Introduction
Colorectal cancer (CRC) is the third most common
cancer and the fourth most common cause of death worldwide
(1). Many countries have
experienced an increase in the incidence of CRC during the past few
decades, and it is expected to continue increasing in the next
decades (1–3). Although there have been improvements
in the rate of early diagnosis, the efficacy of comprehensive
treatment and the rate of surgical resection along with
postoperative survival (4), radical
treatment for metastasis of CRC remains limited. The invasion and
metastasis of colon cancer cells weaken the treatment effect,
leading to a high mortality rate. The key steps involved in
metastasis include detachment of the malignant tumor cells from the
primary tumor, migration to secondary tissue or organ through a
variety of ways and continued growth and proliferation. Metastasis
is closely related to the biological transformation process where
cells gradually lose an epithelial phenotype and obtain a
mesenchymal phenotype, which is known as epithelial-mesenchymal
transition (EMT) (5,6). EMT is characterized by the loss of
epithelial surface markers, particularly E-cadherin, and the
acquisition of mesenchymal markers including vimentin and
N-cadherin (7). Recently, many
studies have shown that EMT is one of the important steps in the
progression of tumors and is a necessary condition for the
increased invasive ability of tumor cells, such as in the
progression of breast (8–12), prostate (13–15),
liver (16) and lung cancer
(17,18). Previous studies have also
demonstrated that the expression of cell adhesion molecules,
including E-cadherin, is reduced in primary tumors, whereas
expression levels are elevated in metastatic foci (19). E-cadherin plays an important role in
embryonic development and morphogenesis (20), and cell proliferation, survival,
invasion and migration (21). The
mechanisms by which E-cadherin is inhibited include gene mutations,
promoter hypermethylation, chromatin remodeling, post-translational
modification and transcriptional repression (22–25).
The major proteins implicated in the transcriptional repression of
E-cadherin include ZEB-1 or ZEB-2 (26), Snail and Slug, Smad interacting
protein 1 (SIP1), and a basic helix-loop-helix (bHLH) protein
called Twist (25,27,28).
Twist was originally described as a key factor in early mesodermal
development of Drosophila, which is also both necessary and
sufficient to drive the development of the embryo (29,30).
Genetic studies have well illustrated the critical role of Twist in
mesodermal development while human studies have shown that Twist
gene mutations can lead to a disease called Saethre-Chotzen
syndrome (SCS) (31). Recently,
more and more research concerning the dysregulation of the Twist
gene in the induction of EMT in diseases has been reported
(32,33). However, there are few studies on the
relationship between the Twist gene and EMT in colon cancer and its
molecular mechanism.
In the present study, we mainly focused on the
expression of E-cadherin and vimentin after upregulation and
downregulation of the Twist gene in SW480, HT29 and HCT116 colon
cancer cell lines in vitro. In addition, we evaluated the
expression of E-cadherin and vimentin genes in primary tumors and
metastatic sites by constructing a heterotopic liver metastasis
model in nude mice with intrasplenic injection of the colon cancer
cell lines (SW480, HT29 or HCT116) transfected with the different
plasmids.
Materials and methods
Transformation, purification and
extraction of DNA plasmids
The pTracer-CMV/BSD-Twist and pTracer-CMV/BSD
plasmids were obtained from our laboratory. The three plasmids
pGenesil1.2-Twist-shRNA, pGenesil1.3-Twist-shRNA and
pGenesil1.2-shRNA were designed and constructed by Wuhan GenSil
Biotechnology Co. Ltd. (Wuhan, China). The plasmids were
successfully constructed, transformed with DH5α, and extracted and
purified from E. coli. The double restriction enzyme
digestion of DNA used the enzymes NheI, BsaI,
SacI and EcoRI, and we performed agarose gel
electrophoresis for confirmation and separation of digested DNA.
All the sequences were confirmed by Beijing Liuhe Huada Gene
Technology, Co., Ltd. (Beijing, China).
Cell culture and transfection
Human colon cancer cell lines SW480 and HT29 were
gifts from the College of Life Science, Nankai University, Tianjin,
China, and the HCT116 cell line was purchased from the Tumor Line
Database of the Chinese Academy of Medical Sciences, Beijing,
China. We used complete medium containing liquid nitrogen and the
cryoprotective agent dimethyl sulfoxide (DMSO) to store the
cryopreserved cultured cells. Lipofectamine™ 2000 reagent
(Invitrogen, Carlsbad, CA, USA) was used to transfect the cells
according to the manufacturer's protocol.
Flow cytometric analysis and MTT
assay
The DNA plasmids coded with green fluorescent
protein (GFP) expressed green fluorescence under an inverted
fluorescence microscope IX70 (Olympus, Tokyo, Japan) 48 h
post-transfection. When cells were in the logarithmic growth phase,
we harvested the cells by trypsinization. A FACS flow cytometer (BD
Biosciences, Bedford, MA, USA) was used to determine the number of
positive cells within a transfected cell population (positive cells
%). A MTT colorimetric assay was applied to test the cell
proliferation and viability.
Fluorescence quantitative real-time
PCR
Total RNA from transfected cells was extracted by
TRIzol reagent (Invitrogen). High capacity cDNA reverse
transcriptase (Invitrogen) was used for cDNA synthesis.
Quantitative real-time PCR was used to determine relative
expression levels of Twist, vimentin and E-cadherin. The relative
expression of mRNA of the three genes was determined and normalized
to the expression of the reference gene GAPDH. The primers used for
PCR amplification were as follows: Twist forward,
5′-GGAGTCCGCAGTCTTACGAG-3′ and reverse, 5′-TCTGGAGGACCTGGTAGAGG-3′;
E-cadherin forward, 5′-GTGTCATCCAACGGGAATGC-3′ and reverse,
5′-TGGCGGCATTGTAGGTGTTC-3′; vimentin, forward,
5′-ATGACCGCTTCGCCAACTAC-3′ and reverse, 5′-CGGGCTTTGTCGTTGGTTAG-3′;
GAPDH forward, 5′-GAAGGTGAAGGTCGGAGTC-3′ and reverse,
5′-GAAGATGGTGATGGGATTTC-3′. The qPCR amplification was performed
for 40 cycles using the Fluorescence Quantitative Real-Time PCR
Machine (MJ, USA); SYBR Select Master Mix (ABI, USA), and it
consisted of three steps: 1 min at 94°C for denaturation, 45 sec at
54°C for primer annealing and 2 min at 72°C for extension. The ΔΔCt
method was used to calculate the relative amount of mRNA.
Transwell migration and invasion
assays
The Transwell migration assay was used to determine
the migration and invasion ability of the different tumor cell
lines. Transwell filters (Corning Inc., Corning, NY, USA) were
coated with EMC Gel (Sigma-Aldrich, St. Louis, MO, USA) [2 mg/l,
dilution with serum-free Dulbecco's modified Eagle's medium (DMEM)
or RPMI-1640 medium] and incubated at 37°C for 2 h. Fetal bovine
serum (FBS) (10%) (as a chemoattractant) was added when EMC became
solidified and FBS-free medium suspension including the harvested
tumor cells were placed into the bottom and the upper compartment
of the chamber (cell concentration, 1×105 cells/ml)
respectively. After 24 and 48 h of incubation in a 37°C atmosphere
and 5% CO2, non-invaded cells were removed from the
upper chamber with a cotton swab and the migrated cells were
stained using hematoxylin and eosin (H&E) (Sigma-Aldrich). In
addition, we counted the number of the cells on the lower side of
the filter under a microscope.
Western blot analysis
Cells transfected for 48 h were lysed on ice for 30
min in a lysis buffer (Thermo Fisher Scientific, Waltham, MA USA).
Equal amounts of protein from the cell extracts were applied to 12%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) gels and transferred to polyvinylidene fluoride (PVDF)
membranes. Membranes were incubated at 4°C with primary antibodies,
including anti-Twist, anti-E-cadherin, anti-vimentin and anti-GAPDH
(1:1,000 dilution ratio; Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA) overnight. The membranes were washed and incubated
at room temperature for 2 h with diluted secondary horseradish
peroxidase (HRP)-conjugated polyclonal antibodies (1:1,000;
dilution ratio; Zhongshan Golden Bridge, Beijing, China). The
blotted proteins were determined using a Bradford protein assay kit
(Beyotime Institute of Biotechnology, Beijing, China).
Building spleen-to-liver mouse model
of metastatic colon cancer and fluorescence RT-qPCR of xenograft
tumors in the spleen and liver of mice
The surgical principle and procedure for xenogenic
spontaneous heterotopic liver metastasis were similar to
descriptions in the literature (34). Female BALB/c nude mice (10–11 weeks
of age) of specific pathogen-free grade were purchased from Beijing
Wei Tong Li Hua Experimental Animal Technology Co. Ltd., and raised
at the Chinese Academy of Medical Sciences Hematology Hospital,
Department of Experimental Animal Center in an aseptic environment.
Mice were fed ad libitum and maintained in a HEPA filter
environment in a cage. All the food and bedding were sterilized by
autoclaving. After the nude mice were anesthetized, a left lateral
flank incision was made to expose the spleen. Tumors were implanted
by intrasplenic injection of 1×106 SW480, HT29 or HCT116
cells using a 27-gauge needle, and the surgical incision was closed
using surgical thread. The mice were euthanized on the 30th day
after intrasplenic injection with the tumor cell lines (35). Liver and spleen specimens were taken
for H&E and immunohistochemical staining and mRNA isolation.
The method of fluorescence RT-qPCR of xenograft tumors in the
spleen and liver of the mice was the same as described above.
Statistical analysis
Data were analyzed using single-factor analysis of
variance (one-way ANOVA) for comparison within groups. Multiple
comparisons were carried out among groups using the least
significant difference method [least significant difference (LSD)].
For analysis of data with an unknown population, the distribution
was carried out by Spearman correlation test. The difference
between the mean values for groups was analyzed by t-test for
normal distribution. Otherwise, the Mann-Whitney U test was used.
Statistical significance was determined at the P<0.05
probability level. The software package SPSS 17.0 (SPSS, Inc.,
Chicago, IL, USA) was used for statistical analysis of all the
experimental data. The results are representative of three
independent experiments.
