Introduction
Breast cancer is highly metastatic, leading to
multiorgan dysfunction and a high mortality rate (1,2), which
can be reduced by early detection and optimized therapeutic options
(3–5). Triple-negative breast cancer (TNBC) is
an aggressive subtype that does not respond to estrogen receptor
(ER)-, progesterone receptor (PR)- and human epidermal growth
factor receptor 2 (HER2)-targeted therapies, and TNBC patient
prognoses are poor (6,7). Surgery, radiotherapy, chemotherapy and
targeted therapy are the standard treatments for breast cancer
(8,9). However, the side-effects associated
with these treatments may include critical alopecia, nephrotoxicity
and hepatotoxicity (10,11). Drug resistance in cancers is also a
serious impediment to treatment and can undermine therapeutic
efficacy (12). Furthermore, target
therapeutic agents, such as trastuzumab, lapatinib and the
anti-angiogenic drug Avastin have demonstrated indeterminate
benefits in clinical settings (13–15).
Under these circumstances, development of new drugs is particularly
compelling and imperative. Although great efforts have been made to
design more powerful drugs for the treatment of breast cancer, the
results remain unsatisfactory. In fact, some herbal drugs in
alternative medicine have been suggested to be a better choice to
improve the current therapeutic strategy.
Glycyrrhizic acid (GA) is the main component of
licorice root extracts. GA contains 8 hydroxyl groups and is highly
hydrophilic; therefore, it is water-soluble and is easily absorbed
and metabolized by the digestive tract. GA constitutes ~2–24% of
the dry weight of licorice root. Licorice root extracts are sweet,
and GA is ~50 times sweeter than sucrose. Therefore, GA is widely
used as an additive in candy and food; it is also used in medicine
and in the beer manufacturing industry (16). GA was recently demonstrated to
induce the apoptosis of human endometrial cancer cells, and
potently inhibited the growth of lung cancer cells via caspase
activation (17). Previous studies
indicate that a mass of compounds induce autophagy for cell death
by various mechanisms (18–21), e.g. glycyrrhetinic acid induces
autophagy in leukemia and myeloma cells by suppression of the mTOR
pathway (22). However, the
potential therapeutic effects of GA on mechanisms associated with
autophagy have not yet been explored in breast cancer.
Mounting evidence indicates that GA-induced cell
apoptosis may be regulated by the caspase cascade in various types
of cancers (23–26). However, few studies have
specifically focused on GA-induced autophagy, particularly in
breast cancer cells (27,28). In the present study, we investigated
the effects of GA on mitochondrial cell death in a highly
aggressive MDA-MB-231 breast cancer model to determine whether GA
provides dual inhibition by enhancing apoptosis and autophagy
caused by damaged mitochondria.
Materials and methods
Materials
GA, N-acetyl-cysteine (NAC), staurosporine
(STS), hydrogen peroxide (H2O2),
N-phenylmaleimide (NPM), 3 methyladenine (3MA), JC-1 and
anti-β-actin (A5411) were purchased from Sigma Co. (St. Louis, MO,
USA). Anti-AIF (113306), anti-BAX (109683), anti-BCL2 (100064),
anti-PARP1 (100573), anti-porin (114187), anti-histone (122148) and
z-VAD-FMK were purchased from GeneTex (ICON-GeneTex Inc., Taipei,
Taiwan); anti-caspase-8 (ab138485), caspase-9 (ab115161),
anti-caspase-3 (ab90437), anti-LC3 (ab48394), anti-beclin 1
(ab62557) and anti-P62 (ab155686) were purchased from Abcam
(Cambridge, UK).
Cell culture
MDA-MB-231 human breast carcinoma cell lines were
cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma Co.)
supplemented with 10% fetal bovine serum (FBS) (v/v), 100 U/ml
penicillin/streptomycin/amphotericin (Sigma Co.) and 0.1 M sodium
bicarbonate. Cells were maintained in a humidified incubator at
37°C with 5% CO2 and passage by 0.25% trypsin-EDTA every
2–3 days.
Cell proliferation assay
Cell viability was analyzed using Cell Counting
Kit-8 (CCK-8) that detects the metabolic activity of cells. At the
end of the various treatments, 10 µl of the CCK-8 reagent was added
to each well (at 1×104 cells/well), and the cells were
then incubated at 37°C for 4 h. Absorbance was recorded using an
ELISA microplate reader at 450 nm.
Cell morphology staining
Cell morphology was studied by Hoechst 33258 and
propidium iodide (PI) double staining for live-cell images. After
treatment, cells (at 1×105 cells/well) were stained with
Hoechst 33258 and PI solution at room temperature for 10 min. Cells
were washed twice with phosphate-buffered saline (PBS) for analysis
by an inverted fluorescence microscope (Carl Zeiss Axiovert M200;
Carl Zeiss, Jena, Germany).
Cell death measurement
Cell survival was assessed by labeling cells with
PI. Briefly, the cells (at 1×105 cells/well) were washed
twice with PBS, re-suspended in a binding buffer and stained with 1
µg/ml of a PI solution (Sigma Co.) for 15 min at room temperature
in the dark. After incubation, the cells were analyzed using flow
cytometry.
Cellular production of reactive oxygen
species (ROS)
Intracellular production of ROS, namely hydrogen
peroxide was measured using DCFH-DA (Enzo Life Sciences, Inc.,
Farmingdale, NY, USA). Cells (at 1×105 cells/well) from
each experimental condition were stained with DCFH-DA (2 µM) at
37°C for 30 min. ROS production of cells was evaluated by flow
cytometry (FACSCalibur; Bioscience, Tokyo, Japan). Values were
expressed relative to the fluorescence signal of the control.
Mitochondrial function assays
Cells (at 1×105 cells/well) from each
experimental condition were stained with JC-1 (10 µg/ml) reagent
and MitoSOX Red (Molecular Probes Inc., Eugene, OR, USA) at 37°C
for 30 min for detection of the mitochondrial membrane potential
and mitochondrial ROS, respectively. Cells were washed twice with
PBS, trypsinized, and collected, and the pellet was re-suspended in
PBS for analysis using flow cytometry.
Immunofluorescence labeling and
immunoblotting assay
The immunoassay was performed as previously
described (28).
