules, CA,
USA).
Cell viability assay
The MHCC97H cells were plated into 96-well plates at
a density of 5,000 cells/well and allowed to adhere for at least 24
h and serum-starved overnight (1% FBS in DMEM) before the beginning
of the experiments. After different treatments, the cell viability
was assessed by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Twenty microliters of 5 mg/ml MTT solution per 100 µl of
growth medium was added into each well after the removal of the
media and incubation followed at 37°C for 4 h. Following, 100 µl
DMSO was added to each well and the optical density (OD) was
assessed at 490 nm on a multifunction microplate reader (POLARstar
OPTIMA; BMG Labtech Ltd., Ortenberg, Germany). The proliferation
rate was calculated according to the following equation:
proliferation rate = (OD sample/OD control) × 100%.
Cell invasion analysis
The MHCC97H cell invasion was assessed by a
chamber-based invasion assay. In brief, the upper surface of a
filter (pore size, 8.0 µm; Millipore, Billerica, MA, USA) was
coated with basement membrane Matrigel (BD Biosciences, Franklin
Lakes, NJ, USA). The cells were suspended in DMEM containing 1%
FBS. Then the cell suspensions (200 µl containing 40,000 cells)
were added to the upper chambers. Simultaneously, 500 µl of DMEM
containing 10% FBS was placed in the lower chambers. The cells were
allowed to migrate for 24 h at 37°C. The non-invading cells were
removed from the upper surface by scraping with a wet cotton swab.
After rinsing with phosphate buffered saline (PBS), the filter was
fixed and stained with crystal violet. The invasion ability was
determined by counting the stained cells on the bottom surface.
Five random fields were captured at a magnification, ×200
(n=3).
Immunofluorescence staining
The cells were fixed in 4% formaldehyde diluted in
PBS for 15 min, permeabilized with 0.3% Triton X-100, treated with
blocking buffer (5% BSA in PBS), and then incubated overnight with
the primary antibody at 4°C. The cells were then incubated with
Red-conjugated secondary antibody from Jackson Immunoresearch
Laboratories (West Grove, PA, USA) for 1 h at room temperature.
Slides were mounted and examined using a Zeiss Instruments
microscope (Carl Zeiss, Oberkochen, Germany).
Statistical analysis
Statistical analysis was performed using SPSS 17.0
software (SPSS, Inc., Chicago, IL, USA). Each experiment was
performed at least three times. Data are presented as the means ±
standard deviation (SD). Differences between the groups were
analyzed by analysis of variance (ANOVA). P<0.05 was considered
to indicate a statistically significant difference.
Results
CCL2 promotes the invasion of HCC
cells via the induction of EMT
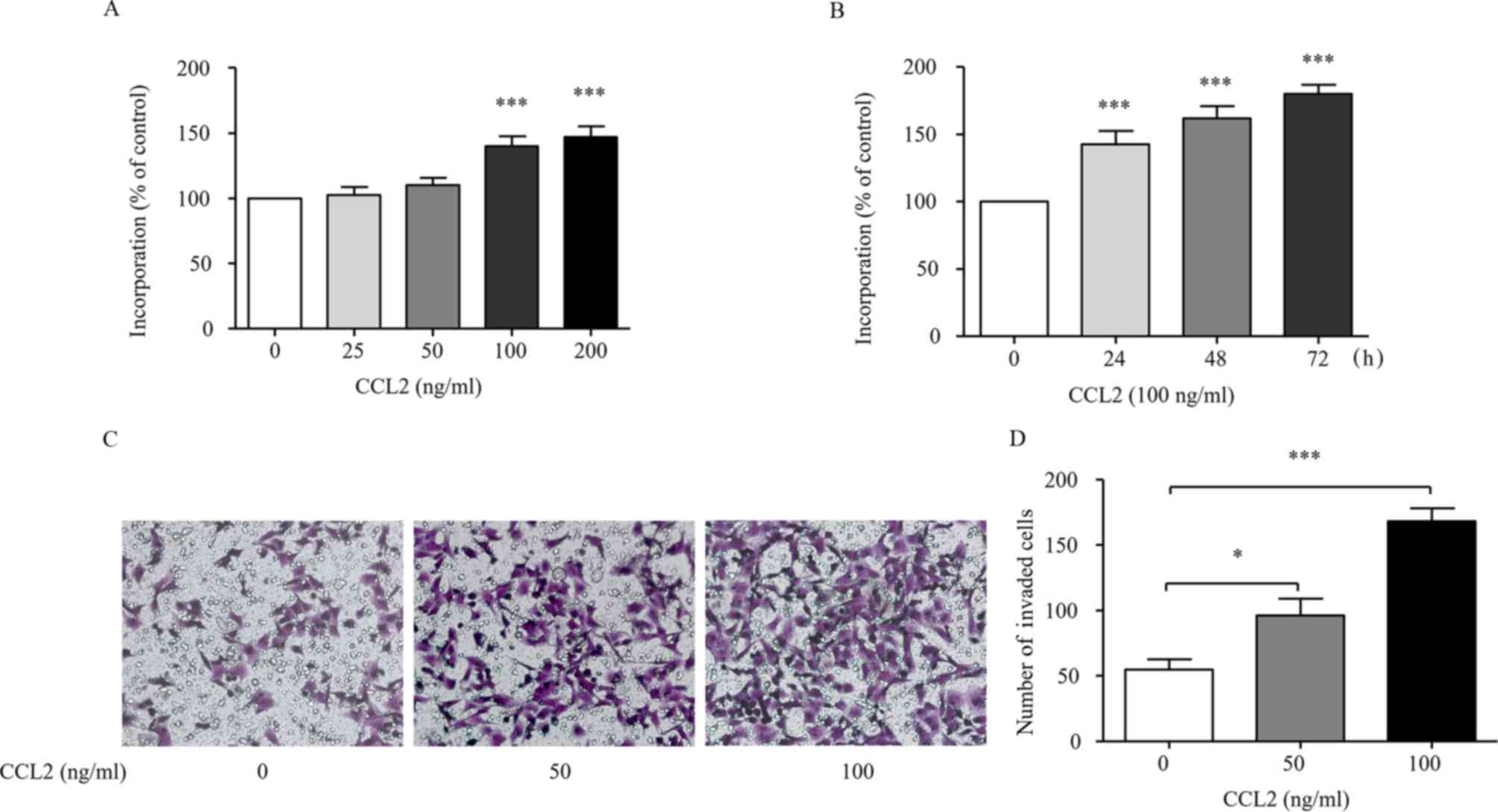
The MHCC97H cells were treated with different
concentrations of CCL2 (0, 25, 50, 100 and 200 ng/ml) at different
time-points (24, 48 and 72 h) and the cell proliferation was
assessed by an MTT assay. Fig. 1A
indicates that CCL2 stimulated MHCC97H proliferation in a
dose-dependent manner. As shown in Fig.
