Introduction
Lung cancer is one of the most deadly malignancies,
as it is the leading cause of cancer-related mortality worldwide.
Lung cancer can be divided into two main subtypes: non-small cell
lung cancer (NSCLC) accounting for almost 80% of lung cancer cases
(1–4), and small cell lung cancer accounting
for ~20% (5,6). Today, treatments for lung cancer
patients rely mainly on radiotherapy and chemotherapy (7). Recent studies have shown that
microRNAs (miRNAs) are of great value in the early diagnosis and
treatment of NSCLC. Therefore, it is important to identify
effective miRNAs and explore their roles and underlying mechanisms
for the diagnosis, prediction and prevention of NSCLC (8).
miRNAs are a type of non-coding RNA (ncRNA), ~21–24
nucleotides in length, that function in the post-transcriptional
regulation of gene expression. Typically miRNAs interact with
specific mRNAs through complementary base-pairing to influence the
translation or stability of the target mRNA molecule (9–11). The
important biological processes involved in miRNAs include the
development, differentiation, apoptosis and proliferation of cells
(9,12,13).
The abnormal expression of miRNAs is usually associated with
inhibition or induction of the progression of cancer. Studies have
shown that miRNAs can function as tumor-suppressor genes or
oncogenes (14,15). miR-16 is widely found in different
species. miR-16 plays important roles in regulating the development
of organisms and cell self-renewal, differentiation and many other
physiological activities. The miRNA microarray analysis of lung
cancer and adjacent normal lung tissues showed the miR-16 is
downregulated in lung cancer tissues (16–18).
miR-16 was found to be downregulated in lung squamous cell
carcinomas and adenocarcinomas, which was correlated with the
expression of cyclin D1 (19).
miR-16 may be a prognostic marker in NSCLC. The level of miR-16 was
found to be associated with the outcome of NSCLC patients (20). Moreover, Ke et al
demonstrated that miR-16 is downregulated in NSCLC tissue samples
and cell lines (21).
Overexpression of miR-16 significantly suppressed cell
proliferation, colony formation, cell migration and cell invasion
in NSCLC cells by targeting hepatoma-derived growth factors
(21). Thus, miR-16 plays an
essential role in NSCLC, and may be associated with NSCLC
progression. It was reported that A549 cells express a lower level
of miR-16 than normal bronchial epithelial cells, and that other
mRNAs are involved in the suppression of NSCLC cell proliferation
and promotion of apoptosis by miR-16 such as wip1 (17). It was reported that overexpression
of miR-16 inhibited cell proliferation and induced apoptosis via
regulating the expression of p27, Bcl-2, Bax and caspase-3 in NSCLC
cells (16). A recent study
demonstrated that miR-16 is a potent inducer of autophagy (22). Overexpression of miR-16 inhibited
the phosphorylation of mTORC1 and p70S6K, inhibiting cell
proliferation and G1/S cell cycle transition in human cervival
carcinoma HeLa cells, and enhanced anticancer drug
camptothecin-induced autophagy and apoptotic cell death in HeLa
cells by targeting Rictor (22).
Autophagy was enhanced by miR-16 overexpression in skeletal muscle
(23). However, the role of miR-16
in NSCLC cell autophagy and the underlying mechanism are still
unclear.
TGF-β1 plays an important role in the induction of
epithelial-to-mesenchymal transition (EMT) (24,25).
Other miRNAs such as miR-19 were found to inhibit the autophagy of
human cardiac fibroblasts by targeting TGF-βRII mRNA during
TGF-β1-induced fibrogenesis (26).
Autophagy is critical for the metastasis of cancer cells through
the induction of EMT and activation of TGF-β signaling plays a key
role in regulating autophagy-induced EMT (27). In erlotinib-resistant lung
adenocarcinoma, cells expressed high basal autophagy-related 3
protein (ATG3) (28). ATG3-mediated
autophagy also plays an important role in apoptotic cell death of
NSCLC cells (28).
In the present study, we hypothesized that ATG3 may
be a target gene of miR-16 playing an important role in the
autophagy of NSCLC cells using bioinformatics tools. To test
whether miR-16 targets ATG3 which is involved in the autophagy of
NSCLC cells, we studied the expression levels of miR-16 and ATG3 in
NSCLC patient tissues, verified the targeting of ATG3 by miR-16 by
luciferase reporter gene system, and investigated the role of
miR-16 in the autophagy and EMT of NSCLC cells. Finally, the
results showed that miR-16 overexpression rescued TGF-β1-mediated
inhibition of autophagy and plays important roles in the EMT of
NSCLC cells.
Materials and methods
NSCLC patients and tissue samples
The present study was approved by the Ethics
Committee of The Second Hospital of Shandong University and was
carried out according to the World Medical Association Declaration
of Helsinki. All patients were enrolled after obtaining written
informed consents. NSCLC tissue samples and their matched adjacent
tissues were collected from 20 patients. Among these patients, 14
were male and 6 were female, with the median age of 49 years.
Cell culture and transfection
The human NSCLC cell lines A549, NCI-H1299 and
HCC827 were purchased from the Cell Bank, Chinese Academy of
Sciences (Shanghai, China), and the 293 cell line and normal human
bronchial epithelial (HBE) cells were purchased from the American
Type Culture Collection (ATCC; Manassas, VA, USA). All cells were
cultured in Dulbeccos modified Eagles medium (DMEM) containing 10%
fetal bovine serum (FBS) (both from Invitrogen, Carlsbad, CA, USA)
and maintained at 37°C in a humidified atmosphere containing 5%
CO2.
Cells were treated with 5 ng/ml TGF-β1 (PeproTech,
Rocky Hill, NJ, USA) for 24 h. The miR-16 mimics and negative
control (NC) were purchased from Biomics Biotechnology (Jiangsu,
China). A549 cells were seeded into a 6-well plate
(2.5×105/well) for 24 h before transfection, and then
were transfected with miR-16 mimics (50 nM) or NC (50 nM) using
Lipofectamine 2000 (Invitrogen) in accordance with the
manufacturer's instructions. After transfection, the cells were
grown in medium without antibiotics for 48 h and then were used for
the following experiments. The expression of miR-16 and ATG3 in
transfected cells was detected by qRT-PCR.
