Introduction
Breast cancer is the most common malignancy in woman
worldwide, and the second leading cause of cancer-related deaths in
females (1). Considering that
metastasis is responsible for 90% of these deaths (2), understanding the mechanisms which
contribute to this endpoint is fundamental in the design of
treatment strategies to alleviate breast cancer mortality. The
pathological progression of breast cancer is well documented
(3). However, the factors which
govern this progression are less characterized (4). Considerable attention has been paid to
the contribution of somatic mutation and epigenetic alterations in
cancer initiation and progression (5–7).
However, the role of tumour-associated microenvironmental changes
may be equally contributive and are only recently gaining impetus
(8).
Hypoxia exemplifies one microenvironmental change
associated with tumourigenesis. In the earliest stages of tumour
development, abnormal proliferation and accumulation of cells can
lead to increased cellular mass, elevated intra-tissue pressure,
insufficient perfusion and subsequent O2 deficiency
(9). In breast cancer patients,
intra-tumour measurements conducted in situ have revealed
substantial levels of O2 deprivation compared to normal
breast tissue (10). Hypoxia in
breast cancer has been associated with poor patient prognosis
(11–13), resistance to chemotherapy (14,15)
and increased metastasis (13,16,17).
How hypoxia influences cancer progression is not
fully defined. It is known that cells respond to reduced
O2 availability by increasing the activity of
hypoxia-inducible factors (HIF-1α and HIF-2α) which, in turn,
mediate global transcriptional changes (18). These transcriptional changes involve
many genes and may alter various cellular processes which
contribute to cancer progression (18). Numerous studies connecting hypoxia
and cancer have been conducted on transformed cells isolated from
animal models, patient tumours and established cancer cell lines
(19). However, these cells harbour
many cancer-associated genetic and epigenetic changes. How hypoxia
affects breast epithelial cells in the earlier stages of
transformation remains less well defined.
In the present study, we used the untransformed
MCF-10A breast epithelial cell line and hypoxic culture conditions
to replicate conditions found within early hyperplastic breast
lesions. Using this model we were able to study the effects of
O2 deprivation independent from the contribution of
cancer-associated genetic and epigenetic changes. We demonstrated
that reduced O2 availability induced a number of changes
consistent with increased metastatic potential. Proliferation and
cell cycle progression were perturbed along with an increase in
apoptosis. A rise in the CD44+CD24−/low cells
coupled with an increased colony forming ability indicated a rise
in the stem cell population. Cells underwent cellular and molecular
changes consistent with epithelial-to-mesenchymal transition (EMT).
Furthermore, hypoxia increased the migratory and invasive
capabilities of these cells. Collectively, these results highlight
the contribution of hypoxic microenvironmental changes in cancer
progression and dissemination.
Materials and methods
Human tissue samples and ethics
statement
Surplus breast tissue initially removed surgically
for diagnostic purposes was used in the present study following
informed patient consent. Archived paraffin-embedded tissue was
obtained from Bristol Royal Infirmary under ethical approval from
the NHS Health Research Authority and UWE Ethics Committee (Ref.
11/SW/0127). All methods were performed in accordance with the NHS
Health Research Authority guidelines and regulations.
Cell culture and hypoxia
MCF-10A cells were purchased from the American
Tissue Culture Collection (ATCC; Manassas, VA, USA) and cultured in
Dulbecco's modified Eagle's medium/Nutrient Mixture F-12 Ham
supplemented with 100 ng/ml cholera toxin (Sigma, St. Louis, MO,
USA), 20 ng/ml epidermal growth factor (EGF) (Thermo Fisher
Scientific, Inc., Waltham, MA, USA), 10 µg/ml insulin, 500 ng/ml
hydrocortisone and 5% heat-inactivated horse serum (all from
Sigma). Experiments were conducted in the aforementioned media
mixture excluding EGF (media was replaced at least 24 h before
experiments). MCF-10A cells were subjected to no >8 passages in
culture before experiments. Whilst control cells were incubated at
37°C in a humidified atmosphere containing 5% CO2 and
~21% O2 (termed normoxia), hypoxic conditions (termed
hypoxia) were induced using an airtight modular incubator chamber
(Billups-Rothenberg, Inc., San Diego, CA, USA). Briefly, the cells
were sealed in the modular incubator chambers with a sterile
phosphate-buffered saline (PBS) reserve to maintain humidity, and
then purged with a reduced O2 gas mixture (1%
O2, 5% CO2 and 94% N2). The
chamber was then sealed and placed in an incubator at 37°C for 72
h.
Immunofluorescence microscopy
Paraffin blocks containing embedded human breast
tissue were sectioned at 4 µm using a microtome (Leica RM2235) and
mounted on Superfrost Plus slides (Thermo Fisher Scientific, Inc.).
Sections were then deparaffinized with Histoclear (National
Diagnostics, Atlanta, GA, USA) and rehydrated using a series of
ethanol concentrations and dH2O. Antigen unmasking was
performed by heating in citrate buffer (pH 6.0) using a water bath
for 30 min (95–100°C) and then allowing the sections to cool to
room temperature (RT) in the buffer. Cultured MCF-10A cells were
fixed with ice cold 4% paraformaldehyde for 20 min and then stored
at 4°C in 70% ethanol. The slides and/or fixed cells were incubated
in blocking serum [goat serum (Vector Laboratories, Burlingame, CA,
USA) diluted in Tris-buffered saline (TBS)] for 30 min at RT, and
then incubated in a primary antibody overnight at 4°C. Antibodies
used were anti-human and are as follows: CA IX (1:20; a kind gift
from Professor M. Ladomery), Ki-67 (RM-9106-S, 1:50; Thermo Fisher
Scientific, Inc.), cleaved caspase-3 (9661s, 1:200; Cell Signaling
Technology, Inc., Beverly, MA, USA), E-cadherin (610181, 1:200),
β-catenin (610153, 1:200) and vimentin (550513, 1:100) (all from BD
Biosciences, Franklin Lakes, NJ, USA). The following day, the
slides and/or cells were washed in TBS and then incubated with
suitable fluorescent-labelled secondary antibodies [Alexa
Fluor-conjugated secondary antibodies (A1101/A11008/A11005, 1:200;
Thermo Fisher Scientific, Inc.)] for 1 h at RT. Subsequently, the
slides and/or cells were washed with TBS, then mounted using
Vectashield Hardset Mounting Media with
4,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). All
images were obtained using a fluorescence microscope (Nikon Eclipse
80i).