Results
mRNA transcription and protein
expression levels of Twist in different colon cancer cell
lines
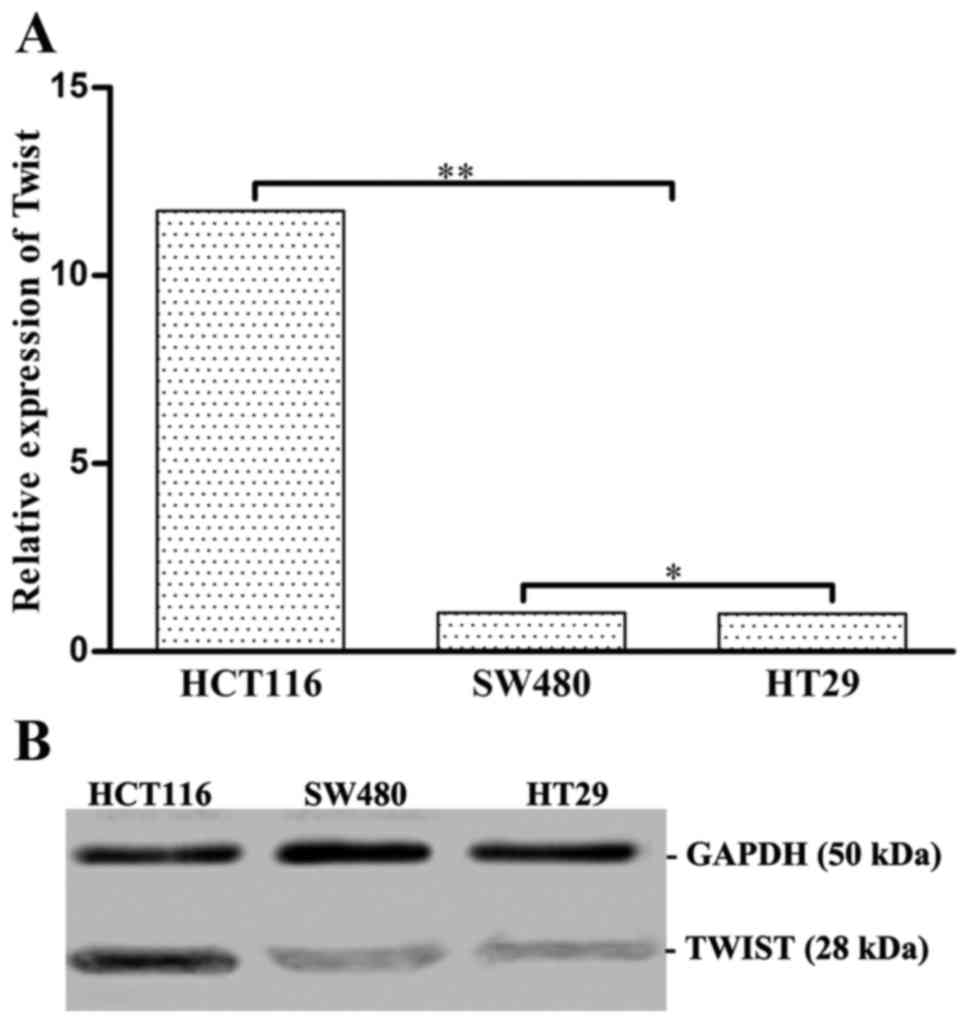
The mRNA transcription copies and protein expression
levels of Twist in different colon cancer cell lines from high to
low were HCT116 → SW480 → HT29 (Fig. 1A
and B). The relative mRNA transcription copies of HCT116, SW480
and HT29 were 11.7, 1.03 and 1, respectively. Least significant
difference (LSD) showed that the difference in mRNA transcription
and protein expression levels of Twist among the groups was
significant (P<0.05).
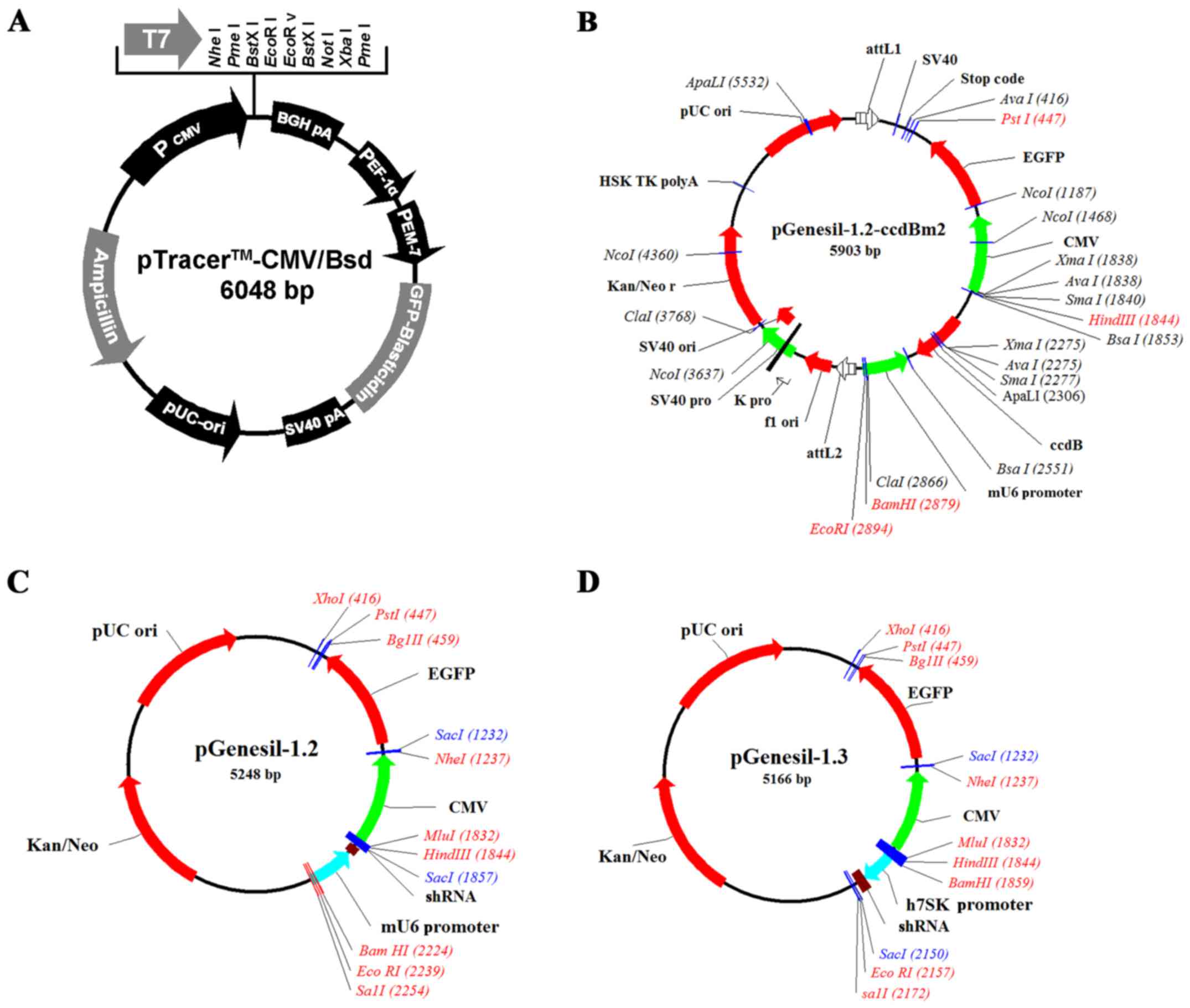
Successful transfection of plasmids in
colon cancer cell lines
The plasmids pTracer-CMV/BSD-Twist, pTracer-CMV/BSD,
pGenesil1.2-Twist-shRNA, pGenesil1.3-Twist-shRNA and
pGenesil1.2-shRNA were successfully transformed with DH5α and
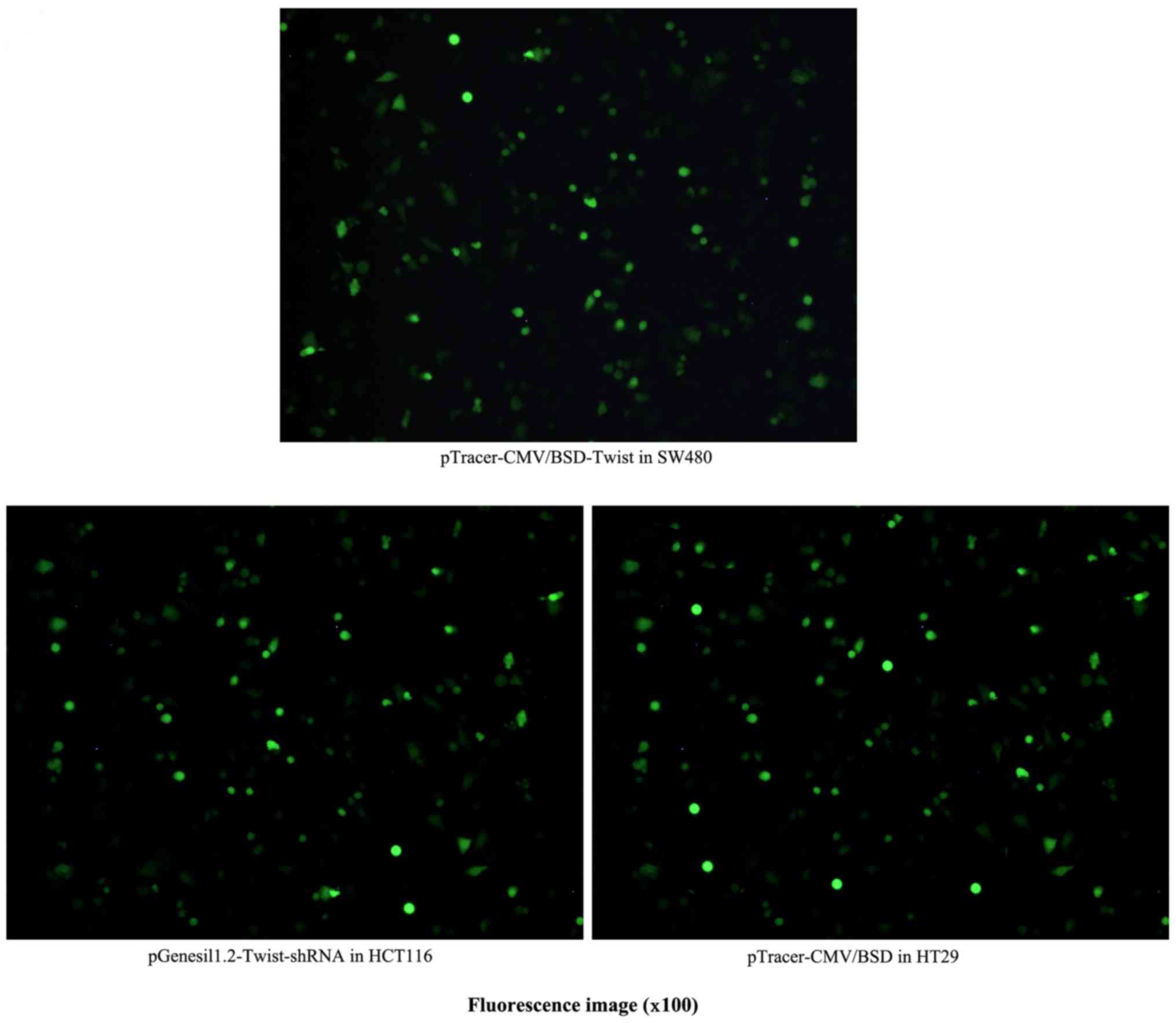
extracted and purified from E. coli (Fig. 2). After transfection of the tumor
cells using Lipofectamine 2000, 48 h later the DNA plasmids coded
with the GFP gene in the colon cancer cell lines expressed green
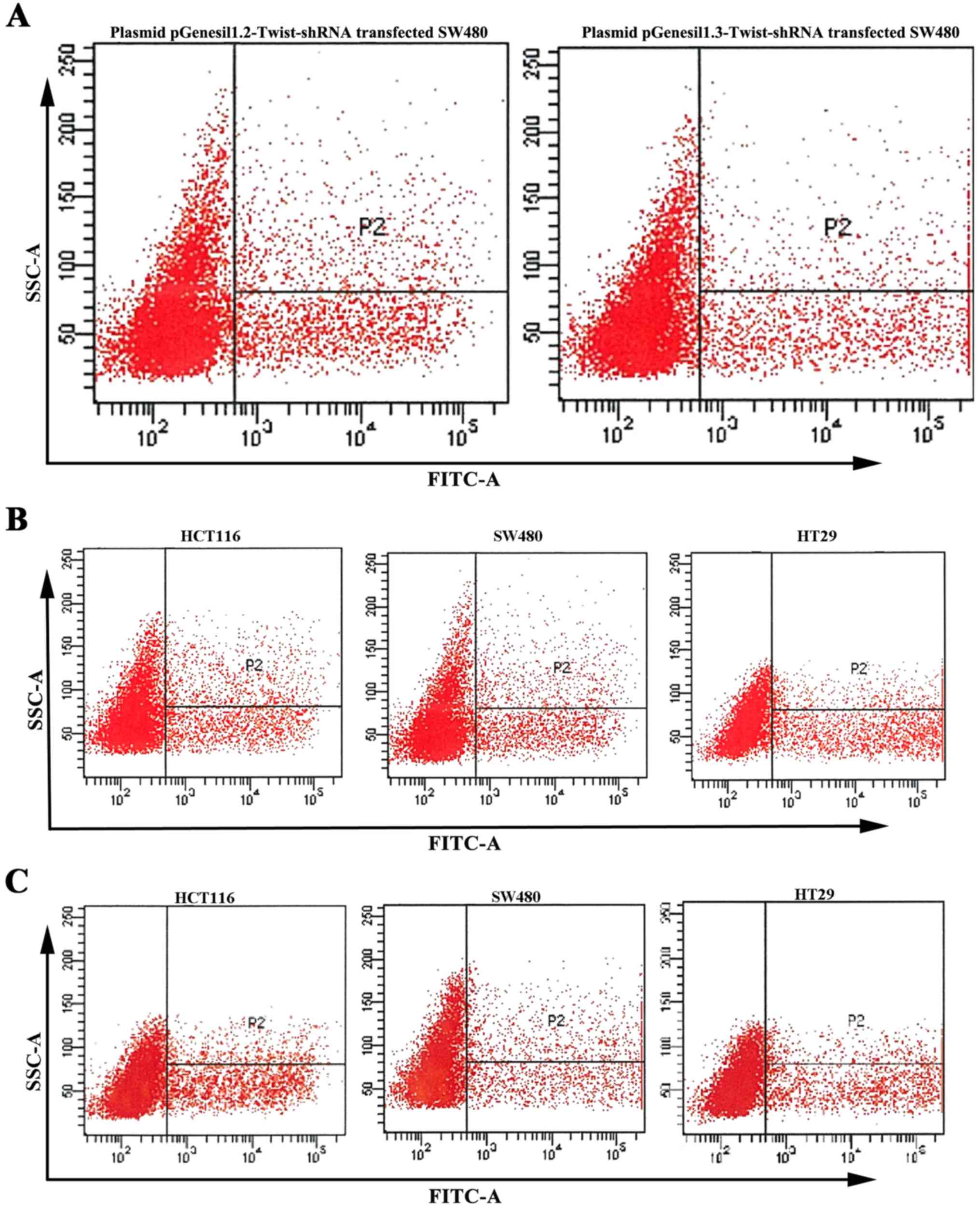
fluorescence under an inverted fluorescence microscope (Fig. 3). We harvested the tumor cells and
