Introduction
Glioblastoma (GBM) is the most aggressive and
deadliest brain malignancy in adults. According to the latest
statistics from the United States National Cancer Institute, it is
estimated that 23,000 Americans will be diagnosed with brain and
other nervous system cancers in 2017 and 16,700 will die from these
cancers (1). Gliomas are divided
into four grades, of which grade four or GBM is the most aggressive
and has the highest incidence (2).
GBM progression cannot be controlled by surgical resection,
radiotherapy, or chemotherapy, which results in a median survival
time of less than 15 months (3).
Thus, new therapeutic approaches are needed.
Omega3-polyunsaturated fatty acids (ω3-PUFAs) and
ω6-polyunsaturated fatty acids (ω6-PUFAs) are essential nutrients
for good health. ω3-PUFAs are long-chain fatty acids that have a
double bond between the third and fourth carbon atoms from the
methyl end of the carbon chain. The Western diet contains a high
amount of ω6-PUFAs, with an ω6-PUFA to ω3-PUFA ratio of
approximately 50:1 (4). A high
ω6-PUFA to ω3-PUFAs ratio contributes to the pathogenesis of many
diseases, including cancer and inflammatory diseases, whereas
increased levels of ω3-PUFAs (a low ω6-PUFA to ω3-PUFA ratio) have
suppressive effects on these diseases (5). For this reason, nutritionists
recommend a diet with an optimal ω6-PUFA to ω3-PUFA ratio of 1:1
(6). We previously demonstrated in
numerous studies that ω3-PUFAs, docosahexaenoic acid (DHA) in
particular, induce cell death in cancer cells through several
mechanisms, including inhibition of Wnt/β-catenin signaling
(7–9), activation of mitogen-activated protein
kinases (10), inhibition of
Cox-2/PGE2 signaling (7,8,11) and
inhibition of NF-κB signaling (11). Recently, we reported that DHA
induces autophagy through p53/AMPK/mTOR signaling (12) and that autophagy induction enhances
apoptosis in human cancer cells (12–14).
More recently, DHA was found to induce degradation of HPV E6/E7
oncoproteins by activating the ubiquitin-proteasome system
(15). However, the anticancer
activity of DHA in GBM cells has not been studied intensively. In
addition, the molecular mechanisms of the anticancer activity of
DHA are not fully understood. Therefore, in the present study, we
investigated the mechanisms underlying the anticancer activity of
DHA on GBM in vitro and in vivo.
Materials and methods
Glioma cell lines and reagent
treatment
Three human (D54MG, U87MG and U251MG) and mouse
(GL261) glioma cell lines were used in the present study. D54MG and
GL261 cell lines were obtained from Dr Yancie Gillespie (University
of Alabama-Birmingham, Birmingham, AL). To note, the D54MG cell
line is misidentified according to the following database:
http://iclac.org/wp-content/uploads/Cross-Contaminations-v8_0.pdf.
This cell line is contaminated with another human glioblastoma cell
line (A-172). U87MG, U251MG and GL261 cells were maintained in
Dulbecco's modified Eagles medium (DMEM; Gibco, Grand Island, NY,
USA), and D54MG was grown in RPMI-1640 medium (Gibco) supplemented
with 10% fetal bovine serum (FBS; Gibco) and antibiotics
(penicillin 10,000 U/ml and streptomycin 10,000 µg/ml; Gibco) in 5%
CO2 at 37°C. Absolute ethanol was used to dissolve DHA
(Cayman Chemical, Ann Arbor, MI, USA). The following antibodies
were used in the present study: poly(ADP-ribose) polymerase (PARP)
(#9542, Rabbit, Polyclonal, 1:2,000), LC3B (#3868, Rabbit IgG,
Monoclonal, 1:5,000), Akt (#9272, Rabbit, Polyclonal, 1:2,000),
p-Akt(Ser473) (#4060, Rabbit IgG, Monoclonal, 1:2,000), mTOR
(#2972, Rabbit, Polyclonal, 1:1,000), p-mTOR(Ser2448) (#2971,
Rabbit, Polyclonal, 1:1,000), AMPK (#2532, Rabbit, Polyclonal,
1:2,000) and p-AMPK(Thr172) (#2535, Rabbit IgG, Monoclonal,
1:2,000). All antibodies were purchased from Cell Signaling
Technology (Beverly, MA, USA). Goat anti-rabbit and goat anti-mouse
secondary antibodies were purchased from Calbiochem (Billerica, MA,
USA).
MTT assay
Cell viability was determined using thiazolyl blue
tetrazolium bromide (MTT; Sigma-Aldrich, St. Louis, MO, USA).
D54MG, U87MG, U251MG and GL261 cells were seeded onto 96-well
tissue culture plates and incubated at 37°C for 18 h. The cells
were incubated with serum-free media for 24 h at 37°C, after which
the cells were treated with the indicated concentrations of DHA for
another 24 h whereas the D54MG cells were incubated for 6 h. The
cells were then incubated with 0.005 mg/ml of MTT dissolved in
serum-free medium for 1 h at 37°C. After a 1-h incubation, the
formazan product, which is indicated by dark blue water-insoluble
crystals, was formed in the event the cells were alive. These
crystals were dissolved in 100 µl dimethyl sulfoxide (DMSO), the
absorbance was then measured at 570 nm, and cell viability was
expressed as a ratio vs. the untreated control cells. Each
experiment was performed in triplicate.
Western blot analysis
Cells extracts were prepared from 100-mm tissue
culture plates. Cell pellets were lysed in cell lysis buffer and
then sonicated for 20 sec. The supernatants were prepared by
centrifugation at 17,000 rpm at 4°C for 20 min. Cell lysates (30
µg) were separated using 6–15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The
separated proteins were then transferred to a PVDF membrane
(Millipore, Billerica, MA, USA), and the membranes were blocked
with 5% skim milk in TBS/T for 1 h at room temperature. The blots
were incubated with the diluted primary antibodies overnight at
4°C, followed by incubation with goat anti-rabbit
peroxidase-conjugated secondary antibody. The bands were then
visualized with the ECL western blotting detection system
(WBKLS0500, Millipore) according to the manufacturers
instructions.
Flow cytometry assay
For the flow cytometry cell sorting (FACS) assay,
both floating and attached cells were collected after the drug
treatment, washed in phosphate-buffered saline (PBS), fixed with
70% ethanol for 24 h, treated with 500 µg/ml of RNase A
(Sigma-Aldrich), and then stained with 50 µg/ml propidium iodide
(PI; Sigma-Aldrich) for 10 min at 37°C. DNA staining with PI was
analyzed with a FACSCalibur flow cytometer (BD Biosciences, San
Diego, CA, USA).