Western blot analyses
Cells were harvested in lysis buffer (10 mM
Tris-HCl, 50 mM sodium chloride, 5 mM EDTA, 15 mM sodium
pyrophosphate, 50 mM sodium fluoride and 100 µM sodium
orthovanadate) supplemented with phosphatase (Roche Applied
Science, Indianapolis, IN, USA) and protease (Sigma) inhibitor
cocktails at 4°C for 30 min. Following collection, the cells were
sonicated on ice using Soniprep 150 (MSE Ltd., London, UK), then
centrifuged at 15,000 × g for 15 min at 4°C and then the
supernatant was collected. The protein concentration was determined
using a Coomassie (Bradford) protein assay kit (Thermo Fisher
Scientific, Inc.). An equal amount of protein from each sample was
separated using 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) gel and transferred onto a
nitrocellulose membrane (GE Healthcare, Little Chalfont, UK). After
blocking with 5% milk powder for 1 h at room temperature, the
membranes were incubated in a primary antibody overnight at 4°C.
The antibodies used were anti-human and are as follows: CA IX
(1:20; a gift from Professor M. Ladomery), E-cadherin (610181,
1:2,000), β-catenin (610153, 1:2,000), vimentin (550513, 1:1,000)
(all from BD Biosciences) and β-actin (MA1-140, 1:10,000; Thermo
Fisher Scientific, Inc.) which was used as a loading control. The
blot membrane was washed, then incubated with horseradish
peroxidase-conjugated secondary antibodies (PI-1000/PI-2000,
1:2,000-1:5,000; Vector Laboratories), and signals were revealed
using a chemiluminescence kit (Thermo Fisher Scientific, Inc.).
Proliferation and apoptosis
scoring
Proliferation and apoptosis were assessed as a
percentage of Ki-67-positive cells and a percentage of cleaved
caspase-3-positive cells, respectively. Briefly, 10 evenly
distributed ×40 fields of view were imaged using a fluorescence
microscope (Nikon Eclipse 80i) for each independent group.
Positively-labeled cells were counted and scored as a percentage of
total cells. Experiments were performed at least in triplicate for
each group.
Flow cytometry
Cells were washed once with Hanks balanced salt
solution (HBSS) (Thermo Fisher Scientific, Inc.), and then
harvested with 0.05% trypsin/0.025% EDTA (Thermo Fisher Scientific,
Inc.). Detached cells were washed with HBSS containing 2% horse
serum (Sigma) (wash buffer), and re-suspended in the wash buffer
(106 cells/100 µl). Anti-human CD24-FITC-conjugated
(560992, 1:25; BD Biosciences) and anti-human CD44-APC-conjugated
(103012, 1:25; BioLegend, Inc., San Diego, CA, USA) antibodies or
the respective isotype controls were added to the cell suspension,
as recommended by the manufacturer, and incubated at 4°C in the
dark for 30 min. Subsequently, the labelled cells were washed in
wash buffer and then analysed on an Accuri C6 cytometer using CFlow
Plus software (both from BD Biosciences).
Cell cycle analysis
Cells were harvested, washed with ice-cold PBS, and
then fixed in 70% ethanol for at least 30 min at 4°C. Before
analysis, the cells were washed again in PBS, then incubated in
staining buffer [100 µg/ml RNase and 50 µg/ml propidium iodide (PI)
(Sigma)] in the dark at 4°C for 30 min. The samples were analysed
by flow cytometry using an Accuri C6 cytometer (BD Biosciences).
CFlow plus software (BD Biosciences) was used to calculate the
percentage of cells in the G0/G1, S and G2/M phases. All studies
were performed in triplicate.
Mammosphere forming assay
Six-well culture plates were coated with
poly(2-hydroxyethyl methacrylate) (Santa Cruz Biotechnology, Santa
Cruz, CA, USA) to obtain an ultra-low adhesion surface. Following
treatment, the cells were trypsinized and mechanically disrupted to
obtain single-cell suspensions. The single-cell suspensions were
then plated at 1×103 in 1 ml MCF-10A medium in the
ultra-low adhesion wells. The cells were left to form spheres for
10 days, and mammospheres were considered cell aggregates >50 µm
in diameter. The mammospheres were imaged, counted and measured
using a phase-contrast inverted microscope (Nikon Eclipse TE300).
Each experiment was repeated in triplicate.
Wound healing assay
Cells were plated in 6-well culture plates, and
wounds were inflicted upon the cell monolayers using a sterile
plastic 200-µl micropipette tip. Phase-contrast microscopy images
were immediately obtained after wounding and again 48 h later using
an inverted microscope (Nikon Eclipse TE300). The experiments were
independently performed in triplicate, and the migration distance
under each condition was assessed by analyzing the images using
ImageJ software (National Institutes of Health, Rockville, MD,
USA).
Transwell invasion assay
Transwell inserts (Millipore, Billerica, MA, USA)
containing polycarbonate filters with 8-µm pores were used in the
assay. The inserts were coated with 50 µl of Matrigel matrix (1
mg/ml) according to the manufacturer's recommendations (Thermo
Fisher Scientific, Inc.). The cells were seeded in the upper
chambers of the inserts at a density of 2×105 cells in 1
ml serum-free MCF-10A medium. MCF-10A medium (2 ml) containing
serum was placed in the lower chambers. Following 72 h of
treatment, the cells on the upper surface of the membrane were
removed using a methanol coated cotton swab. The cells on the lower
chamber were fixed in 4% paraformaldehyde and stained with
hematoxylin (Sigma). For each membrane, the number of cells was
counted in 10 evenly distributed ×40 fields of view using a light
microscope (Nikon Eclipse 80i). Each experiment was repeated in
triplicate.
Statistical analysis
Data for each group are presented as the mean ± SD.
Statistical analyses were performed using SPSS for Windows, version
20.0 (IBM SPSS, Inc., Chicago, IL, USA). Values of P<0.05 were
deemed statistically significant.