1B, CCL2 time-dependently stimulated MHCC97H proliferation,
indicating that CCL2 could effectively stimulate MHCC97H
proliferation. These findings were consistent with the results that
CCL2 promoted non-small cell lung cancer A549 cell viability
(20). The MHCC97H cells were
treated with or without CCL2 for 24 h and then the invasion ability
was assessed via a Transwell assay. The results demonstrated that
50 or 100 ng/ml of CCL2 significantly increased the invasion
ability of MHCC97H cells (P<0.05, Fig. 1C and D). These results reveal that
CCL2 promotes cell invasion.
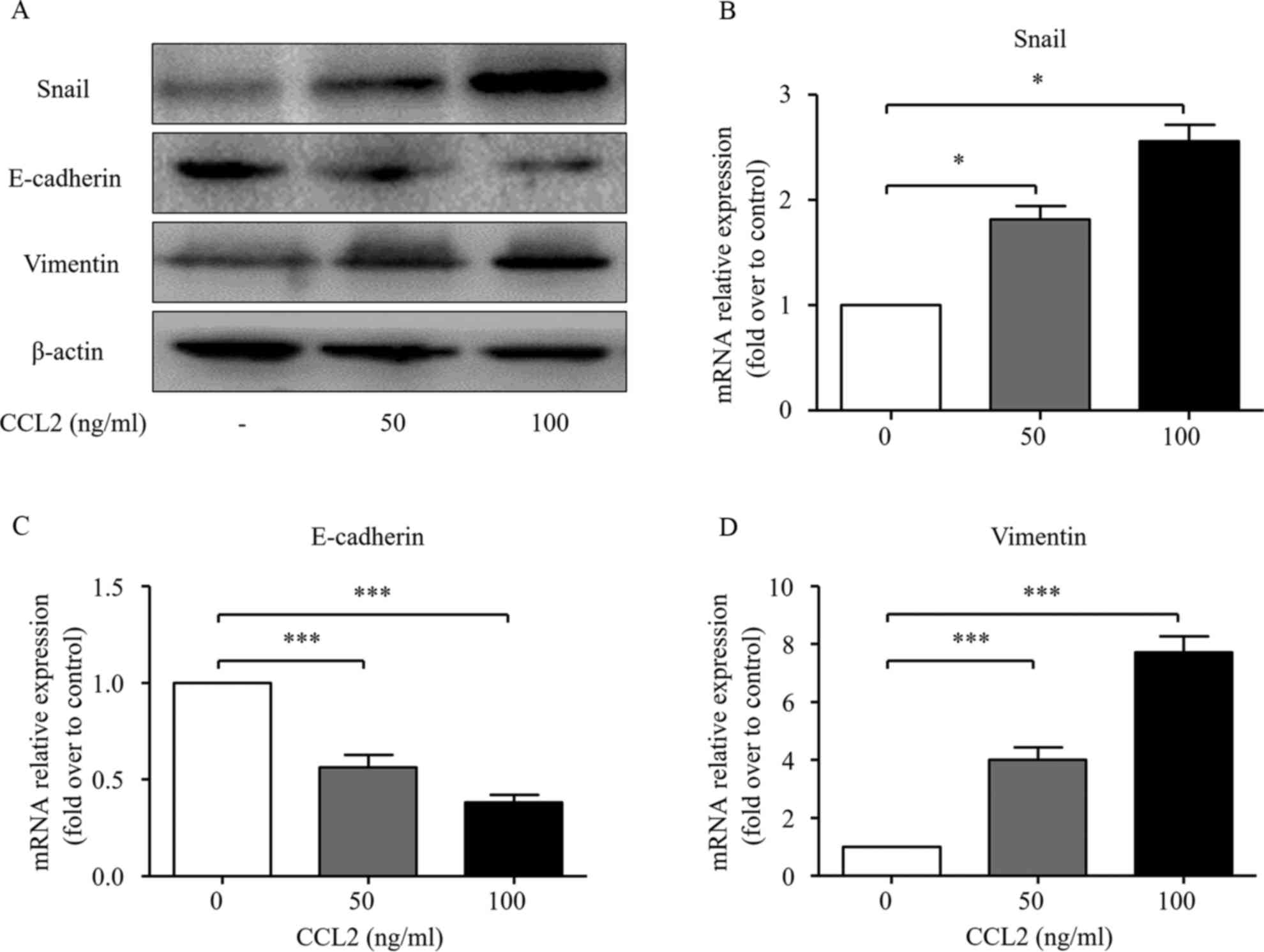
EMT has been confirmed to play an important role in
cancer progression, which is characterized by the loss of cell-cell
adhesion with diminished expression of epithelial markers such as
E-cadherin and increased expression of mesenchymal markers such as
vimentin and EMT transcription factors such as Snail (21). To further investigate the possible
role of the CCL2/CCR2 axis on EMT process, the expression of Snail,
E-cadherin and vimentin was detected via western blot analysis and
real-time PCR at both the protein and mRNA levels. As shown in
Fig. 2, both 50 and 100 ng/ml of
CCL2 significantly decreased the expression of E-cadherin at both
the mRNA and protein levels. Simultaneously, the expression levels
of Snail and vimentin at both the mRNA and protein levels were
significantly increased after the CCL2 treatment.
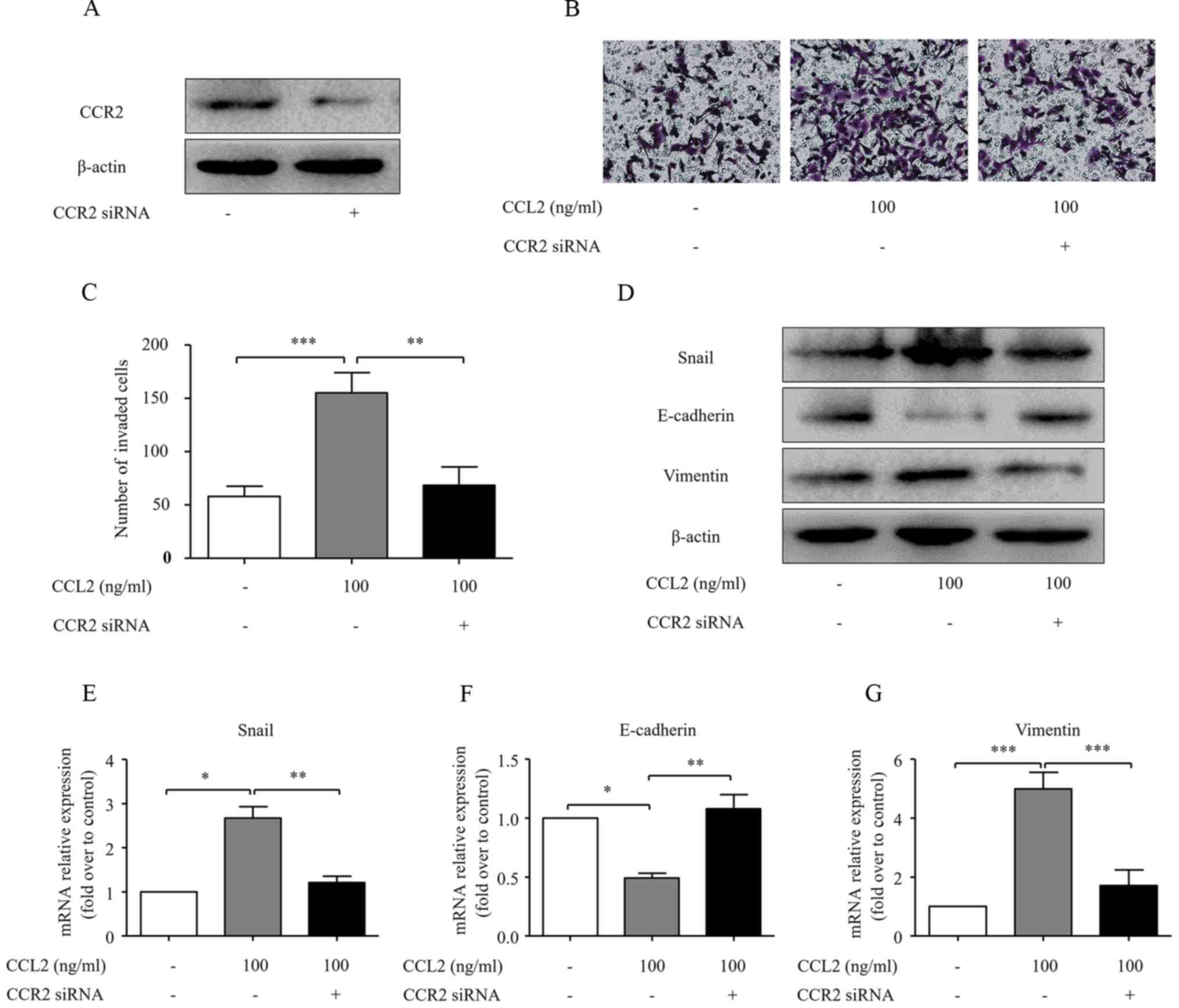
CCL2 promotes the invasion ability and
EMT of HCC cells in a CCR2-dependent manner
Although CCL2 influenced the EMT process and cell
invasive ability, the underlying mechanisms remain largely unclear.