TUNEL assay
For the TUNEL assay in vitro, D54MG cells
were seeded onto glass coverslips in 24-well plates and then grown
for 24 h. The cells were then incubated with serum-free media for
24 h at 37°C, and then the cells were treated with 30 µM DHA for
another 2 h. Cells were washed with 1X PBS and processed with TUNEL
using the DeadEnd Fluorometric TUNEL system (Promega, Fitchburg,
WI, USA). Cells were mounted with mounting solution (Dako A/S,
Copenhagen, Denmark) with DAPI. The number of TUNEL-positive
(green) and DAPI-positive cells (blue nuclear stain) was visually
counted. All samples were analyzed with at least two biological
replicates, and three images from each replicate were captured
using a 10X objective for counting the TUNEL- and DAPI-positive
cells. The percentage of TUNEL-positive cells was calculated as
(number of TUNEL-positive cells/total number of cells) × 100.
Transfection
Cells grown to 80% confluency were switched to and
incubated with serum-free media for 24 h. Transient transfection
was then performed with the GFP-LC3 expression vector (a kind gift
from Dr Tamotsu Yoshimori, National Institute of Genetics, Mishima,
Japan) as recommended by the vendor. After 24 h of transfection,
the cells were exposed to the indicated concentrations of DHA for 1
h. The transfected cells were then observed under a fluorescence
microscope, and fluorescence imaging was performed to detect the
punctate pattern of GFP-LC3B.
Tumorigenicity experiments
Transgenic (tg) mice were kindly provided by Dr J.X.
Kang (Harvard University, Cambridge, MA, USA). Control C57BL/6 mice
were purchased from Central Lab Animal Inc., Seoul, Korea. The
control and fat-1 tg mice were kept under specific
pathogen-free conditions and received care according to the
guidelines of the Institutional Animal Care and Use Committee of
Chungnam National University which approved the protocol of the
animal research. Both control and fat-1 tg mice were
6-week-old male mice. Each mouse was subcutaneously injected with
2×106 cells (mouse glioma GL261 cells) in a total volume
of 100 µl. The day of implantation was designated as day 0. The
tumor size was measured with a caliper every other day. The tumor
size was calculated as length × wide, and the tumor volume was
