Introduction
The efficacy of epidermal growth factor
receptor-tyrosine kinase inhibitors (EGFR-TKIs), such as gefitinib
and erlotinib, in non-small cell lung cancer (NSCLC) therapy has
been widely demonstrated (1).
Patients with EGFR-sensitizing mutations treated with EGFR-TKIs
have a significantly longer progression-free survival (PFS)
compared with those treated with standard chemotherapy (1,2).
However, despite an excellent initial response, drug resistance
eventually develops in the majority of patients, limiting the mean
drug-response duration to less than 1 year (3).
The estrogen receptor (ER) pathway is among the most
well-studied pathways in breast cancer. Data on ER signaling in
lung cancer have increased in recent decades. In the Women's Health
Initiative, more than 16,000 postmenopausal women with an intact
uterus and no breast cancer history were randomly allocated to
supplemental estrogen and progesterone or no hormone replacement
therapy (HRT). After 5.6 years of study and 2.4 years of follow-up,
the hazard ratio for lung cancer incidence in the HRT group was
1.28 (P=0.12), and the hazard ratio for death in patients with
NSCLC and death specifically from NSCLC was 1.61 (P=0.02) and 1.87
(P=0.004), respectively (4).
Furthermore, a prospective cohort study confirmed an increased,
dose-dependent lung cancer risk among women who received HRT
(5). Commonly expressed in patients
with NSCLC, human NSCLC cell lines, and mouse models, ERβ is the
major functional receptor of lung cancer (6,7).
Evidence supports an interaction between EGFR and
ERβ pathways in the development of lung cancer. Aromatase is a
candidate prognostic factor in patients with lung adenocarcinoma,
particularly in those with EGFR mutations, and may also be a
beneficial therapeutic target in these patients (8). The combination of anastrozole and
gefitinib compared with either drug alone maximally inhibits cell
proliferation, induces apoptosis, and affects downstream signaling
pathways (9). Fulvestrant adds to
the effects of EGFR inhibitors, including synergy in the
EGFR-mutant, erlotinib-resistant H1975 cell line. Tumor stability
is achieved in human tumor xenografts with either fulvestrant or
EGFR inhibitors, but tumors regress significantly when both
pathways are inhibited (10).
However, the function of the full-length ERβ protein, ERβ1, in
resistance to EGFR-TKIs in NSCLC is still unknown.
Therefore, the present study proposed that ERβ1
activation accelerates the resistance to EGFR-TKIs in NSCLC.
Secondary TKI-resistant cell lines, PC9-ER and Hcc827-ER, and
xenografts were established to investigate the function of ERβ1 in
secondary resistance progression and efficiency by targeting ERβ1
to reverse resistance. Furthermore, the correlations between ERβ1
and follow-up outcome after EGFR-TKI therapy were analyzed in 53
patients with advanced NSCLC and in tumors biopsy confirmed with
EGFR-sensitive mutants. The results established the activation and
potential therapeutic effects of ERβ1 in NSCLC with resistance to
EGFR-TKIs via the bypass signals of extracellular signal-regulated
protein kinases 1 and 2 (ERK1/2) and Akt pathways.
Materials and methods
Cells and reagents
The human NSCLC cell lines Hcc827 and PC9 were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). These cells were cultured under a humidified
atmosphere of 5% CO2 at 37°C in RPMI-1640 medium
(11835–030; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS). Erlotinib was
kindly provided by Roche (Mannheim, Germany) and fulvestrant
(129453-61-8) was purchased from Cayman Chemical (Ann Arbor, MI,
USA). Erlotinib-resistant Hcc827-ER and PC9-ER cells were selected
from a subculture that had acquired resistance to erlotinib using
the following procedure. Cultured Hcc827 and PC9 cells were
maintained in a medium containing 0.2 µM erlotinib for 7 days.
After exposure to erlotinib, they were washed and cultured in
drug-free medium for 14 days. Upon an increase in viable cells,
they were seeded in a medium with an increasing concentration of
erlotinib, from 0.4 to 4 µM, on 24-well culture plates for
subcloning until a single clone was obtained. Then, EGFR mutations
were detected by ARMS EGFR Mutation Detection kit (Amoy Dx,
Shenzhen, China). Mutations of the PC9-ER Hcc827-ER were found to
include E746-A750del. The cells were transfected with siRNA-ERβ1
using Lipofectamine 2000 according to the manufacturers protocol.
siRNA-ERβ1 oligos were ordered from Invitrogen (Shanghai, China):
siRNA-ERβ1 sense sequence, CAGAUACUCUUUU AGACCATT; antisense
sequence, UGGUCUAAAAGAGU AUCGTG), and a scrambled siRNA.
Ethical approval
All procedures performed in studies involving human
participants were in accordance with the ethical standards of the
Institutional and/or National Research Committee and with the 1964
Helsinki Declaration and its later amendments or comparable ethical
standards. All applicable international, national and/or
institutional guidelines for the care and use of animals were
followed.
Cell viability and proliferation
analysis
For short-term cell viability assays, 3,000 cells
were seeded in triplicate into 96-wells for 1 day, and then
incubated for 5 days with various concentrations of erlotinib and
fulvestrant. Nine duplicate wells were used for each group. At the
end of the culture period, the viability of the cells was measured
using the Cell Counting Kit-8 (CCK-8) assay according to the
manufacturer's instructions. In brief, 90 µl of fresh serum-free
medium and 10 µl of CCK-8 reagent were added to each well after
decanting the old medium, and the culture was continued at 37°C for
2 h. The optical density at 450 nm was measured using a microplate
reader (Promega, Madison, WI, USA).
For long-term colony formation assays,
100,000-200,000 cells were plated/3-cm well the day before the
start of treatment. The cells were retreated with fresh media with
or without erlotinib and fulvestrant every 3 days until the
appropriate confluence, as estimated by the control conditions, was
reached. Each experiment was performed in triplicate. The plates
were stained with 0.2% crystal violet/10% formalin, and the cell
number was estimated under a microscope.