Results
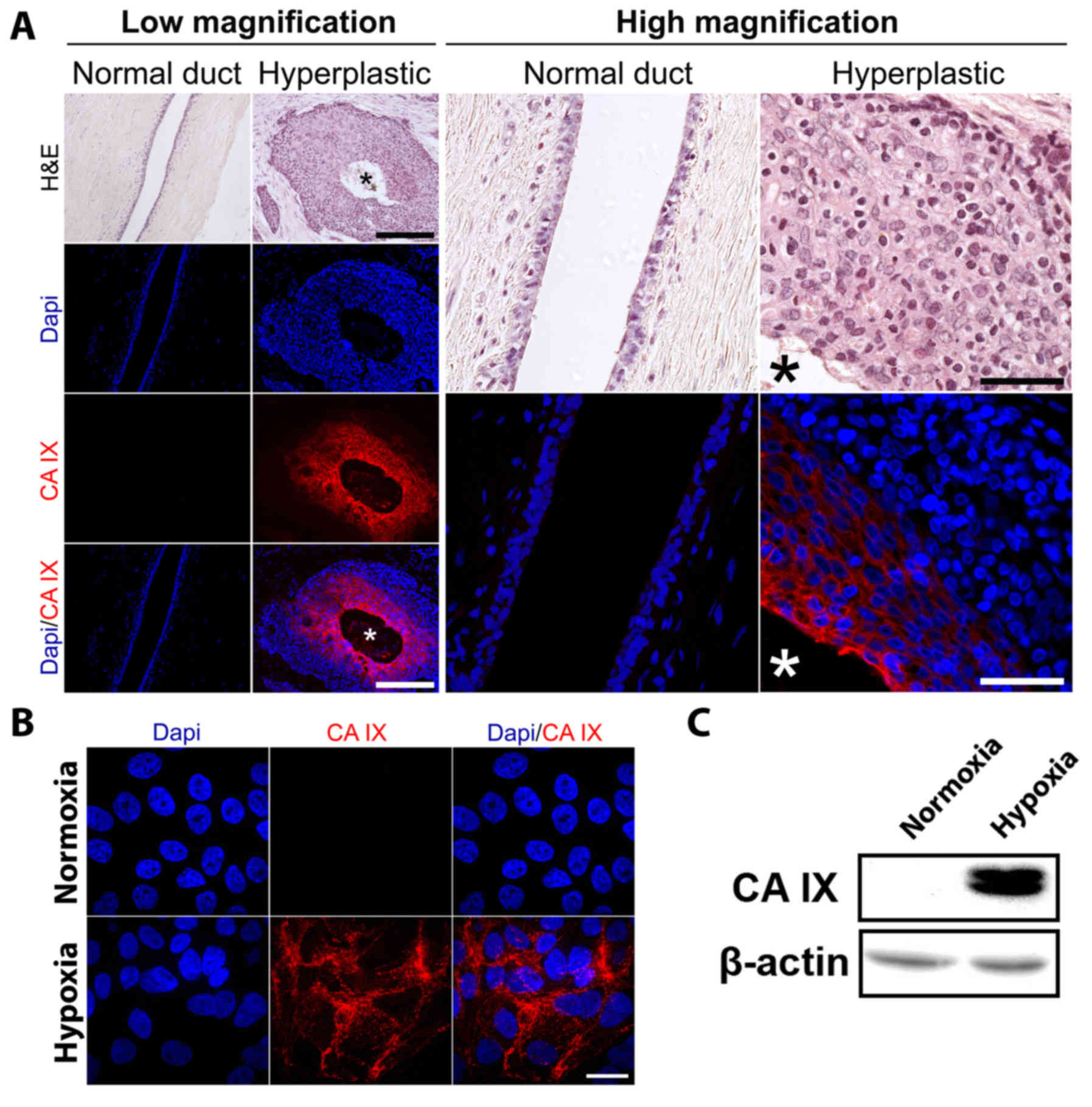
Hypoxic conditions induce upregulation
of carbonic anhydrase IX (CA IX)
CA IX is a downstream target of HIF-1α and a robust
marker of hypoxia (20). To assess
the consequence of abnormal breast cell propagation on
intracellular O2 levels, sections of hyperplastic breast
tissue were labeled for CA IX and compared to control tissue
(Fig. 1A). Whilst CA IX expression
was undetectable in control tissue, upregulation was prominent
within hyperplastic tissue with the highest expression observed
within the center of lesions corresponding to areas with the most
limited access to blood supply.
To model hypoxic conditions found within the breast
tumour microenvironment and delineate associated consequences,
MCF-10A cells were cultured in hypoxic conditions (1%
O2) for 72 h and compared to cells cultured in normoxia
(21% O2). Previous studies have suggested a level of ~1%
O2 is found within the breast tumour microenvironment
(21). Whilst control MCF-10A cells
cultured in normoxia displayed undetectable levels of CA IX
expression, MCF-10A cells cultured under hypoxic conditions
displayed an increase in CA IX expression detected by both
fluorescence microscopy (Fig. 1B)
and western blot analysis (Fig.
1C).
Hypoxia reduces proliferation, induces
apoptosis and perturbs cell cycle progression
Increased cell division and evasion of cell death
are both prominent features in most tumours (22). To assess the effects of hypoxia on
cell division, proliferation was analysed by monitoring changes in
Ki-67 expression (Fig. 2A). A
statistically significant reduction in the percentage of Ki-67
positive cells was observed in MCF-10A cells cultured under hypoxic
conditions in comparison to those cultured in normoxia (2.50±1.28
compared to 29.53±3.89% respectively; P<0.05) (Fig. 2B). To assess the effects of hypoxia
on cell death, apoptosis was analysed using cleaved caspase-3
expression (Fig. 2C). A
statistically significant increase in the percentage of cleaved
caspase-3 positive cells was observed in MCF-10A cells cultured
under hypoxic conditions compared with those cultured in normoxia
(3.91±1.12 compared to 0.35±0.18% respectively; P<0.05)
(Fig. 2D).