calculated as 0.5 × length × (width)2.
Immunohistochemistry
After deparaffinization and antigen retrieval, the
tissues were blocked with Dako Protein Block, stained with TUNEL
using the DeadEnd Fluorometric TUNEL System (Promega), LC3B
(1:3,000; Cell Signaling, Technology), p-AktS473 (1:50;
Cell Signaling, Technology), and p-AMPKT172 (1:100; Cell
Signaling, Technology) primary antibodies, visualized using an
anti-rabbit IgG conjugated secondary antibody with Texas Red and
counterstained with DAPI. These fluorescently stained tissues were
observed under a fluorescence microscope as previously described
using the DP Controller software (Olympus) for image acquisition.
Final images were a non-contrast-adjusted merge of the two
channels.
Statistical analysis
Statistical analyses were performed as recommended
by independent analysis. These included the unpaired Students
t-test. All values are expressed as mean ± SD and statistical
significance was indicated by P<0.05 (*P<0.05, **P<0.01
and ***P<0.001, respectively).
Results
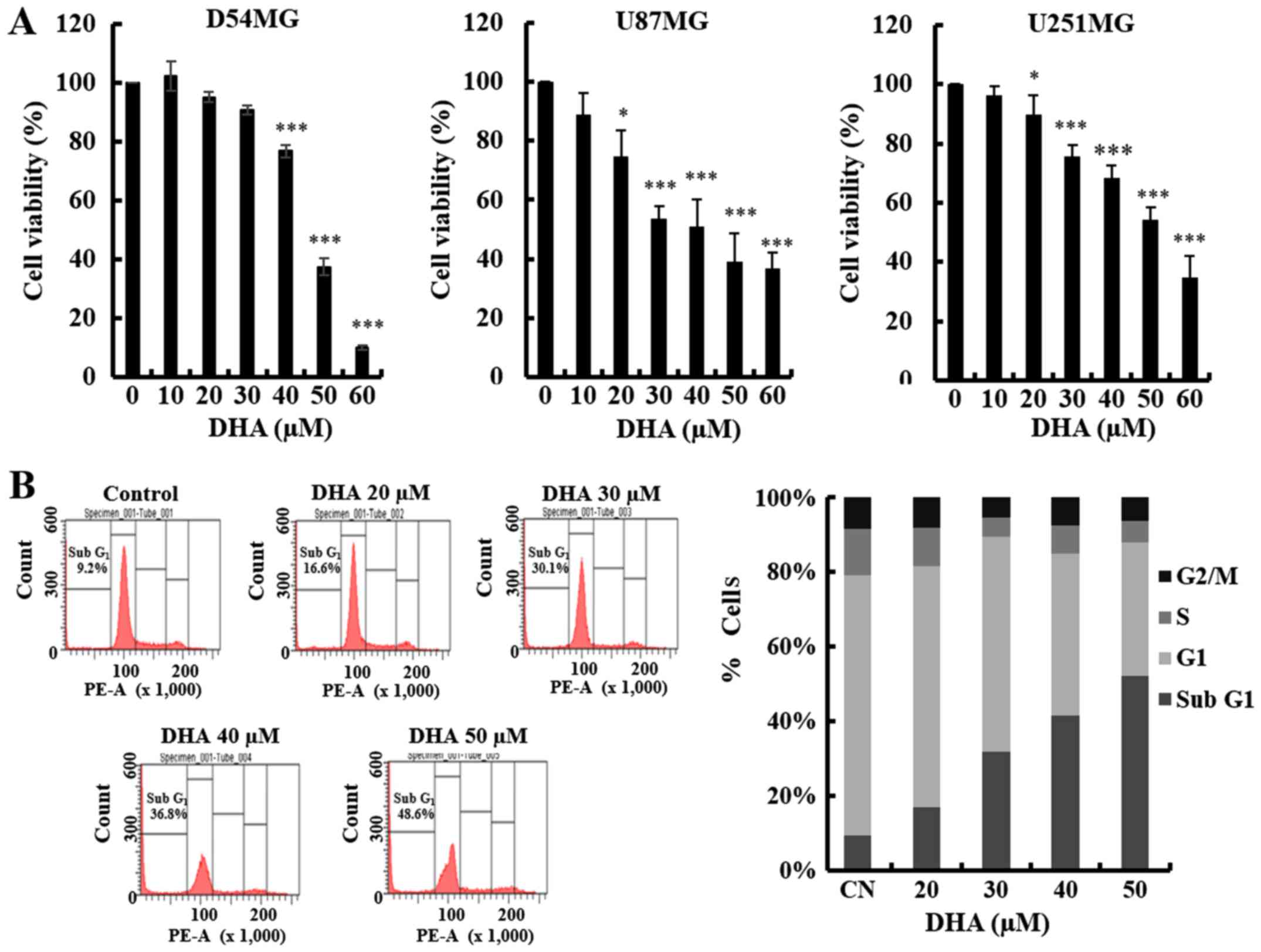
DHA induces a cytotoxic effect on GBM
cells
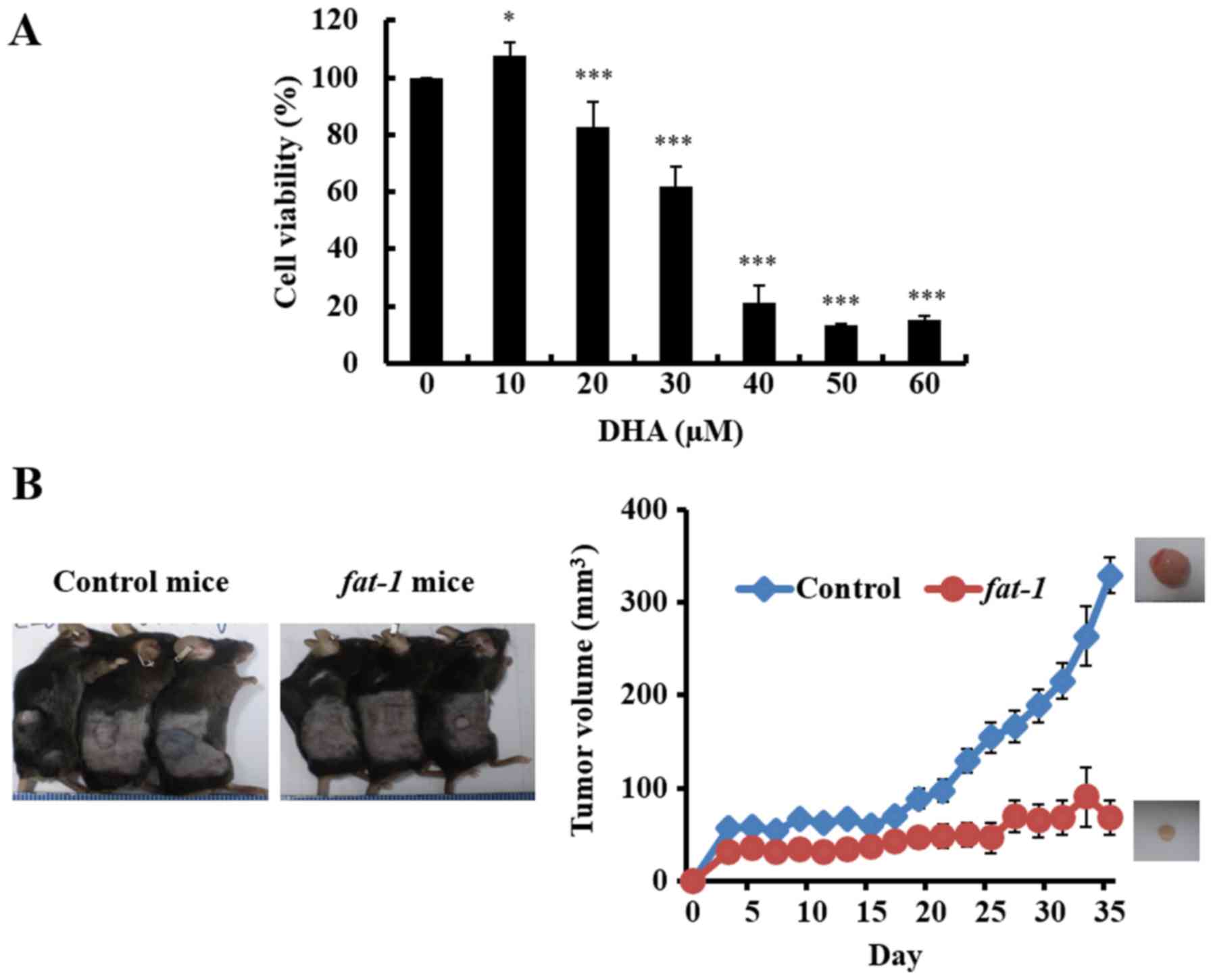
To investigate the effects of DHA on the growth of
GBM cell lines, D54MG, U87MG, U251MG and GL261, the cells were
incubated with various concentrations of DHA for 24 h, or 6 h in
the case of D54MG cells. DHA decreased the viability of each GBM
cell line in a dose-dependent manner, as determined by the MTT
assay (Fig. 1A). A decrease in cell
viability was observed at DHA concentrations of 20 µM or greater.
These findings support the hypothesis that DHA exerts a significant
influence on GBM cell proliferation.
DHA induces apoptosis and autophagy in
GBM cells
According to recent reports, DHA induces apoptosis
in various types of cancer cells by activating both intrinsic and
extrinsic pathways (16,17). To assess whether DHA also induces
apoptosis in GBM cells, we first analyzed the sub-G1 population of
DHA-treated D54MG cells by flow cytometry. As shown in Fig. 1B, DHA induced an increase in the
proportion of cells in the sub-G1 phase. To determine whether the
observed reduction in cell viability was caused by apoptosis, we
assessed the ability of DHA to induce apoptosis in human GBM cells.