Quantitative real-time PCR
PC9, PC9-ER and Hcc827-ER cells were plated in
60-mm-diameter cell culture dishes. Total RNA was isolated from the
cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA)
according to the manufacturers instructions. Complementary DNA
(cDNA) was synthesized from total RNA with Oligo(dT), Enzyme Mix
and primers (Invitrogen). The primer sequences used were as
follows: forward primer, 5′-GTCAGGCATGCGAGTAACAA-3′, reverse
primer, 5′-GGGAGCCCTCTTTGCTTTTA-3′. For amplification, cDNA was
initially denatured at 95°C for 10 min, 40 cycles of 95°C for 15
sec, 60°C for 35 sec, and 72°C for 40 sec (ABI PRISM 7300 Sequence
Detection system). For each PCR reaction, a cDNA standard curve was
used to generate relative expression changes in ER mRNA levels,
which were normalized to the β-actin gene.
Western blotting
The cells were detached using trypsin, washed three
times with phosphate-buffered saline, treated with lysis buffer [25
mM Tris-HCl pH 7.4, 1% Triton X-100, 150 mM NaCl, 5%
ethylenediaminetetraacetic acid, 10 mM NaF, 1 mM
phenylmethylsulfonyl fluoride (PMSF), and 10 mg of aprotinin and
leupeptin], and incubated for 30 min on ice. Lung cancer tissues
were lysed in PMSF, followed by homogenization and determination of
the concentration of protein. The lysate was centrifuged for 10 min
at 12,000 rpm, and the supernatant was collected. The
concentrations of protein were measured using the Bradford method
(Bio-Rad, Hercules, CA, USA). A 50 µg aliquot of protein/lane was
electrophoresed on an 8–12% sodium dodecyl sulfate-polyacrylamide
gel and electroblotted on polyvinylidene fluoride membranes
(Millipore, Billerica, MA, USA). The transferred membranes were
blocked with 5% non-fat dry skimmed milk in Tris-buffered saline
(TBST) (25 mM Tris-HCl pH 7.4, 125 mM NaCl, 0.05% Tween-20) and
incubated at 4°C overnight with the appropriate primary antibodies,
which were specific for the following proteins: ERβ1 (MCA1974ST;
1:500) (Serotec Biologicals, Raleigh, NC, USA). After being washed
with TBST, the membranes were incubated with a horseradish
peroxidase-labeled secondary antibody (1:2,000) for 1 h at 37°C
before detection using ECL Plus Western Blotting Detection Reagents
(Pierce, Rockford, IL, USA). Glyceraldehyde 3-phosphate
dehydrogenase was used as an internal control for protein loading
and analysis.
Tumor xenograft model
PC9-ER (5.0×106 cells/mouse) cells were
subcutaneously implanted into the posterior flank of 4-week old NOD
SCID female mice. The tumor size was monitored as previously
described (10). When the average
tumor size reached 50 mm3, erlotinib (10 mg/kg) and/or
fulvestrant (10 mg/kg) were subcutaneously administered twice per
week. After 22 days, the mice were sacrificed and the tumors were
collected and divided into two groups. One group was fixed in 10%
formalin and embedded in paraffin for pathological examinations and
immunohistochemical analysis, and the other group was stored in
liquid nitrogen, and then at −80°C for further use (western blot
analysis). The aforementioned measurements were performed by two
qualified technicians in a double-blinded manner, and the mean of
the two scores was obtained.
Patient selection and
immunohistochemistry
The present study included 53 Chinese patients with
advanced NSCLC who received EGFR-TKI therapy at the Tongji Hospital
between July 2012 and December 2015. All diagnoses were
histologically confirmed and evaluated as stage IV according to the
current Tumor-Node-Metastasis (TNM) Staging System (IASLC 2009).
Only patients with sufficient tissue for both EGFR mutation
analysis and ERβ1 immunohistochemical staining were enrolled after
obtaining appropriate approval from the Institutional Review Board
(IRB) (IRB ID no. 20141101). Full consent was obtained from
patients involved in the present study. Clinicopathological
information from the patients was collected, and the database was
tabulated in an anonymous manner. Responses were classified using
standard Response Evaluation Criteria in Solid Tumors, version 1.1.
The PFS was assessed from the first day of EGFR-TKI treatment until
radiologic progression or death. Immunohistochemical staining was
performed using an ERβ1 antibody, which was confirmed to be
specific (11). Cells positive for
ERβ1 appeared yellow or yellowish brown in the nucleus or
cytoplasm, or they contained yellowish brown granules. A
pathological examination and semi-quantitation based on the
staining intensity and proportion of positive cells were performed
as previously described (12,13).
Statistical analysis
Data are expressed as mean ± standard deviation from
three independent experiments. Comparisons among groups were
performed with the analysis of variance. A P-value of <0.05 was
considered to indicate a statistically significant result.
Statistical tests were performed using SPSS 13.0 (SPSS, Inc.,
Chicago, IL, USA).
Results
Expression of ERβ1 is upregulated
following resistance to erlotinib, with β-estradiol dependence
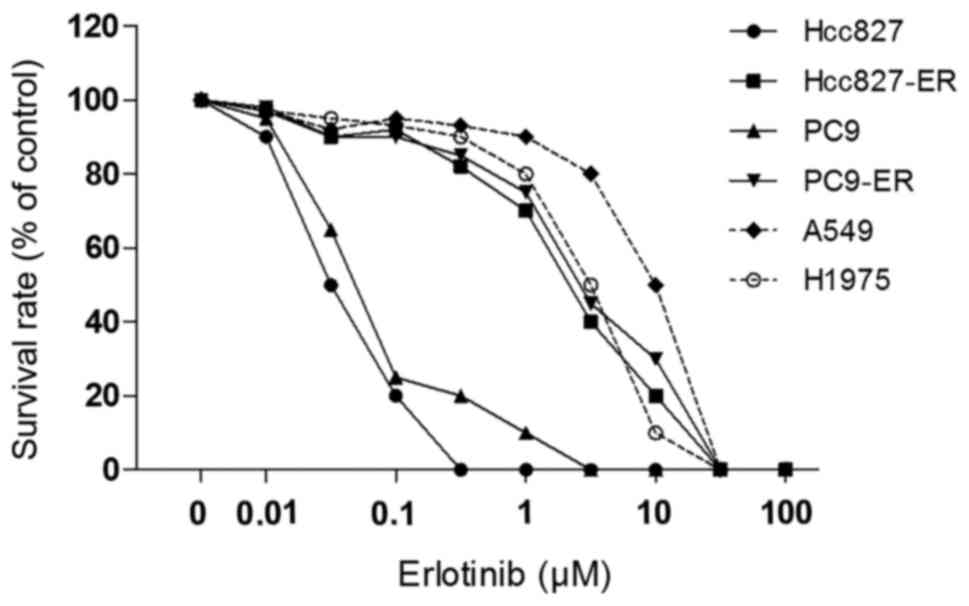
First, the effect of erlotinib on six NSCLC cell
lines was compared using the CCK-8 assay. Four of the six NSCLC
cell lines showed considerable resistance to erlotinib.