Caspase activity
Caspase-8, −9 and −3 activities were measured by the
colorimetric activity assay kits (Chemicon International, Temecula,
CA, USA). The assay is based on cleavage of the chromogenic
substrates, DEVD-pNA and LEHD-pNA, by caspase-3 and −9,
respectively. Cells (at 1×104 cells/well) were lysed
with the chilled lysis buffer on ice for 10 min and centrifuged for
5 min at 10,000 × g. Supernatant of each treatment was transferred
to a fresh tube and equal protein concentration was adjusted to
that of the positive control before incubating with the caspase
inhibitor in a 96-well microplate for 10 min. Caspase substrate
solution with specific peptide substrate was then added into the
supernatant and incubated for 2 h at 37°C before measuring by ELISA
reader at 405 nm.
Cell death detection by Annexin
V-FITC/PI double staining
Analysis of the cell death of each condition was
performed using an Alexa Fluor Annexin V/Dead Cell Apoptosis kit
(Molecular Probes Inc.) according to the manufacturer's protocol.
Briefly, cells (at 1×105 cells/well) were harvested
after treatment. Cells were then washed twice with cold binding
buffer, resuspended in binding buffer and stained with 5 µl each of
Annexin V-FITC and PI solution for 15 min at room temperature in
the dark. After incubation, 1 ml binding buffer was added, and
cells were analyzed by flow cytometry.
Statistical analysis
The intensity of bands in western blot analyses or
fluorescence was quantified using ImageJ software (NIH) and Zeiss
Zen software. The intensity value for each protein was normalized
against the intensity of the loading control for that same sample.
The values after normalizing to the loading control in the control
groups were set as 1.0. Data presented are the mean ± standard
error of the mean (SEM) from at least 3 independent experiments and
were analyzed using a Students t-test with two-tailed distribution
between groups as indicated in the graphs. All calculations were
performed using GraphPad Prism 6.01 version.
Results
Induction of cell death by GA in
MDA-MB-231 cells
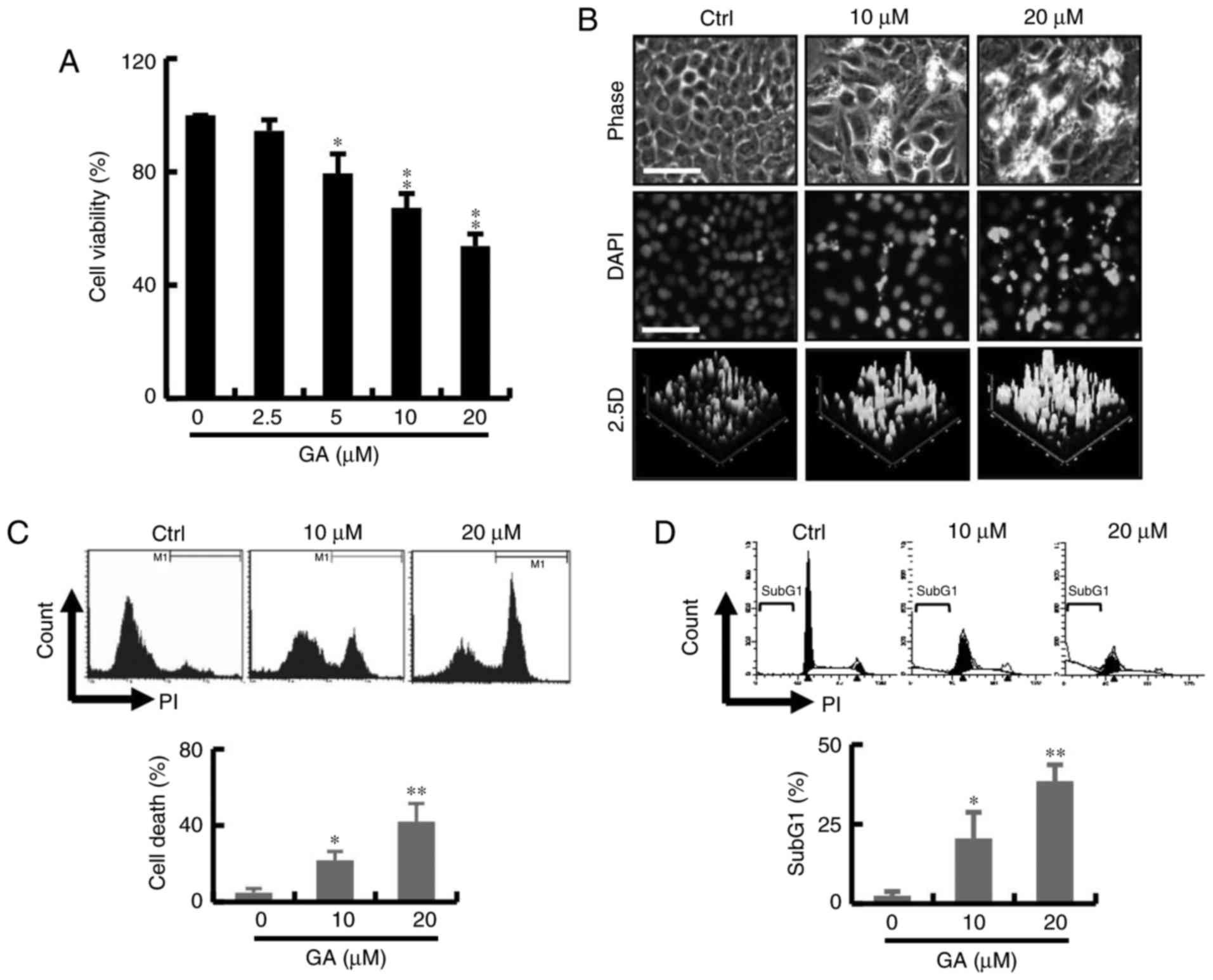
The effect of GA on cell survival is shown in
Fig. 1A. The cytotoxicity of the GA
was measured by CCK-8 assay. As shown in Fig. 1A, GA exerted a significant cytotoxic
effect at concentrations 2.5–20 mM at 24 h and the IC50
value was found to be 20 µM. These results indicated that the GA
treatment significantly reduced cancer cell viability in a
dose-dependent manner. To further evaluate the cytotoxic effect of
GA-induced breast cancer cell death, cells were stained with DAPI
after exposure to GA for 24 h. Significant change in blebbing, cell
shrinkage, DNA condensation was observed in the MDA-MB-231 breast
cancer cells; an increase in fluorescence intensity was observed by
2.5 D reconstruction image after treatment with GA (Fig. 1B). As shown in Fig. 1C, GA markedly increased the
percentage of PI-positive cells by live-cell fluorescent. In
addition, cell cycle analyses showed an increase in the subG1 phase
population at 24 h in the GA treatment groups. The increased subG1
population of GA-treated breast cells at 24 h coincided with that
of the cell viability assay (Fig.