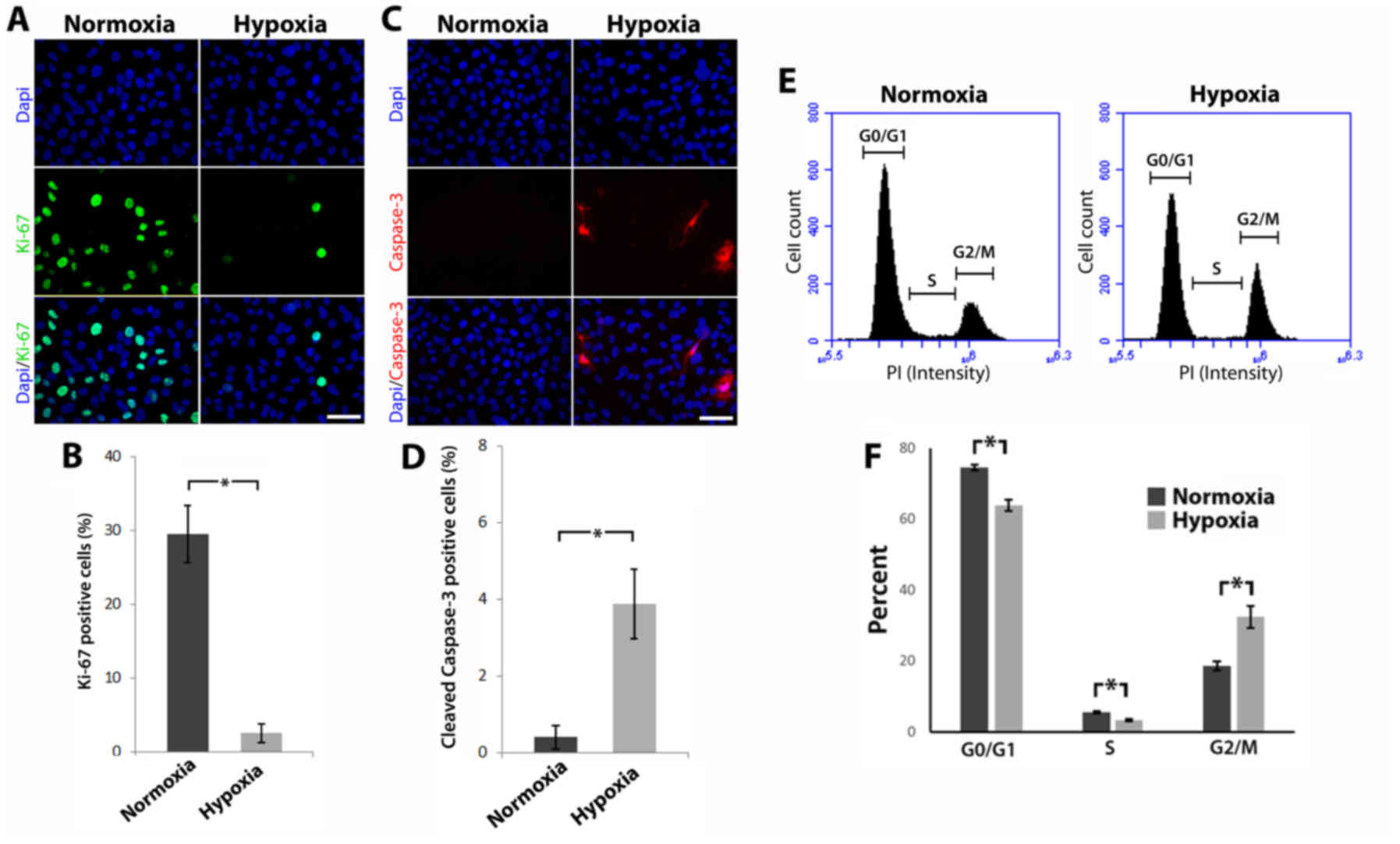 | Figure 2.Hypoxia reduces proliferation,
perturbs cell cycle progression and induces apoptosis. (A) Cells
were labeled for Ki-67 using fluorescent immunohistochemistry
(scale bar, 25 µm). (B) Percentage of Ki-67 positive cells (error
bars ± standard deviation, *P≤0.05, Mann-Whitney U test, n≥3). (C)
Cells were labeled for cleaved caspase-3 using fluorescent
immunohistochemistry (scale bar, 25 µm). (D) Percentage of cleaved
caspase-3 positive cells (error bars ± standard deviation, *P≤0.05,
Mann-Whitney U test, n≥3). (E) Cell cycle distribution was
evaluated using flow cytometry. (F) Graph displays the cell cycle
phase expressed as a percentage of total cells (error bars ±
standard deviation, *P≤0.05, Mann-Whitney U test, n≥3). |
Given the decrease in proliferation and increase in
apoptosis in MCF-10A cells cultured in hypoxia and the link between
these parameters and cell cycle progression, cell cycle
distribution analysis was performed using PI staining and flow
cytometry (Fig. 2E). ‘Gating’ was
performed in analyses to include live cells in the G0/G1, S or G2/M
phases whilst excluding debris and/or necrotic cells. MCF-10A cells
cultured in normoxia had the following distribution: G0/G1 phase,
74.27±0.81%; S phase, 5.63±0.32%; and G2/M phase, 18.57±1.30%,
whilst MCF-10A cells cultured in hypoxia had this distribution:
G0/G1 phase, 63.83±1.63%; S phase, 3.33±0.25%; and G2/M phase,
32.40±3.15%. As shown in Fig. 2F,
MCF-10A cells cultured in hypoxic conditions had a statistically
significant decrease in the percentage of cells in the G0/G1 and S
phases (P<0.05), compared to MCF-10A cells cultured in normoxia.
Conversely, a statistically significant increase in the percentage
of cells in the G2/M phase (P<0.05) was observed in MCF-10A
cells cultured in hypoxia compared to MCF-10A cells cultured in
normoxia. Collectively, these data revealed that hypoxia can
regulate cell growth and can block cell cycle progression in the
G2/M phase.
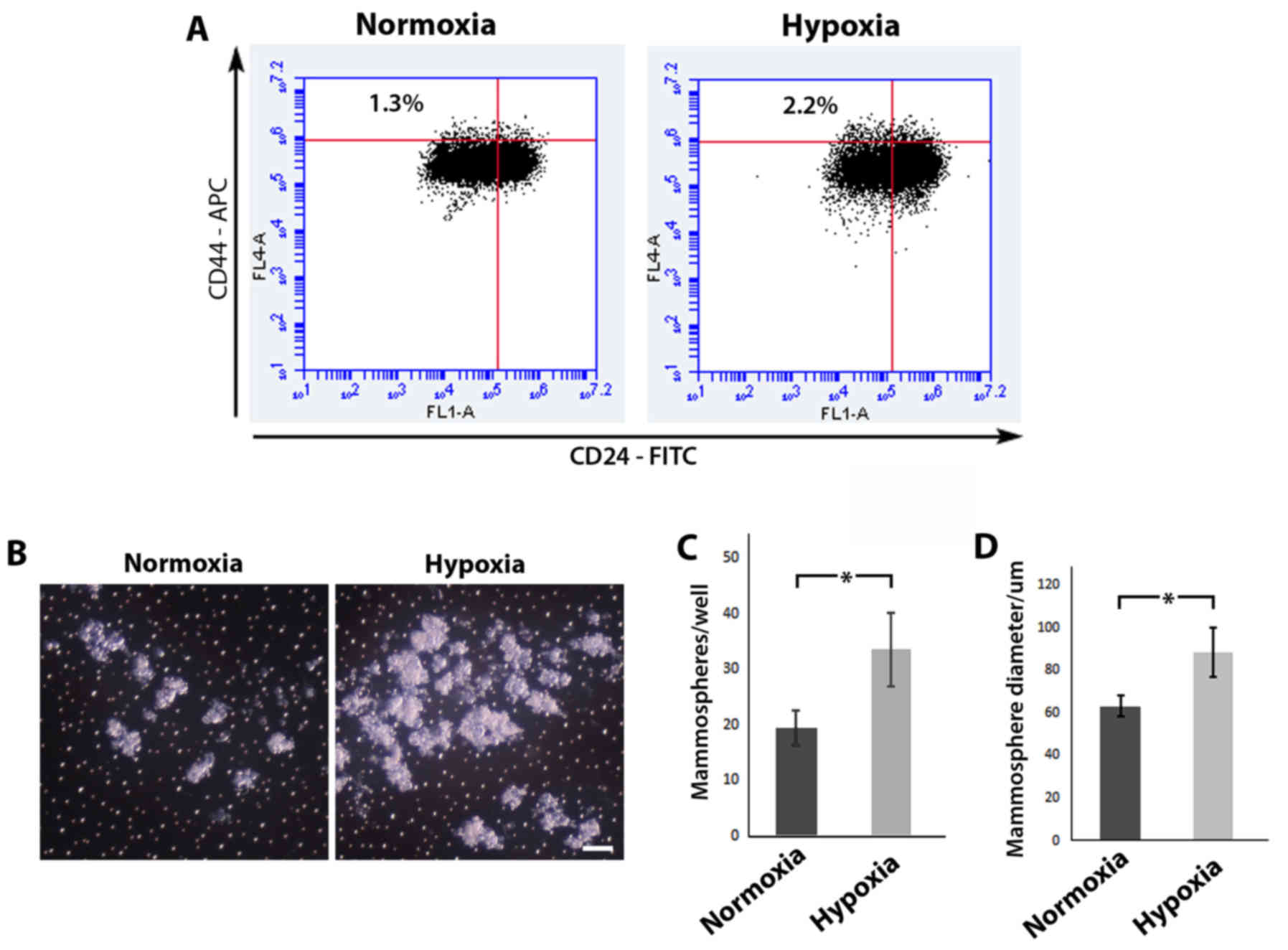
Hypoxia increases the stem cell
population
Previous studies have utilized the cell surface
markers CD44 and CD24 to distinguish a
CD44+CD24−/low sub-population which is
enriched for stem cells/cancer stem cells (23,24).