CCR2 knockdown was carried out in the present study to investigate
the role of CCR2 in CCL2-induced HCC invasion. Transfection of CCR2
siRNA significantly reduced CCR2 expression (Fig. 3A). Transfection of CCR2 siRNA
reversed the effect of CCL2 on MHCC97H cell invasion (P<0.05;
Fig. 3B and C). Furthermore, the
CCL2-induced downregulation of E-cadherin and the upregulation of
Snail and vimentin in HCC cells were abolished upon knockdown of
CCR2 (Fig. 3D-G). Thus, these data
reveal that CCL2 promotes HCC progression through its receptor
CCR2.
CCL2 promotes the activation of the Hh
pathway in HCC cells in a CCR2-dependent manner
Several studies have revealed that the Hh pathway
may be a treatment target for HCC (14,22).
Our results revealed that CCL2 significantly increased the
transcription of the Hh pathway-related genes, including SMO and
Gli-1 in MHCC97H cells. However, the levels of SHH and Ptch
remained unchanged, compared to the normal controls (Fig. 4A-C). CCR2 knockdown was carried out
in the present study, to investigate the role of CCR2 in
CCL2-induced activation of the Hh pathway. Transfection of CCR2
siRNA significantly decreased the CCL2-induced expression of SMO
and Gli-1 both at the protein and mRNA levels, but there was no
effect on Ptch and SHH expression levels (Fig. 4D-F). These results indicated that Hh
signaling was activated in MHCC97H cells under the CCL2 treatment.
Upon activation, Gli-1 proteins translocate from the cytoplasm to
the nucleus and activate the transcription of target genes
(23,24). Additionally, as demonstrated by
immunofluorescence, the nuclear translocation of Gli-1 was enhanced
as an effect of CCL2 treatment, while transfection of CCR2 siRNA
obviously decreased the nuclear translocation of Gli-1 (Fig. 4G). Collectively, these findings
indicate that CCL2 activates the Hh pathway in a CCR2-dependent
manner.
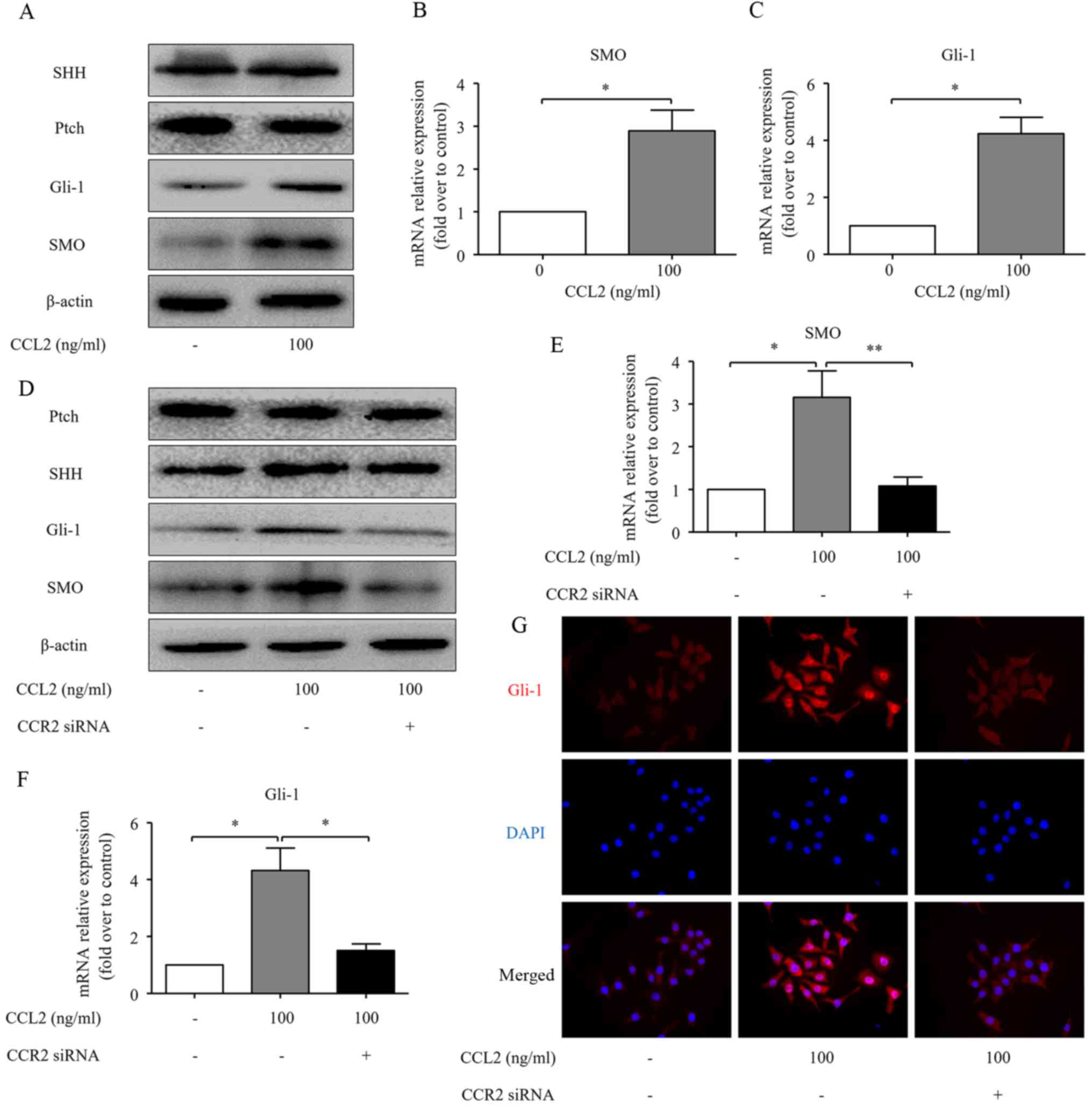 | Figure 4.CCL2/CCR2 axis activates the Hh
pathway in human HCC cells. (A) The expression of SHH, Ptch, SMO
and Gli-1 proteins in the MHCC97H cells was evaluated by western
blot analysis following treatment with the CCR2 ligand CCL2 for 48
h. (B and C) The expression of SMO and Gli-1 at mRNA levels was
assessed by real-time RT-PCR for the same cells. (D) CCR2 siRNA
diminished the effects of CCL2 on the expression of Ptch, SHH, SMO
and Gli-1 at the protein level in MHCC97H cells, as determined by