Dose-response curves to erlotinib for all cell lines assessed by
the colony formation assay are presented in Fig. 1. Among the six NSCLC lines, A549,
H1975, PC9-ER and Hcc827-ER showed 100- to 200-fold greater
resistance to erlotinib compared with the PC9 and Hcc827 cells. The
drug sensitivity of all six human cancer cell lines to erlotinib
and fulvestrant was examined using the CCK-8 assay, and the
IC50 values are presented in Table I. The IC50 values of
Hcc827 to erlotinib and fulvestrant were 0.03±0.01 and 7.93±0.87,
and the IC50 values of PC-9 were 0.05±0.002 and
8.05±1.22, respectively. A549, H1975, PC9-ER and Hcc827-ER cells
had 100-fold or greater resistance to erlotinib (Table I).
 | Table I.Sensitivities to erlotinib and
fulvestrant as assessed by CCK-8 assay in Hcc827 (19-Del),
Hcc827-ER (19-Del), PC9 (19-Del), PC9-ER (19-Del), A549 (WT) and
H1975 (L858R/T790M) cells. |
Table I.
Sensitivities to erlotinib and
fulvestrant as assessed by CCK-8 assay in Hcc827 (19-Del),
Hcc827-ER (19-Del), PC9 (19-Del), PC9-ER (19-Del), A549 (WT) and
H1975 (L858R/T790M) cells.
|
| IC50
values (µM) |
|---|
|
|
|
|---|
| Cell lines | Erlotinib | Fulvestrant |
|---|
| Hcc827 |
0.03±0.01 |
7.93±0.87 |
| Hcc827-ER |
7.57±0.67 |
15.76±2.33 |
| PC9 |
0.05±0.002 |
8.05±1.22 |
| PC9-ER |
8.42±0.23 |
18.66±1.51 |
| A549 |
10.06±1.03 |
34.02±0.52 |
| H1975 |
6.23±1.50 |
22.04±3.61 |
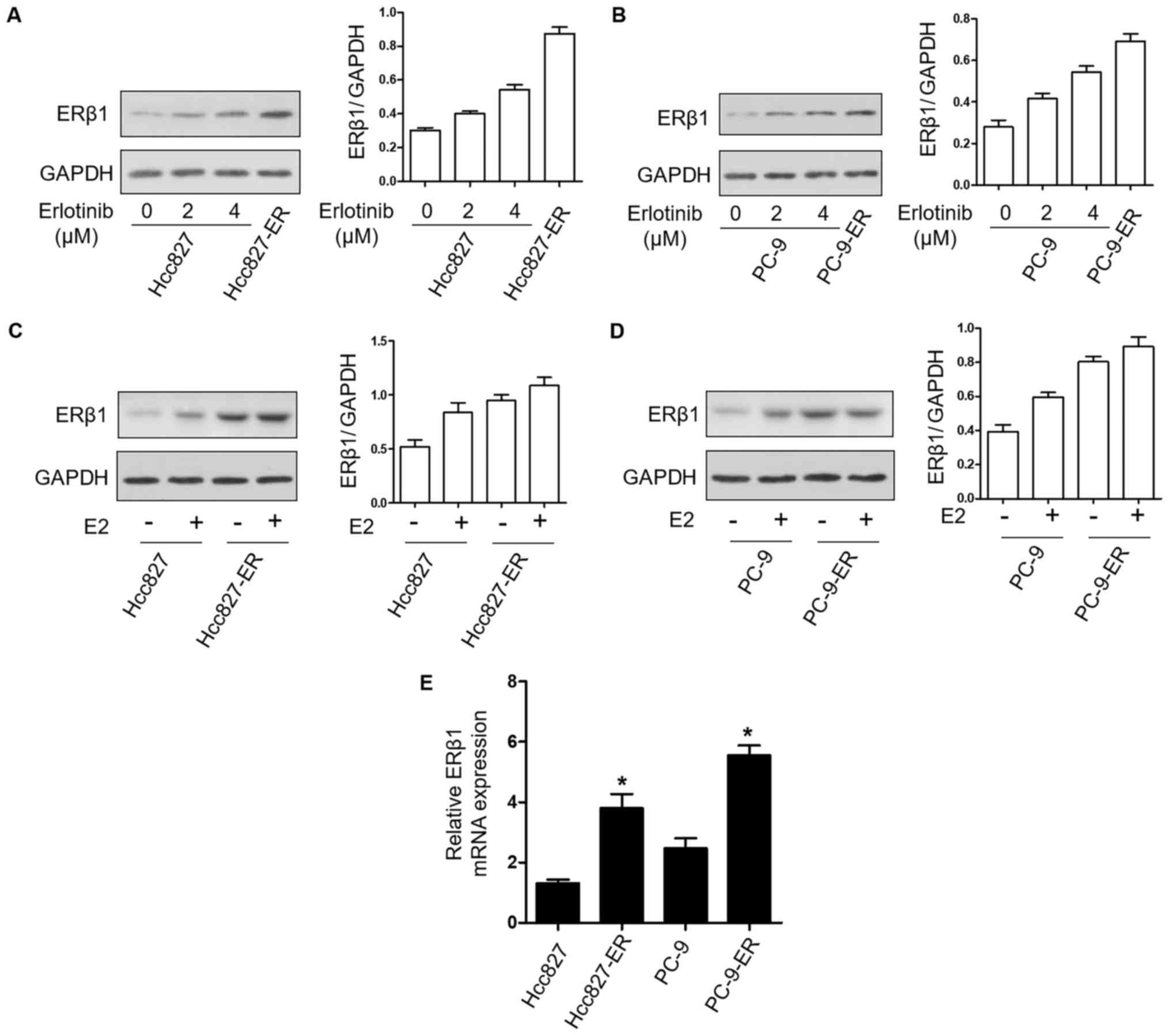
To determine the effects of erlotinib resistance on
the expression of ERβ1 protein in NSCLC cell lines, the expression
of ERβ1 was examined by western blotting in PC9 and Hcc827 cell
lines treated with the indicated concentrations of erlotinib and in
PC9-ER and Hcc827-ER cells. ERβ1 had higher expression with
increasing erlotinib concentrations; the expression was the highest
in the PC9-ER and Hcc827-ER cells (Fig.