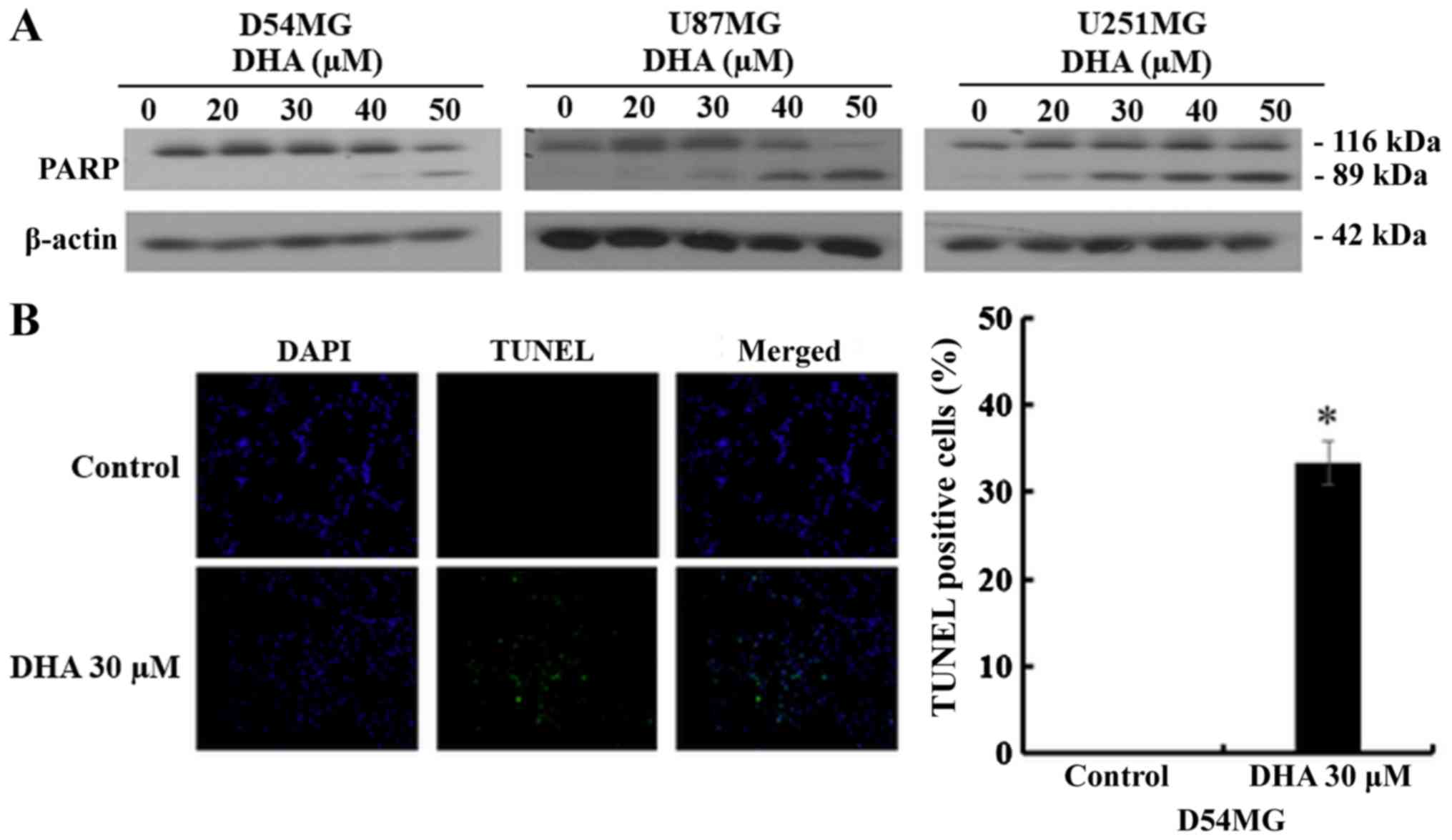
DHA led to proteolytic cleavage of poly(ADP-ribose) polymerase
(PARP), which is widely used as an apoptosis marker (Fig. 2A). Furthermore, TUNEL assay was
performed to detect apoptotic nuclear DNA breaks in D54MG cells;
after DHA treatment, the number of TUNEL-positive cells increased
significantly compared with the untreated control, in which no
apoptotic activity was observed (Fig.
2B). These results demonstrated that DHA induced apoptosis in
GBM cells.
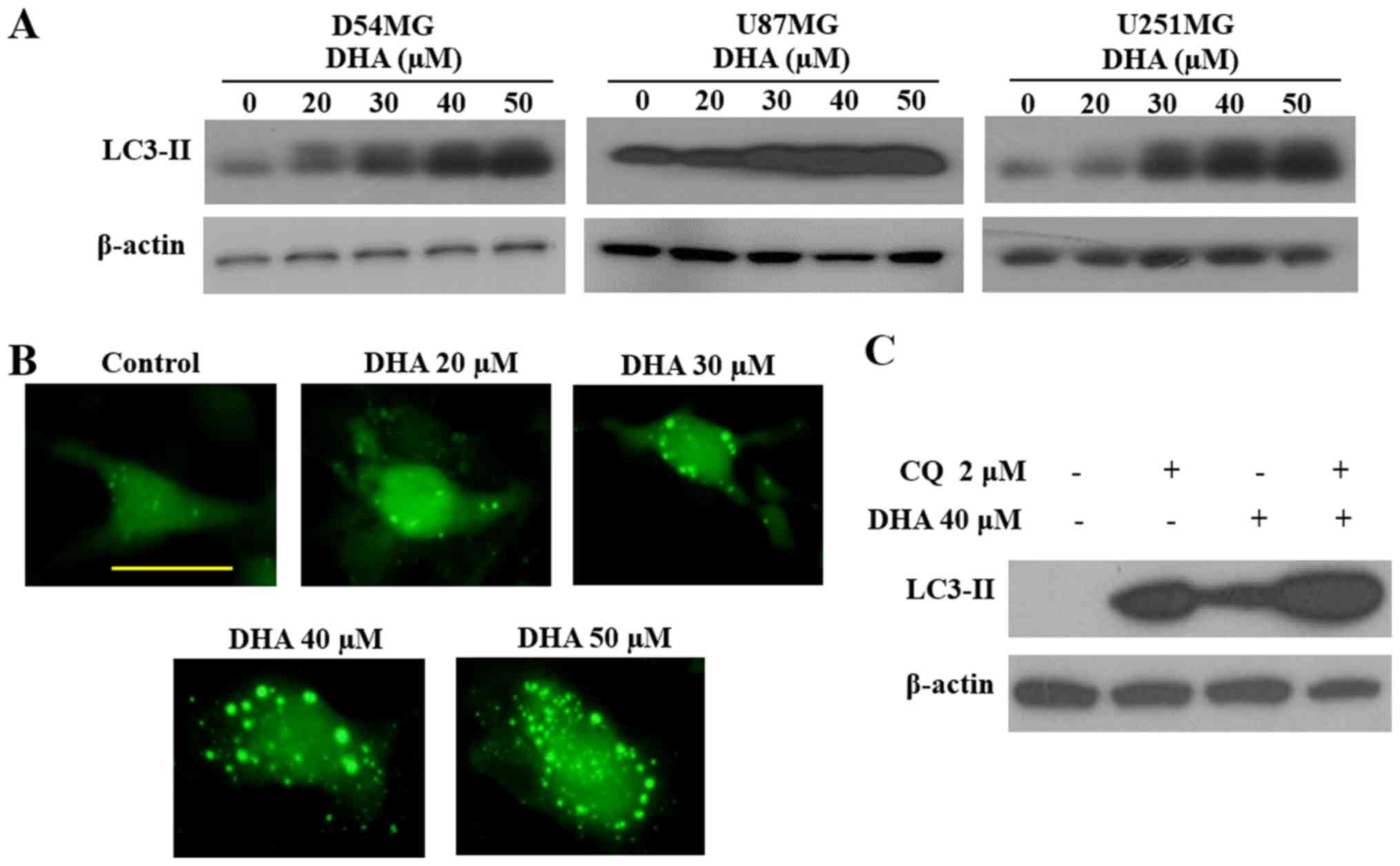
To evaluate autophagy as another possible mechanism
for induction of GBM cell death by DHA, we next performed
autophagic cell death experiments. DHA treatment increased the
level of LC3B-II, an autophagy marker, in a dose-dependent manner
(Fig. 3A). This observation was
confirmed in DHA-treated D54MG cells transfected with GFP-LC3B,
which showed a greater number of GFP-LC3B puncta compared with
untreated cells (Fig. 3B).