1D).
Treatment with GA significantly
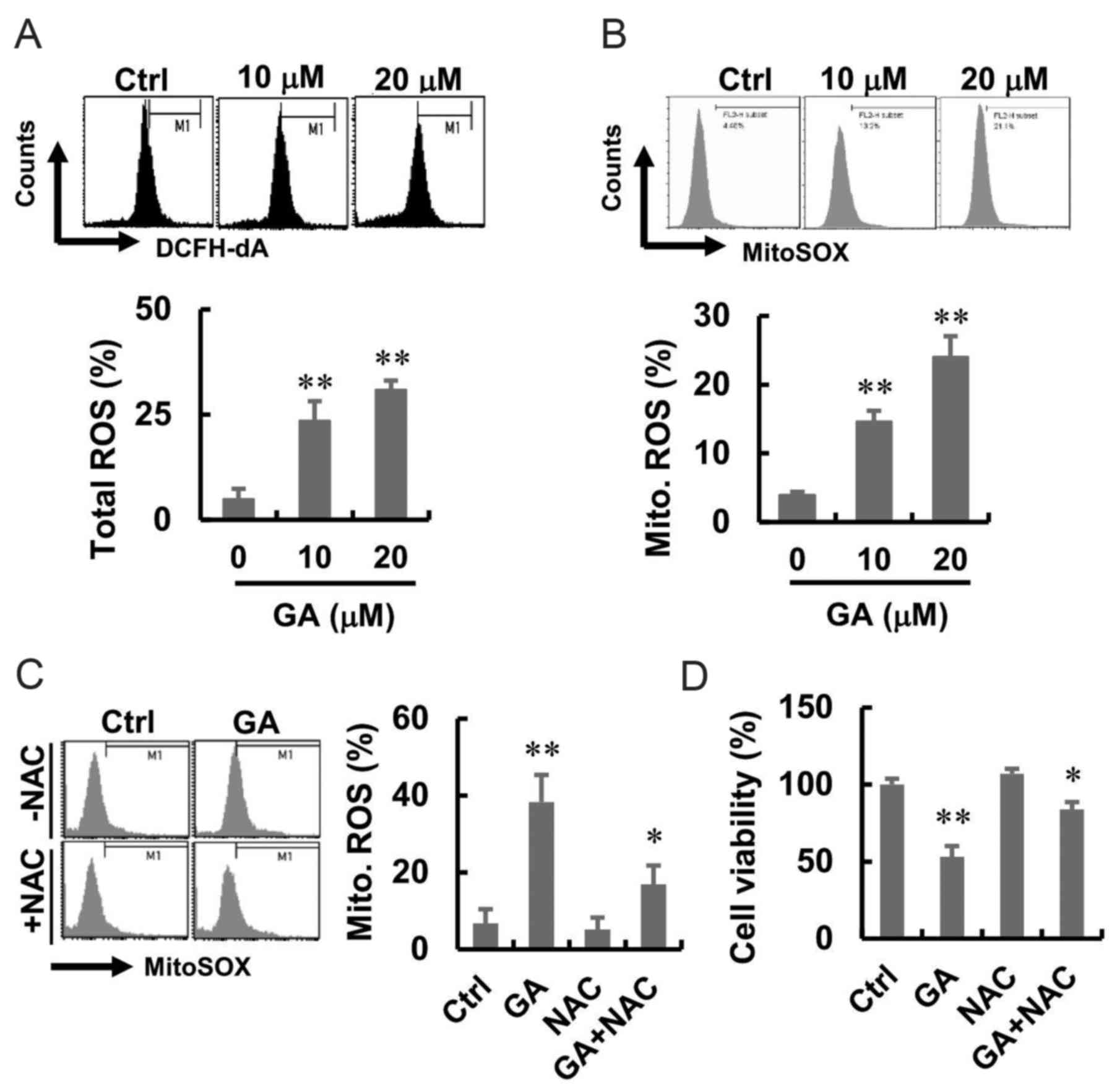
elevates the generation of ROS
To investigate the effect of GA on intracellular ROS
generation, MDA-MB-231 cells were treated with GA at different
concentrations and stained with fluorescent probe DCFH-DA and
MitoSOX, which detect hydroperoxide and mitochondrial ROS,
respectively. Cells following GA treatment showed higher green and
red fluorescence compared with that in the normal control group
(Fig. 2A and B). A flow cytometric
analysis was performed to quantify the effect of GA-induced
intracellular and mitochondrial ROS generation, and the results are
presented in Fig. 2A and B. GA
induced a significant shift in green and red fluorescence, whereas
following GA, enhanced total ROS and mitochondrial ROS production
were well apparent. To investigate whether ROS were involved in the
GA-induced cell death, we introduced NAC, an inhibitor that blocks
ROS generation, before GA treatment. The results showed that NAC
alone had no effect on the mitochondrial ROS. However, NAC
treatment blocked the GA-induced mitochondrial ROS in MDA-MB-231
cells (Fig. 2C). Moreover, there
was increased in cell viability after treatment with GA and NAC
(Fig. 2D).