2A and B). In addition, in the PC9 and PC9-ER cells as well as
in the Hcc827 and Hcc827-ER cells, treatment with 10 nM E2
upregulated the expression of ERβ1 (Fig. 2C and D). Additionally, ERβ1 mRNA
levels were also higher in the resistant cells compared to levels
noted in the parental cell lines (Fig.
2E). These results indicated that the expression of ERβ1 was
upregulated after resistance to erlotinib, with β-estradiol
dependence.
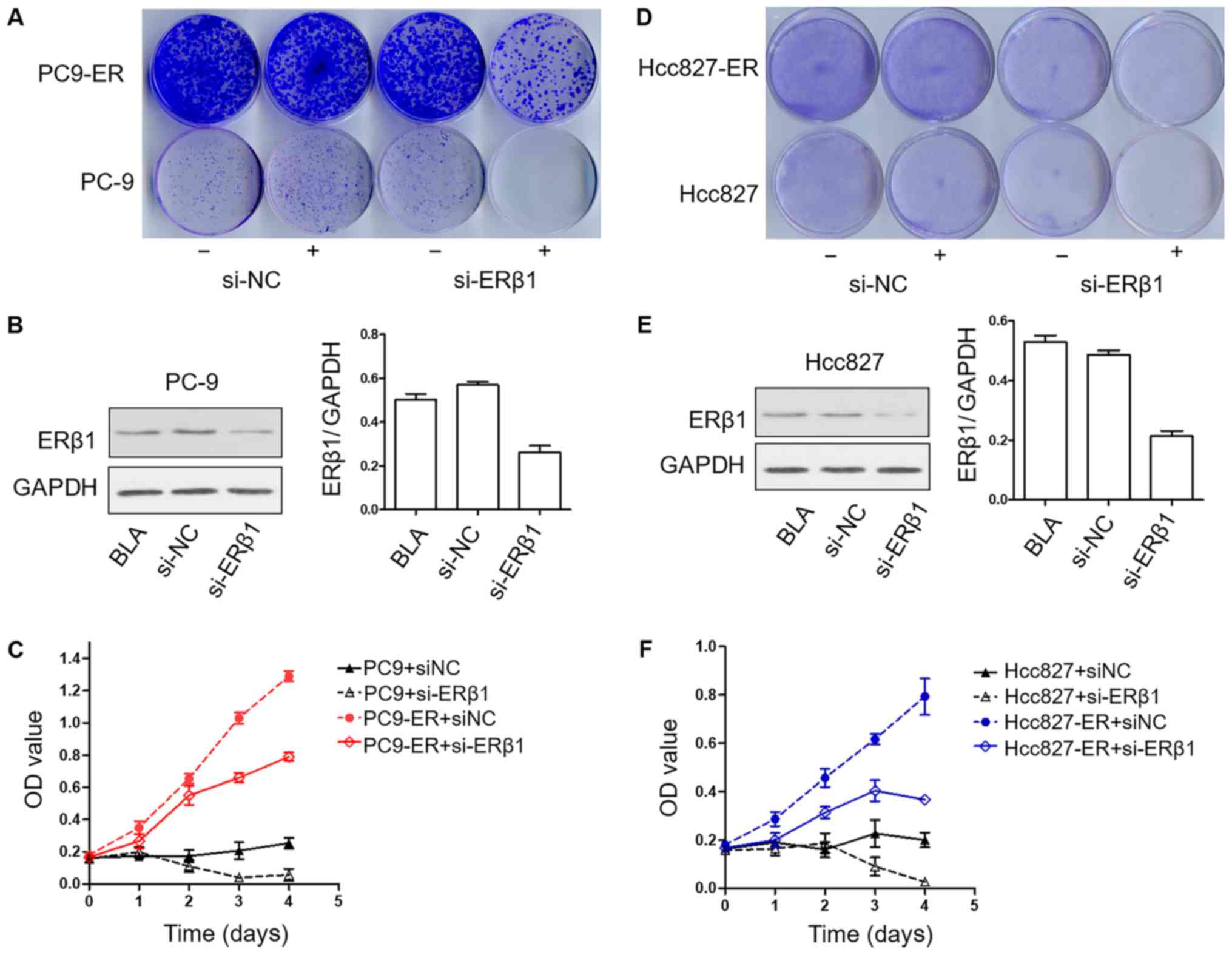
PC9-ER and Hcc827-ER cell lines are
sensitive to erlotinib after downregulation of the expression of
ERβ1
To determine the effects of ERβ1 knockdown on the
development of resistance to erlotinib in long-term colony
formation assays, PC9, PC9-ER, Hcc827 and Hcc827-ER cells were
cultured in erlotinib in the presence or absence of siRNA-NC or
siRNA-ERβ1 until colonies were formed. Strikingly, in this
long-term assay, ERβ1 knockdown strengthened the effects to
erlotinib in the PC9-ER and Hcc827-ER cells. Notably, the knockdown
of ERβ1 was also effective when induced in PC9 and Hcc827 cells,
that is, after initial erlotinib treatment for 14 days (Fig. 3A, B, D and F). To confirm the
biological effects of ERβ1 knockdown, viability of the PC9, PC9-ER,
Hcc827 and Hcc827-ER cells was assessed using CCK-8 assay after
treatment as indicated. Downregulation of ERβ1 induced both PC9-ER
and Hcc827-ER cells to grow slower when compared to the control
group. In addition, ERβ1 knockdown induced the lowest proliferation
in PC9 and Hcc827 groups (Fig. 3C and
F), showing that the proliferation of both sensitive and
resistant cell lines was suppressed by erlotinib after the
downregulation of the expression of ERβ1.