Quantitative real-time polymerase
chain reaction (qRT-PCR)
Total RNA was extract from the tissues and cells
using the RNeasy/miRNeasy kit (Qiagen, Hilden, Germany). Total RNA
(1 µg) was reverse-transcribed to cDNA using SuperScript II reverse
transcriptase (Invitrogen) according to the manufacturer's
instructions. PCR analysis was performed using Applied Biosystems
7500 Sequence Detection system [Applied Biosystems (ABI) Foster
City, CA, USA] using SYBR Premix Ex Taq GC kit (Takara, Tokyo,
Japan). The following primers were used: miR-16,
5′-TCGGCGTAGCAGCACGTAAAT-3′ (sense) and
5′-GTATCCAGTGCAGGGTCCGAGGT-3′ (antisense); U6,
5′-CTCGCTTCGGCAGCACA-3′ (sense) and 5′-AACGCTTCACGAATTTGCGT-3′
(antisense); ATG3, 5′-CCAACATGGCAATGGGCTAC-3′ (sense) and
5′-ACCGCCAGCATCAGTTTTGG-3′ (antisense); GAPDH,
5′-CGACCACTTTGTCAAGCTCA123-3′ (sense) and
5′-AGGGGAGATTCAGTGTGGTG-3′ (antisense). The gene expression was
normalized to the level of GAPDH using the relative ΔΔCt
method.
Acridine orange (AO) staining and
TEM
Formation of acidic vesicular organelles (AVO) is a
characteristic feature of autophagic cells (29,30).
AO staining was performed as previously described (31) to detect AVO in miR-16
mimic-transfected cells after treatment with TGF-β1. Briefly, cells
grown overnight were transfected with miR-16 mimics/NC, exposed to
5 ng/ml TGF-β1 for 24 h in slide cultures, and then incubated with
AO (1 µg/ml) for 15 min for staining. Another set of TGF-β1
unexposed cells was used as control. After washing thrice with
phosphate-buffered saline (PBS), pH 7.4, all slides containing the
AO-stained cells were detected by flow cytometric analysis (Beckman
Coulter, Miami, FL, USA) after AO staining. On the basis of
fluorescence intensity, formation of AVO in autophagic cells can be
determined. Transmission electron microscopy (TEM) was also
performed for the observation of autophagosomes. LysoTracker
staining was performed by LysoTracker Red kit (Beyotime, Shanghai,
China) according to the manufacturers instructions. For detection
of hydrolytic activity of cathepsin D, cells were fixed with 4%
polyformaldehyde, blocked with 1% bovine serum albumin, and fixed
with rabbit primary antibody (1:100; AF1645; Beyotime) overnight
and incubated with the goat-anti rabbit secondary antibody (1:500;
A-11034; Invitrogen). The images were captured using a confocal
microscope (Leica TCS SP5; Leica Biosystems, Wetzlar, Germany).
Luciferase reporter assay and
electrophoretic mobility-shift assay (EMSA)
The wild-type and mutant ATG3 3′UTR sequences were
amplified by PCR and ligated into the pMIR-REPORT luciferase vector
(Ambion, Austin, TX, USA) to yield pMIR-ATG3 3′UTR (ATG3 3′UTR).
293 cells are easy to transfect. In the present study, we used 293T
cells to detect whether miR-9 directly targets on E-cadherin. The
293 cells were seeded into 6-well plates and cotransfected with
wild-type/mutant ATG3 3′UTR and miR-16 mimics/NC using
Lipofectamine 2000. After 24 h, cells were harvested, lysed and
luciferase reporter assay was carried out according to the
manufacturer's instructions (Promega, Madison, WI, USA).
Interaction of miR-16 and ATG3 was detected by LightShift
Chemiluminescent RNA EMSA kit (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the manufacturers instructions.
Western blotting
Total proteins were extracted from the NSCLC tissues
and cell lines using ice-cold radioimmunoprecipitation assay (RIPA)
buffer. Protein concentration was quantified using Bio-Rad DC
Protein Assay kit (Bio-Rad, Hercules, CA, USA). Protein (50–100 µg)
was separated on sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) gel, and transferred to nitrocellulose
membranes (Millipore, Billerica, MA, USA). The membranes were
blocked with 5% non-fat milk in TBS-Tween and incubated with the
primary antibody against ATG3 (1:500; sc-100508), LC3B (1:200;
sc-271625), E-cadherin (1:200; sc-71009), V-cadherin (1:200;
sc-52751) and GAPDH (1:1,000; sc-47724) overnight at 4°C, followed
by incubation with HPR-conjugated secondary antibody (all from
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 60 min at
room temperature. Polyclonal anti-GAPDH was used as an internal
control. Then, the blots were visualized using the ECL detection
system (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
Cell invasion assay
Cell invasion was performed using Transwell inserts
with a 8-µm membrane (Corning Inc., Corning, NY, USA) precoated
with Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). Cells
(1×105/well) were seeded in the upper chamber, and
medium containing 10% FBS was added to the lower chamber. Following
incubation for 24 h, the non-invaded cells were removed with a swab
cotton and the invaded cells on the surface of the membrane were
fixed in ethanol, stained with haematoxylin, imaged and counted
using microscopy.
Statistical analysis
All experiments were performed at least three times.
All statistical analysis was performed using SPSS version 19.0
(SPSS, Inc., Chicago, IL, USA) using the Student's t-test. Data are
presented as the mean ± SD. P<0.05 was considered to indicate a
statistically significant result.