Using flow cytometric analysis (Fig.
3A), we revealed that MCF-10A cells cultured under hypoxic
conditions displayed a higher percentage of cells in the
CD44+CD24−/low sub-population in comparison
to MCF-10A cells cultured in normoxia (2.2% in comparison to 1.3%,
respectively). Mammosphere assays have been previously used as a
surrogate reporter of stem cell activity (25,26),
and an increase in the number and/or size of formed colonies are
indicative of an expanded stem cell population. MCF-10A cells grown
in normoxia or hypoxia were seeded in low adhesion culture vessels,
left to form spheres in normoxia and then compared (Fig. 3B). Following re-oxygenation, MCF-10A
cells cultured in hypoxia displayed a statistically significant
increase in both mammosphere forming efficiency (33.50±6.56/well
compared to 19.50±3.11/well, respectively; P<0.05) (Fig. 3C) and mammosphere size (88.78±11.57
compared to 63.49±4.77 µm respectively, P<0.05) (Fig. 3D) in comparison to MCF-10A cells
initially cultured in normoxia. Collectively, these results suggest
that hypoxic conditions lead to an expansion of the stem cell
population.
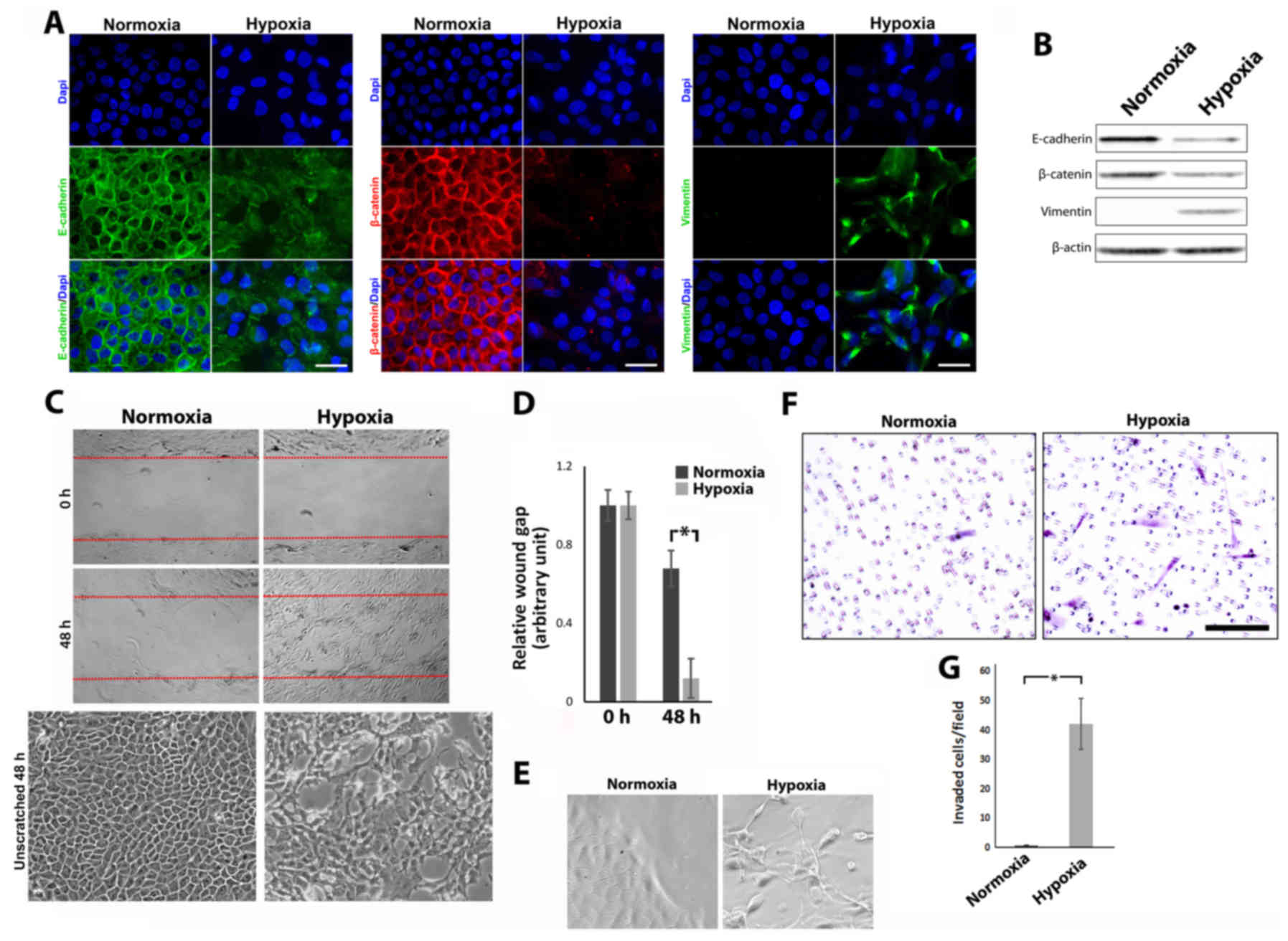
Hypoxia induces EMT, increases
migration and invasion
EMT is a process in which epithelial cells lose
epithelial characteristics and acquire mesenchymal properties. It
is recognized as an important event in the progression and
dissemination of cancer (27).