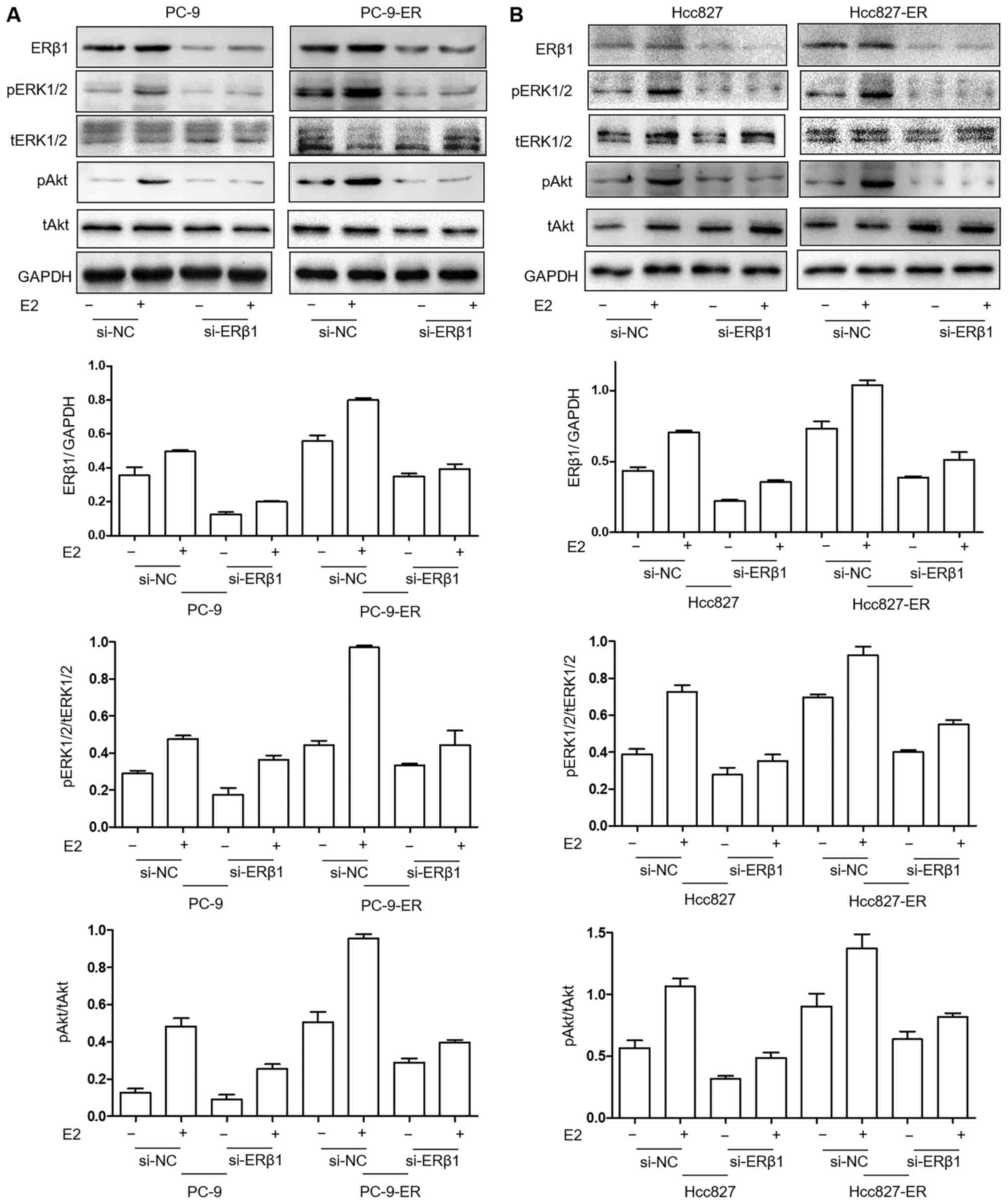
ERK1/2 and Akt pathways are decreased
following the silencing of the expression of ERβ1 in PC9-ER and
Hcc827-ER cell lines
In an effort to investigate the signaling involved
in the re-sensitization to erlotinib treatment after ERβ1 silencing
in PC9-ER cells, the expression levels of tERK1/2 (4695), pERK1/2
(4376) (both from Cell Signaling Technology, Inc., Danvers, MA,
USA), tAkt (BS1810) and pAkt (BS4007) (both from Bioworld
Technology, Inc., Nanjing, China) were evaluated in PC9, PC9-ER,
Hcc827 and Hcc827-ER cells. The levels of pERK1/2 and pAkt were
decreased by siRNA-ERβ1 treatment in the PC9-ER and Hcc827-ER
cells. Levels were the lowest in the PC9 (Fig. 4A) and Hcc827 (Fig. 4B) cells. In addition, the lack of E2
strengthened the downregulation of ERK1/2 and Akt pathways by
ERβ1-knockdown.
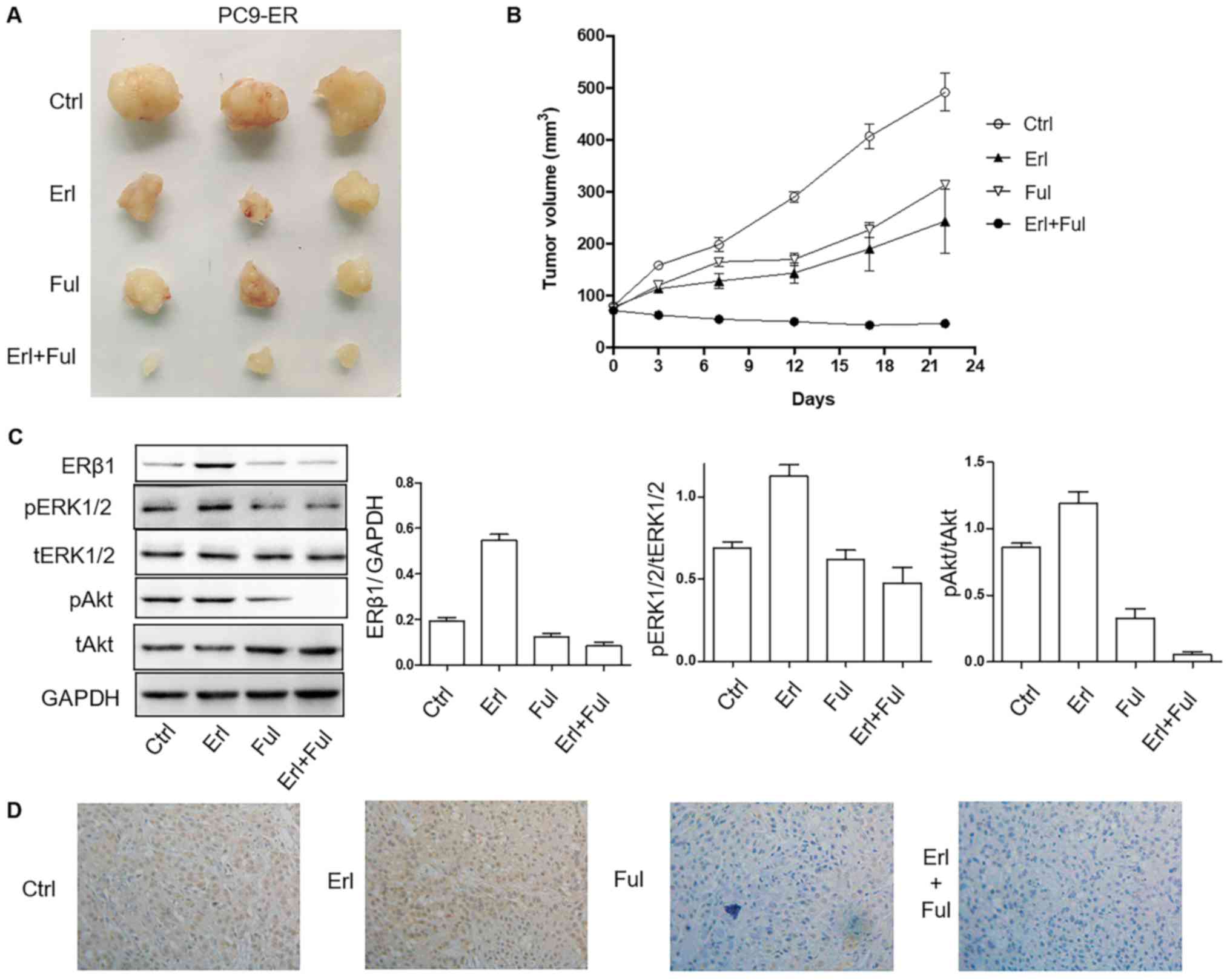
Co-treatment of erlotinib and
fulvestrant results in better tumor inhibition efficiency in PC9-ER
cell-derived xenografts compared with treatment of each agent
alone
NOD SCID mice harboring tumors derived from human
NSCLC PC9-ER cells were randomly assigned to a vehicle control,
erlotinib 10 mg/kg, fulvestrant 10 mg/kg (subcutaneously) groups
for a total of 22 days. Erlotinib and fulvestrant alone delayed
tumor growth. However, treatment with both erlotinib and