GA induces caspase-independent cell
death in MDA-MB-231 cells
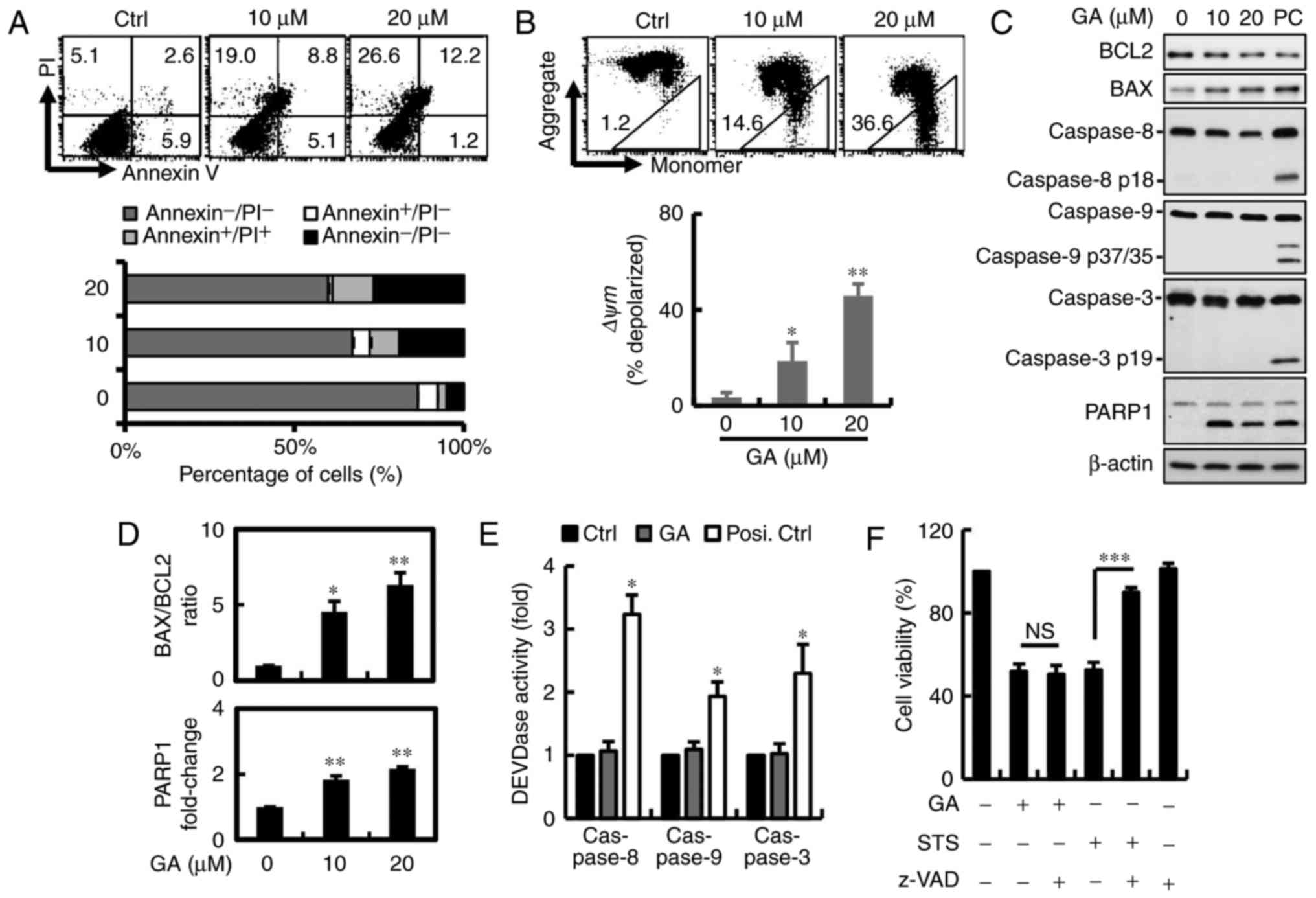
Annexin V/PI double-staining was used to detect the
PS out-flipping phenomenon, one of the clearest characteristics of
apoptotic cell death. To examine whether inhibition of GA-induced
cell proliferation was associated with apoptosis, cells were
detected after 24 h of GA exposure by flow cytometry. GA reduced
the number of viable MDA-MB-231 breast cancer cells from 87 to 60%
in a dose-dependent manner. However, we found that the cell
population shifted directly to PI+ quadrants without
going through the PI−/Annexin V+ transition.
This observation suggests that cell death induced by GA differs
from apoptosis in MDA-MB-231 cells, apparently relying mainly on
necrosis or autophagy (Fig. 3A).
Mitochondrial integrity or the loss of Δψm has been linked to
initiation of apoptosis and multiple cell death, including
autophagy and necrosis. To determine Δψm loss, breast cancer cells
were stained with JC-1 when harvested and subjected to flow
cytometric analysis. JC-1 concentrates in the mitochondria as red
fluorescent aggregates at high membrane potentials and is converted
to green monomers when Δψm is lost. Red fluorescence in MDA-MB-231
breast cancer cells after GA treatment was reduced from 99 to 64%,
indicating mitochondrial membrane potential (MMP) depolarization in
breast cancer cells (Fig. 3B). To
determine whether GA-induced apoptosis, characterized mainly by
activation of caspase-3-associated proteins leading to cleavage of
PARP1, protein expression was measured by western blotting
(Fig. 3C). Along with decreased
levels of PARP1 and a significantly increased BAX/BCL2 ratio was
observed after GA treatment (Fig.
3D). No changes in cleaved caspase-3, −9 and −8 expression and
activity were found in MDA-MB-231 breast cancer cells (Fig. 3C and E). To corroborate that
GA-induced cell death was independent of caspase, the cells were
pre-treated with 1 mM pan-caspase inhibitor z-VAD-FMK for 2 h. No
significant recovery in cell viability was observed after inhibitor
pre-treatment in the MDA-MB-231 cells, indicating that GA induced
cell death in a caspase-independent manner (Fig. 3F).
Roles of apoptosis-inducing factor
(AIF) proteins in GA-induced MDA-MB-231 cell death
To further investigate the cellular mechanism of
GA-induced cell death, the protein levels of AIF were detected. AIF
is a FAD-dependent oxidoreductase that has a vital role in
oxidative phosphorylation and is independent of the caspase
pathway. We investigated whether the translocation of AIF occurred
in the GA-induced cell death of MDA-MB-231 cells. As shown in
Fig. 4A and B, addition of GA
resulted in a significant increase in overlap coefficient between
nuclear AIF. AIF translocation occurred and subsequently appeared
in the nuclear fraction of MDA-MB-231 cells treated with GA for 24
h. We also confirmed and quantified the cellular distribution of
AIF. Immunoblotting revealed that AIF mostly resided in the
mitochondria of the cancer cells (Fig.
4C). To investigate whether the AIF pathway is involved in the
GA-induced cell death, we introduced NPM, an inhibitor that blocks
AIF activation, before GA treatment. The results showed that NPM
combined with GA had a significant increased on the expression of
AIF and PARP1 (Fig. 4D). After the
treatment of NPM, JC-1 gathered in the mitochondrial matrix and
produced red fluorescence (Fig.
4E). As shown in the results of flow cytometry, GA caused a
dramatic reduction in red fluorescence which indicated a loss of
mitochondrial membrane potential (ΔΨm) and damage of mitochondria
in the MDA-MB-231 cells (Fig. 4F).
The ratio of red and green fluorescence represented the level of
depolarization in mitochondria: NPM protected MDA-MB-231 cells
against the damage caused by GA treatment (Fig. 4G). Moreover, there was increased
cell viability after treatment with NPM, indicating that AIF may be
the major factor involved in GA-induced cell death (Fig. 4H). Furthermore, NAC treatment also
blocked the GA-induced AIF translocation to the nucleus, suggesting
that ROS may be the regulator involved in this process (Fig. 4I).