used FACS flow cytometry to determine the number of GFP-positive
cells among the transfected cells (positive cells %). The GFP
expression of pGenesil 1.2-Twist-shRNA reached 21.2% while GFP
expression of pGenesil1.3-Twist-shRNA reached only 19.8% in the
SW480 cells. Thus, we used pGenesil 1.2-Twist-shRNA for further
experiments (Fig. 4A). The
transfection efficiency of pGenesil 1.2-Twist-shRNA analyzed by
CellQuest software in the HCT116, HT29 and SW480 cells was 23.4,
30.3 and 21.2%, respectively (Fig.
4B), and the transfection efficiency of pTracer-CMV/BSD-Twist
in the HCT116, HT29 and SW480 cells was 22.3, 22.7 and 21.6%,
respectively (Fig. 4C).
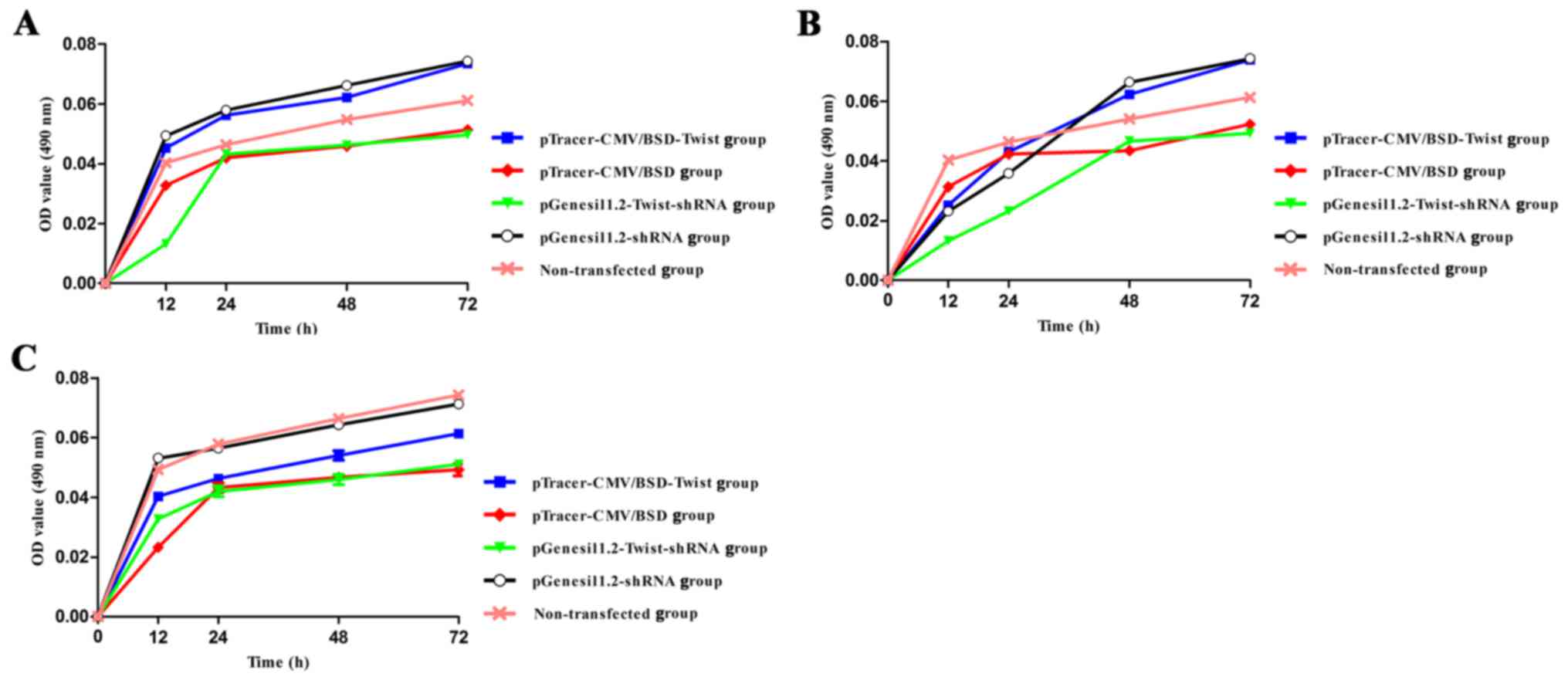
MTT proliferation assay results for
the transfected cell lines
As shown in Fig.
5A-C, the proliferation and viability of the three cell lines
were not affected after the transfection of the recombinant
plasmids. The difference in cell proliferation of the plasmid
pTracer-CMV/BSD-Twist and pGenesil1.2-Twist-shRNA-transfected
groups was statistically insignificant compared with that of the
plasmid pTracer-CMV/BSD, pGenesil1.2-shRNA and negative control
groups (P>0.05).