MCF-10A cells cultured in hypoxia or normoxia were labeled for
E-cadherin, β-catenin and vimentin using fluorescent
immunohistochemistry (Fig. 4A).
MCF-10A cells cultured in hypoxic conditions displayed a loss of
total and membrane bound E-cadherin, a loss of total and membrane
bound β-catenin (although no nuclearization was apparent)
concomitant with an upregulation of vimentin expression.
Collectively, these expression changes along with characteristic
changes noticed in cell shape are indicative of EMT (28,29).
Total levels of protein expression, as detected by western
blotting, confirmed global changes (Fig. 4B). Migratory and invasive
capabilities are further traits acquired by cells to allow cancer
metastasis (30). Scratch wound
assays were used to assess the migratory ability of MCF-10A cells
cultured in hypoxia vs. normoxia (Fig.
4C). MCF-10A cells cultured under hypoxic conditions displayed
an increase in migratory ability. Comparative unscratched areas
demonstrate highly polarized and tightly packed cells following
normoxic culture conditions whilst following hypoxia, cells lose
their tightly packed formation and appear more sporadic.
Collectively, this suggests that wound closure is due to increased
migratory abilities rather than an increase in cell number.
Quantitative analysis of wound gap closure (Fig. 4D) revealed a statistically
significant increase in gap closure and thus a higher rate of
migration in MCF-10A cells cultured under hypoxic conditions
(relative gap remaining after 48 h in normoxic culture conditions
0.68±0.091 compared to 0.12±0.1 in cells cultured under hypoxic
conditions; P<0.05). Higher magnification of MCF-10A cells at
the leading edge of migration revealed the extent of changes in
cell shape and a more ‘mesenchymal/fibroblastic’ appearance of
cells cultured in hypoxia as opposed to normoxia (Fig. 4E). A Matrigel Transwell invasion
assay was used to compare and analyze the invasive capacity of
MCF-10A cells cultured in hypoxia vs. normoxia. Whilst cells
cultured in normoxia displayed a limited ability to move across the
Matrigel barrier, cells cultured in hypoxic conditions were readily
observed on the bottom of the insert (Fig. 4F). Quantification of these cells
revealed a significant increase in the number of invaded cells in
cells cultured under hypoxic conditions in comparison to those
cultured in normoxia (42.2±8.57/field of view compared to
0.77±0.05/field of view respectively; P<0.05). Collectively,
these results revealed that O2 deprivation in MCF-10A
cells can lead to changes consistent with EMT, increased migratory
ability and increased invasive capabilities.
Discussion
As metastasis is responsible for ~90% of
cancer-related deaths (2),
underscoring the mechanisms that contribute to cancer dissemination
are vital in our understanding of the disease and may thus help
expose potential preventative strategies. Whilst considerable
attention has been paid to the contribution of genetic and
epigenetic alterations in this pathological process (5–7), the
importance of tumour microenvironmental changes are beginning to be
exposed (8). In the present study,
we used hypoxic conditions to replicate an important
microenvironmental change associated with tumourigenesis. MCF-10A
cells were exposed to low O2 levels to replicate
conditions found within the earlier stages of breast
tumourigenesis. O2 deprivation led to some changes not
immediately associated with tumourigenesis, such as decreased
proliferation, cell cycle arrest and increased apoptosis. In
contrast, hypoxia did induce other changes more consistent with a
progression towards metastatic disease, such as an increase in
‘stemness’, induction of EMT and increased migratory and invasive
capabilities.
Previous studies have linked hypoxia with reduced
proliferation in breast cancer cell lines and ductal carcinoma
in situ (31), a precursor
of invasive ductal carcinoma. HIF-1α expression has been observed
at this early stage of breast tumour development (31,32),
and has been revealed to both inhibit transcription (33), and promote degradation (34) of c-MYC, an essential regulator of
cellular growth and the cell cycle (35). Cell cycle progression was attenuated
at the G2/M phase in our model and may represent a further
mechanism for our observed reductions in proliferation. Previous
studies have linked hypoxia with several G2/M checkpoint regulators
and blockage of cell cycle progression at this phase (36,37)
and this has been reported to contribute to increased chemo- and/or
radio-resistance in some tumours (38,39).
The induction of apoptosis in our model may also be explained
through HIF-1α expression. HIF-1α has previously been
reported to promote apoptosis (40,41).
This, in part, could be explained by stabilization of p53 by HIF-1α
(42) and/or by increased
transcription of HIF-1α targets which are pro-apoptotic such as
NIP3 (43).
Given that sustaining proliferative signaling and
resisting cell death are ‘hallmarks’ of cancer (22), reduced proliferation, cell cycle
arrest and increased apoptosis induced by hypoxia may seem
disadvantageous for tumourigenesis. However, previous studies have
demonstrated that hypoxia can exert a selective pressure whereby
cells with accumulated genetic alterations, such as the loss of p53
(44), gain a selective advantage
and constitute the tumour (44,45).
Furthermore, it is known that hypoxia can increase mutation
frequency and lead to genomic instability (46). This is most likely due to the
effects of hypoxia on the DNA damage response exerted through both
HIF-1α dependent (47) and HIF-1α
independent means (48).
Collectively, these changes can lead to the generation of cells
which possess the genetic and epigenetic adaptations essential for
tumour progression into metastatic disease.
Hypoxia in our model also induced an increase in the
stem cell population. The link between stem cells and cancer is
well documented (49,50), and cancer stem cells are reportedly
responsible for initiating metastatic growth in various cancers
including breast (23,51,52).
Previous studies have reported a less differentiated phenotype
and/or an increase in stemness induced by hypoxia in breast tumour
tissue (31,53,54).
Increases in stemness can, in part, be explained by the ability of
HIF-1α and HIF-2α to induce various transcriptional programs, some
of which include pluripotency factors (55,56).
The consequence of hypoxia-induced increases in stem cell numbers
in early neoplastic lesions and the contribution of this to
metastatic disease may be two-fold. First, an increase in stem cell
numbers due to hypoxia along with the increased rate of mutation
may increase the chance of oncogenic mutations occurring within
stem cell populations leading to cancer stem cells. Second, hypoxia
leading to an increase in the number of cancer stem cells may lead
to an increase in metastatic potential. These reasons may
contribute to why hypoxia is linked to increased metastatic disease
(13,16,17).
A more direct involvement of hypoxia in metastasis
is elucidated from the induction of EMT observed in our model along
with the increased migratory and invasive behavior of these cells.