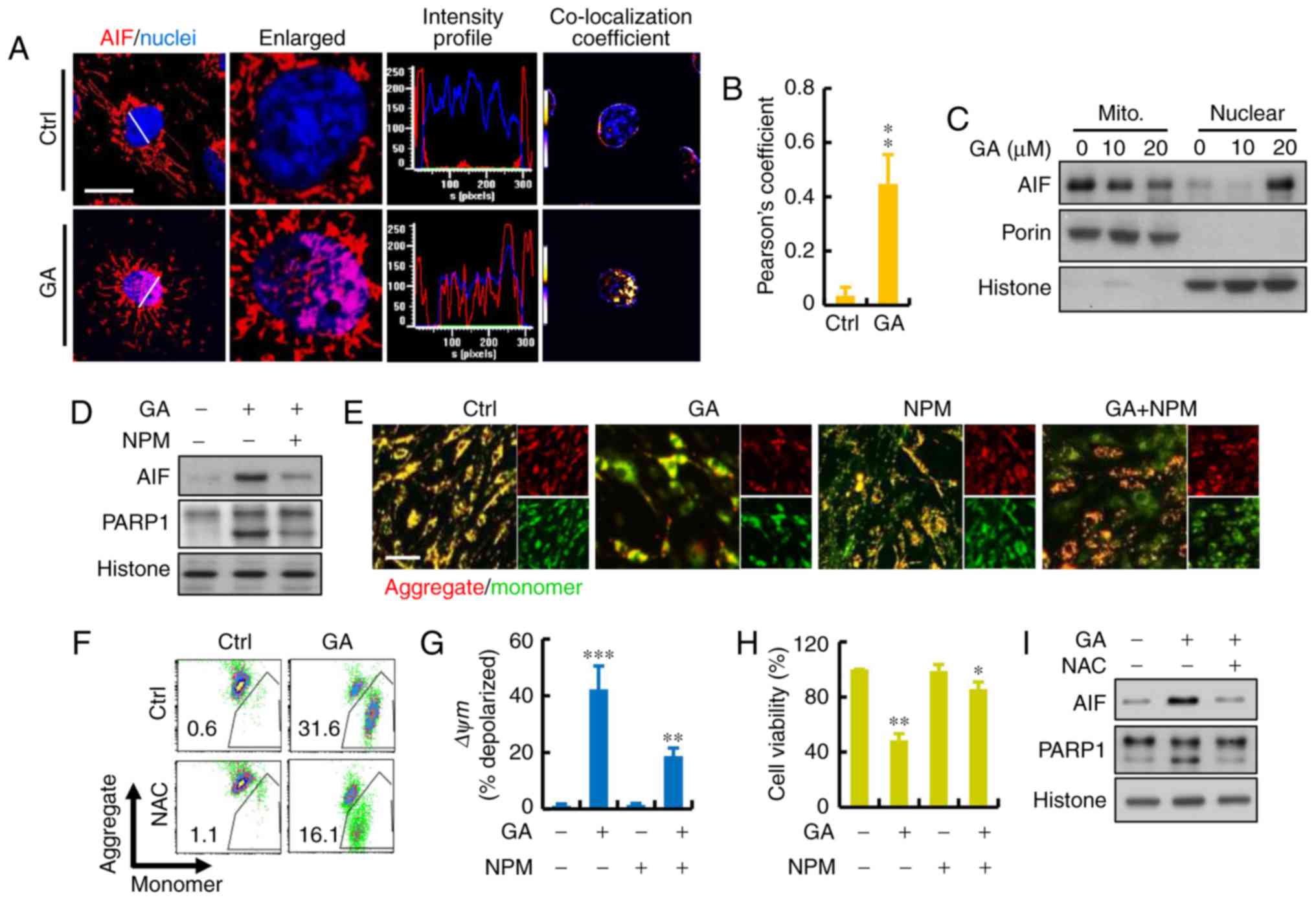 | Figure 4.Role of AIF protein in GA-induced
MDA-MB-231 cell death. (A) Representative immunofluorescence of AIF
(red) and nuclei (blue) using DAPI staining. Calculated and
quantification of Pearson's co-localization in the enlarged images,
the overlapping of the fluorescence (purple) increased in
GA-treated cells. Histograms demonstrate the fluorescence intensity
profiles along the lines indicated in the upper row images. Scare
bar, 10 µm. (B) Calculated and quantification of Pearson's
co-localization coefficients between DAPI and AIF for each group.
The coefficients were generated using ImageJ software. (C) AIF
release, revealed by western blot analysis in cytosolic and nuclear
fractions, was detected in GA-treated breast cancer cells. (D)
MDA-MB-231 cell death induced by GA in the presence or absence of
AIF inhibitor N-phenylmaleimide (NPM) as measured by
immunoblotting. (E) MMP was measured using JC-1 fluorescence
imaging in MDA-MB-231 cells. The JC-1 monomer was represented by
green fluorescence, the JC-1 aggregate image was represented by red
fluorescence, and the merged images were the combined of the green
and red images. Control cells showed strong aggregated red
fluorescence indicative of normal membrane potential. Scare bar,
100 µm. (F and G) Change in MMP in GA-pretreated MDA-MB-231 cells
by flow cytometry. Fluorescence intensity shifted from the higher
level to the lower one indicating the loss of MMP. Mitochondrial
depolarization is indicated by an increase in the red fluorescence
intensity ratio. (H) MDA-MB-231 cell death induced by GA in the
presence or absence of AIF inhibitor NPM measured by CCK-8 assay.