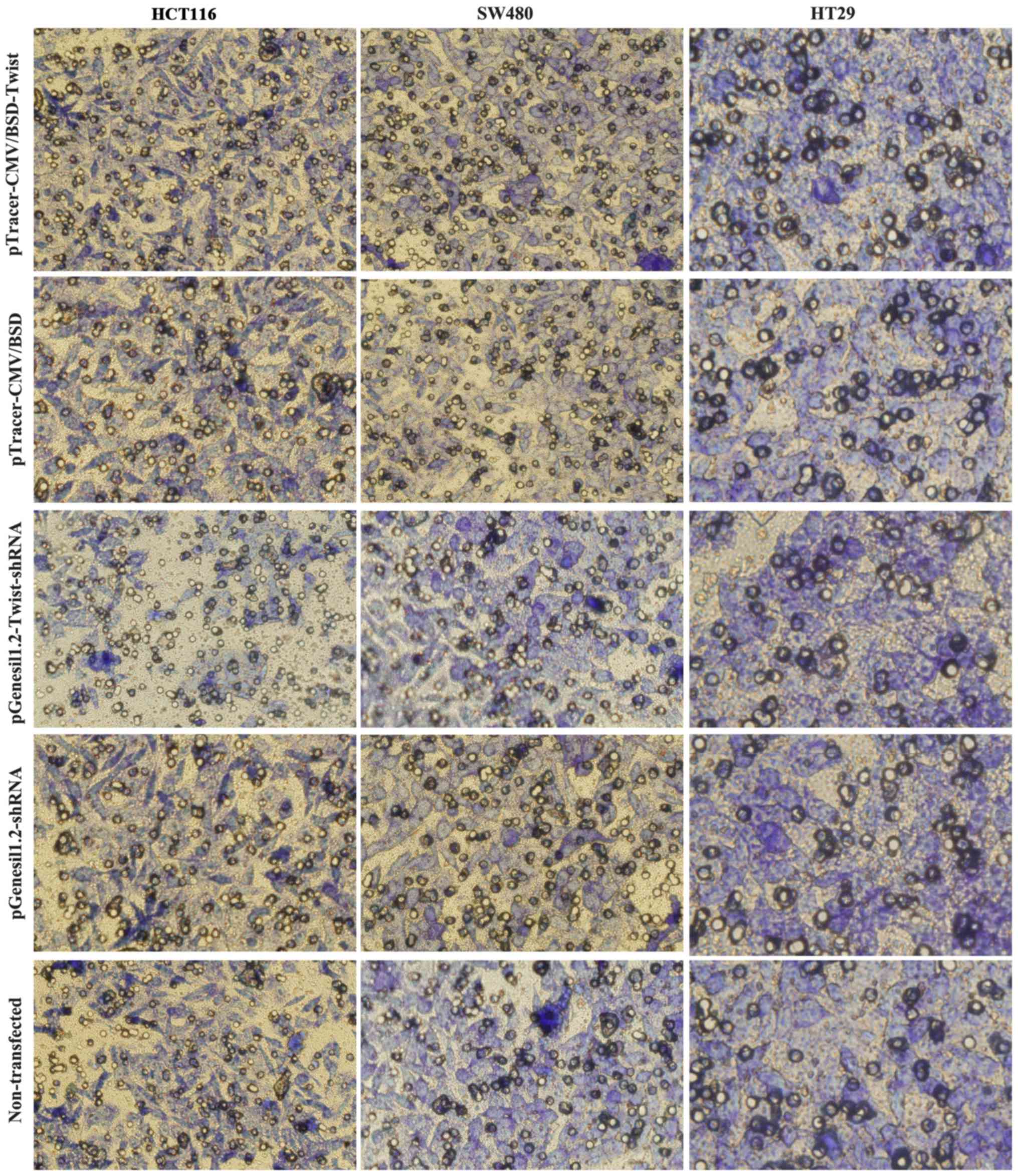
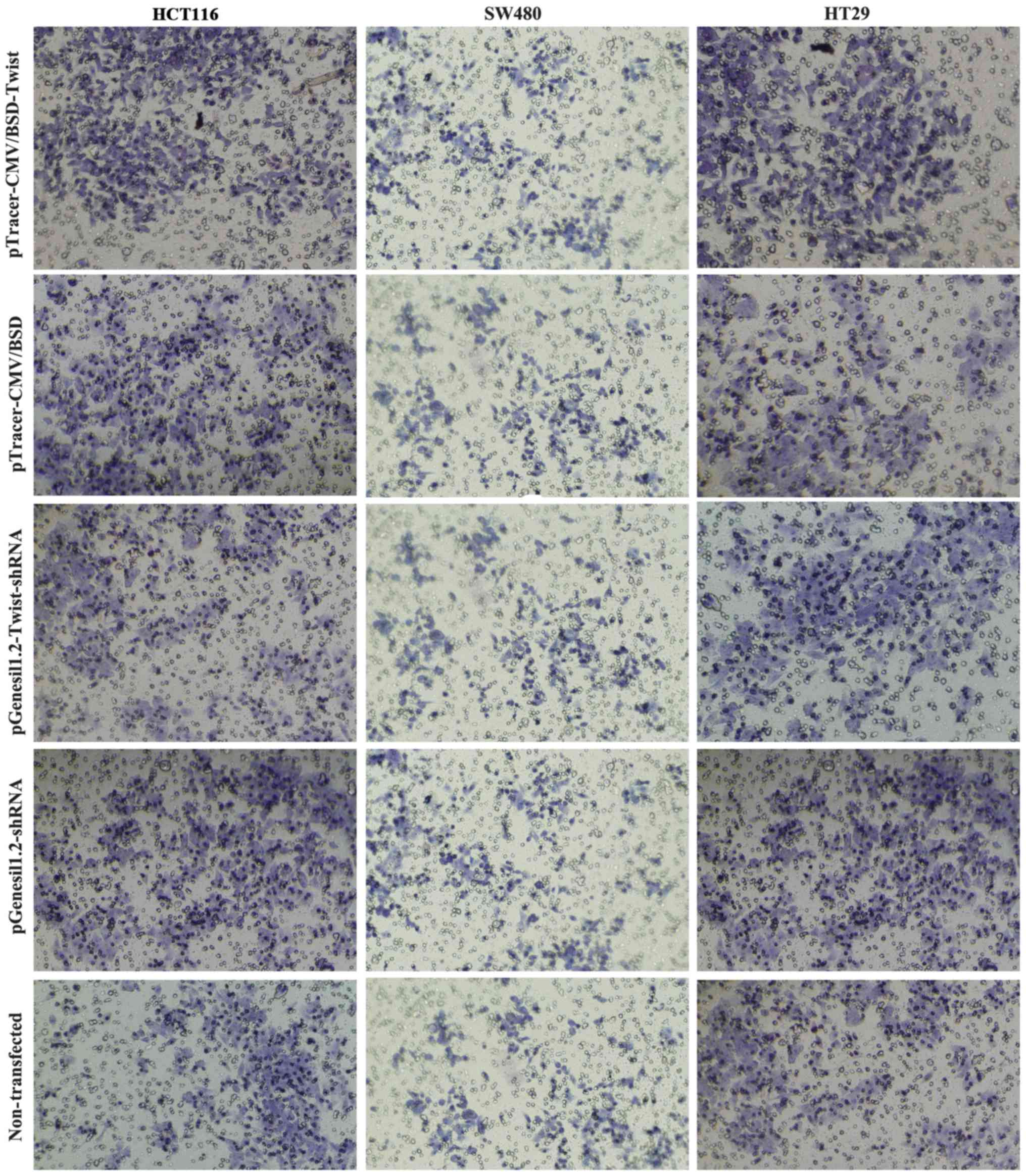
Transwell migration and invasion assay
results
The total number of migrated cells was ~900–1,200
cells, which showed that all three malignant cell lines had
elevated spontaneous Transwell migration. The Transwell migration
assay results (Figs. 6 and 8A-C) showed that the number of migrated
cells in all three cell lines transfected with the
pTracer-CMV/BSD-Twist was increased compared with that in the
control groups, and the difference was statistically significant
(P<0.01). The number of migrated cells in the HCT116 cell line
transfected with pGenesil1.2-Twist-shRNA was decreased compared
with that in the control group, and the difference was
statistically significant (P<0.01), but there was no
statistically significant difference between the plasmid
pGenesil1.2-Twist-shRNA-transfected group and the control groups in
the SW480 and HT29 cell lines (P>0.05). The Transwell invasion
assay results (Figs. 7 and 8D-F) showed that cell invasion in all
three cell lines in the plasmid pTracer-CMV/BSD-Twist-transfected
groups was higher than that in all other groups (P<0.01),
whereas cell invasion only in HCT116 in the
pGenesil1.2-Twist-shRNA-transfected group was comparatively lower
than that in all other groups (P<0.01). The results above
indicated that after transfection with pGenesil1.2-Twist-shRNA
plasmid, the migration and invasion ability of the HCT116 cell line
was significantly inhibited (P<0.01).
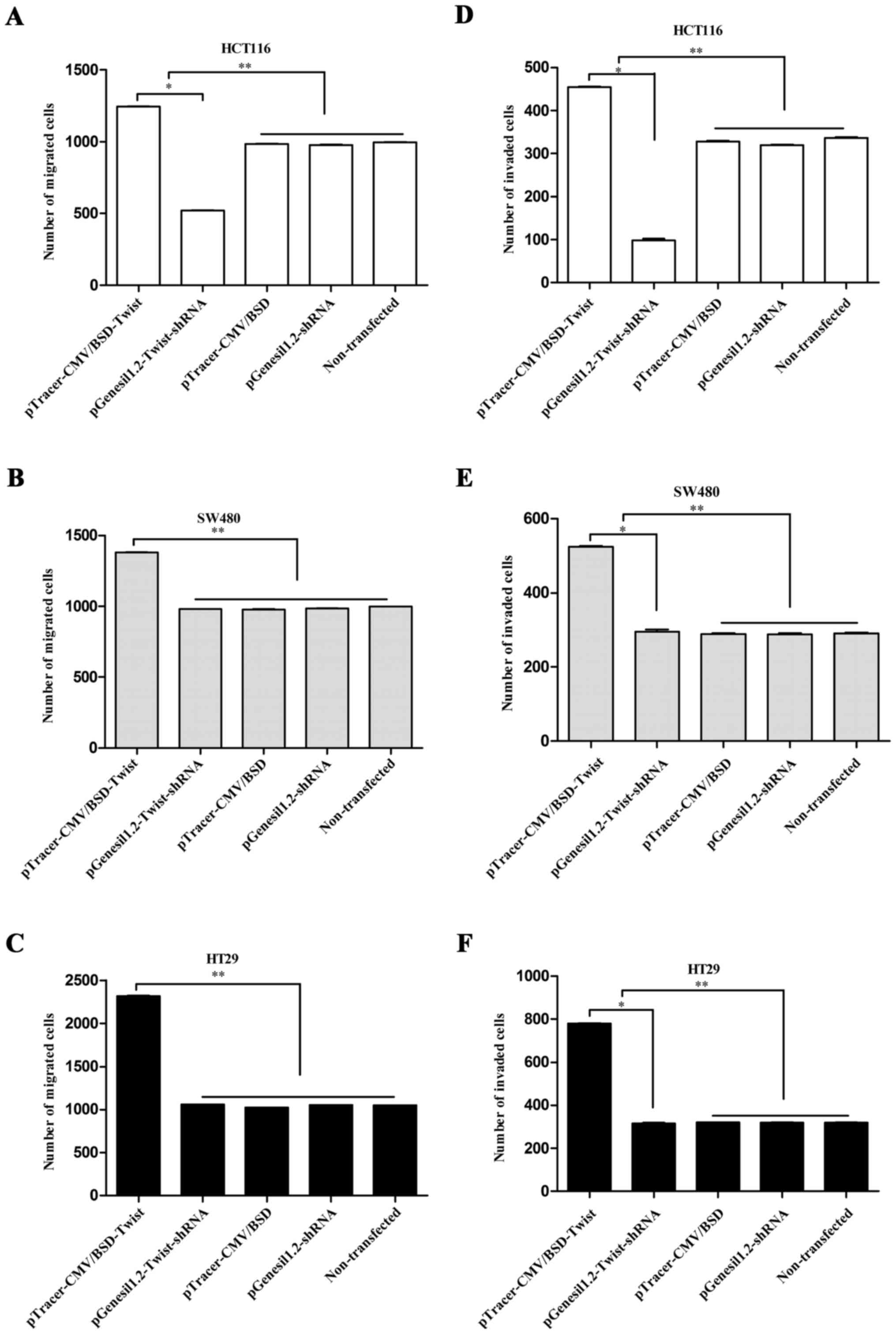 | Figure 8.(A) The Transwell migrated number of
HCT116 cells through the insert in the plasmid
pTracer-CMV/BSD-Twist-transfected, plasmid
pGenesil1.2-Twist-shRNA-transfected and non-transfected group was
1,245.45±2.32, 521.76±2.85 and 996.21±1.32. (B) The Transwell
migrated number of SW480 cells through the insert in the plasmid
pTracer-CMV/BSD-Twist-transfected, plasmid
pGenesil1.2-Twist-shRNA-transfected and non-transfected group was
1,380.21±1.13, 979.43±1.42 and 998.54±1.32. (C) The Transwell
migrated number of HT29 cells through the insert in the plasmid
pTracer-CMV/BSD-Twist-transfected, plasmid
pGenesil1.2-Twist-shRNA-transfected and non-transfected group was
2,320.32±1.10, 1,058.54±1.54 and 1,047.76±1.65. (D) The Transwell
invaded number of HCT116 cells through the insert in the plasmid
pTracer-CMV/BSD-Twist-transfected, plasmid
pGenesil1.2-Twist-shRNA-transfected and non-transfected group was
454.24±1.55, 98.23±3.74 and 335.98±2.47. (E) The Transwell invaded
number of SW480 cells through the insert in the plasmid
pTracer-CMV/BSD-Twist-transfected, plasmid
pGenesil1.2-Twist-shRNA-transfected and non-transfected group was
524.12±2.3, 295.12±6.3 and 290.76±2.4. (F) The Transwell invaded
number of HT29 through the insert in plasmid pTracer-CMV/BSD-Twist
transfected, plasmid pGenesil1.2-Twist-shRNA transfected and
non-transfected group was 780.12±1.6, 315.32±4.5 and 319.89±1.5,
respectively. The data obtained by counting the average numbers of
cells from four different fields are represented as the means ± SEM
(*P<0.05 and **P<0.01). |
Twist promotes EMT in tumor cells in
vitro
After transfection of the tumor cells for 48 h,
fluorescence qRT-PCR results (Fig.