Moreover, autophagic flux assays in cells pretreated with
chloroquine, an inhibitor of lysosomal acidification, provided
further confirmation that DHA treatment increased LC3-II expression
(Fig. 3C). Altogether, these
observations indicate that DHA induces autophagy in GBM cells.
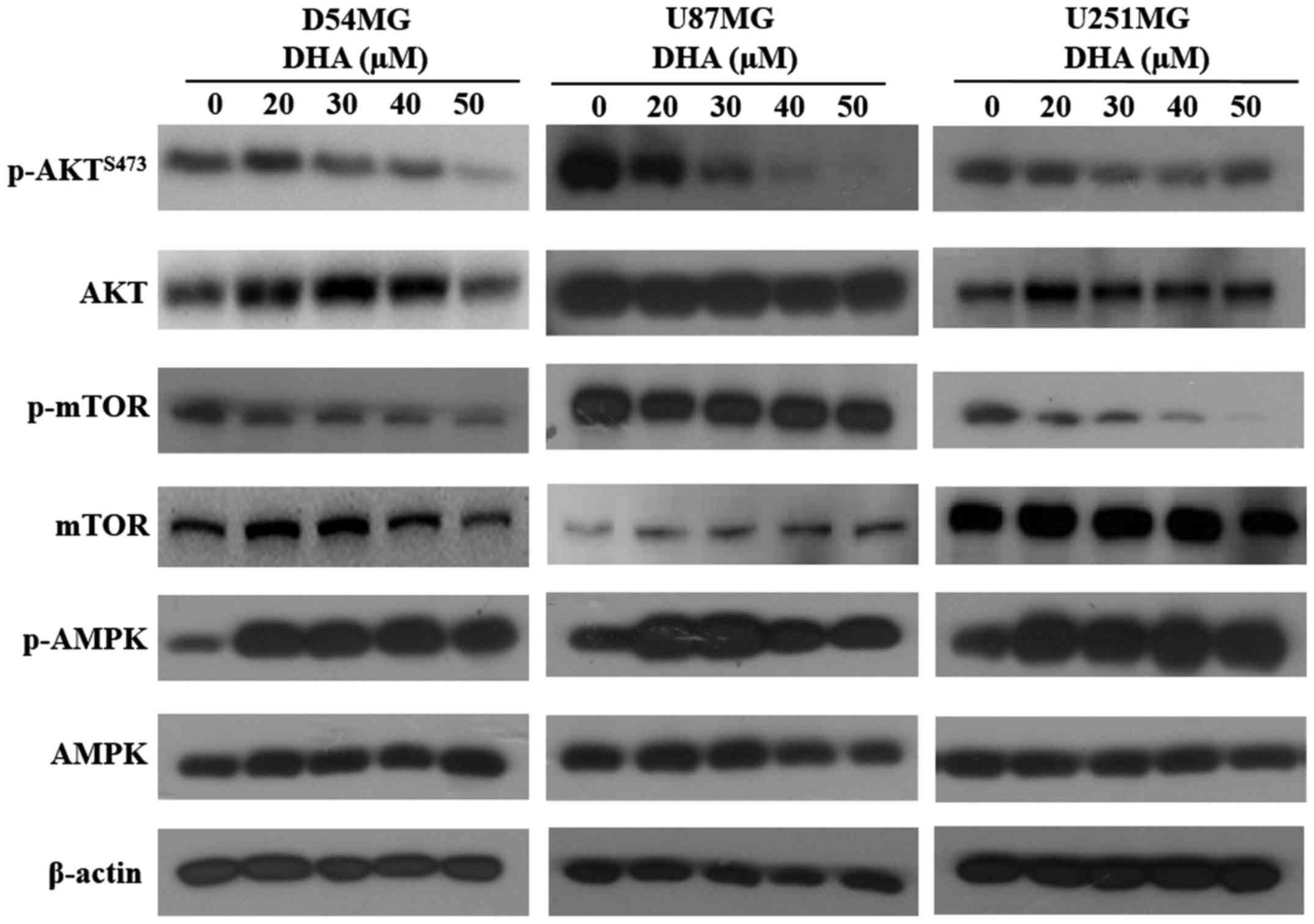
DHA inhibits mTOR signaling through
inhibition of Akt and activation of AMPK in GBM cells
The mTOR pathway is one of the most important
pathways that regulate both apoptosis and autophagy. This pathway
is also directly linked to the Akt survival pathway, leading us to
question whether the simultaneous induction of apoptosis and
autophagy by DHA in GBM cells was mediated by this signaling
pathway. To test this, we first examined the effect of DHA on Akt
expression. Treatment of GBM cells with DHA led to a marked
decrease in p-AktSer473 levels and a gradual decrease in
p-mTOR levels. We also measured AMPK levels, and observed a
significant increase in p-AMPK levels after DHA treatment (Fig. 4). These results indicate that DHA
suppresses the mTOR pathway in GBM cells through inhibition of Akt
and activation of AMPK.
ω3-PUFAs suppress tumor growth in
vivo
To evaluate the relevance of these findings in
vivo, we employed fat-1 tg mice as a model of elevated
ω3-PUFA concentration in tissue. fat-1 tg mice carry a
ω3-desaturase gene from Caenorhabditis elegans that converts
ω6-PUFAs to ω3-PUFAs, resulting in a significant increase in the
concentration of ω3-PUFA and a decrease in the concentration of
ω6-PUFAs throughout the body (18).
A mouse model of GBM was created using the mouse glioma cell line
GL261, which was chosen because this cell line is derived from a
GBM from C57BL/6 mice and forms tumors in these mice.
Before implantation of GL261 cells into mice, the
effect of DHA on GL261 cells was confirmed in vitro.
Consistent with the results obtained in human GBM cells, DHA
significantly decreased the viability of GL261 cells (Fig. 5A).
GL261 cells were implanted into fat-1 tg mice
and control mice (C57/BL6 genetic background), and the
tumorigenicity of the inoculated cells in these mice was examined.