Results
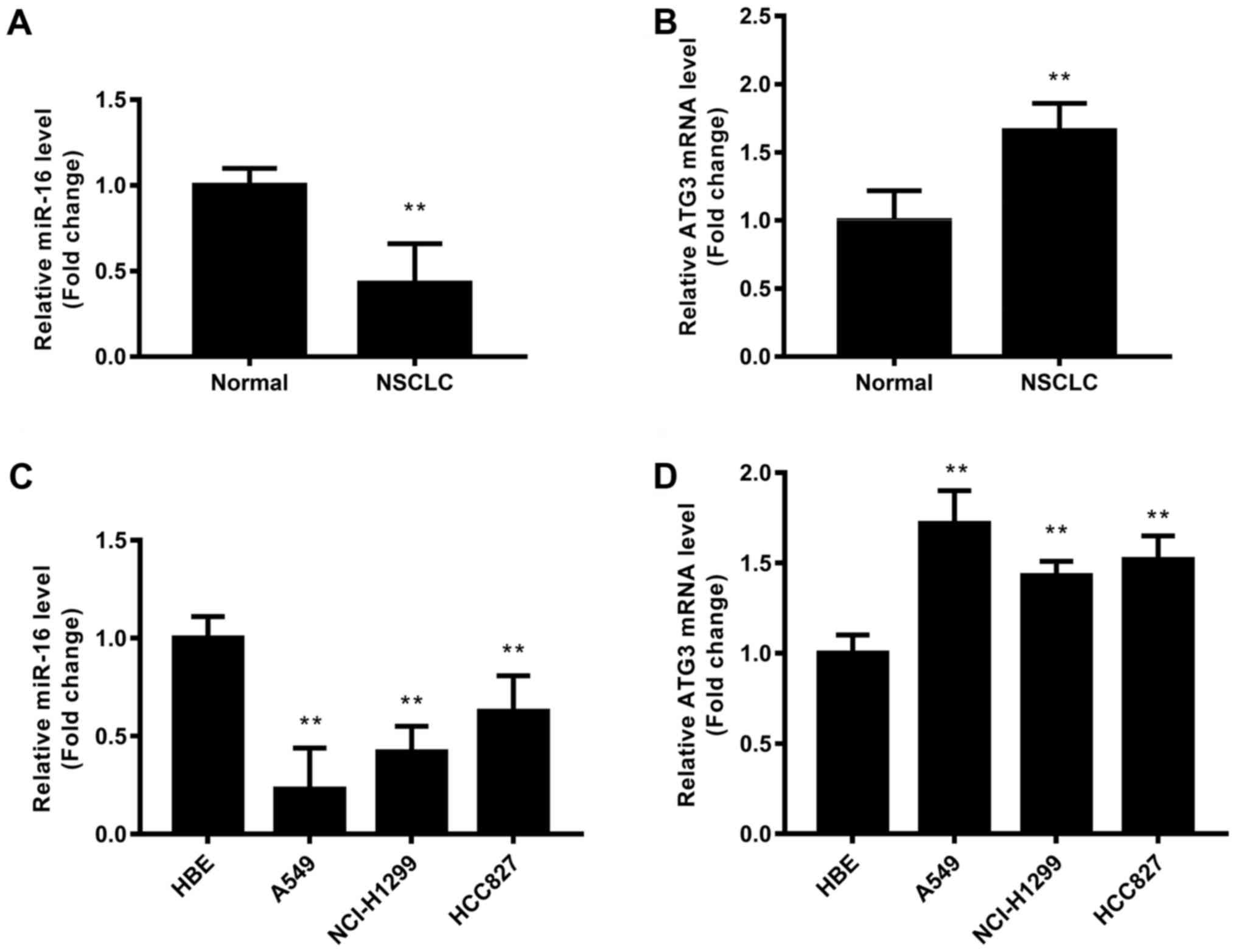
miR-16 is downregulated in NSCLC
tissues, and ATG3 is upregulated in NSCLC tissues
NSCLC and adjacent tissues pairs were collected
(n=20), and then the expression levels of miR-16 and ATG3 mRNA were
detected by qRT-PCR (Fig. 1). The
results revealed that miR-16 was significantly downregulated in the
NSCLC tissues compared with that noted in the normal tissues
(P<0.01) (Fig. 1A). ATG3 mRNA in
NSCLC tissues was significantly upregulated (P<0.01) (Fig. 1B). Thus, miR-16 was significantly
downregulated and ATG3 was significantly upregulated in the NSCLC
patient tissues.
In order to test the roles of miR-16 and ATG3 in
NSCLC, NSCLC cell lines including A549, HCI-H1299 and HCC827 were
used. The normal HBE cell line was used as a control. The results
showed that miR-16 expression was significantly downregulated in
all three NSCLC cell lines, while ATG3 mRNA was significantly
upegulated in all three NSCLC cell lines, compared with the HBE
cells (Fig. 1C and D). It was
suggested that miR-16 expression was inversely correlated with ATG3
expression in the NSCLC cell lines. A549 that was found to have the
lowest endogenous miR-16 expression and highest endogenous ATG3
expression was selected for the following experiments.