western blot analysis. (E and F) CCR2 siRNA diminished the effects
of CCL2 on the expression of SMO and Gli-1 at mRNA level in the
MHCC97H cells, as determined by real-time RT-PCR. (G)
Immuofluorescence staining of Gli-1 in the MHCC97H cells under
normal control or 100 ng/ml CCL2 stimulation or CCR2 siRNA combined
with 100 ng/ml CCL2 stimulation for 48 h. Red represents Gli-1
staining. Blue represents nuclear DNA staining by DAPI. Column,
mean (n=3); bar, SD; *P<0.05; **P<0.01. CCL2, chemokine (C-C
motif) ligand 2; CCR2, C-C chemokine receptor type 2; Hh, Hedgehog;
HCC, hepatocellular carcinoma; SHH, sonic Hh homolog; Ptch, Patched
1; SMO, smoothened. |
SMO mediates CCL2-induced EMT and cell
invasion in HCC cells
Since the activation of the CCL2/CCR2 axis
simultaneously induces HCC cell invasion and Hh pathway activation,
we hypothesized that there is a cross-talk between the CCL2/CCR2
axis and the Hh pathway and this cross-talk contributes to HCC cell
invasion. To test this hypothesis, we investigated the relationship
between the CCL2/CCR2-induced cell invasion and Hh pathway
activation using an SMO antagonist, cyclopamine, as an inhibitor of
the Hh pathway (25). Notably,
pretreatment with cyclopamine significantly decreased HCC cell
invasion (Fig. 5A and B), reversed
the downregulation of E-cadherin and the upregulation of Snail and
vimentin and reduced the expression of SMO and Gli-1 even in the
presence of CCL2 (Fig. 5C). The
Snail, SMO, Gli-1, E-cadherin and vimentin mRNA levels were
consistent with the protein changes (Fig. 5D-H). In addition, immunofluorescence
staining of these treated cells also ascertained that CCL2 induced
Gli-1 expression in the nucleus of MHCC97H cells, while
pretreatment with cyclopamine markedly decreased the nuclear
translocation of Gli-1 (Fig. 5I).
Therefore, we concluded that the inhibition of the Hh pathway
activation via blocking the function of SMO eliminates cell
invasion and EMT induced by CCL2. These findings reveal that there
is a cross-talk between the CCL2/CCR2 axis and the Hh pathway
activation, which is mediated by an increased SMO expression.
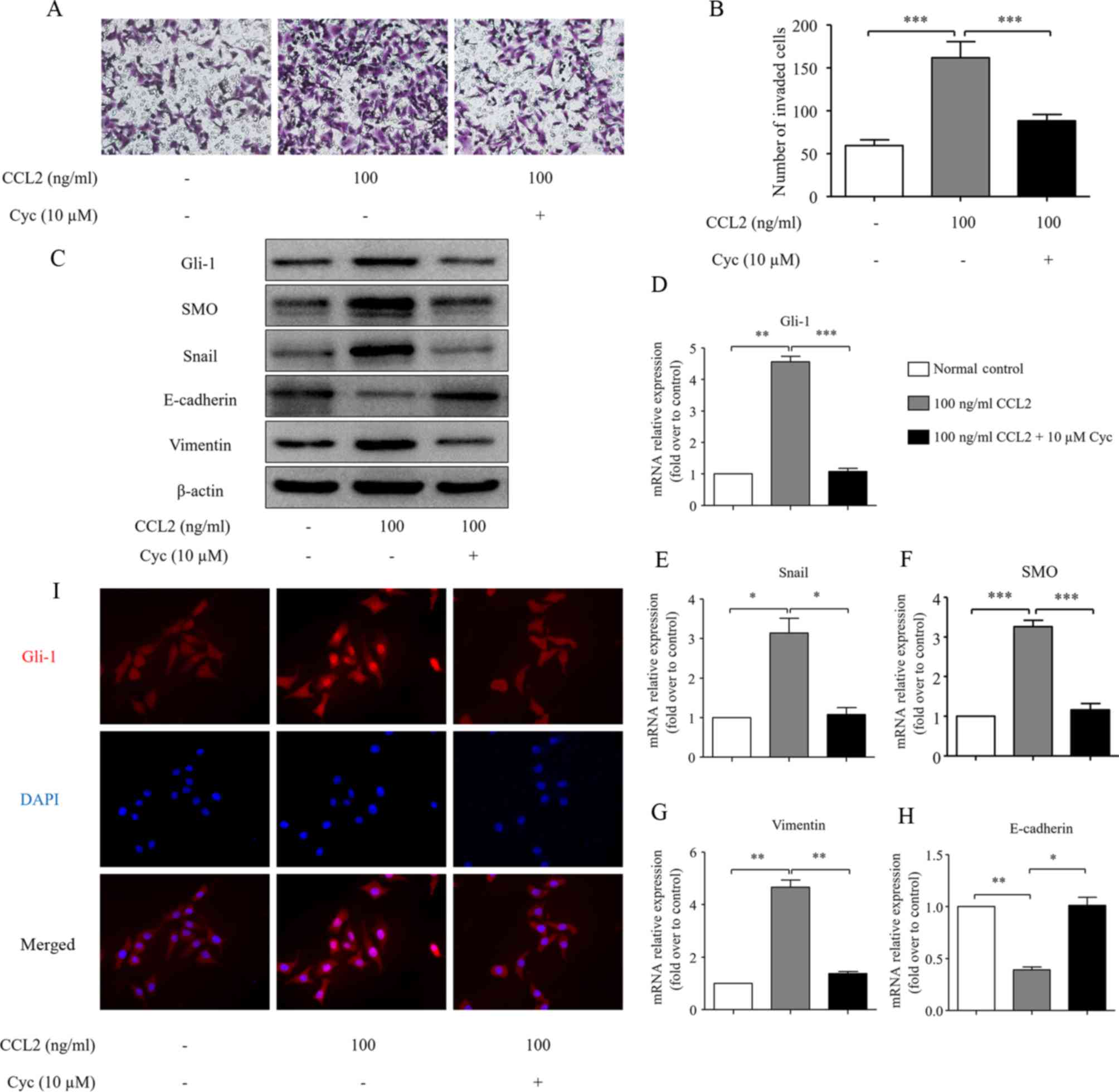 | Figure 5.The effects of Cyc on CCL2-induced
HCC cell invasion and EMT. (A) The effects of Cyc (10 µM) on HCC
cell invasion. After treatment with Cyc for 24 h, the cells were
seeded into a Matrigel-coated invasion chamber with or without CCL2
for 24 h. The number of cells was counted under a light microscope.