As shown in Fig. 5B, there was a
marked difference in tumor volume between fat-1 tg (n=5) and
control mice (n=5). After 35 days, the average tumor volume was 75%
lower in the fat-1 tg mice than the tumor volume noted in
the control mice (Fig. 5C). To
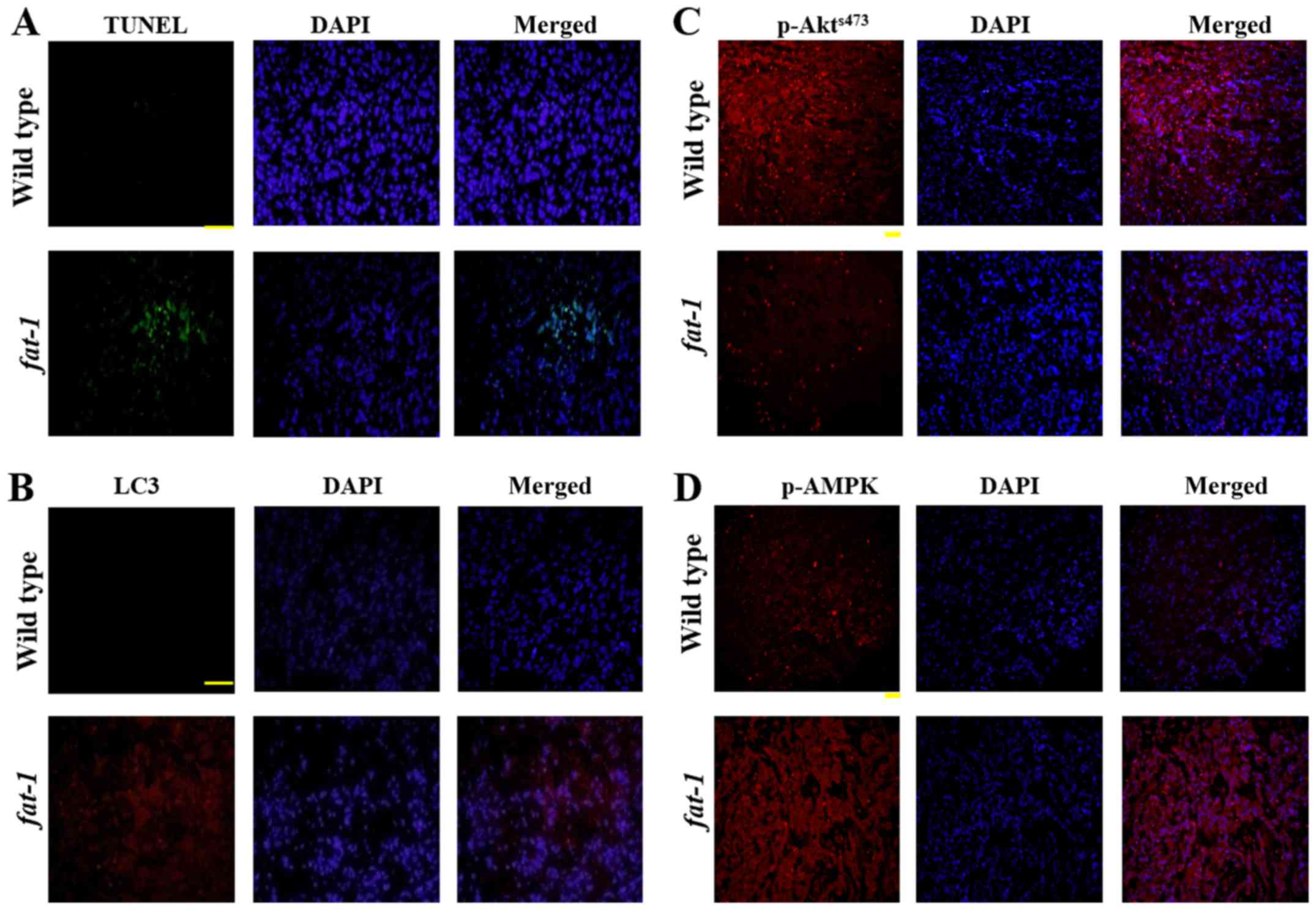
determine whether apoptosis and autophagy were also related to the
inhibition of tumor growth observed in vivo, we performed
TUNEL assays and measured LC3 levels in the tumor tissues. Both the
proportion of TUNEL-positive cells and the level of LC3B were
higher in the tumor tissue derived from fat-1 tg mice than
in tumor tissue derived from wild-type mice (Fig. 6A and B), indicating that both
apoptosis and autophagy are involved in the reduction of tumor
growth in fat-1 tg mice. Subsequently, to determine whether
Akt and AMPK were also related to the increase in apoptosis and
autophagy in these mice, the levels of Akt and AMPK in tumor tissue
were analyzed by immunohistochemistry. As shown in Fig. 6C and D, the level of
p-AktSer473 was reduced and the level of p-AMPK was
elevated in tumor tissue derived from fat-1 tg mice,
compared with tumor tissue derived from the control mice. These
data demonstrate that ω3-PUFAs induce both apoptosis and autophagy
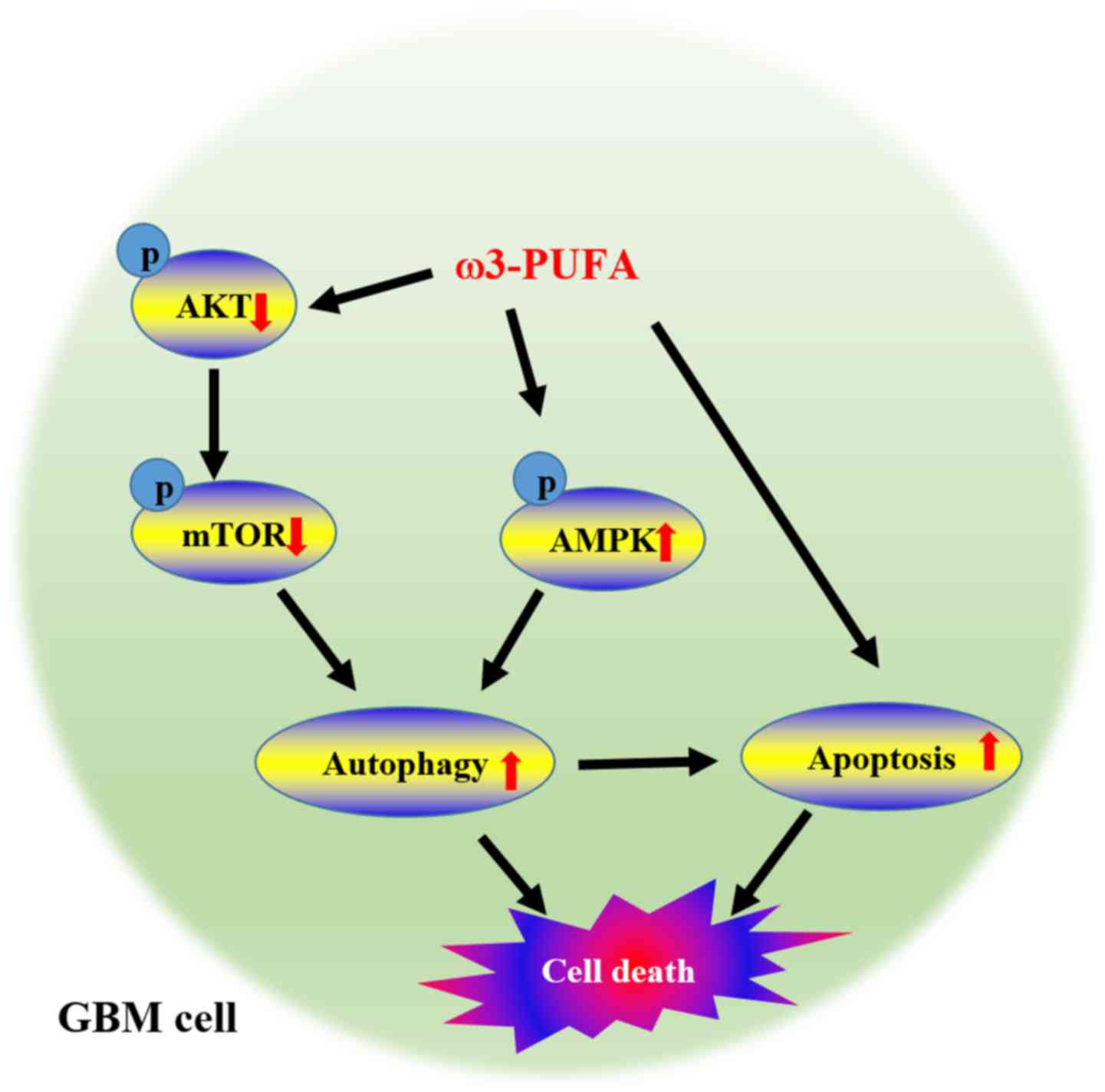
in vivo by regulating AMPK and Akt/mTOR signaling (Fig. 7).