fulvestrant elicited significantly reduced tumor volume compared
with all other treatments (P<0.001), with minimal tumors after
several weeks of therapy (Fig. 5A and
B). The expression of pERK and pAkt in tumor tissues was lower
in the single-drug group, and it was the lowest in the
co-inhibition group (Fig. 5C).
Consistent with the observations concerning tumor size, the
combination (erlotinib plus fulvestrant) led to the inhibition of
the expression of ERβ1 in tumor tissues (Fig. 5D).
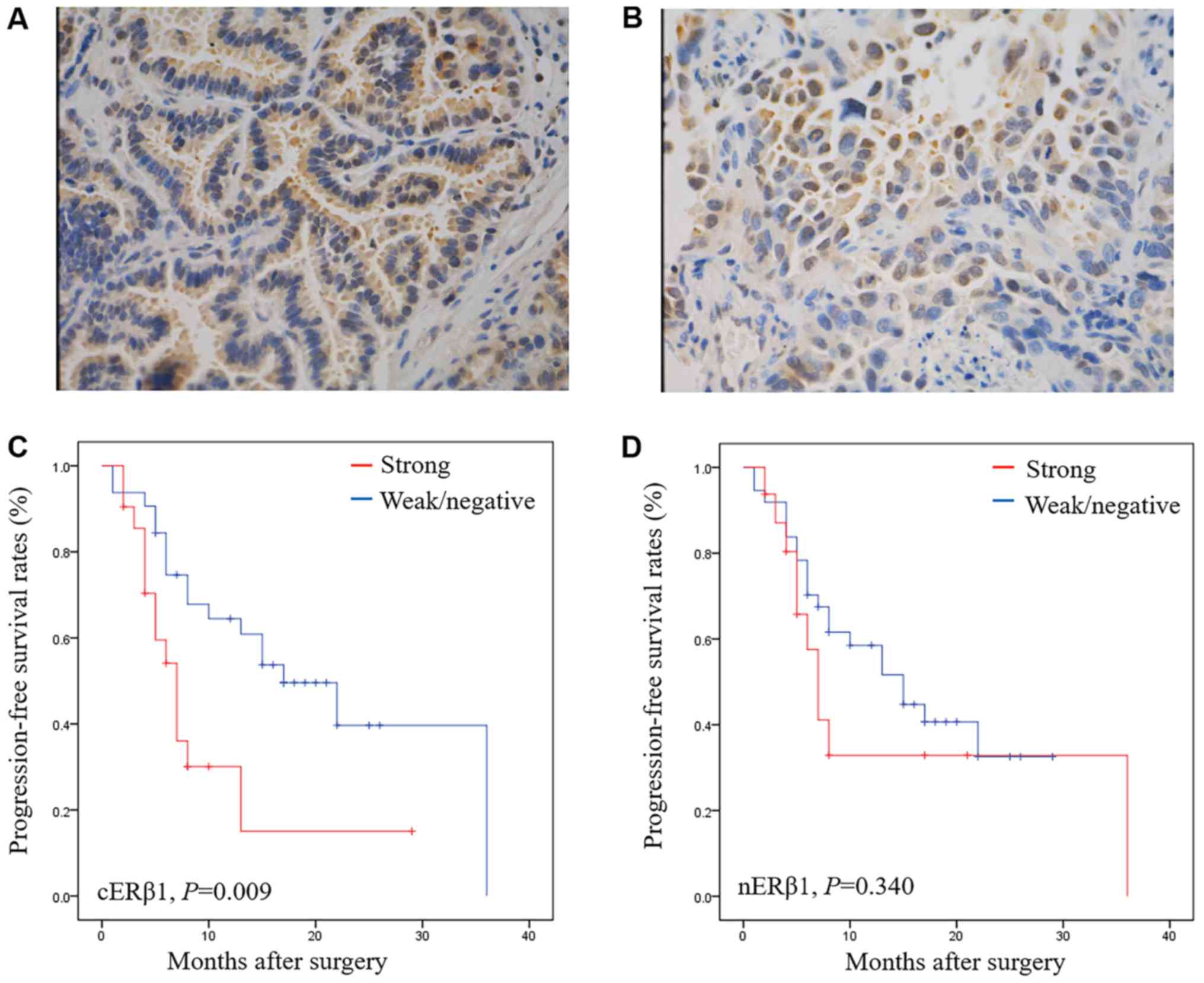
Strong expression of cERβ1 is related
to a shorter PFS in EGFR-TKI-treated patients
A total of 53 patients with NSCLC treated with
EGFR-TKIs were analyzed. Most patients were never/light smokers
(40, 75.47%) and had adenocarcinoma (46, 86.79%). A total of 48
patients (90.57%) carried EGFR-sensitizing mutations (19-Del or
L858R). The expression of ERβ1 was positive in 79.24% (42/53) of
the patients with different intracellular distribution patterns,
including nuclear (nERβ1) and cytoplasmic (cERβ1) ERβ1 (Fig. 6A and B). No significant correlations
were observed between the expression of ERβ1 and EGFR mutations
(P=0.093) or sex (P=0.370). Moreover, neither nuclear nor
cytoplasmic expression of ERβ was associated with sex (P=0.586, and
P=0.105, respectively) or any other clinicopathological
characteristic (data not shown).