9A-C) and western blot results (Fig. 9D-F) showed higher mRNA transcription
and protein expression of Twist and vimentin in the
pTracer-CMV/BSD-Twist-transfected groups compared with those in the
other groups (P<0.01). In addition, E-cadherin mRNA and protein
expression levels were decreased (P<0.01) in all three colon
cancer cell lines after Twist gene transfection. Correlation
analysis showed that Twist and vimentin mRNA transcription and
protein levels were positively correlated (P<0.01), while
negatively correlated (P<0.01) with E-cadherin.
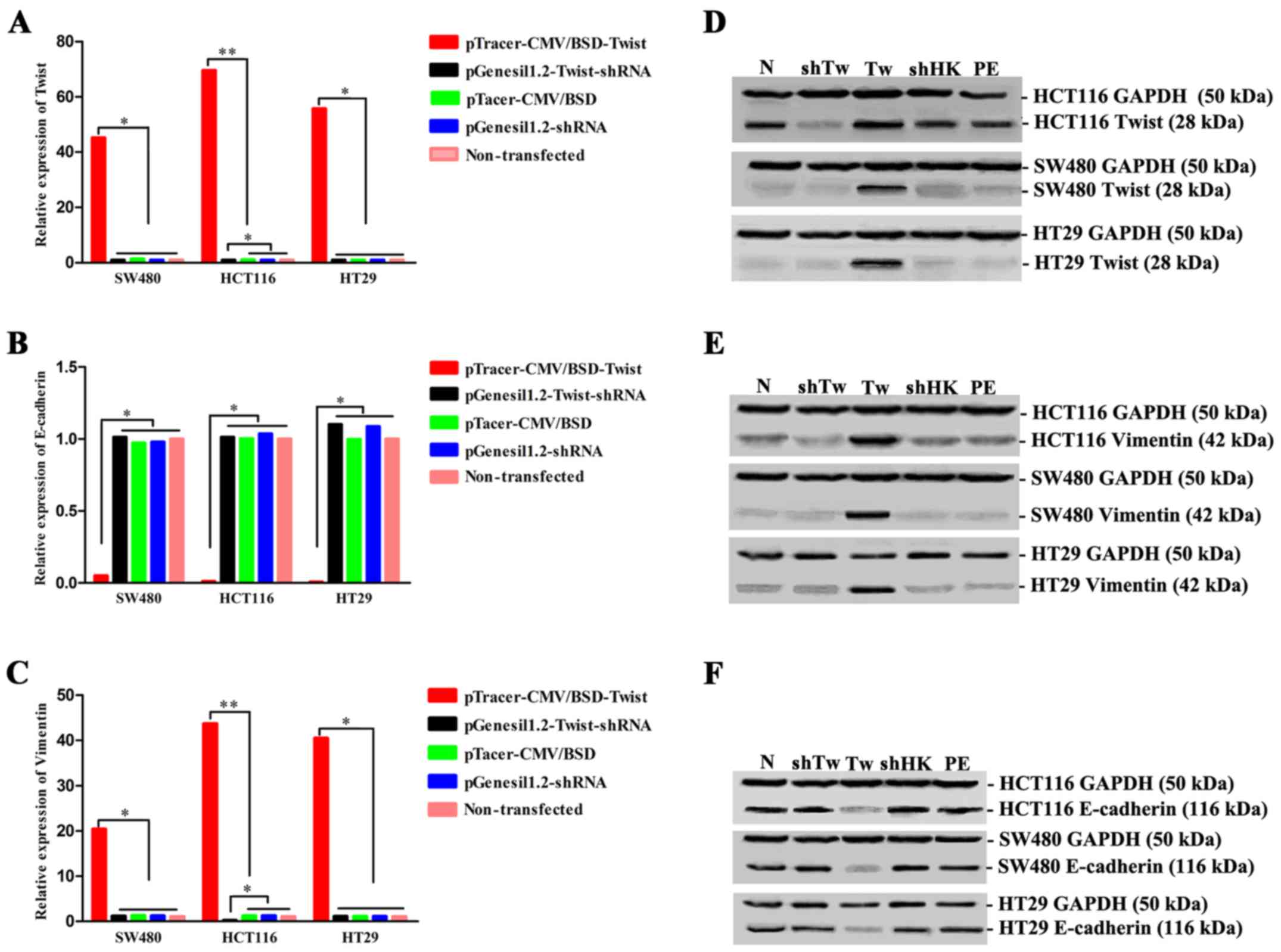 | Figure 9.Twist promotes the process of EMT in
tumor cells and the Twist gene was successfully silenced by
Twist-shRNA in the HCT116 cell line in vitro. The
fluorescence real-time PCR results detecting mRNA transcription
level of (A) Twist, (B) E-cadherin and (C) vimentin in
pTracer-CMV/BSD-Twist-, pGenesil1.2-Twist-shRNA-, pTracer-CMV/BSD-,
pGenesil1.2-shRNA-transfected and non-transfected groups in three
colon cancer cell lines. Protein expression of (D) Twist, (E)
vimentin and (F) E-cadherin by western blot analysis in each
transfection group in the three colon cancer cell lines (N,
non-transfection group; ShTw, pGenesil1.2-Twist-shRNA-transfected
group; Tw, pTracer-CMV/BSD-Twist-transfected group; ShHK,
pGenesil1.2-shRNA-transfected group; and PE,
pTracer-CMV/BSD-transfected group) (*P<0.05 and
**P<0.01). |
Silencing of the Twist gene by
Twist-shRNA in the HCT116 cell line
After transfection of the tumor cells for 48 h, the
qRT-PCR results (Fig. 9A-C) and
western blot analysis (Fig. 9D-F)
showed that in the SW480 and HT29 cell lines, the mRNA
transcription and protein expression levels of Twist, vimentin and
E-cadherin in the pGenesil1.2-Twist-shRNA groups were the same as
the control groups (P>0.05). However, the mRNA transcription and
protein expression levels of Twist and vimentin were decreased only
in the HCT116 cell line compared with those in the control groups
(P<0.01); the difference in E-cadherin mRNA transcription and
protein expression level was statistically insignificant
(P>0.05) between the pGenesil1.2-Twist-shRNA transfected and
control groups.