As mentioned previously, EMT is an important event in the
progression and dissemination of cancer (27). Previous studies have linked hypoxia,
EMT, increased migration and invasion to various cancer cell lines
including breast (57–59). This most likely occurs through
HIF-1α- and HIF-2α-related transcriptional changes (60). In the present study we demonstrated
that hypoxia-induced EMT, increased migratory and invasive behavior
in untransformed cells. Given that metastasis occurs in the later
stages of cancer progression following an accumulation of genetic
and epigenetic alterations, the significance of this finding
remains unclear. However, a mechanistic dissection of the roles of
HIF-1α and HIF-2α isoforms at this early stage of transformation
and the relevance of their true input warrant further investigation
and should be the basis of future experiments.
To conclude, the present study provided evidence
that tumour-associated microenvironmental changes have a
substantial role alongside genetic and epigenetic alterations in
the progression of breast cancer. Hypoxia can occur in the earliest
stages of tumourigenesis and influence various cellular processes
associated with metastatic potential. Although the present study
uses a simplistic approach to delineate the contributions of the
hypoxic microenvironment from the myriad of genetic and epigenetic
alterations found in human tumours; in reality, understanding the
interactions between these co-contributors may elucidate the true
factors driving metastasis in human disease.
Acknowledgements
We thank Dr Muhammed Sohail at Bristol Royal
Infirmary for overseeing the cataloging and processing of human
breast tissue samples. We also thank Mr. David Corry, Dr Jeff Davey
and Dr Natasha McGuire for their technical support and Mr. Paul
Kendrick for assistance in histology.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mehlen P and Puisieux A: Metastasis: A
question of life or death. Nat Rev Cancer. 6:449–458. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rosen PP: Rosen's Breast Pathology. 3rd.
LWW; pp. 171–242. 2008
|
|
4
|
Thompson A, Brennan K, Cox A, Gee J,
Harcourt D, Harris A, Harvie M, Holen I, Howell A, Nicholson R, et
al: Evaluation of the current knowledge limitations in breast
cancer research: A gap analysis. Breast Cancer Res. 10:R262008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome - biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Network CGA: Cancer Genome Atlas Network:
Comprehensive molecular portraits of human breast tumours. Nature.
490:61–70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bissell MJ and Hines WC: Why don't we get
more cancer? A proposed role of the microenvironment in restraining
cancer progression. Nat Med. 17:320–329. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vaupel P, Höckel M and Mayer A: Detection
and characterization of tumor hypoxia using pO2 histography.
Antioxid Redox Signal. 9:1221–1235. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vaupel P: Prognostic potential of the
pre-therapeutic tumor oxygenation status. Adv Exp Med Biol.
645:241–246. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Van den Beucken T, Koch E, Chu K,
Rupaimoole R, Prickaerts P, Adriaens M, Voncken JW, Harris AL,
Buffa FM, Haider S, et al: Hypoxia promotes stem cell phenotypes
and poor prognosis through epigenetic regulation of DICER. Nat
Commun. 5:52032014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schindl M, Schoppmann SF, Samonigg H,
Hausmaninger H, Kwasny W, Gnant M, Jakesz R, Kubista E, Birner P
and Oberhuber G; Austrian Breast and Colorectal Cancer Study Group,
: Overexpression of hypoxia-inducible factor 1alpha is associated
with an unfavorable prognosis in lymph node-positive breast cancer.
Clin Cancer Res. 8:1831–1837. 2002.PubMed/NCBI
|
|
14
|
Samanta D, Gilkes DM, Chaturvedi P, Xiang
L and Semenza GL: Hypoxia-inducible factors are required for
chemotherapy resistance of breast cancer stem cells. Proc Natl Acad
Sci USA. 111:pp. E5429–E5438. 2014; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
O'Reilly EA, Gubbins L, Sharma S, Tully R,
Guang MH, Weiner-Gorzel K, McCaffrey J, Harrison M, Furlong F, Kell
M, et al: The fate of chemoresistance in triple negative breast
cancer (TNBC). BBA Clin. 3:257–275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hussain SA, Ganesan R, Reynolds G, Gross
L, Stevens A, Pastorek J, Murray PG, Perunovic B, Anwar MS,
Billingham L, et al: Hypoxia-regulated carbonic anhydrase IX
expression is associated with poor survival in patients with
invasive breast cancer. Br J Cancer. 96:104–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hiraga T, Kizaka-Kondoh S, Hirota K,
Hiraoka M and Yoneda T: Hypoxia and hypoxia-inducible factor-1
expression enhance osteolytic bone metastases of breast cancer.
Cancer Res. 67:4157–4163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Semenza GL: Hypoxia-inducible factors in
physiology and medicine. Cell. 148:399–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kunz M and Ibrahim SM: Molecular responses
to hypoxia in tumor cells. Mol Cancer. 2:232003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Potter C and Harris AL: Hypoxia inducible
carbonic anhydrase IX, marker of tumour hypoxia, survival pathway
and therapy target. Cell Cycle. 3:164–167. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Höckel M and Vaupel P: Tumor hypoxia:
Definitions and current clinical, biologic, and molecular aspects.
J Natl Cancer Inst. 93:266–276. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 100:pp.
3983–3988. 2003; View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghebeh H, Sleiman GM, Manogaran PS,
Al-Mazrou A, Barhoush E, Al-Mohanna FH, Tulbah A, Al-Faqeeh K and
Adra CN: Profiling of normal and malignant breast tissue show
CD44high/CD24low phenotype as a predominant stem/progenitor marker
when used in combination with Ep-CAM/CD49f markers. BMC Cancer.
13:2892013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dontu G, Abdallah WM, Foley JM, Jackson
KW, Clarke MF, Kawamura MJ and Wicha MS: In vitro propagation and
transcriptional profiling of human mammary stem/progenitor cells.
Genes Dev. 17:1253–1270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Diaz-Guerra E, Lillo MA, Santamaria S and
Garcia-Sanz JA: Intrinsic cues and hormones control mouse mammary
epithelial tree size. FASEB J. 26:3844–3853. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blanco D, Vicent S, Elizegi E, Pino I,
Fraga MF, Esteller M, Saffiotti U, Lecanda F and Montuenga LM:
Altered expression of adhesion molecules and epithelial-mesenchymal
transition in silica-induced rat lung carcinogenesis. Lab Invest.
84:999–1012. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yoshida R, Kimura N, Harada Y and Ohuchi
N: The loss of E-cadherin, α- and β-catenin expression is
associated with metastasis and poor prognosis in invasive breast
cancer. Int J Oncol. 18:513–520. 2001.PubMed/NCBI
|
|
30
|
Friedl P and Wolf K: Tumour-cell invasion
and migration: Diversity and escape mechanisms. Nat Rev Cancer.