At the time of data collection (Jan 1, 2016), 31
patients (58.59%) presented with progressive disease. Notably,
patients with strong expression of cERβ1 had a poorer PFS after
EGFR-TKI treatment (P=0.009) compared with those with weak or
without expression (P=0.009) (Fig. 6C
and D). Strong nERβ1 immunoreactivity was not significantly
associated with worse PFS (P=0.340). When categorized by positive
or negative expression of ERβ1, no statistical significance was
achieved in the PFS in the nuclear or cytoplasmic expression
subgroup. By Cox multivariate analysis, strong expression of cERβ1
was found to be independent factors predicting worse prognosis
[P=0.019, (hazard ratio) HR=2.547, 95% confidence interval (CI)
1.165–5.564], which were in agreement with our Kaplan-Meier plots
(Table II).
| Table II.PFS by Cox univariate and multivariate
analyses in the NSCLC patients. |
Table II.
PFS by Cox univariate and multivariate
analyses in the NSCLC patients.
|
| Univariate | Multivariate |
|---|
|
|
|
|
|---|
| Variable | P-value | HR (95% CI) | P-value | HR (95% CI) |
|---|
| cERβ1 (strong vs.
weak/neg) | 0.014 | 6.052
(1.212–5.489) | 0.019 | 2.547
(1.165–5.564) |
| nERβ1 (strong vs.
weak/neg) | 0.356 | 0.853
(0.659–3.190) | 0.532 | 1.295
(0.575–2.917) |
| Differentiation
(trend) | 0.210 | 1.574
(0.301–1.301) | 0.858 | 0.866
(0.179–4.198) |
| TNM stage
(trend) | 0.287 | 1.136
(0.870–1.605) | 0.685 | 1.145
(0.594–2.210) |
| Age (linear,
years) | 0.486 | 0.486
(0.940–1.030) | 0.769 | 1.008
(0.953–1.067) |
| Sex (male vs.
female) | 0.994 | 0.000
(0.455–2.185) | 0.726 | 0.844
(0.328–2.175) |
Discussion
In the present study, secondary resistant cell lines
were constructed, and it was demonstrated that ERβ1 upregulation
occurred following the development of resistance to EGFR-TKIs,
accompanied by the activation of ERK1/2 and Akt signals. Fulvetrant
plus erlotinib reversed TKI resistance in vitro and in
vivo, revealing a new approach to overcome EGFR-TKI resistance.
Consistently, patients given EGFR-TKI therapy exhibited a longer
PFS in subpopulations with lower expression of cERβ1. The present
study discovered a new mechanism of EGFR-TKI resistance from a
gender perspective.
The aim of the present study was to investigate the
mechanism of acquired resistance to EGFR-TKIs and explore
strategies to overcome the resistance to EGFR-TKIs from a gender
perspective. Following stimulation with increasing concentrations
of erlotinib, the expression of ERβ1 in PC9 and Hcc827 cells was
found to be gradually upregulated, consistent with the progressive
resistance. Established with the stepwise escalation of EGFR-TKI
concentration, PC9-ER, and Hcc827-ER cells had 100-fold or greater
resistance compared to sensitive sublines respectively, as
previously described (14). In
addition, following knockdown of ERβ1, pERK1/2 and pAkt were
decreased in the PC9-ER and Hcc827-ER cells. These findings suggest
that ERK1/2 and Akt pathways are activated following
resistance-induced ERβ1 upregulation. EGFR-TKIs may alter ERβ1
expression through the ERK1/2 and Akt pathways, the bidirectional
signaling between ERβ1 and EGFR. Several mechanisms are believed to
be responsible for acquired resistance to EGFR-TKIs, including
secondary EGFR T790M and minor mutations, MET amplification,
activation of MET/HGF axis, acquisition of an
epithelial-to-mesenchymal transition signature, and transformation
from nNSCLC to small cell lung cancer (SCLC) (14). In addition, the present study
demonstrated that ERβ1 upregulation following resistance with
activation of the ERK1/2 and Akt pathways, may be another mechanism
in EGFR-TKI resistance.
The present study revealed that a combination of
erlotinib and a novel ERβ inhibitor fulvestrant significantly
inhibited the growth of PC9-ER xenografts in nude mice. Additional
treatment of fulvestrant could overcome secondary resistance to
EGFR-TKIs in vivo. Strategies for overcoming EGFR-TKI
resistance include targeting T790M EGFR or other receptor TKs,
alternatively activated bypass pathway molecules, and various
downstream signaling molecules. The present study demonstrated a
better efficiency by co-treatment of erlotinib and fulvestrant
compared with alternatives, and complementary application of
fulvestrant reversed resistance to erlotinib in vitro and
in vivo. A combination with fulvestrant may provide a
prolonged effectiveness of EGFR-TKIs within the range of tolerated
toxicity. Previous studies also revealed that a combination of
anastrozole and gefitinib compared with either drug alone can
maximally inhibit cell proliferation, induce apoptosis and affect
downstream signaling pathways (14). Fulvestrant adds to the effects of
EGFR inhibitors, including synergy in the EGFR-mutant,
erlotinib-resistant H1975 cell line. Tumor stability is achieved in
human tumor xenografts with either fulvestrant or EGFR inhibitors,
but tumors regress significantly when both pathways are inhibited
(10). However, more targeted
applications with different genetic abnormalities in a
heterogeneous tumor population warrant further exploation. The
targeted inhibition strategy from a gender perspective provides a
new idea for the clinical treatment of EGFR-TKI resistance.