Discussion
Compelling evidence from many studies has
demonstrated that DHA induces apoptosis and protects against a
variety of cancers, including lung cancer, via multiple targets
(8,13). Although the anticancer mechanisms of
ω3-PUFAs in several cancers have been reported, the relevant
mechanisms are still unclear in the case of brain cancer. In the
present study, we used in vitro experiments and in
vivo mouse models to evaluate the anticancer activity of
ω3-PUFAs against GBM. DHA treatment induced dose- and
time-dependent growth inhibition in three human GBM cell lines. Of
these three cell lines, D54MG cells were selected for further
experiments as the viability of D54MG cells was reduced by only
~20% after DHA treatment. The observations that DHA induced an
increase in the concentration of cleaved PARP in D54MG cells, with
a concomitant increase in the sub-G1 cell population, as
demonstrated by cell cycle analysis using flow cytometry, confirmed
that DHA induced apoptotic cell death in D54MG cells (Figs. 1 and 2). Additionally, using the TUNEL assay, a
common method for detecting DNA fragmentation resulting from
apoptotic signaling cascades, the proportion of TUNEL-positive
D54MG cells was shown to increase after DHA treatment (Fig. 2B). Gobeil et al (19) reported the possible implication of
lysosomal enzymes in the necrotic cleavage of PARP-1. They observed
two major PARP-1 fragments (a C-terminal 55 kDa active and a
N-terminal 62 kDa inactive fragment) after necrotic inducers like
H2O2 in Jurkat T cells. In our data, only an
89-kDa fragment was observed after DHA treatment (Fig. 2A) in the GBM cells. These results
strongly indicate that DHA induced apoptotic cell death, not
necrotic cell death in the GBM cells.
Autophagy and apoptosis have been shown to be
interconnected by crosstalk between several molecular nodes. The
functional relationship between apoptosis and autophagy is complex;
in some circumstances, autophagy constitutes a stress adaptation to
avoid cell death, whereas in other circumstances, it constitutes an
alternative cell death pathway. Both autophagy and apoptosis can be
triggered by a common upstream signal (20). Jing et al (13) reported that autophagy and apoptosis
occur simultaneously and act cooperatively to induce cell death
after DHA treatment. To determine whether autophagy is also induced
by DHA in GBM cells, we first examined the levels of LC3-II, an
autophagy marker. DHA increased the expression levels of LC3-II in
three GBM cell lines (Fig. 3A).
These results were confirmed conclusively in D54MG cells
transfected with GFP-LC3, which showed a higher number of GFP-LC3
puncta after exposure to DHA (Fig.
3B). Moreover, autophagic flux assays in cells pretreated with
chloroquine, a lysosomal inhibitor, confirmed that DHA markedly
increased LC3-II expression (Fig.
3C). These results clearly demonstrated that DHA induces GBM
cell death by both apoptosis and autophagy.
The Akt/mTOR pathway is one of the most frequently
dysregulated pathways in several types of cancers. Several
selective inhibitors of Akt and mTOR have been investigated, both
in preclinical tumor models and in various clinical trials;
however, these inhibitors have been found not to improve survival
significantly because of feedback activation of the Akt/mTOR
pathway (21). Several other
studies have shown that DHA inhibits the Akt/mTOR pathway in
various cancer cells, including prostate cancer cells (14,22).
We therefore assessed whether inhibition of Akt/mTOR signaling is
also related to apoptotic and autophagic cell death in GBM by
testing the effect of DHA exposure on Akt and mTOR levels in three
GBM cell lines. DHA treatment caused a dose-dependent decrease in
p-AktSer473 and p-mTOR levels, suggesting that
DHA-induced autophagy is related to the inhibition of mTOR through
downregulation of p-Akt.
mTOR receives signals from nutrients, growth
factors, and many cellular kinases, including AMPK. Phosphorylation
of AMPK activates downstream signaling that triggers mTOR
inhibition and autophagy (23,24).
Jing et al (13) reported
that DHA inhibits mTOR through AMPK activation. Our data confirmed
that DHA increases the level of p-AMPK and that DHA-induced
autophagy is related to inhibition of mTOR through activation of
AMPK.
To support these in vitro data, we performed
in vivo experiments using fat-1 tg mice, implanting
GL261 mouse glioma cells into fat-1 tg mice and wild-type
mice. fat-1 tg mice ubiquitously express a C. elegans
ω3-desaturase, leading to a significant increase in the
ω3-PUFA/ω6-PUFA ratio across all organs and tissues (18). This mouse model was selected as the
endogenous source of ω3-PUFAs provides a consistent ω3-PUFA
concentration and eliminates potential dietary variations
associated with long-term feeding of ω3-PUFAs. A significant
reduction in GL261 primary tumor growth was observed in the
fat-1 tg mice (Fig. 5B), and
tumor tissue derived from fat-1 tg mice exhibited an
increase in the proportion of TUNEL-positive cells (Fig. 6A) and LC3-II levels (Fig. 6B) compared with tumor tissue derived
from wild-type mice. These findings provide important in
vivo evidence for growth retardation of GL261 cells by ω3-PUFAs
via induction of apoptosis and autophagy. Moreover, we confirmed
that p-Akt levels were reduced (Fig.