(B) The number of invaded cells was quantified by counting the
cells from 5 random fields at ×200 magnification. The data are
representative of 3 independent experiments. (C) The EMT-related
molecules Snail, E-cadherin and vimentin, and the Hh
pathway-related proteins SMO and Gli-1 protein levels were measured
via western blot analysis. (D-H) The EMT-related molecules Snail,
E-cadherin and vimentin, and the Hh pathway-related proteins SMO
and Gli-1 mRNA levels were analyzed by real-time RT-PCR. (I)
Immuofluorescence staining of Gli-1 in MHCC97H cells under normal
control or 100 ng/ml CCL2 stimulation or 10 µM Cyc combined with
100 ng/ml CCL2 stimulation for 48 h. Red represents Gli-1 staining.
Blue represents nuclear DNA staining by DAPI. Column, mean (n=3);
bar, SD; *P<0.05, **P<0.01, ***P<0.001. Cyc, cyclopamine;
CCL2, chemokine (C-C motif) ligand 2; HCC, hepatocellular
carcinoma; EMT, epithelial-mesenchymal transition; Hh, Hedgehog;
SMO, smoothened. |
Gli-1 mediates CCL2-induced EMT and
cell invasion in HCC cells
Previous studies have demonstrated that Gli-1
induces mobility and invasion of HCC cells and is an important
regulator of EMT (18). In the
present study, since we found that the activation of CCR2 by CCL2
simultaneously induces tumor cell invasion and Hh pathway
activation, we hypothesized that Gli-1 is a critical mediator of
the CCL2-induced EMT and cell invasion of HCC cells. In order to
further confirm the effect of Gli-1 on CCL2-induced invasion of HCC
cells, we investigated the role of Gli-1 in the CCL2/CCR2
axis-induced invasion and Hh pathway activation using siRNA
targeting Gli-1. Transfection of Gli-1 siRNA significantly reduced
the Gli-1 protein level (Fig. 6A).
Furthermore, Transwell assays revealed that transfection with Gli-1
siRNA significantly inhibited the invasion ability of HCC cells
induced by CCL2 (Fig. 6B and C).
The knockdown of Gli-1 blocked the CCL2-induced increase in Snail
and vimentin and reduction of E-cadherin protein levels (Fig. 5D). These data demonstrate that CCL2
promotes HCC invasion and EMT by modulating Gli-1-dependent Snail,
vimentin and E-cadherin expression.
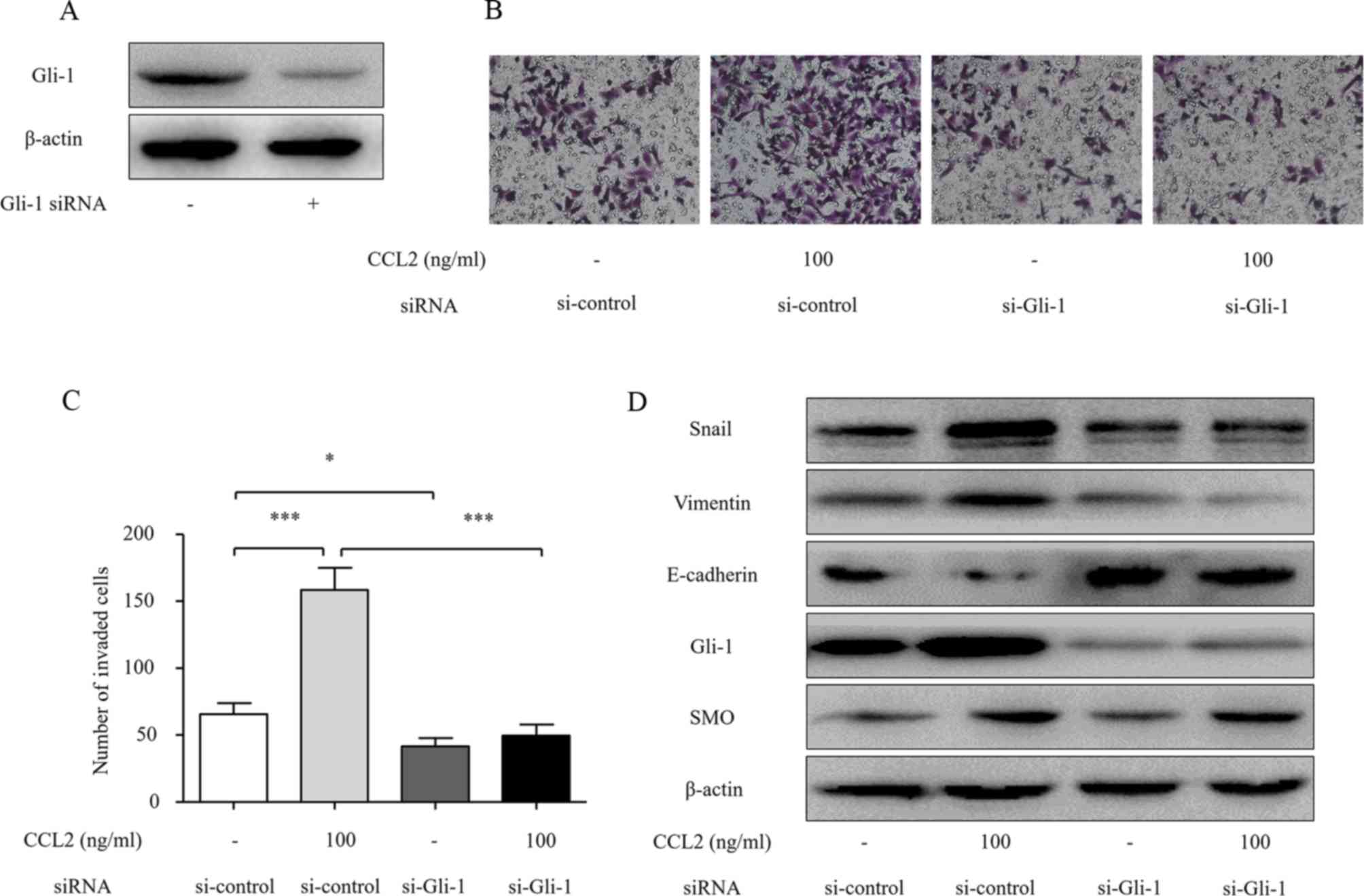 | Figure 6.Gli-1 siRNA abolishes the effects of
CCL2-mediated invasion and EMT in HCC cells. (A) Knockdown of Gli-1
by siRNA for 48 h was confirmed by western blot analysis. (B) The
effect on cell invasion in response to Gli-1 knockdown. After
transfection with siRNA for 48 h, the cells were seeded into a
Matrigel-coated invasion chamber with or without CCL2 for 24 h. The
number of cells was counted under a light microscope. (C) The
number of invaded cells was quantified by counting the cells from 5
random fields at ×200 magnification. The data are representative of
3 independent experiments. (D) The effects of Gli-1 siRNA on the
expression of Snail, SMO, Gli-1, E-cadherin and vimentin. After
transfection of the cells with siRNA for 48 h, Snail, SMO, Gli-1,
E-cadherin and vimentin expression levels were determined by
western blot analysis. Column, mean (n=3); bar, SD; *P<0.05,
**P<0.01, ***P<0.001. CCL2, chemokine (C-C motif) ligand 2;
EMT, epithelial-mesenchymal transition; HCC, hepatocellular
carcinoma; SMO, smoothened. |
Discussion
Chemokines are a family of small, secreted proteins
that have pleiotropic roles in inflammation-related pathological
diseases, including cancer. CCL2 is broadly expressed in a variety
of tissue types and acts as a potent chemoattractant to recruit
monocytes and macrophages to the sites of inflammation. Recent
studies have revealed that a high expression of CCL2 is associated
with unfavorable patient prognosis in various types of cancer
(4–6). CCL2 is overexpressed in human liver
cancer and is an independent prognostic indicator in patients with
HCC. Blockade of the CCL2/CCR2 axis by knockdown of CCR2 or with a
CCR2 antagonist inhibits malignant growth and metastasis, reduces
postsurgical recurrence and enhances survival (9). CCL2 is secreted by different cell
types and is involved in various aspects of liver pathogenesis,
including acute liver injury, chronic HBV/HCV infection, cirrhosis
and tumorigenesis (26–28). CCR2, the only known receptor for
CCL2, has been found to regulate cell growth, angiogenesis,
invasion and metastasis (29).