Patients with strong expression of cERβ1 had a
poorer PFS after EGFR-TKI treatment. No statistical significance
was achieved in the median PFS between strong nERβ expression and
weak/negative expression. Numerous published reports have examined
the ER status in relation to the survival of patients with NSCLC
(11). High cERβ1 staining was
identified as a negative prognostic factor for lung cancer,
independent of other prognostic factors (15). nERβ1 positivity was observed in the
majority of lung cancer cases (15,16)
and found to be a favorable prognostic indicator in various
studies. Moreover, lack of nuclear ERβ or the loss of EGFR
expression were reported to be independent prognosis markers
associated with shorter overall survival (17). However, most studies used antibodies
to total ERβ that could not distinguish different ERβ isoforms. In
addition, previous studies involving ERβ and EGFR signals rarely
identified the difference in ER subtypes.
In conclusion, ERβ1 activation may accelerate
EGFR-TKI resistance. Co-targeting ERβ1 can re-sensitize resistant
cell lines. Anti-estrogen treatment may be a potential strategy
with which to reverse TKI resistance.
Acknowledgements
The present study was funded by the National Natural
Science Foundation of China (NSFC), (grant nos. 81272590 and
81402163), the Natural Science Foundation of Hubei Province (grant
no. 2014CFB152) and the Wuhan Municipal Human Resources and Social
Security Bureau (grant no. 2011415).
References
|
1
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al North-East Japan Study Group, : Gefitinib or chemotherapy for
non-small-cell lung cancer with mutated EGFR. N Engl J Med.
362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosell R, Carcereny E, Gervais R,
Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R,
Pallares C, Sanchez JM, et al Spanish Lung Cancer Group in
collaboration with Groupe Français de Pneumo-Cancérologie and
Associazione Italiana Oncologia Toracica, : Erlotinib versus
standard chemotherapy as first-line treatment for European patients
with advanced EGFR mutation-positive non-small-cell lung cancer
(EURTAC): A multicentre, open-label, randomised phase 3 trial.
Lancet Oncol. 13:239–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lin JJ, Cardarella S, Lydon CA, Dahlberg
SE, Jackman DM, Jänne PA and Johnson BE: Five-year survival in
EGFR-mutant metastatic lung adenocarcinoma treated with EGFR-TKIs.
J Thorac Oncol. 11:556–565. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bouchardy C, Benhamou S, Schaffar R,
Verkooijen HM, Fioretta G, Schubert H, Vinh-Hung V, Soria JC,
Vlastos G and Rapiti E: Lung cancer mortality risk among breast
cancer patients treated with anti-estrogens. Cancer. 117:1288–1295.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chlebowski RT, Schwartz AG, Wakelee H,
Anderson GL, Stefanick ML, Manson JE, Rodabough RJ, Chien JW,
Wactawski-Wende J, Gass M, et al: Women's Health Initiative
Investigators: Oestrogen plus progestin and lung cancer in
postmenopausal women (Women's Health Initiative trial): A post-hoc
analysis of a randomised controlled trial. Lancet. 374:1243–1251.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tang H, Liao Y, Xu L, Zhang C, Liu Z, Deng
Y, Jiang Z, Fu S, Chen Z and Zhou S: Estrogen and insulin-like
growth factor 1 synergistically promote the development of lung
adenocarcinoma in mice. Int J Cancer. 133:2473–2482. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Słowikowski BK, Lianeri M and Jagodziński
PP: Exploring estrogenic activity in lung cancer. Mol Biol Rep.
44:35–50. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kohno M, Okamoto T, Suda K, Shimokawa M,
Kitahara H, Shimamatsu S, Konishi H, Yoshida T, Takenoyama M, Yano
T, et al: Prognostic and therapeutic implications of aromatase
expression in lung adenocarcinomas with EGFR mutations. Clin Cancer
Res. 20:3613–3622. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen L, Li Z, Shen S, Niu X, Yu Y, Li Z,
Liao M, Chen Z and Lu S: The synergistic effect of EGFR tyrosine
kinase inhibitor gefitinib in combination with aromatase inhibitor
anastrozole in non-small cell lung cancer cell lines. Lung Cancer.
78:193–200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Garon EB, Pietras RJ, Finn RS, Kamranpour
N, Pitts S, Márquez-Garbán DC, Desai AJ, Dering J, Hosmer W, von
Euw EM, et al: Antiestrogen fulvestrant enhances the
antiproliferative effects of epidermal growth factor receptor
inhibitors in human non-small-cell lung cancer. J Thorac Oncol.
8:270–278. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Siegfried JM and Stabile LP: Estrongenic
steroid hormones in lung cancer. Semin Oncol. 41:5–16. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang H, Liao Y, Chen G, Xu L, Zhang C, Ju
S and Zhou S: Estrogen upregulates the IGF-1 signaling pathway in
lung cancer through estrogen receptor-β. Med Oncol. 29:2640–2648.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu C, Liao Y, Fan S, Tang H, Jiang Z,
Zhou B, Xiong J, Zhou S, Zou M and Wang J: G protein-coupled
estrogen receptor (GPER) mediates NSCLC progression induced by
17β-estradiol (E2) and selective agonist G1. Med Oncol.
32:1042015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shien K, Toyooka S, Yamamoto H, Soh J,
Jida M, Thu KL, Hashida S, Maki Y, Ichihara E, Asano H, et al:
Acquired resistance to EGFR inhibitors is associated with a
manifestation of stem cell-like properties in cancer cells. Cancer
Res. 73:3051–3061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stabile LP, Dacic S, Land SR, Lenzner DE,
Dhir R, Acquafondata M, Landreneau RJ, Grandis JR and Siegfried JM:
Combined analysis of estrogen receptor beta-1 and progesterone
receptor expression identifies lung cancer patients with poor
outcome. Clin Cancer Res. 17:154–164. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luo Z, Wu R, Jiang Y, Qiu Z, Chen W and Li
W: Overexpression of estrogen receptor beta is a prognostic marker
in non-small cell lung cancer: A meta-analysis. Int J Clin Exp Med.
8:8686–8697. 2015.PubMed/NCBI
|
|
17
|
Mauro LV, Dalurzo M, Carlini MJ, Smith D,
Nuñez M, Simian M, Lastiri J, Vasallo B, Bal de Kier Joffé E,
Pallotta MG, et al: Estrogen receptor β and epidermal growth factor
receptor as early-stage prognostic biomarkers of non-small cell
lung cancer. Oncol Rep. 24:1331–1338. 2010.PubMed/NCBI
|