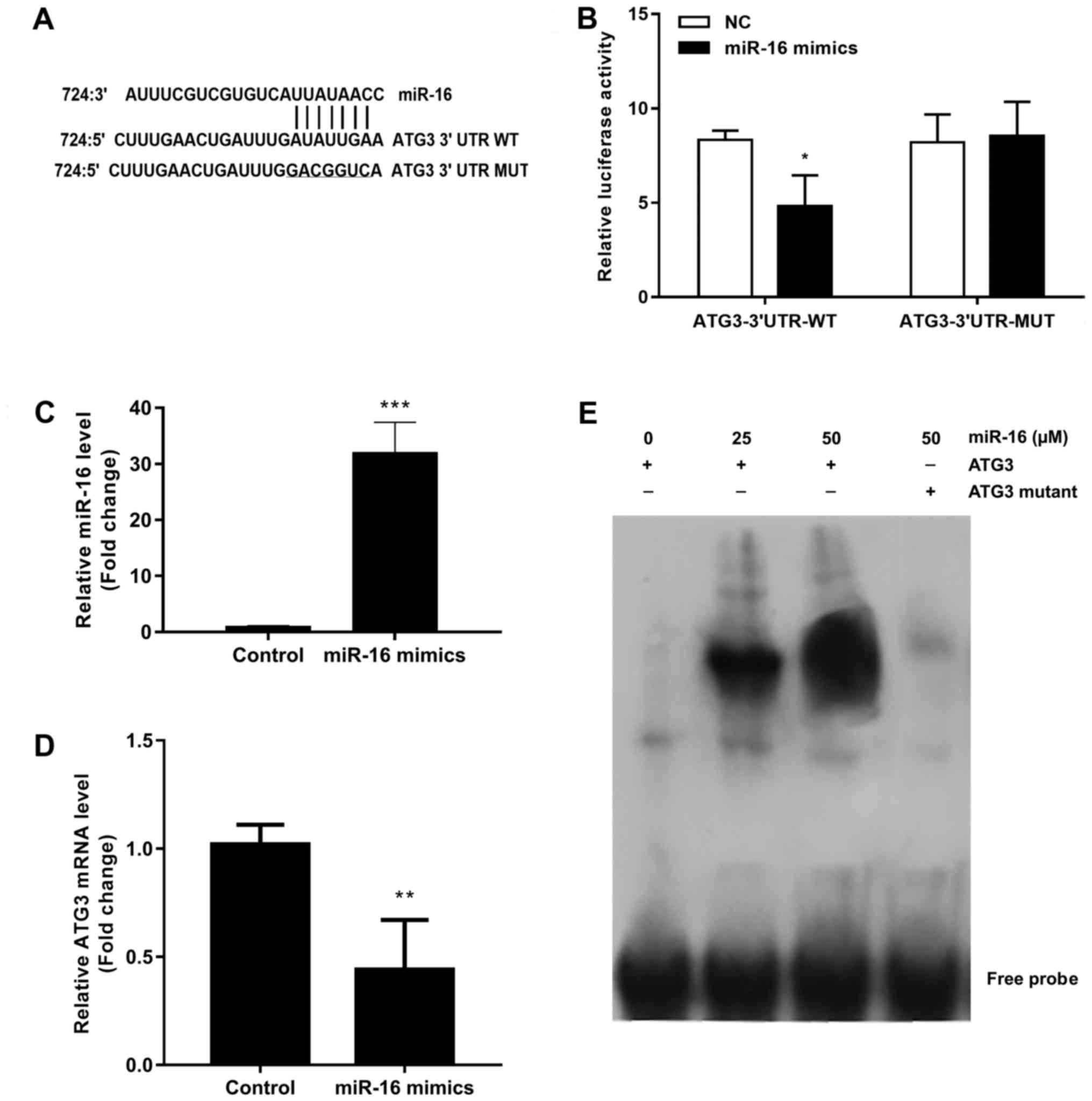
ATG3 is a direct target of miR-16
To identify whether ATG3 is a direct target gene of
miR-16, we predicted the interaction site of miR-16 and ATG3 3′UTR.
The predicted binding sequences of wild-type or mutant ATG3 3′UTR
are shown in Fig. 2A, and they were
transfected into 293 cells together with miR-16 mimics or NC. The
results of the luciferase reporter assay showed that the
transcriptional activity of wild-type ATG3 3′UTR was significantly
decreased by miR-16 mimics (Fig.
2B). The transcriptional activity of mutant ATG3 3′URT was not
affected by miR-16 mimics (Fig.
2B). The role of miR-16 in ATG3 was confirmed by transfecting
miR-16 mimics into A549 cells. The results of the qRT-PCR analysis
demonstrated that miR-16 was significantly upregulated in the
miR-16 mimic-transfected cells (Fig.
2C), and the expression of ATG3 was also significantly
downregulated in the cells transfected with the miR-16 mimics
(Fig. 2D). The interaction of
miR-16 and ATG3 was confirmed by EMSA (Fig. 2E).
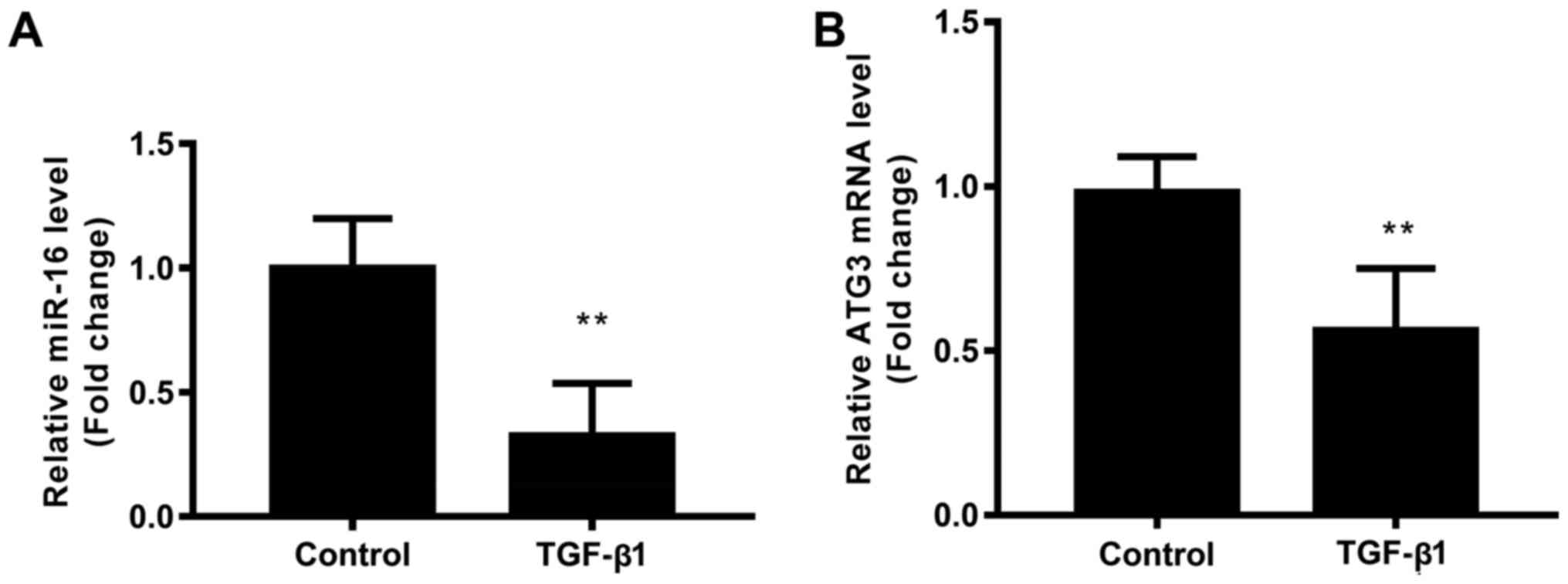
TGF-β1 inhibits the expression of
miR-16 and ATG3
To investigate the effect of miR-16 on
TGF-β1-modulated NSCLC cell autophagy, the expression of miR-16 and
ATG3 in A549 cells in the presence of TGF-β1 were detected. Results
showed that TGF-β1 significantly downregulated the expression of
miR-16 (Fig. 3A), and downregulated
the expression of ATG3 mRNA (Fig.