However, the role of the CCL2/CCR2 axis in HCC cell invasion and
its molecular mechanisms remain poorly understood. Our results
revealed that CCL2 promotes HCC invasion in a dose-dependent
manner.
EMT occurs as a key step during embryonic
morphogenesis and is now implicated in the progression of primary
tumors towards metastases. In recent years, numerous studies have
ascertained that EMT plays important roles in the progression of
human cancer (30). The zinc-finger
transcriptional repressor Snail reportedly contributes to EMT in
HCC and plays a key role in tumorigenesis, differentiation,
migration and invasiveness (31).
In addition, recent research has revealed that Snail expression is
negatively related to tumor differentiation, which is an
independent factor predictive of survival in HCC patients (32). Loss of E-cadherin and gain of
vimentin are known to play key roles in the EMT process of various
human types of cancer, including HCC (18). Previous studies have demonstrated
that CCL2 induces this phenomenon via the EMT-related genes
(8,33). In the present study, our results
revealed that CCL2 induced upregulation of the EMT transcription
factor Snail and the mesenchymal marker vimentin, however, the
epithelial marker E-cadherin was downregulated after treatment with
CCL2 (Fig. 2). However, the EMT
process and invasion ability were reversed after transfection with
CCR2 siRNA, which indicated that the CCL2-induced HCC invasion and
EMT are CCR2 receptor-dependent (Fig.
3).
Several studies have demonstrated that multiple
components of Hh signaling are affiliated with EMT, invasion and
metastasis in cancer cells (34–37).
In the present study, our results indicated that CCL2 significantly
induced SMO and Gli-1 expression. The immunofluorescence staining
results also confirmed that CCL2 induced Gli-1 translocation into
the nucleus of MHCC97H cells. However, Ptch and SHH remained
unchanged compared to normal controls. Based on previously
published studies and our findings, we thus hypothesized that a
cross-talk exists between the CCL2/CCR2 axis and the Hh pathway,
which is critical for CCL2-induced cancer cell invasion and EMT.
This hypothesis is further supported by the fact that knockdown of
CCR2 by siRNA prevented the activation of the Hh pathway and the
invasion by CCL2, suggesting that CCR2 is the key factor in the
CCL2-mediated activation of the Hh pathway and pro-invasion
ability.
The Hh signaling pathway, which is normally
quiescent in adult liver, has been shown to be very active in HCC
(38). Without binding to Hh
ligands, Ptch holds SMO, a seven transmembrane spanning protein, in
an inactive state and thus prohibits signaling to downstream genes.
Once activated, SMO initiates the transcriptional activity of
downstream targets and Gli-1 proteins translocate from the
cytoplasm to the nucleus and activate the transcription of target
genes, which in turn regulate cell proliferation, differentiation,
apoptosis and invasion (39–41).
Therefore, the expression of SMO and Gli-1 is presumed to be a
marker of the Hh pathway activation.
Cyclopamine, an SMO antagonist, especially binds to
SMO heptahelical bundle to inhibit its activity so as to suppress
Hh signaling. We exposed HCC cells to cyclopamine in the presence
of CCL2. Cyclopamine markedly reduced the tumor invasion and
reversed the EMT progress induced by CCL2. Consistent with previous
results, blocking SMO function with cyclopamine decreased the
expression of the transcription factor Gli-1. We also observed that
the nuclear Gli-1 expression was downregulated via
immunofluorescence (Fig. 5). It is
known that Gli-1 is a critical mediator of the Hh pathway in cancer
cell invasion and metastasis, which was consistent with our results
that Gli-1 siRNA inhibited EMT progress by suppressing Snail and
vimentin expression and enhancing E-cadherin expression.
Subsequently we treated the HCC cell lines with siRNA specific to
Gli-1 in the presence of CCL2. Prior silencing of Gli-1 with siRNA
abolished CCL2-induced Snail and vimentin upregulation, E-cadherin
reduction, as well as HCC invasion and EMT. However, Gli-1 siRNA
could not interrupt the CCL2-mediated increase in SMO.
Collectively, these findings reveal that it is probable that CCL2
induced EMT and cell invasion via the activation of SMO and Gli-1
expression (Fig. 6).
In some preclinical cancer models, targeting of CCL2
is an effective therapeutic approach. The neutralizing antibody
carlumab (formerly named CNTO 888) is a human immunoglobulin G1κ
monoclonal antibody that binds with high affinity and specificity
to human CCL2 and has entered clinical trials for the treatment of
prostate cancer (42). Li et
al findings (9) revealed that
there was no marked malignant progression observed after treatment
with a CCR2 antagonist in their postsurgical recurrence animal
models and that animals receiving CCR2 inhibition had a much longer
survival rate, suggesting that CCL2 or CCR2 may be a potential
targeting strategy in cancer treatment. However, its safety and
efficacy need more clinical trial support.
The present study has certain limitations. Firstly,
we only analyzed Snail, E-cadherin and vimentin expression. The
absence of analysis of Slug as well as other epithelial and
mesenchymal markers (N-cadherin and ZO-1), in addition to the
absence of confocal analysis of E-cadherin and/or vimentin staining
in the cells is one of the limitations of the present study.