3:362–374. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Helczynska K, Kronblad A, Jögi A, Nilsson
E, Beckman S, Landberg G and Påhlman S: Hypoxia promotes a
dedifferentiated phenotype in ductal breast carcinoma in situ.
Cancer Res. 63:1441–1444. 2003.PubMed/NCBI
|
|
32
|
Bos R, Zhong H, Hanrahan CF, Mommers EC,
Semenza GL, Pinedo HM, Abeloff MD, Simons JW, van Diest PJ and van
der Wall E: Levels of hypoxia-inducible factor-1 alpha during
breast carcinogenesis. J Natl Cancer Inst. 93:309–314. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Koshiji M, Kageyama Y, Pete EA, Horikawa
I, Barrett JC and Huang LE: HIF-1alpha induces cell cycle arrest by
functionally counteracting Myc. EMBO J. 23:1949–1956. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang H, Gao P, Fukuda R, Kumar G,
Krishnamachary B, Zeller KI, Dang CV and Semenza GL: HIF-1 inhibits
mitochondrial biogenesis and cellular respiration in VHL-deficient
renal cell carcinoma by repression of C-MYC activity. Cancer Cell.
11:407–420. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schmidt EV: The role of c-myc in cellular
growth control. Oncogene. 18:2988–2996. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Amellem O, Löffler M and Pettersen EO:
Regulation of cell proliferation under extreme and moderate
hypoxia: The role of pyrimidine (deoxy)nucleotides. Br J Cancer.
70:857–866. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hasvold G, Lund-Andersen C, Lando M,
Patzke S, Hauge S, Suo Z, Lyng H and Syljuåsen RG: Hypoxia-induced
alterations of G2 checkpoint regulators. Mol Oncol. 10:764–773.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sullivan R, Paré GC, Frederiksen LJ,
Semenza GL and Graham CH: Hypoxia-induced resistance to anticancer
drugs is associated with decreased senescence and requires
hypoxia-inducible factor-1 activity. Mol Cancer Ther. 7:1961–1973.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wouters A, Pauwels B, Lardon F and
Vermorken JB: Review: Implications of in vitro research on the
effect of radiotherapy and chemotherapy under hypoxic conditions.
Oncologist. 12:690–712. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Carmeliet P, Dor Y, Herbert JM, Fukumura
D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R,
Maxwell P, et al: Role of HIF-1alpha in hypoxia-mediated apoptosis,
cell proliferation and tumour angiogenesis. Nature. 394:485–490.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Volm M and Koomägi R: Hypoxia-inducible
factor (HIF-1) and its relationship to apoptosis and proliferation
in lung cancer. Anticancer Res. 20:1527–1533. 2000.PubMed/NCBI
|
|
42
|
Ravi R, Mookerjee B, Bhujwalla ZM, Sutter
CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL and Bedi A:
Regulation of tumor angiogenesis by p53-induced degradation of
hypoxia-inducible factor 1alpha. Genes Dev. 14:34–44.
2000.PubMed/NCBI
|
|
43
|
Bruick RK: Expression of the gene encoding
the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad
Sci USA. 97:pp. 9082–9087. 2000; View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Graeber TG, Osmanian C, Jacks T, Housman
DE, Koch CJ, Lowe SW and Giaccia AJ: Hypoxia-mediated selection of
cells with diminished apoptotic potential in solid tumours. Nature.
379:88–91. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Semenza GL: Hypoxia, clonal selection, and
the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol.
35:71–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Reynolds TY, Rockwell S and Glazer PM:
Genetic instability induced by the tumor microenvironment. Cancer
Res. 56:5754–5757. 1996.PubMed/NCBI
|
|
47
|
Sendoel A, Kohler I, Fellmann C, Lowe SW
and Hengartner MO: HIF-1 antagonizes p53-mediated apoptosis through
a secreted neuronal tyrosinase. Nature. 465:577–583. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bindra RS and Glazer PM: Genetic
instability and the tumor microenvironment: Towards the concept of
microenvironment-induced mutagenesis. Mutat Res. 569:75–85. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Clevers H: The cancer stem cell: Premises,
promises and challenges. Nat Med. 17:313–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Visvader JE and Stingl J: Mammary stem
cells and the differentiation hierarchy: Current status and
perspectives. Genes Dev. 28:1143–1158. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Oskarsson T, Batlle E and Massagué J:
Metastatic stem cells: Sources, niches, and vital pathways. Cell
Stem Cell. 14:306–321. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Velasco-Velázquez MA, Popov VM, Lisanti MP
and Pestell RG: The role of breast cancer stem cells in metastasis
and therapeutic implications. Am J Pathol. 179:2–11. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Keith B and Simon MC: Hypoxia-inducible
factors, stem cells, and cancer. Cell. 129:465–472. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Conley SJ, Gheordunescu E, Kakarala P,
Newman B, Korkaya H, Heath AN, Clouthier SG and Wicha MS:
Antiangiogenic agents increase breast cancer stem cells via the
generation of tumor hypoxia. Proc Natl Acad Sci USA. 109:pp.
2784–2789. 2012; View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mathieu J, Zhang Z, Nelson A, Lamba DA,
Reh TA, Ware C and Ruohola-Baker H: Hypoxia induces re-entry of
committed cells into pluripotency. Stem Cells. 31:1737–1748. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang C, Samanta D, Lu H, Bullen JW, Zhang
H, Chen I, He X and Semenza GL: Hypoxia induces the breast cancer
stem cell phenotype by HIF-dependent and ALKBH5-mediated
m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. 113:pp.
E2047–E2056. 2016; View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Muñoz-Nájar UM, Neurath KM, Vumbaca F and
Claffey KP: Hypoxia stimulates breast carcinoma cell invasion
through MT1-MMP and MMP-2 activation. Oncogene. 25:2379–2392. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Krishnamachary B, Zagzag D, Nagasawa H,
Rainey K, Okuyama H, Baek JH and Semenza GL: Hypoxia-inducible
factor-1-dependent repression of E-cadherin in von Hippel-Lindau
tumor suppressor-null renal cell carcinoma mediated by TCF3,
ZFHX1A, and ZFHX1B. Cancer Res. 66:2725–2731. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lester RD, Jo M, Montel V, Takimoto S and
Gonias SL: uPAR induces epithelial-mesenchymal transition in
hypoxic breast cancer cells. J Cell Biol. 178:425–436. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Semenza GL: The hypoxic tumor
microenvironment: A driving force for breast cancer progression.
Biochim Biophys Acta. 1863:382–391. 2016. View Article : Google Scholar : PubMed/NCBI
|