3B), indicating the miR-16 and ATG3 play an important role in
TGF-β1-modulated NSCLC cell function.
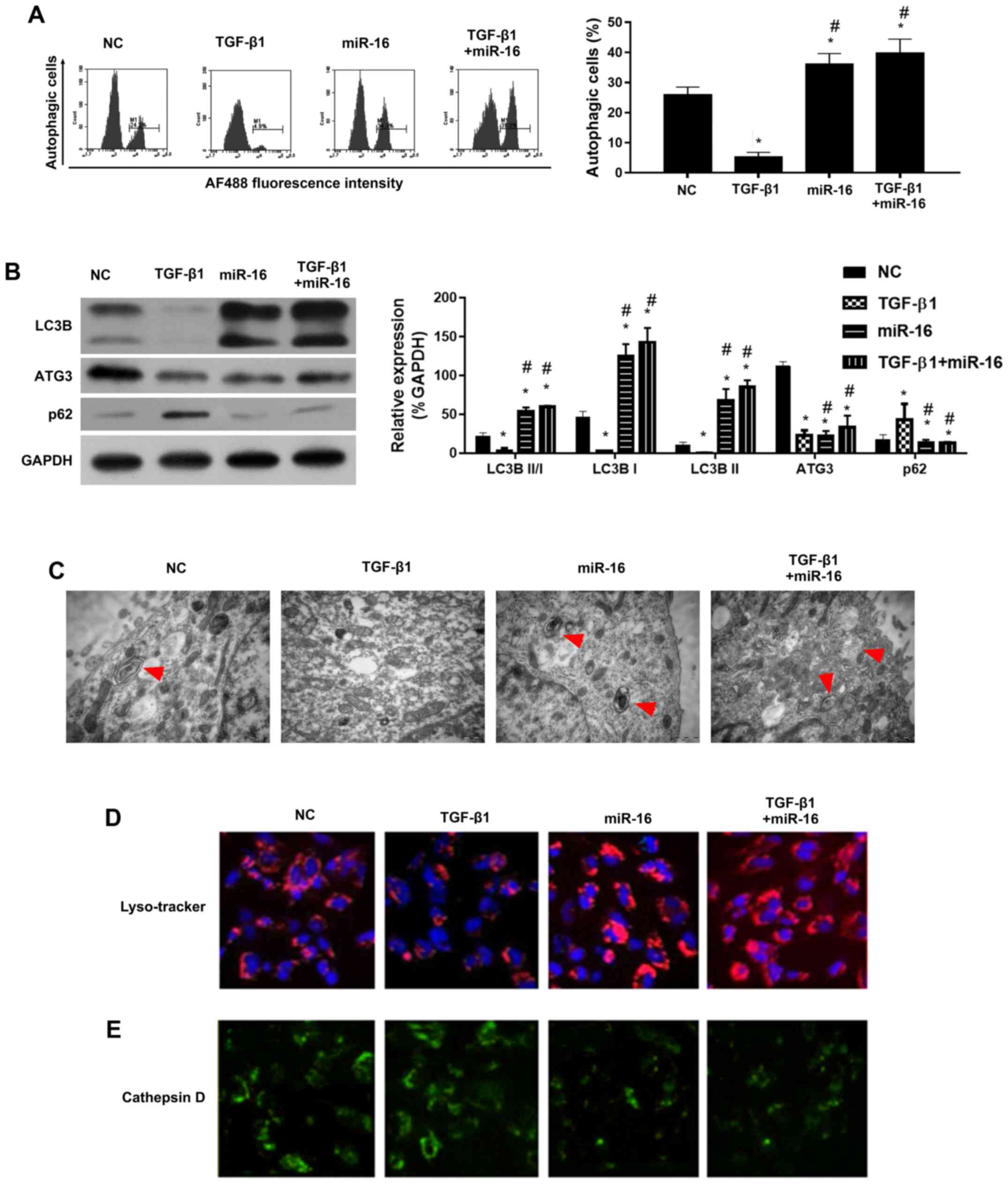
miR-16 mimics rescue TGF-β1-mediated
inhibition of autophagy in NSCLC cells
Recently, intense attention has been paid to the
roles of autophagy in cancer development. We investigated whether
TGF-β1 induced autophagy in the NSCLC cells. Cell autophagy was
detected using AO staining and flow cytometry, western blotting and
EMT. AO staining showed that the percentage of autophagic cells was
reduced by TGF-β1 compared with the NC, and miR-16 mimics increased
the percentage of autophagic cells (Fig. 4A).
Furthermore, we investigated whether there was
autophagic flux in the TGF-β1 and miR-16 mimic-treated cells. As
well-known and vital proteins in autophagic flux, ATG3 and LC3BI/II
were detected as the representative proteins of autophagy by
western blotting. Results showed a reduction in LC3BII/I levels in
the TGF-β1-treated cells, and miR-16 mimics rescued the levels of
LC3BII/I (Fig. 4B). TEM results
confirmed the inhibition of autophagy in the presence of TGF-β1,
and miR-16 enhanced autophagy in the presence of TGF-β1 (Fig. 4C).