(I) MDA-MB-231 cell death induced by GA in the presence or absence
of ROS inhibitor NAC as measured by immunoblotting; *P<0.05,
**P<0.01 and ***P<0.001. GA, glycyrrhizic acid; NAC,
N-acetyl-cysteine; NPM, N-phenylmaleimide. |
Autophagy and ROS are involved in
GA-mediated cell death
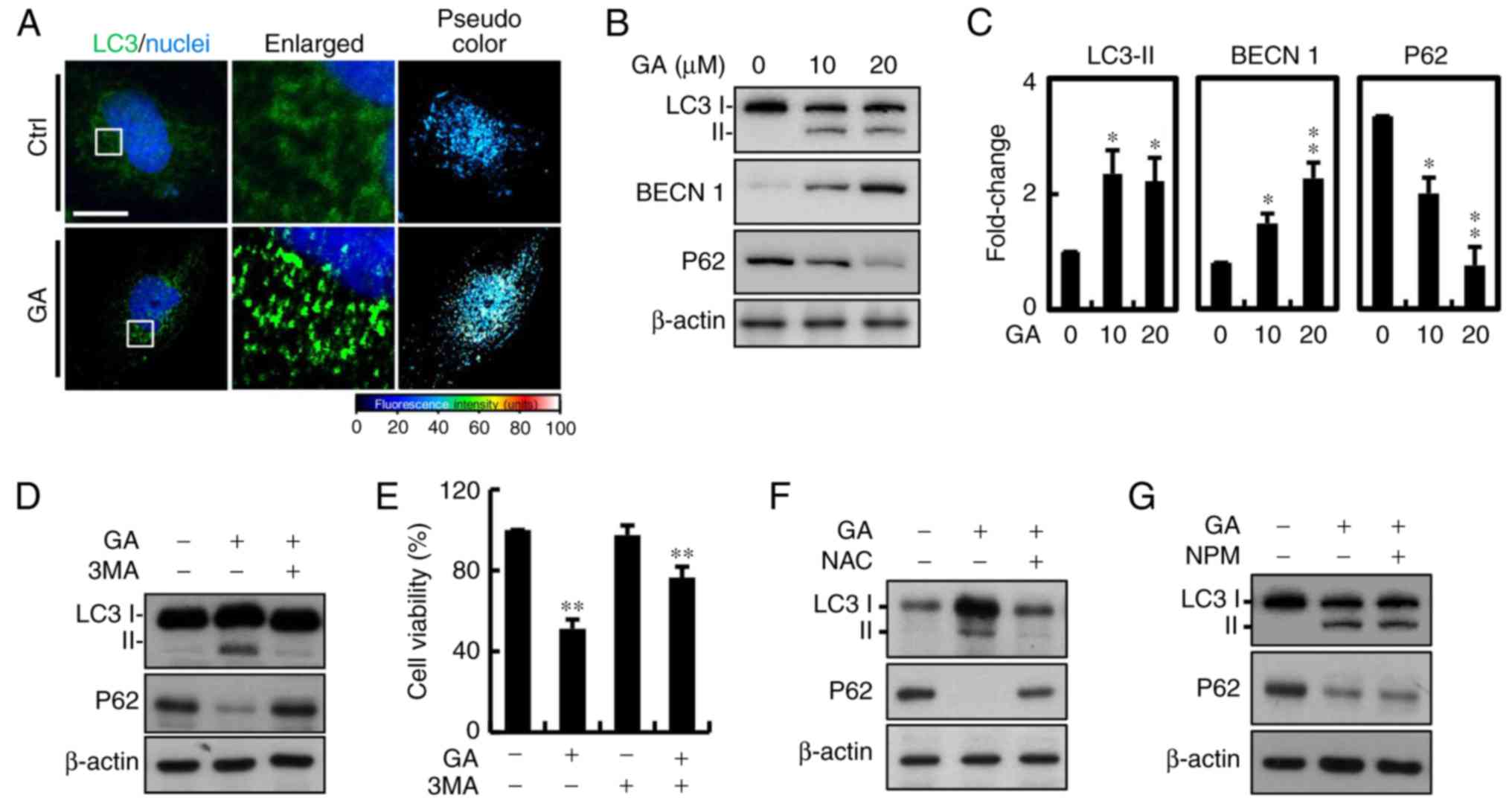
Autophagic protein LC-3 is a component of
double-membrane vesicle formation into autophagosomes. To explore
whether autophagy was involved in GA-induced cell death, the
fluorescence expression was utilized. A large number of bubbles
were observed in the GA-exposed cells after 24 h (Fig. 5A). Moreover, a number of
autophagosomes or lysosomes containing segregated were observed in
the GA-treated cells. Conversion from cytosolic LC3-I to LC3-II,
the membrane-binding form, was analyzed by western blotting. The
protein expression of LC3-II was significantly increased in the
MDA-MB-231 breast cancer cells and p62 was downregulated, which is
one of the selective substrates for autophagy. These results
suggested that AIF and LC3-II may be involved in GA-induced
autophagy (Fig. 5B and C).
Autophagy is a major process of organelle and protein degradation.
An increasing number of studies have shown that autophagy may be a
new target for anticancer therapy. To further confirm that
autophagy was involved in GA-induced cell death, 2 mM 3
methylamphetamine (3MA), an autophagy blocker that inhibits PI3K
class III protein, was added. After addition of 3MA, the increase
in LC3 was significantly inhibited compared with the control cells
(Fig. 5D). Considering all of the
above results, we conclude that GA-induced cell death in MDA-MB-231
cells was mainly due to autophagy. Moreover, there was increased in
cell viability after treatment with 3MA, indicating that autophagy
may be the major factor involved in GA-induced cell death (Fig. 5E). We further determined the
involvement of ROS in GA-induced autophagy, and protein expression
of associated signaling molecules (Fig.
5F). NAC reversed the effects of GA on protein expression of
LC3 and P62. However, treatment with AIF inhibitor NPM showed no
significant effects (Fig. 5G).
These results suggested that the anticancer effects of GA on breast
cancer cells were mediated by ROS-dependent mitochondrial
dysfunction with the involvement of AIF and LC3 signaling
pathway.
Discussion
The present study demonstrated that glycyrrhizic
acid (GA), the main component of licorice root extracts, is
cytotoxic to human breast cancer cell line MDA-MB-231. This effect
appears to involve promotion of apoptosis, autophagy and ROS
generation via mitochondria. A recent study showed that GA caused a
dose-dependent activation of NF-κB as well as ROS generation in
HepG2 cells (29). In contract,
Thirugnanam et al reported that the GA-induced apoptosis in
prostate cancer cells was dependent of caspase-3 activation
(26). Additional evidence shows
that DNA damage-inducing agents trigger caspase-independent
apoptosis in various cancer cell lines (23,24).
The present study using MDA-MB-231 cells showed that caspase
activity was not required for GA-induced apoptosis. zVAD-fmk did
not suppress GA-induced apoptosis. Moreover, GA did not induce
caspase activation (Fig. 3C). In
the present study, our results suggest that GA-induced apoptosis in
MDA-MB-231 cells is mediated by a caspase-independent
mechanism.
Autophagy or autophagocytosis is a metabolic process
involving degradation of cellular components by lysosomes that
responds to metabolic stress (30,31).
This tightly regulated process helps cells maintain balance and
subsequent recycling of cellular organelles and proteins. It is a
key mechanism by which starving cells allocate nutrients away from
unnecessary processes (32). This
mechanism is also associated with progression of certain diseases
such as atherosclerosis, neurodegenerative disease and cancers. The
two major processes involved are Atg-regulated and
chaperone-mediated autophagy (33).