6C) and p-AMPK levels were elevated (Fig. 6D) in tumor tissue from fat-1
tg mice, compared with tumor tissue from wild-type mice. Thus, the
present study clearly demonstrates that ω3-PUFAs, including DHA,
induce GBM cell death through repression of mTOR via Akt inhibition
and AMPK activation (Fig. 7). In
summary, this study provides important information on the mechanism
underlying the antitumorigenic activity of ω3-PUFAs and encouraging
preclinical evidence for a possible strategy for the
chemoprevention and treatment of GBM. Therefore, it is possible
that ω3-PUFAs, being non-toxic, may represent safe chemopreventive
and therapeutic agents for GBM patients.
Acknowledgements
This study was supported by the National Research
Foundation of Korea (NRF) grant funded by the Korea government
(MSIP) (NRF-2015R1D1A1A01056887) and research fund of Chungnam
National University (2014). We thank Dr Jing X. Kang at the Harvard
Medical School for providing the Fat-1 transgenic mice.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Holland EC: Glioblastoma multiforme: The
terminator. Proc Natl Acad Sci USA. 97:pp. 6242–6244. 2000;
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Preusser M, de Ribaupierre S, Wöhrer A,
Erridge SC, Hegi M, Weller M and Stupp R: Current concepts and
management of glioblastoma. Ann Neurol. 70:9–21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Simopoulos AP: Human requirement for N-3
polyunsaturated fatty acids. Poult Sci. 79:961–970. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Simopoulos AP: Omega-3 fatty acids in
inflammation and autoimmune diseases. J Am Coll Nutr. 21:495–505.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Simopoulos AP: The importance of the ratio
of omega-6/omega-3 essential fatty acids. Biomed Pharmacother.
56:365–379. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Song KS, Jing K, Kim JS, Yun EJ, Shin S,
Seo KS, Park JH, Heo JY, Kang JX, Suh KS, et al:
Omega-3-polyunsaturated fatty acids suppress pancreatic cancer cell
growth in vitro and in vivo via downregulation of Wnt/Beta-catenin
signaling. Pancreatology. 11:574–584. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lim K, Han C, Dai Y, Shen M and Wu T:
Omega-3 polyunsaturated fatty acids inhibit hepatocellular
carcinoma cell growth through blocking beta-catenin and
cyclooxygenase-2. Mol Cancer Ther. 8:3046–3055. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lim K, Han C, Xu L, Isse K, Demetris AJ
and Wu T: Cyclooxygenase-2-derived prostaglandin E2 activates
beta-catenin in human cholangiocarcinoma cells: Evidence for
inhibition of these signaling pathways by omega 3 polyunsaturated
fatty acids. Cancer Res. 68:553–560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jeong S, Jing K, Kim N, Shin S, Kim S,
Song KS, Heo JY, Park JH, Seo KS, Han J, et al: Docosahexaenoic
acid-induced apoptosis is mediated by activation of
mitogen-activated protein kinases in human cancer cells. BMC
Cancer. 14:4812014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yun EJ, Song KS, Shin S, Kim S, Heo JY,
Kweon GR, Wu T, Park JI and Lim K: Docosahexaenoic acid suppresses
breast cancer cell metastasis by targeting
matrix-metalloproteinases. Oncotarget. 7:49961–49971. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim N, Jeong S, Jing K, Shin S, Kim S, Heo
JY, Kweon GR, Park SK, Wu T, Park JI, et al: Docosahexaenoic acid
induces Cell death in human non-small cell lung cancer cells by
repressing mTOR via AMPK activation and PI3K/Akt inhibition. Biomed
Res Int. 2015:2397642015.PubMed/NCBI
|
|
13
|
Jing K, Song KS, Shin S, Kim N, Jeong S,
Oh HR, Park JH, Seo KS, Heo JY, Han J, et al: Docosahexaenoic acid
induces autophagy through p53/AMPK/mTOR signaling and promotes
apoptosis in human cancer cells harboring wild-type p53. Autophagy.
7:1348–1358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shin S, Jing K, Jeong S, Kim N, Song KS,
Heo JY, Park JH, Seo KS, Han J, Park JI, et al: The omega-3
polyunsaturated fatty acid DHA induces simultaneous apoptosis and
autophagy via mitochondrial ROS-mediated Akt-mTOR signaling in
prostate cancer cells expressing mutant p53. BioMed Res Int.
2013:5686712013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jing K, Shin S, Jeong S, Kim S, Song KS,
Park JH, Heo JY, Seo KS, Park SK, Kweon GR, et al: Docosahexaenoic
acid induces the degradation of HPV E6/E7 oncoproteins by
activating the ubiquitin-proteasome system. Cell Death Dis.
5:e15242014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zajdel A, Wilczok A, Latocha M, Tarkowski
M, Kokocińska M and Dzierzewicz Z: Polyunsaturated fatty acids
potentiate cytotoxicity of cisplatin in A549 cells. Acta Pol Pharm.
71:1060–1065. 2014.PubMed/NCBI
|
|
17
|
Gleissman H, Johnsen JI and Kogner P:
Omega-3 fatty acids in cancer, the protectors of good and the
killers of evil? Exp Cell Res. 316:1365–1373. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kang JX, Wang J, Wu L and Kang ZB:
Transgenic mice: Fat-1 mice convert n-6 to n-3 fatty acids. Nature.
427:5042004. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gobeil S, Boucher CC, Nadeau D and Poirier
GG: Characterization of the necrotic cleavage of poly(ADP-ribose)
polymerase (PARP-1): Implication of lysosomal proteases. Cell Death
Differ. 8:588–594. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jhanwar-Uniyal M, Albert L, McKenna E,
Karsy M, Rajdev P, Braun A and Murali R: Deciphering the signaling
pathways of cancer stem cells of glioblastoma multiforme: Role of
Akt/mTOR and MAPK pathways. Adv Enzyme Regul. 51:164–170. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schley PD, Jijon HB, Robinson LE and Field
CJ: Mechanisms of omega-3 fatty acid-induced growth inhibition in
MDA-MB-231 human breast cancer cells. Breast Cancer Res Treat.
92:187–195. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Faivre S, Kroemer G and Raymond E: Current
development of mTOR inhibitors as anticancer agents. Nat Rev Drug
Discov. 5:671–688. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shaw RJ: Glucose metabolism and cancer.
Curr Opin Cell Biol. 18:598–608. 2006. View Article : Google Scholar : PubMed/NCBI
|