Although ATG3 was found to be a target gene of
miR-16 and ATG3 was downregulated by miR-16 mimics (Fig. 4B), miR-16 mimics increased the
autophagy, indicating that ATG3 may not be involved in the
TGF-β1-mediated inhibition of autophagy.
Taken together, our results indicated that miR-16
mimics rescued TGF-β1-mediated inhibition of autophagy in NSCLC
cell lines. LysoTracker staining (Fig.
4D) and staining of cathepsin D (Fig. 4E) were also performed. Results
showed that TGF-β1 inhibited LysoTracker staining, but the
LysoTracker staining was increased following autophagic stimuli by
miR-16 mimics (Fig. 4D). In
contrast, cathepsin D was upregulated by TGF-β1, but was inhibited
following autophagic stimuli by miR-16 mimics (Fig. 4E). There results strengthened the
role of miR-16 in autophagy in the presence of TGF-β1 in NSCLC cell
lines.
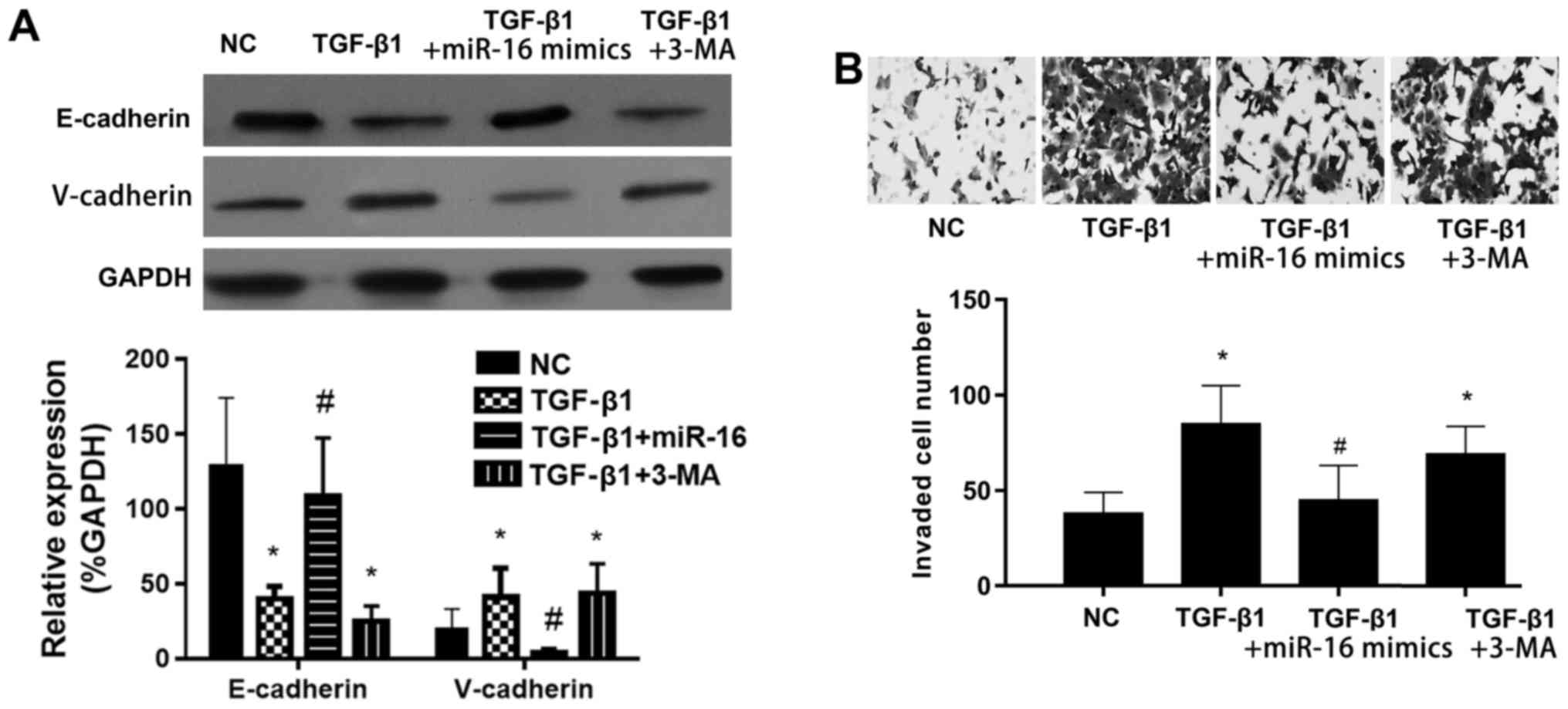
miR-16 mimics suppress the
TGF-β1-induced EMT in NSCLC cells via activation of autophagy
miR-16 mimics reversed the TGF-β1-inhibited
expression of the epithelial marker E-cadherin, while it suppressed
the TGF-β1-induced expression of the mesenchymal marker V-cadherin
(Fig. 5A). The effect of miR-16
mimics was inhibited by autophagy inhibitor 3-methyladenine (3-MA).
The cell invasion was significantly induced by TGF-β1, and this
effect was significantly inhibited by miR-16 mimics (Fig. 5B). The effect of miR-16 mimics was
also inhibited by autophagy inhibitor 3-MA. These results indicated
that miR-16 mimics inhibited the TGF-β1-induced EMT in NSCLC cells
via activation of autophagy.
Discussion
In the present study, we detected the expression of
miR-16 in NSCLC and adjacent normal tissues, predicted the binding
site of ATG3 to miR-16 and confirmed the target relationship
between miR-16 and ATG3 by luciferase reporter gene system. We also
determined the role of miR-16 in the autophagy of NSCLC cells by
transfection of miR-16 mimics into NSCLC cells.
Abnormalities in miR-16 levels have been reported in
various types of cancers, suggesting that miR-16 is involved in
tumor formation and progression. The miRNA microarray analysis of
lung cancer and adjacent normal lung tissues showed that miR-16 was
downregulated in lung cancer tissues (16–18).
miR-16 plays a different role in the proliferation of different
types of tumor cells by regulating a variety of mRNA target genes.