Microphagy sequesters damaged organelles other than proteins in a
double-membrane autophagosome. Autophagosomes derived from the
elongation of small membrane structures are known as autophagosome
precursors. The outer membrane of the autophagosome fuses with a
lysosome in the cytoplasm to form an autolysosome, in which cell
contents undergo acidic degradation (34,35).
As already mentioned earlier, PARP1 belongs to a
family of nuclear enzymes, modulating DNA repair, transcriptional
regulation, chromatin modification and genomic stability through
polyADP-ribosylation (36,37). PARP1 activation leads to ATP
depletion, thereby inducing necrosis and apoptosis (38). However, PARP1 can be degraded
through multiple downstream proteins, including DNA-dependent
protein kinase, ubiquitination and hydrolysis via the proteasome or
by the caspase pathway (39).
Notably, PARP1 activation interfaces with signalling pathways known
to promote autophagy (40). It is
well documented that GA-induced cancer cell apoptosis occurs
through a mitochondrial pathway. We also observed that GA decreased
the levels of ΔΨm in MDA-MB-231 cells (Fig. 3B) in a dose-dependent manner.
Furthermore, upregulation of BAX, reduction of mitochondrial
membrane potentials, enhancement of AIF and LC3 expression, and
activation of autophagy were observed in the present study.
In turn, LC3 and AIF activation led to DNA damage,
and eventually, induction of autophagic cell death. DNA damage was
found in MDA-MB-231 cells after treatment with GA, and
downregulation of PARP1 inhibited DNA synthesis and repair; thus,
enhanced cytotoxicity through inhibition of DNA repair processes is
another possible mechanism explaining the cytotoxic effect of GA.
In addition, the greatly increased level of LC3 protein may have
contributed to autophagic cell death in the MDA-MB-231 breast
cancer cells and reduced cell survival even over long-term
treatment. Moreover, autophagosomes were observed through LC3
staining, which may have been activated by excess ROS production in
the GA-treated cells. To explore the molecular mechanism of
autophagy in GA-induced dell death, an inhibitor of autophagy, 3MA,
was added. 3MA partially reversed conversion of LC3, corroborating
that autophagy was involved in the GA-induced cell death. These
results confirmed the hypothesis that mitochondrial dysfunction
occurred via facilitation of apoptosis and autophagy potentially
regulated by increased ROS levels in GA-treated breast cancer
cells. Based on the antitumor activity profiles of GA treatment
in vitro and the absence of cytotoxicity, we believe that GA
has strong therapeutic value for use against breast cancers.
In conclusion, the present study demonstrated that
GA induced apoptotic and autophagic cell death, and inhibited cell
growth in vitro in breast adenocarcinoma MDA-MB-231 cells.
We conclude that GA-induced mitochondrial apoptosis and autophagy
were mainly caused by AIF and LC-3. Our findings suggest that GA
warrants further investigation as a promising complementary
therapeutic drug for the treatment of human breast cancer.
Acknowledgements
The present study was funded by grants
(103–2314-B-442-002-MY3) from the Ministry of Science and
Technology, Taiwan, and RB15001 and RB16001 from Show Chwan
Memorial Hospital, Taiwan.
References
|
1
|
Ertas IE, Sayhan S, Karagoz G and Yildirim
Y: Signet-ring cell carcinoma of the breast with uterine metastasis
treated with extensive cytoreductive surgery: A case report and
brief review of the literature. J Obstet Gynaecol Res. 38:948–952.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Novakovic S, Hocevar M, Zgajnar J, Besic N
and Stegel V: Detection of telomerase RNA in the plasma of patients
with breast cancer, malignant melanoma or thyroid cancer. Oncol
Rep. 11:245–252. 2004.PubMed/NCBI
|
|
3
|
A new therapy standard in metastatic
breast cancer - gemzar/paclitaxel. Longer survival by optimized
combination therapy. Krankenpfl J. 42:2352004.(In German).
PubMed/NCBI
|
|
4
|
Engel J, Ludwig MS, Schubert-Fritschle G,
Tretter W and Hölzel D: Cancer prevention and the contribution of
cancer registries. J Cancer Res Clin Oncol. 127:331–339. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stickeler E: Prognostic and predictive
markers for Treatment Decisions in Early Breast Cancer. Breast
Care. 6:193–198. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andergassen U, Kölbl AC, Mumm JN, Mahner S
and Jeschke U: Triple-negative breast cancer: New therapeutic
options via signalling transduction cascades. Oncol Rep.
37:3055–3060. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rakha EA, El-Sayed ME, Green AR, Lee AH,
Robertson JF and Ellis IO: Prognostic markers in triple-negative
breast cancer. Cancer. 109:25–32. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hoge AF, Asal N, Owen W and Anderson P:
Histologic and staging classification of breast cancer:
Implications for therapy. South Med J. 75:1329–1334. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bazel S, Ferry K, Shoarinejad F,
Laurykleintop L, Lange M, Tachovsky T, Longo S, Tucker S and
Alhadeff J: Analysis of breast-tissue cathepsin-d isoforms from
patients with breast-cancer, benign breast disease and from normal
controls. Int J Oncol. 5:847–853. 1994.PubMed/NCBI
|
|
10
|
Wu S, Dahut WL and Gulley JL: The use of
bisphosphonates in cancer patients. Acta Oncol. 46:581–591. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sopkova V and Mechl Z: Cisplatin
(platidiame) in the treatment of patients with disseminated breast
cancer. Vopr Onkol. 32:36–38. 1986.(In Russian). PubMed/NCBI
|
|
12
|
Kolacinska A, Chalubinska J, Zawlik I,
Szymanska B, Borowska-Garganisz E, Nowik M, Fendler W, Kubiak R,
Pawlowska Z, Morawiec Z, et al: Apoptosis-, proliferation, immune
function-, and drug resistance-related genes in ER positive, HER2
positive and triple negative breast cancer. Neoplasma. 59:424–432.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Conley SJ, Gheordunescu E, Kakarala P,
Newman B, Korkaya H, Heath AN, Clouthier SG and Wicha MS:
Antiangiogenic agents increase breast cancer stem cells via the
generation of tumor hypoxia. Proc Natl Acad Sci USA. 109:pp.
2784–2789. 2012; View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shimoyama S: Unraveling trastuzumab and
lapatinib inefficiency in gastric cancer: Future steps (Review).