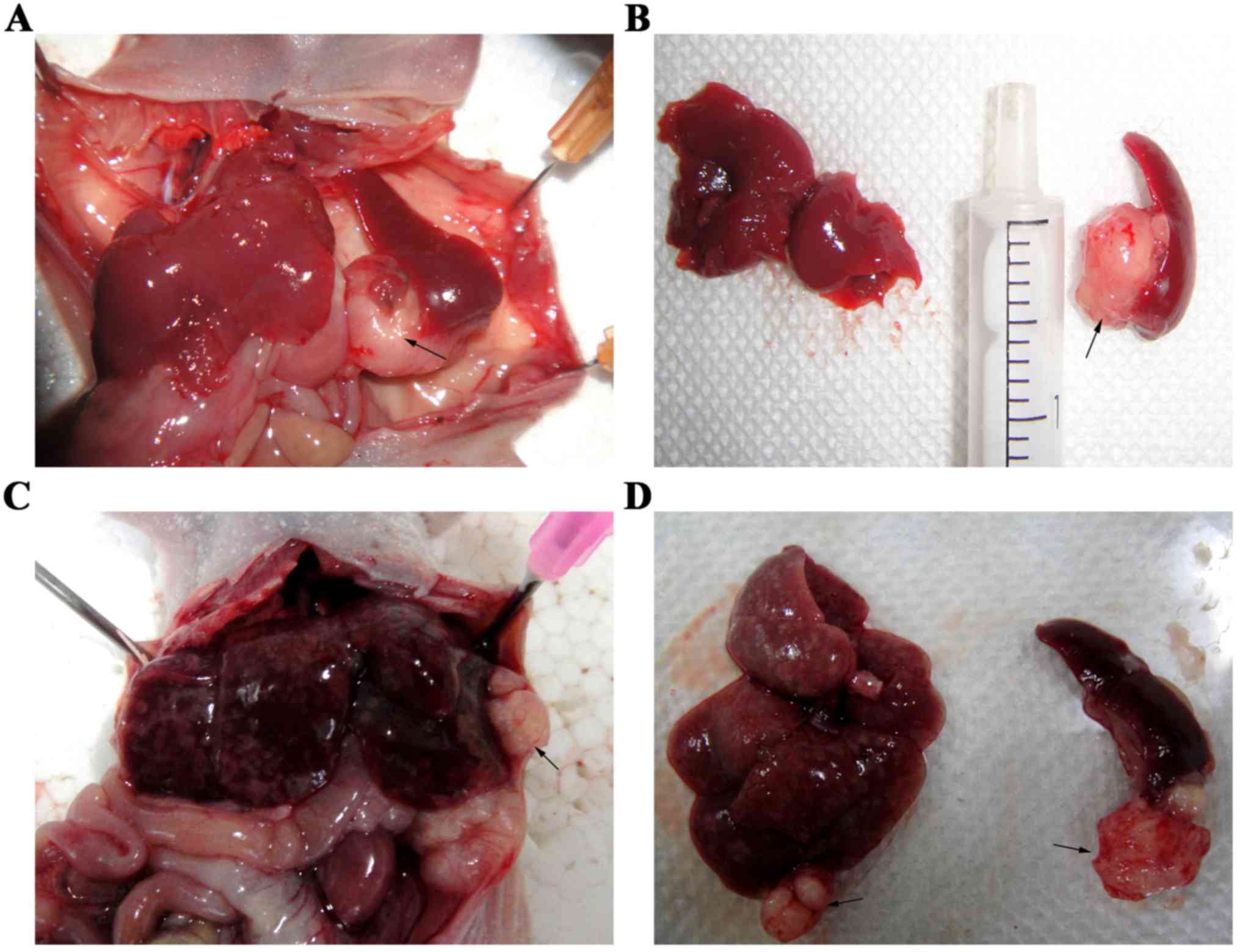
Successful construction of the
xenogenic spontaneous heterotopic liver metastasis model
A spleen-to-liver colorectal cancer metastasis model
was successfully constructed. On the 30th day of intrasplenic
injection with SW480, HT29 and HCT116 cell lines, the animals were
euthanized and examined for tumor growth (only the animals with
primary tumor and secondary metastatic lesions were used for the
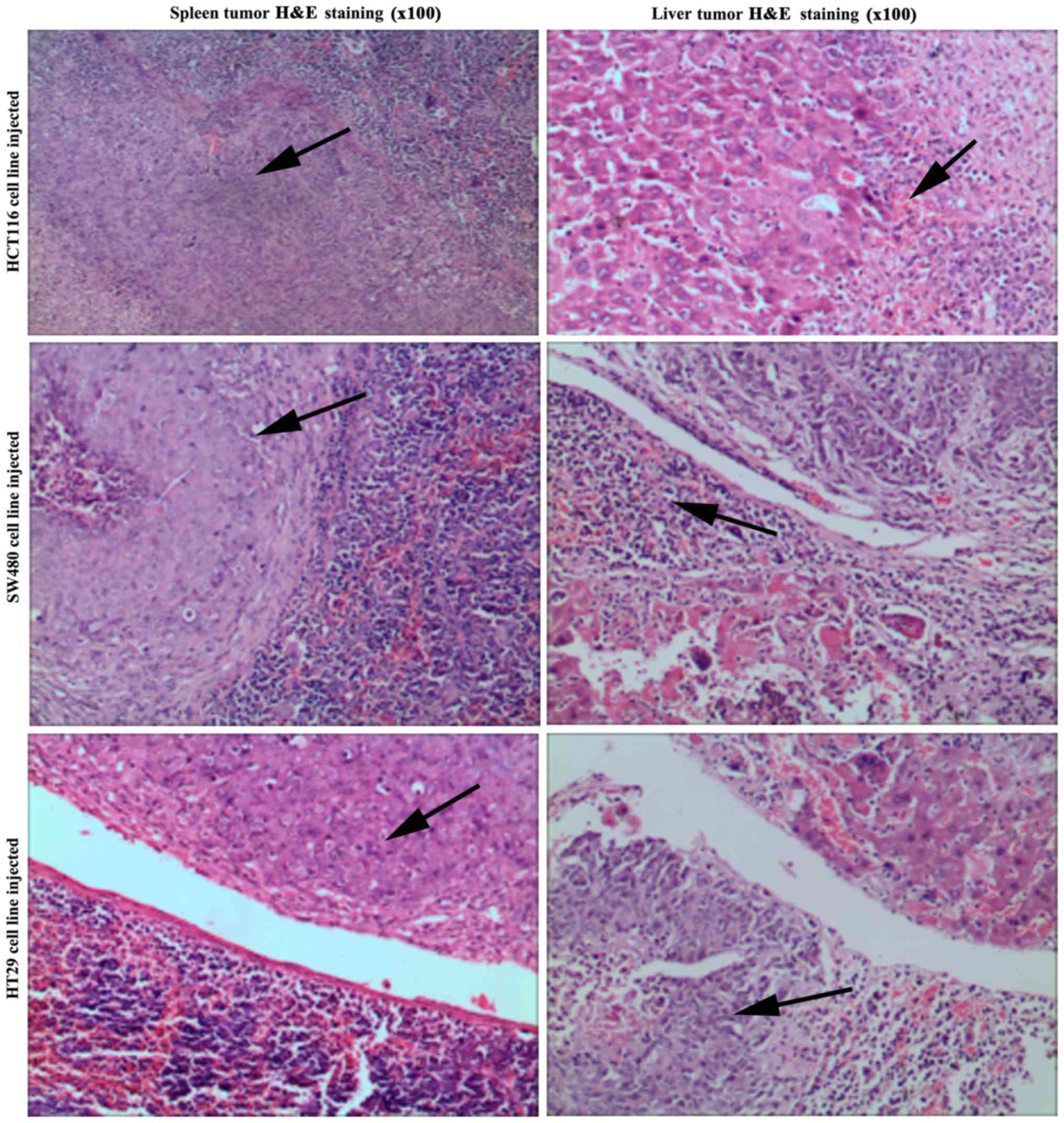
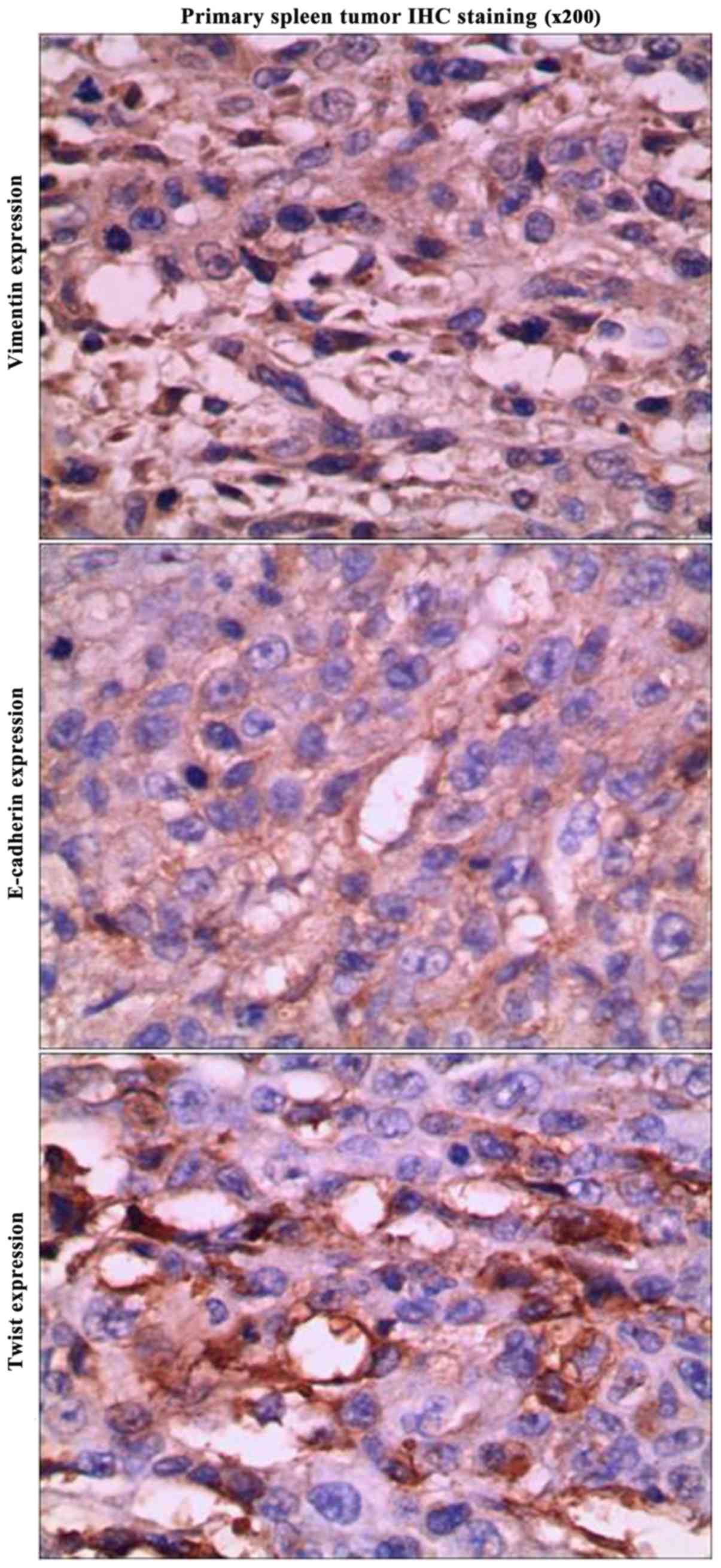
experiment) (Fig. 10). H&E and
immunohistochemical staining results of spleen and liver tumor
tissue demonstrated the presence and location of colon cancer cells
in the liver and spleen tissue sections (Figs. 11 and 12), which demonstrated the successful
construction of the xenogenic spontaneous heterotopic liver
metastasis model.
Experimental groups of the
intrasplenic injection model
The experimental groups were characterized as
follows: Tw, liver or spleen tissues in the model injected with
pTracer-CMV/BSD-Twist plasmid-transfected cell lines (HCT116, SW480
and HT29); ShTw, liver or spleen tissues in the model injected with
pGenesil1.2-Twist-shRNA plasmid-transfected cell lines; control,
liver or spleen tissues in the model injected with non-transfected
cancer cells; normal, normal tissue (liver and spleen).