For example, miR-16 was downregulated in lung squamous cell
carcinomas and adenocarcinomas, which correlates with cyclin D1
(19). miR-16 may be a prognostic
marker in NSCLC. Patients with normal levels of miR-16 had the best
outcome while those with high levels had the worst (20). Moreover, Ke et al
demonstrated that miR-16 is downregulated in NSCLC tissue samples
and cell lines (21). Our results
revealed that miR-16 was significantly downregulated and ATG3 was
significantly upregulated in patients with NSCLC.
Overexpression of miR-16 was found to significantly
suppress cell proliferation and colony formation, and cell
migration and invasion in NSCLC cells by targeting hepatoma-derived
growth factors (21). Thus, miR-16
plays an essential role in NSCLC, and may be associated with NSCLC
progression. A previous study found that A549 cells express lower
level of miR-16 than normal bronchial epithelial cells, and
indicated that mRNAs such as wip1 are involved in miR-16-mediated
suppression of NSCLC cell proliferation and promotion of apoptosis
(17). It was reported that
overexpression of miR-16 inhibited cell proliferation and induced
apoptosis by regulating the expression of p27, Bcl-2, Bax and
caspase-3 in NSCLC cells (16). A
recent study demonstrated that miR-16 is a potent inducer of
autophagy (22). Overexpression of
miR-16 inhibited the phosphorylation of mTORC1 and p70S6K,
inhibiting cell proliferation and G1/S cell cycle transition in
human cervival carcinoma HeLa cells, and enhanced anticancer drug
camptothecin-induced autophagy and apoptotic cell death in HeLa
cells by targeting Rictor (22).
Autophagy was found to be enhanced by miR-16 overexpression in
skeletal muscle (23). In the
present study, we found that TGF-β1 inhibited the autophagy of
NSCLC cells, and miR-16 mimics rescued the TGF-β1-mediated
inhibition of autophagy.
TGF-β1 plays an important role in the induction of
EMT (24,25). Other miRNAs such as miR-19 were
found to inhibit the autophagy of human cardiac fibroblasts by
targeting TGF-βRII mRNA during TGF-β1-induced fibrogenesis
(26). Autophagy is critical for
the metastasis of cancer cells through the induction of EMT and
activation of TGF-β signaling plays a key role in regulating
autophagy-induced EMT (27). In
breast cancer, the autophagy agonist rapamycin increased
TGF-β1-induced protective effects and formation of
cancer-associated fibroblast phenotypes, while autophagy inhibitor
3-MA, Atg5 knockdown or TGF-β type I receptor kinase inhibitor
LY-2157299 blocked TGF-β1-induced effects, indicating that
TGF-β/Smad autophagy was involved in TGF-β1-induced protective
effects and formation of cancer-associated fibroblast phenotype in
the tumor microenvironment (32).
In the present study, we found that miR-16 mimics inhibited
TGF-β1-induced EMT in NSCLC cells via activation of autophagy.
Erlotinib-resistant lung adenocarcinoma cells were
found to expressed high basal ATG3 (28). ATG3-mediated autophagy also plays an
important role in apoptotic cell death of NSCLC cells (28). The results suggested that
upregulation of ATG3 by miR-16 mimics was inhibited by TGF-β1.
miR-16 mimic transfection did not further inhibit the expression of
ATG3 in the presence of TGF-β1. Thus, ATG3 may not be involved in
TGF-β1-mediated inhibition of autophagy. miR-16 may promote
autophagy via targeting an unknown gene in the presence of TGF-β1.
The molecular mechanism underlying the rescue of TGF-β1-inhibited
autophagy by miR-16 warrants further study and must be confirmed
using additional cell lines.
In conclusion, our results indicated that miR-16
mimics inhibited TGF-β1-induced EMT via activation of autophagy in
NSCLC cell lines.
References
|
1
|
Pan X, Zhang X, Sun H, Zhang J, Yan M and
Zhang H: Autophagy inhibition promotes 5-fluorouraci-induced
apoptosis by stimulating ROS formation in human non-small cell lung
cancer A549 cells. PLoS One. 8:e566792013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chiu LY, Hu ME, Yang TY, Hsin IL, Ko JL,
Tsai KJ and Sheu GT: Immunomodulatory protein from Ganoderma
microsporum induces pro-death autophagy through Akt-mTOR-p70S6K
pathway inhibition in multidrug resistant lung cancer cells. PLoS
One. 10:e01257742015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hsin IL, Sheu GT, Jan MS, Sun HL, Wu TC,
Chiu LY, Lue KH and Ko JL: Inhibition of lysosome degradation on
autophagosome formation and responses to GMI, an immunomodulatory
protein from Ganoderma microsporum. Br J Pharmacol. 167:1287–1300.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hsin IL, Ou CC, Wu TC, Jan MS, Wu MF, Chiu
LY, Lue KH and Ko JL: GMI, an immunomodulatory protein from
Ganoderma microsporum, induces autophagy in non-small cell lung
cancer cells. Autophagy. 7:873–882. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu L, Pickle LW, Ghosh K, Naishadham D,
Portier K, Chen HS, Kim HJ, Zou Z, Cucinelli J, Kohler B, et al:
Predicting US- and state-level cancer counts for the current
calendar year: Part II: Evaluation of spatiotemporal projection
methods for incidence. Cancer. 118:1100–1109. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen HS, Portier K, Ghosh K, Naishadham D,
Kim HJ, Zhu L, Pickle LW, Krapcho M, Scoppa S, Jemal A, et al:
Predicting US- and state-level cancer counts for the current
calendar year: Part I: Evaluation of temporal projection methods
for mortality. Cancer. 118:1091–1099. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seifi-Najmi M, Hajivalili M, Safaralizadeh
R, Sadreddini S, Esmaeili S, Razavi R, Ahmadi M, Mikaeili H,
Baradaran B, Shams-Asenjan K, et al: SiRNA/DOX lodeded chitosan
based nanoparticles: Development, characterization and in vitro
evaluation on A549 lung cancer cell line. Cell Mol Biol. 62:87–94.