Mol Clin Oncol. 2:175–181. 2014.PubMed/NCBI
|
|
15
|
Noguchi E, Kamio T, Kamio H, Miura H,
Tamaki M, Nishizawa M, Aoyama K, Oochi T and Kameoka S: Efficacy of
lapatinib monotherapy on occult breast cancer presenting with
cutaneous metastases: A case report. Oncol Lett. 8:2448–2452.
2014.PubMed/NCBI
|
|
16
|
Ming LJ and Yin AC: Therapeutic effects of
glycyrrhizic acid. Nat Prod Commun. 8:415–418. 2013.PubMed/NCBI
|
|
17
|
Huang RY, Chu YL, Jiang ZB, Chen XM, Zhang
X and Zeng X: Glycyrrhizin suppresses lung adenocarcinoma cell
growth through inhibition of thromboxane synthase. Cell Physiol
Biochem. 33:375–388. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He SQ, Gao M, Fu YF and Zhang YN:
Glycyrrhizic acid inhibits leukemia cell growth and migration via
blocking AKT/mTOR/STAT3 signaling. Int J Clin Exp Pathol.
8:5175–5181. 2015.PubMed/NCBI
|
|
19
|
Afnan Q, Kaiser PJ, Rafiq RA, Nazir LA,
Bhushan S, Bhardwaj SC, Sandhir R and Tasduq SA: Glycyrrhizic acid
prevents ultraviolet-B-induced photodamage: A role for
mitogen-activated protein kinases, nuclear factor kappa B and
mitochondrial apoptotic pathway. Exp Dermatol. 25:440–446. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park JM, Park SH, Hong KS, Han YM, Jang
SH, Kim EH and Hahm KB: Special licorice extracts containing
lowered glycyrrhizin and enhanced licochalcone A prevented
Helicobacter pylori-initiated, salt diet-promoted gastric
tumorigenesis. Helicobacter. 19:221–236. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li S, Zhu JH, Cao LP, Sun Q, Liu HD, Li
WD, Li JS and Hang CH: Growth inhibitory in vitro effects of
glycyrrhizic acid in U251 glioblastoma cell line. Neurol Sci.
35:1115–1120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cao B, Li J, Zhou X, Juan J, Han K, Zhang
Z, Kong Y, Wang J and Mao X: Clioquinol induces pro-death autophagy
in leukemia and myeloma cells by disrupting the mTOR signaling
pathway. Sci Rep. 4:57492014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nurdin SU, Le Leu RK, Young GP, Stangoulis
JC, Christophersen CT and Abbott CA: Analysis of the anti-cancer
effects of cincau extract (Premna oblongifolia Merr) and other
types of non-digestible fibre using faecal fermentation
supernatants and Caco-2 cells as a model of the human colon.
Nutrients. 9:pii: E3552017. View Article : Google Scholar
|
|
24
|
Pang L, Zhao X, Liu W, Deng J, Tan X and
Qiu L: Anticancer effect of ursodeoxycholic acid in human oral
squamous carcinoma HSC-3 cells through the caspases. Nutrients.
7:3200–3218. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chueh FS, Hsiao YT, Chang SJ, Wu PP, Yang
JS, Lin JJ, Chung JG and Lai TY: Glycyrrhizic acid induces
apoptosis in WEHI-3 mouse leukemia cells through the caspase- and
mitochondria-dependent pathways. Oncol Rep. 28:2069–2076. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thirugnanam S, Xu L, Ramaswamy K and
Gnanasekar M: Glycyrrhizin induces apoptosis in prostate cancer
cell lines DU-145 and LNCaP. Oncol Rep. 20:1387–1392.
2008.PubMed/NCBI
|
|
27
|
Zafar R and Neerja: Momordica charantia -
a review. Hamdard Med. 34:49–61. 1991.PubMed/NCBI
|
|
28
|
Li CJ, Chu CY, Huang LH, Wang MH, Sheu LF,
Yeh JI and Hsu HY: Synergistic anticancer activity of triptolide
combined with cisplatin enhances apoptosis in gastric cancer in
vitro and in vivo. Cancer Lett. 319:203–213. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hsiang CY, Lin LJ, Kao ST, Lo HY, Chou ST
and Ho TY: Glycyrrhizin, silymarin, and ursodeoxycholic acid
regulate a common hepatoprotective pathway in HepG2 cells.
Phytomedicine. 22:768–777. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tu YF, Kaipparettu BA, Ma Y and Wong LJ:
Mitochondria of highly metastatic breast cancer cell line
MDA-MB-231 exhibits increased autophagic properties. Biochim
Biophys Acta. 1807:1125–1132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shen M, Duan WM, Wu MY, Wang WJ, Liu L, Xu
MD, Zhu J, Li DM, Gui Q, Lian L, et al: Participation of autophagy
in the cytotoxicity against breast cancer cells by cisplatin. Oncol
Rep. 34:359–367. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Abounit K, Scarabelli TM and McCauley RB:
Autophagy in mammalian cells. World J Biol Chem. 3:1–6. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kaushik S, Kiffin R and Cuervo AM:
Chaperone-mediated autophagy and aging: A novel regulatory role of
lipids revealed. Autophagy. 3:387–389. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Richard V, Kindt N and Saussez S:
Macrophage migration inhibitory factor involvement in breast cancer
(Review). Int J Oncol. 47:1627–1633. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Richard V, Kindt N, Decaestecker C, Gabius
HJ, Laurent G, Noël JC and Saussez S: Involvement of macrophage
migration inhibitory factor and its receptor (CD74) in human breast
cancer. Oncol Rep. 32:523–529. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Krishnakumar R and Kraus WL: The PARP side
of the nucleus: Molecular actions, physiological outcomes, and
clinical targets. Mol Cell. 39:8–24. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gerö D, Szoleczky P, Chatzianastasiou A,
Papapetropoulos A and Szabo C: Modulation of poly(ADP-ribose)
polymerase-1 (PARP-1)-mediated oxidative cell injury by ring finger
protein 146 (RNF146) in cardiac myocytes. Mol Med. 20:313–328.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Z, Wang F, Tang T and Guo C: The role
of PARP1 in the DNA damage response and its application in tumor
therapy. Front Med. 6:156–164. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Arun B, Akar U, Gutierrez-Barrera AM,
Hortobagyi GN and Ozpolat B: The PARP inhibitor AZD2281 (Olaparib)
induces autophagy/mitophagy in BRCA1 and BRCA2 mutant breast cancer
cells. Int J Oncol. 47:262–268. 2015. View Article : Google Scholar : PubMed/NCBI
|