Secondly, despite that we detected Gli-1 expression via western
blot analysis and immunofluorescence staining, the absence of Gli-1
reporter luciferase assays in response to CCL2 performed in order
to demonstrate that there is an activation of the Hh pathway is
another limitation of the present study.
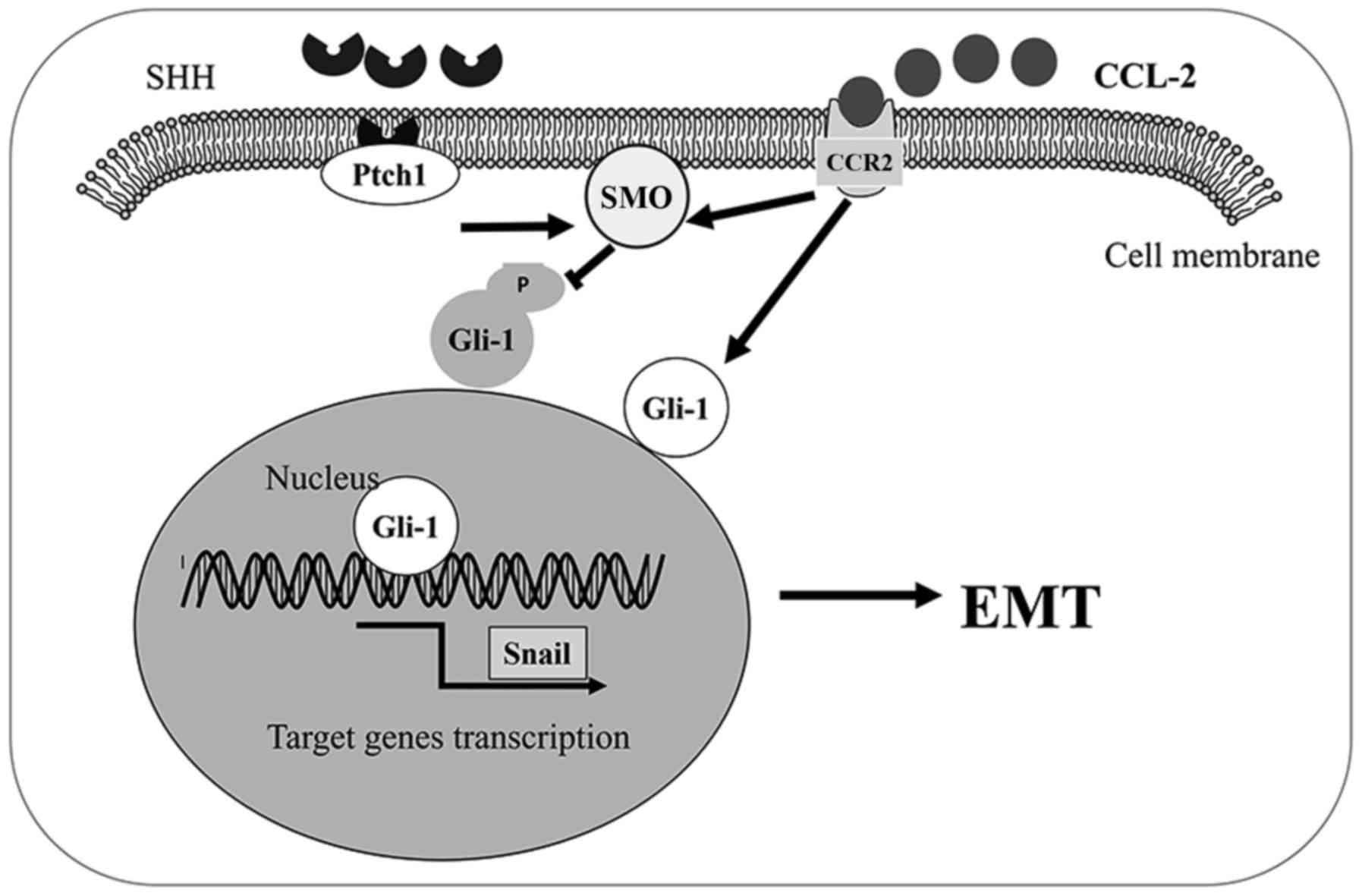
In conclusion, we found that the CCL2/CCR2 axis
induced HCC invasion and EMT in vitro through the activation
of the Hh pathway. The CCL2/CCR2 axis induced invasion and tumor
cell EMT through the increase of SMO and Gli-1 expression. Our
results revealed that the link between CCR2 and the Hh pathway
plays an important role in HCC progression. Therefore, the
CCL2/CCR2 axis may represent a promising therapeutic target to
prevent HCC progression (Fig.
7).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu Q, Wang Z, Chen X, Duan W, Lei J, Zong
L, Li X, Sheng L, Ma J, Han L, et al: Stromal-derived
factor-1α/CXCL12-CXCR4 chemotactic pathway promotes perineural
invasion in pancreatic cancer. Oncotarget. 6:4717–4732. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Z, Xie H, Zhou L, Liu Z, Fu H, Zhu Y,
Xu L and Xu J: CCL2/CCR2 axis is associated with postoperative
survival and recurrence of patients with non-metastatic clear-cell
renal cell carcinoma. Oncotarget. 7:51525–51534. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang Y, Zhai C, Chang Y, Zhou L, Shi T,
Tan C, Xu L and Xu J: High expression of chemokine CCL2 is
associated with recurrence after surgery in clear-cell renal cell
carcinoma. Urol Oncol. 34:238.e19–238.e26. 2016. View Article : Google Scholar
|
|
6
|
Zhang J, Patel L and Pienta KJ: CC
chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis
and metastasis. Cytokine Growth Factor Rev. 21:41–48. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lim SY, Yuzhalin AE, Gordon-Weeks AN and
Muschel RJ: Targeting the CCL2-CCR2 signaling axis in cancer
metastasis. Oncotarget. 7:28697–28710. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rao Q, Chen Y, Yeh CR, Ding J, Li L, Chang
C and Yeh S: Recruited mast cells in the tumor microenvironment
enhance bladder cancer metastasis via modulation of ERβ/CCL2/CCR2
EMT/MMP9 signals. Oncotarget. 7:7842–7855. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li X, Yao W, Yuan Y, Chen P, Li B, Li J,
Chu R, Song H, Xie D, Jiang X and Wang H: Targeting of
tumour-infiltrating macrophages via CCL2/CCR2 signalling as a
therapeutic strategy against hepatocellular carcinoma. Gut.