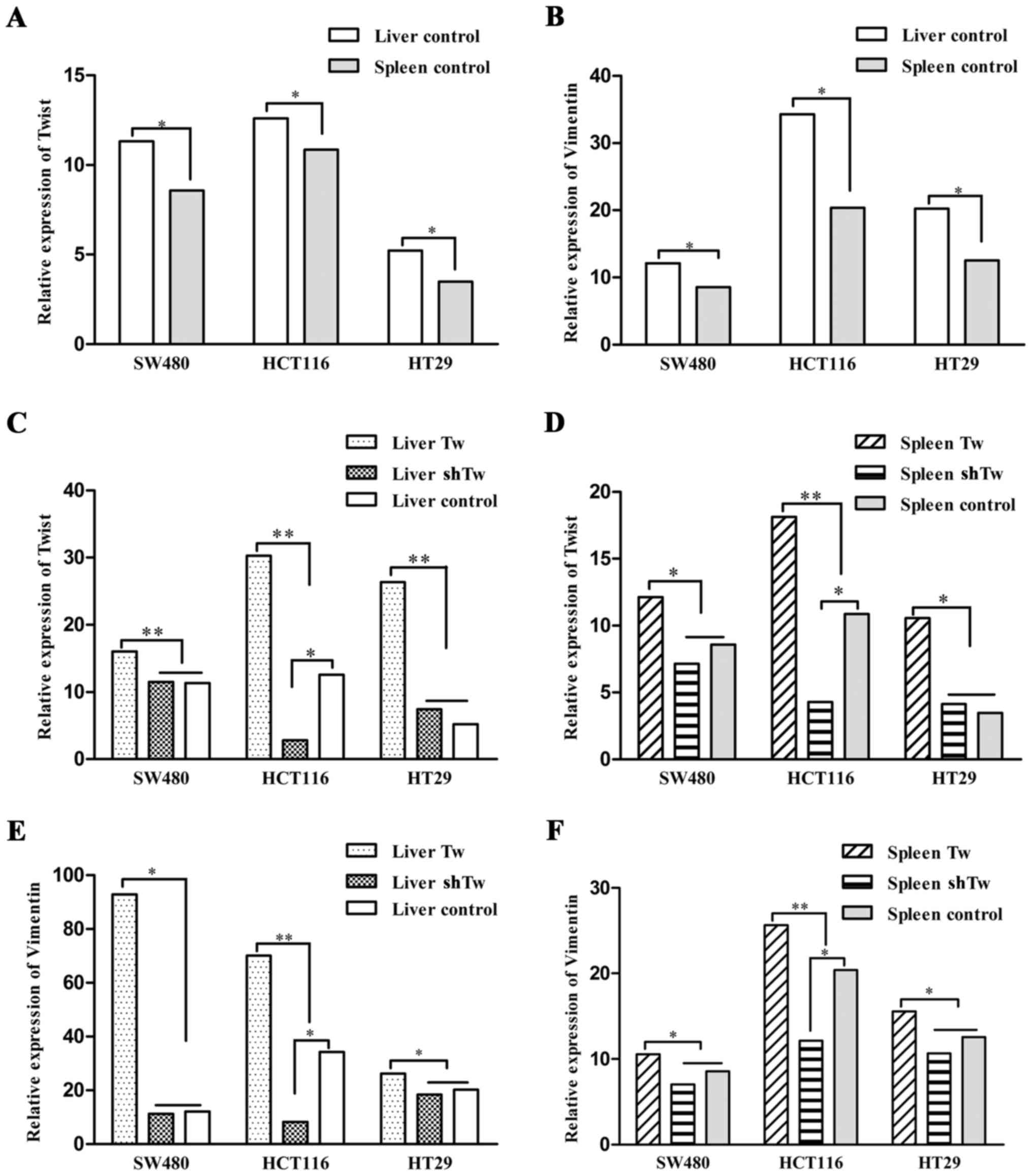
mRNA transcription level of Twist and
vimentin in the liver and spleen tissues
Fluorescence qRT-PCR analysis (Fig. 13A and B) showed that Twist and
vimentin mRNA transcription levels were higher in the liver than in
the spleen of the model after injection of non-transfected cells
(P<0.05). Twist and vimentin mRNA transcription levels in both
the liver and spleen were higher in the model injected with the
three cell lines transfected with pTracer-CMV/BSD-Twist (Tw)
compared with the levels in the cells transfected with
pGenesil1.2-Twist-shRNA (shTw) (P<0.05). However, compared with
the liver and spleen tissues of the non-transfected cell groups,
only the liver and spleen tissues of the model injected with
plasmid pGenesil1.2-Twist-shRNA-transfected (shTw) HCT116 cells
appeared to have a prominently inhibited mRNA transcription level
of Twist (Fig. 13C and D) and
vimentin (Fig. 13E and F).
Discussion
Colon cancer is one of the most common diseases
worldwide, and its incidence is increasing (36,37).
There are two biological processes that are essential for the
progression of this disease, despite the diversity of etiology and
pathophysiology. One of these processes is epithelial-mesenchymal
transition (EMT), which has a significant role in the development
of the human body, is involved in the onset and progression of
colon cancer, and has an important role in tissue fibrosis
(38–42). Cells that experience EMT show many
morphologic and characteristic changes, such as the loss of cell
adhesion, repression of E-cadherin and increased cell motility
(43,44). At the same time, activation or
elevated expression of tyrosine kinases, upregulation of
N-cadherin, vimentin, fibronectin, zinc-finger domain proteins and
basic helix-loop helix domain protein Twist1 expression are all
related to a mesenchymal-like phenotype (45–48).
In basic research, these proteins are often used as markers for
EMT, indicating strong motility and invasiveness of the cells.
In vitro and in vivo evidence suggests that EMT in
primary invasive breast, lung, prostate, liver and pancreatic
cancer, and other cancers and secondary transfer plays an important
role. The other process closely related to normal tissue and organ
development is a reverse phenomenon, called the
mesenchymal-epithelial transition (MET). MET is also involved in
the initial step of colorectal carcinogenesis. Research shows that
it may play an important role in the regeneration of colonic mucosa
(49,50). Currently, the induction and
regulation of these complex, reversible biological processes is not
fully understood. The significant influence of EMT and MET in
treatment is widely recognized; however, the exact description of
these biological phenomena is still unclear. Although the
understanding of the mechanisms and developmental processes of EMT
and MET have great clinical importance, data on their role in the
induction and regulation, as well as their role in the pathogenesis
of colonic diseases, are scarce in the scientific literature.
Therefore, the present study selected the Twist gene, which may
have a close relationship to tumor progression and metastasis. We
evaluated Twist expression and EMT markers (E-cadherin, vimentin)
after induction and inhibition (knockdown) of the Twist gene in
colon cancer cell lines by transfection of the plasmids
pTracer-CMV/BSD-Twist and pGenesil-Twist-shRNA, respectively. In
the in vitro study, after we transfected human colon cancer
cell lines SW480, HCT116 and HT29 with pTracer-CMV/BSD-Twist and
pGenesil1.2-Twist-shRNA, we found differences in Twist expression
in all the cell lines transfected with the Twist overexpression and
silencing plasmids. Expression of Twist was higher in the HCT116
cells in comparison to the SW480 and HT29 cells. Upregulation of
Twist gene expression in the three cell lines induced high
expression of vimentin molecules and low expression of E-cadherin
molecules, hence promoting the EMT-enhanced metastatic ability of
the tumor cells (48,51,52).
Twist-shRNA effectively silenced Twist gene expression in the
HCT116 cell line, inhibiting vimentin expression, reversing
epithelial-mesenchymal transition, promoting mesenchymal-epithelial
transition, and effectively inhibiting colon cancer cell migration
and invasion. The mRNA and protein expression analyses showed that
Twist expression was negatively correlated (P<0.01) with
E-cadherin expression where it was positively correlated
(P<0.01) with vimentin expression. The Transwell migration and
invasion assays showed an increased capability for invasion in the
three cell lines transfected with the Twist overexpression plasmid.
However, only HCT116 showed decreased invasion capability after
transfection with Twist-shRNA. Thus, the results confirmed that the
Twist gene plays a vital role during the processes of cell EMT,
epithelial invasion and metastasis.
One of the most typical features of cancer is
metastatic progression. The process of metastasis is not only
related to the characteristics of the primary tumor cells but is
also related to the tumor microenvironment, which usually presents
as extreme conditions, such as hypoxia and acidosis, elevated tumor
interstitial fluid pressure, extracellular matrix and isolation
from the host organ by stromal barriers (53). Moreover, the progression of
metastases also depends on the ability of target tissues and
stromal cells to cause metastasis (54). Injecting cancer cells directly into
the liver parenchyma (53) or
portal vein (55) has been used to
induce tumors in the liver. However, neither approach allows for
selection of metastatic cells from injected cells, which may be the
potential reason for the variation in the number of mice that
develop an intrahepatic tumor in this type of experimental model
(56). Previous studies have
described the implantation of a primary tumor into the cecum
(55) or spleen (56) subsequent release into the portal
system. The main advantage of such a model is that only some
animals develop disseminated tumors and/or at different locations
of primary or metastatic tumors, which leads to an increase in the
number of experimental animals needed. There is evidence to support
that when tumor cells are implanted in the spleen, more uniform
tumor development is achieved compared with direct injection of the
same cell line into the portal vein, which demonstrates the
importance of metastatic selection of the cells (57). Some metastatic animal models are
available for research and therapeutic development. Metastatic
cells can easily spread from splenic tumors to the liver through
the portal system, which however may also directly mediate
implantation of the cells in hepatic tissue after intrasplenic
injection. Thus, it is important to distinguish these events in the
course of study. Nude mice present with hepatic tumors in 100% of
the mice used with regard to liver metastases originating from
primary spleen tumors (58). The
spleen is a suitable injection site with low-mortality surgery,
while the portal system is the most similar to the clinical
situation, which may be the most relevant route for the
dissemination of tumor cells into the liver. In addition, the
progression of liver metastases cannot be tracked by external
observation, so a model that can be used to assess the spread of
the disease at its early stage is what we needed. Comprehensively
considering the above, the spleen-to-liver colorectal cancer model
established in nude mice was used in our research to study the
expression of Twist in primary tumors (in spleen) and metastatic
tumors (in liver), to reveal the relationship between the Twist
gene and colon cancer progression and metastasis. We found that
Twist expression was much higher in the metastatic lesions than in
the primary tumor. The high expression of Twist in the liver may
indicate that the invasiveness and metastatic potential of the
cancer cells remained high. RNA interference of the Twist gene
effectively silenced Twist gene expression in the spleen and liver
in mice injected with the HCT116 cell line, which suggests that the
expression status of the EMT-related transcription factor Twist may
play an important role in the implantation of metastatic foci.
In summary, from the experimental findings of the
present study, we conclude that upregulation of the Twist gene
promotes EMT molecular events and enhances the metastatic ability
of tumor cells, while Twist-shRNA effectively silences Twist gene
expression in the HCT116 cell line, promoting
mesenchymal-epithelial transition and effectively inhibiting colon
cancer cell migration and invasion. Activation and deactivation of
Twist may be used as a prognostic factor and as a cancer
therapeutic target.
Acknowledgements
The present study was supported by a grant from
‘Identification of Specific Markers and Drug Discovery for Cancer
Stem Cells by Single Cell Analysis’, Program of International
S&T Cooperation (no. 2014DFA30450).