2016.PubMed/NCBI
|
|
8
|
Li J, Yi W, Jiang P, Sun R and Li T:
Effects of ambroxol hydrochloride on concentrations of paclitaxel
and carboplatin in lung cancer patients at different administration
times. Cell Mol Biol. 62:85–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rajewsky N: microRNA target predictions in
animals. Nat Genet. 38 Suppl 38:S8–S13. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Valencia-Sanchez MA, Liu J, Hannon GJ and
Parker R: Control of translation and mRNA degradation by miRNAs and
siRNAs. Genes Dev. 20:515–524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rosa A and Brivanlou AH: MicroRNAs in
early vertebrate development. Cell Cycle. 8:3513–3520. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harfe BD: MicroRNAs in vertebrate
development. Curr Opin Genet Dev. 15:410–415. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fattore L, Costantini S, Malpicci D,
Ruggiero CF, Ascierto PA, Croce CM, Mancini R and Ciliberto G:
MicroRNAs in melanoma development and resistance to target therapy.
Oncotarget. 8:22262–22278. 2017.PubMed/NCBI
|
|
15
|
Lin X, Khalid S, Qureshi MZ, Attar R,
Yaylim I, Ucak I, Yaqub A, Fayyaz S, Farooqi AA and Ismail M: VEGF
mediated signaling in oral cancer. Cell Mol Biol. 62:64–68. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang W, Chen J, Dai J, Zhang B, Wang F and
Sun Y: MicroRNA-16-1 inhibits tumor cell proliferation and induces
apoptosis in A549 non-small cell lung carcinoma cells. Oncol Res.
24:345–351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gu Y, Wang XD, Lu JJ, Lei YY, Zou JY and
Luo HH: Effect of mir-16 on proliferation and apoptosis in human
A549 lung adenocarcinoma cells. Int J Clin Exp Med. 8:3227–3233.
2015.PubMed/NCBI
|
|
18
|
Fan L, Qi H, Teng J, Su B, Chen H, Wang C
and Xia Q: Identification of serum miRNAs by nano-quantum dots
microarray as diagnostic biomarkers for early detection of
non-small cell lung cancer. Tumour Biol. 37:7777–7784. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bandi N, Zbinden S, Gugger M, Arnold M,
Kocher V, Hasan L, Kappeler A, Brunner T and Vassella E: miR-15a
and miR-16 are implicated in cell cycle regulation in a
Rb-dependent manner and are frequently deleted or down-regulated in
non-small cell lung cancer. Cancer Res. 69:5553–5559. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Navarro A, Diaz T, Gallardo E, Viñolas N,
Marrades RM, Gel B, Campayo M, Quera A, Bandres E, Garcia-Foncillas
J, et al: Prognostic implications of miR-16 expression levels in
resected non-small-cell lung cancer. J Surg Oncol. 103:411–415.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ke Y, Zhao W, Xiong J and Cao R:
Downregulation of miR-16 promotes growth and motility by targeting
HDGF in non-small cell lung cancer cells. FEBS Lett. 587:3153–3157.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang N, Wu J, Qiu W, Lyu Q, He J, Xie W,
Xu N and Zhang Y: MiR-15a and miR-16 induce autophagy and enhance
chemosensitivity of camptothecin. Cancer Biol Ther. 16:941–948.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee DE, Brown JL, Rosa ME, Brown LA, Perry
RA Jr, Wiggs MP, Nilsson MI, Crouse SF, Fluckey JD, Washington TA,
et al: microRNA-16 is downregulated during insulin resistance and
controls skeletal muscle protein accretion. J Cell Biochem.
117:1775–1787. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeng YE, Yao XH, Yan ZP, Liu JX and Liu
XH: Potential signaling pathway involved in
sphingosine-1-phosphate-induced epithelial-mesenchymal transition
in cancer. Oncol Lett. 12:379–382. 2016.PubMed/NCBI
|
|
25
|
Zeng Y, Yao X, Chen L, Yan Z, Liu J, Zhang
Y, Feng T, Wu J and Liu X: Sphingosine-1-phosphate induced
epithelial- mesenchymal transition of hepatocellular carcinoma via
an MMP-7/syndecan-1/TGF-β autocrine loop. Oncotarget.
7:63324–63337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zou M, Wang F, Gao R, Wu J, Ou Y, Chen X,
Wang T, Zhou X, Zhu W, Li P, et al: Autophagy inhibition of
hsa-miR-19a-3p/19b-3p by targeting TGF-β R II during TGF-β1-induced
fibrogenesis in human cardiac fibroblasts. Sci Rep. 6:247472016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo
Y, Song Z, Zheng Q and Xiong J: Autophagy promotes hepatocellular
carcinoma cell invasion through activation of
epithelial-mesenchymal transition. Carcinogenesis. 34:1343–1351.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee JG and Wu R: Combination
erlotinib-cisplatin and Atg3-mediated autophagy in erlotinib
resistant lung cancer. PLoS One. 7:e485322012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo JY and White E: Autophagy, metabolism,
and cancer. Cold Spring Harb Symp Quant Biol. 81:73–78. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rabinowitz JD and White E: Autophagy and
metabolism. Science. 330:1344–1348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chakrabarti M, Klionsky DJ and Ray SK:
miR-30e blocks autophagy and acts synergistically with
proanthocyanidin for inhibition of AVEN and BIRC6 to increase
apoptosis in glioblastoma stem cells and glioblastoma SNB19 cells.
PLoS One. 11:e01585372016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu FL, Mo EP, Yang L, Du J, Wang HS,
Zhang H, Kurihara H, Xu J and Cai SH: Autophagy is involved in
TGF-β1-induced protective mechanisms and formation of
cancer-associated fibroblasts phenotype in tumor microenvironment.
Oncotarget. 7:4122–4141. 2016. View Article : Google Scholar : PubMed/NCBI
|