66:157–167. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shih YT, Wang MC, Zhou J, Peng HH, Lee DY
and Chiu JJ: Endothelial progenitors promote hepatocarcinoma
intrahepatic metastasis through monocyte chemotactic protein-1
induction of microRNA-21. Gut. 64:1132–1147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li X, Wang Z, Ma Q, Xu Q, Liu H, Duan W,
Lei J, Ma J, Wang X, Lv S, et al: Sonic hedgehog paracrine
signaling activates stromal cells to promote perineural invasion in
pancreatic cancer. Clin Cancer Res. 20:4326–4338. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li X, Ma Q, Xu Q, Liu H, Lei J, Duan W,
Bhat K, Wang F, Wu E and Wang Z: SDF-1/CXCR4 signaling induces
pancreatic cancer cell invasion and epithelial-mesenchymal
transition in vitro through non-canonical activation of Hedgehog
pathway. Cancer Lett. 322:169–176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Diao Y, Azatyan A, Rahman MF, Zhao C, Zhu
J, Dahlman-Wright K and Zaphiropoulos PG: Blockade of the Hedgehog
pathway downregulates estrogen receptor alpha signaling in breast
cancer cells. Oncotarget. 7:71580–71593. 2016.PubMed/NCBI
|
|
14
|
Wang Y, Han C, Lu L, Magliato S and Wu T:
Hedgehog signaling pathway regulates autophagy in human
hepatocellular carcinoma cells. Hepatology. 58:995–1010. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Villavicencio EH, Walterhouse DO and
Iannaccone PM: The sonic hedgehog-patched-gli pathway in human
development and disease. Am J Hum Genet. 67:1047–1054. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lei J, Ma J, Ma Q, Li X, Liu H, Xu Q, Duan
W, Sun Q, Xu J, Wu Z and Wu E: Hedgehog signaling regulates hypoxia
induced epithelial to mesenchymal transition and invasion in
pancreatic cancer cells via a ligand-independent manner. Mol
Cancer. 12:662013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng X, Zeng W, Gai X, Xu Q, Li C, Liang
Z, Tuo H and Liu Q: Role of the Hedgehog pathway in hepatocellular
carcinoma (Review). Oncol Rep. 30:2020–2026. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Q, Liu Z, Xu M, Xue Y, Yao B, Dou C,
Jia Y, Wang Y, Tu K, Zheng X and Yao Y: PCAF inhibits
hepatocellular carcinoma metastasis by inhibition of
epithelial-mesenchymal transition by targeting Gli-1. Cancer Lett.
375:190–198. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
An J, Xue Y, Long M, Zhang G, Zhang J and
Su H: Targeting CCR2 with its antagonist suppresses viability,
motility and invasion by downregulating MMP-9 expression in
non-small cell lung cancer cells. Oncotarget. 8:39230–39240.
2017.PubMed/NCBI
|
|
21
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Che L, Yuan YH, Jia J and Ren J:
Activation of sonic hedgehog signaling pathway is an independent
potential prognosis predictor in human hepatocellular carcinoma
patients. Chin J Cancer Res. 24:323–331. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kasper M, Regl G, Frischauf AM and Aberger
F: GLI transcription factors: Mediators of oncogenic Hedgehog
signalling. Eur J Cancer. 42:437–445. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Y, Yang W, Yang Q and Zhou S: Nuclear
localization of GLI1 and elevated expression of FOXC2 in breast
cancer is associated with the basal-like phenotype. Histol
Histopathol. 27:475–484. 2012.PubMed/NCBI
|
|
25
|
Chen JK, Taipale J, Cooper MK and Beachy
PA: Inhibition of Hedgehog signaling by direct binding of
cyclopamine to Smoothened. Genes Dev. 16:2743–2748. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sahin H, Trautwein C and Wasmuth HE:
Functional role of chemokines in liver disease models. Nat Rev
Gastroenterol Hepatol. 7:682–690. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mandrekar P, Ambade A, Lim A, Szabo G and
Catalano D: An essential role for monocyte chemoattractant
protein-1 in alcoholic liver injury: Regulation of proinflammatory
cytokines and hepatic steatosis in mice. Hepatology. 54:2185–2197.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baeck C, Wehr A, Karlmark KR, Heymann F,
Vucur M, Gassler N, Huss S, Klussmann S, Eulberg D, Luedde T, et
al: Pharmacological inhibition of the chemokine CCL2 (MCP-1)
diminishes liver macrophage infiltration and steatohepatitis in
chronic hepatic injury. Gut. 61:416–426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qian BZ, Li J, Zhang H, Kitamura T, Zhang
J, Campion LR, Kaiser EA, Snyder LA and Pollard JW: CCL2 recruits
inflammatory monocytes to facilitate breast-tumour metastasis.
Nature. 475:222–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wan Z, Pan H, Liu S, Zhu J, Qi W, Fu K,
Zhao T and Liang J: Downregulation of SNAIL sensitizes
hepatocellular carcinoma cells to TRAIL-induced apoptosis by
regulating the NF-κB pathway. Oncol Rep. 33:1560–1566. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang M, Dong X, Zhang D, Chen X and Zhu
X: High expression of Snail and NF-κB predicts poor survival in
Chinese hepatocellular carcinoma patients. Oncotarget. 8:4543–4548.
2017.PubMed/NCBI
|
|
33
|
Lee CC, Ho HC, Su YC, Lee MS, Hung SK and
Lin CH: MCP1-induced epithelial-mesenchymal transition in head and
neck cancer by AKT activation. Anticancer Res. 35:3299–3306.
2015.PubMed/NCBI
|
|
34
|
Yang Z, Koehler AN and Wang L: A novel
small molecule activator of nuclear receptor SHP inhibits HCC cell
migration via suppressing Ccl2. Mol Cancer Ther. 15:2294–2301.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang J, Peng Y, Liu Y, Yang J, Huang M and
Tan W: AT-101 inhibits hedgehog pathway activity and cancer growth.
Cancer Chemother Pharmacol. 76:461–469. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mimeault M, Rachagani S, Muniyan S,
Seshacharyulu P, Johansson SL, Datta K, Lin MF and Batra SK:
Inhibition of hedgehog signaling improves the anti-carcinogenic
effects of docetaxel in prostate cancer. Oncotarget. 6:3887–3903.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zuo M, Rashid A, Churi C, Vauthey JN,
Chang P, Li Y, Hung MC, Li D and Javle M: Novel therapeutic
strategy targeting the Hedgehog signalling and mTOR pathways in
biliary tract cancer. Br J Cancer. 112:1042–1051. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang S, He J, Zhang X, Bian Y, Yang L,
Xie G, Zhang K, Tang W, Stelter AA, Wang Q, et al: Activation of
the hedgehog pathway in human hepatocellular carcinomas.
Carcinogenesis. 27:1334–1340. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chuang PT and McMahon AP: Vertebrate
Hedgehog signalling modulated by induction of a Hedgehog-binding
protein. Nature. 397:617–621. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cai H, Li H, Li J, Li X, Li Y, Shi Y and
Wang D: Sonic hedgehog signaling pathway mediates development of
hepatocellular carcinoma. Tumour Biol. 37:16199–16205. 2016.
View Article : Google Scholar
|
|
41
|
Huang XH, Chen JS, Wang Q, Chen XL, Wen L,
Chen LZ, Bi J, Zhang LJ, Su Q and Zeng WT: miR-338-3p suppresses
invasion of liver cancer cell by targeting smoothened. J Pathol.
225:463–472. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pienta KJ, Machiels JP, Schrijvers D,
Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA,
Takimoto C, et al: Phase 2 study of carlumab (CNTO 888), a human
monoclonal antibody against CC-chemokine ligand 2 (CCL2), in
metastatic castration-resistant prostate cancer. Invest New Drugs.
31:760–768. 2013. View Article : Google Scholar : PubMed/NCBI
|