References
|
1
|
Arnold M, Sierra MS, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global patterns and trends in
colorectal cancer incidence and mortality. Gut. 66:683–691. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brabletz T, Hlubek F, Spaderna S,
Schmalhofer O, Hiendlmeyer E, Jung A and Kirchner T: Invasion and
metastasis in colorectal cancer: Epithelial-mesenchymal transition,
mesenchymal-epithelial transition, stem cells and β-catenin. Cells
Tissues Organs. 179:56–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Haggar FA and Boushey RP: Colorectal
cancer epidemiology: Incidence, mortality, survival, and risk
factors. Clin Colon Rectal Surg. 22:191–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Costi R, Leonardi F, Zanoni D, Violi V and
Roncoroni L: Palliative care and end-stage colorectal cancer
management: The surgeon meets the oncologist. World J
Gastroenterol. 20:7602–7621. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Serrano-Gomez SJ, Maziveyi M and Alahari
SK: Regulation of epithelial-mesenchymal transition through
epigenetic and post-translational modifications. Mol Cancer.
15:182016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xue C, Plieth D, Venkov C, Xu C and
Neilson EG: The gatekeeper effect of epithelial-mesenchymal
transition regulates the frequency of breast cancer metastasis.
Cancer Res. 63:3386–3394. 2003.PubMed/NCBI
|
|
10
|
Wang Y and Zhou BP: Epithelial-mesenchymal
transition in breast cancer progression and metastasis. Chin J
Cancer. 30:603–611. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hayashida T, Jinno H, Kitagawa Y and
Kitajima M: Cooperation of cancer stem cell properties and
epithelial-mesenchymal transition in the establishment of breast
cancer metastasis. J Oncol. 2011:591427. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y and Zhou BP: Epithelial-mesenchymal
transition - a hallmark of breast cancer metastasis. Cancer Hallm.
1:38–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sethi S, Macoska J, Chen W and Sarkar FH:
Molecular signature of epithelial-mesenchymal transition (EMT) in
human prostate cancer bone metastasis. Am J Transl Res. 3:90–99.
2010.PubMed/NCBI
|
|
14
|
Yan B, Jiang N and Niu YJ: Epithelial
mesenchymal transition in prostate cancer: Advances in current
research. Zhonghua Nan Ke Xue. 21:847–851. 2015.PubMed/NCBI
|
|
15
|
Khan MI, Hamid A, Adhami VM, Lall RK and
Mukhtar H: Role of epithelial mesenchymal transition in prostate
tumorigenesis. Curr Pharm Des. 21:1240–1248. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van Zijl F, Zulehner G, Petz M, Schneller
D, Kornauth C, Hau M, Machat G, Grubinger M, Huber H and Mikulits
W: Epithelial-mesenchymal transition in hepatocellular carcinoma.
Future Oncol. 5:1169–1179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nurwidya F, Takahashi F, Murakami A and
Takahashi K: Epithelial mesenchymal transition in drug resistance
and metastasis of lung cancer. Cancer Res Treat. 44:151–156. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Soltermann A, Tischler V, Arbogast S,
Braun J, Probst-Hensch N, Weder W, Moch H and Kristiansen G:
Prognostic significance of epithelial-mesenchymal and
mesenchymal-epithelial transition protein expression in non-small
cell lung cancer. Clin Cancer Res. 14:7430–7437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ikeguchi M, Makino M and Kaibara N:
Clinical significance of E-cadherin-catenin complex expression in
metastatic foci of colorectal carcinoma. J Surg Oncol. 77:201–207.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Roy F and Berx G: The cell-cell
adhesion molecule E-cadherin. Cell Mol Life Sci. 65:3756–3788.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu X and Chu KM: E-cadherin and gastric
cancer: Cause, consequence, and applications. BioMed Res Int.
2014:637308. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Saito T, Oda Y, Kawaguchi K, Sugimachi K,
Yamamoto H, Tateishi N, Tanaka K, Matsuda S, Iwamoto Y, Ladanyi M,
et al: E-cadherin mutation and Snail overexpression as alternative
mechanisms of E-cadherin inactivation in synovial sarcoma.
Oncogene. 23:8629–8638. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shargh SA, Sakizli M, Khalaj V, Movafagh
A, Yazdi H, Hagigatjou E, Sayad A, Mansouri N, Mortazavi-Tabatabaei
SA and Khorshid HR Khorram: Downregulation of E-cadherin expression
in breast cancer by promoter hypermethylation and its relation with
progression and prognosis of tumor. Med Oncol. 31:2502014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yan HB, Wang XF, Zhang Q, Tang ZQ, Jiang
YH, Fan HZ, Sun YH, Yang PY and Liu F: Reduced expression of the
chromatin remodeling gene ARID1A enhances gastric cancer cell
migration and invasion via downregulation of E-cadherin
transcription. Carcinogenesis. 35:867–876. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hanrahan K, O'Neill A, Prencipe M, Bugler
J, Murphy L, Fabre A, Puhr M, Culig Z, Murphy K and Watson RW: The
role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in
mediating docetaxel-resistant prostate cancer. Mol Oncol.
11:251–265. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rosivatz E, Becker I, Specht K, Fricke E,
Luber B, Busch R, Höfler H and Becker KF: Differential expression
of the epithelial-mesenchymal transition regulators snail, SIP1,
and twist in gastric cancer. Am J Pathol. 161:1881–1891. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jurgens G, Wieschaus E, Nussleinvolhard C
and Kluding H: Mutations affecting the pattern of the larval
cuticle in Drosophila melanogaster: II. Zygotic loci on the third
chromosome. Dev Genes Evol. 193:267–282. 1984.
|
|
30
|
Sandmann T, Girardot C, Brehme M,
Tongprasit W, Stolc V and Furlong EE: A core transcriptional
network for early mesoderm development in Drosophila melanogaster.
Genes Dev. 21:436–449. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reardon W and Winter RM: Saethre-Chotzen
syndrome. J Med Genet. 31:393–396. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zang C, Liu X, Li B, He Y, Jing S, He Y,
Wu W, Zhang B, Ma S, Dai W, et al: 6/STAT3/TWIST inhibition
reverses ionizing radiation-induced EMT and radioresistance in
esophageal squamous carcinoma. Oncotarget. 8:11228–11238.
2017.PubMed/NCBI
|
|
33
|
Fan Q, Qiu MT, Zhu Z, Zhou JH, Chen L,
Zhou Y, Gu W, Wang LH, Li ZN, Xu Y, et al: Twist induces
epithelial-mesenchymal transition in cervical carcinogenesis by
regulating the TGF-β/Smad3 signaling pathway. Oncol Rep.
34:1787–1794. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu HK, Kim JS, Lee HJ, Ahn JH, Lee SK,
Hong SW and Yoon Y: Suppression of colorectal cancer liver
metastasis and extension of survival by expression of
apolipoprotein(a) kringles. Cancer Res. 64:7092–7098. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lavilla-Alonso S, Abo-Ramadan U, Halavaara
J, Escutenaire S, Tatlisumak T, Saksela K, Kanerva A, Hemminki A
and Pesonen S: Optimized mouse model for the imaging of tumor
metastasis upon experimental therapy. PLoS One. 6:e268102011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Center MM, Jemal A and Ward E:
International trends in colorectal cancer incidence rates. Cancer
Epidemiol Biomarkers Prev. 18:1688–1694. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thukkani N, Williams JL and Sonnenberg A:
Epidemiologic characteristics of patients with inflammatory bowel
disease undergoing colonoscopy. Inflamm Bowel Dis. 17:1333–1337.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li M, Luan F, Zhao Y, Hao H, Zhou Y, Han W
and Fu X: Epithelial-mesenchymal transition: An emerging target in
tissue fibrosis. Exp Biol Med. 241:1–13. 2016. View Article : Google Scholar
|
|
42
|
Tennakoon AH, Izawa T, Kuwamura M and
Yamate J: Pathogenesis of type 2 epithelial to mesenchymal
transition (EMT) in renal and hepatic fibrosis. J Clin Med. 5:pii:
E42015. View Article : Google Scholar
|
|
43
|
Avizienyte E, Brunton VG, Fincham VJ and
Frame MC: The SRC-induced mesenchymal state in late-stage colon
cancer cells. Cells Tissues Organs. 179:73–80. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bujko M, Kober P, Mikula M, Ligaj M,
Ostrowski J and Siedlecki JA: Expression changes of cell-cell
adhesion-related genes in colorectal tumors. Oncol Lett.
9:2463–2470. 2015.PubMed/NCBI
|
|
45
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ribatti D: Epithelial-mesenchymal
transition in morphogenesis, cancer progression and angiogenesis.
Exp Cell Res. 353:1–5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Steinestel K, Eder S, Schrader AJ and
Steinestel J: Clinical significance of epithelial-mesenchymal
transition. Clin Transl Med. 3:172014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rubio D, Garcia S, De la Cueva T, Paz MF,
Lloyd AC, Bernad A and Garcia-Castro J: Human mesenchymal stem cell
transformation is associated with a mesenchymal-epithelial
transition. Exp Cell Res. 314:691–698. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sipos F and Műzes G: Isolated lymphoid
follicles in colon: Switch points between inflammation and
colorectal cancer? World J Gastroenterol. 17:1666–1673. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Birchmeier W and Behrens J: Cadherin
expression in carcinomas: Role in the formation of cell junctions
and the prevention of invasiveness. Biochim Biophys Acta.
1198:11–26. 1994.PubMed/NCBI
|
|
52
|
Christofori G and Semb H: The role of the
cell-adhesion molecule E-cadherin as a tumour-suppressor gene.
Trends Biochem Sci. 24:73–76. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fukumura D and Jain RK: Tumor
microvasculature and microenvironment: Targets for
anti-angiogenesis and normalization. Microvasc Res. 74:72–84. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nakagawa H, Liyanarachchi S, Davuluri RV,
Auer H, Martin EW Jr, de la Chapelle A and Frankel WL: Role of
cancer-associated stromal fibroblasts in metastatic colon cancer to
the liver and their expression profiles. Oncogene. 23:7366–7377.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Satoh Y, Esche C, Gambotto A, Shurin GV,
Yurkovetsky ZR, Robbins PD, Watkins SC, Todo S, Herberman RB, Lotze
MT, et al: Local administration of IL-12-transfected dendritic
cells induces antitumor immune responses to colon adenocarcinoma in
the liver in mice. J Exp Ther Oncol. 2:337–349. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bouvet M, Tsuji K, Yang M, Jiang P, Moossa
AR and Hoffman RM: In vivo color-coded imaging of the interaction
of colon cancer cells and splenocytes in the formation of liver
metastases. Cancer Res. 66:11293–11297. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Thalheimer A, Otto C, Bueter M, Illert B,
Gattenlohner S, Gasser M, Fein M, Germer CT and Waaga-Gasser AM:
Tumor cell dissemination in a human colon cancer animal model:
Orthotopic implantation or intraportal injection? Eur Surg Res.
42:195–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yamamoto N, Yang M, Jiang P, Xu M,
Tsuchiya H, Tomita K, Moossa AR and Hoffman RM: Determination of
clonality of metastasis by cell-specific color-coded
fluorescent-protein imaging. Cancer Res. 63:7785–7790.
2003.PubMed/NCBI
|