Introduction
Ovarian cancer is a common gynecological cancer that
causes a large number of deaths in women worldwide (1,2).
Epithelial ovarian cancer is the most common type, accounting for
over 75% of ovarian malignancies. The symptoms for patients with
early (International Federation of Gynecology and Obstetrics; FIGO
I–II) and advanced (FIGO III–IV) stages of disease are
significantly different (2).
Despite this, 80% of epithelial ovarian cancer cases are diagnosed
at an advanced stage. Chemotherapy is currently the standard
treatment for epithelial ovarian cancer patients. Cisplatin is
among the most widely used chemotherapeutic agents and has
demonstrated significant efficacy against ovarian cancer. However,
the majority of ovarian cancer patients acquire resistance to
cisplatin during therapy, which represents a major obstacle to the
clinical application of cisplatin (3). Therefore, the cellular and molecular
mechanisms of cisplatin in ovarian cancer need to be elucidated.
Bcl-2, the founding member of the Bcl-2 protein family, has
anti-apoptotic activity and is overexpressed in many types of
cancers. There is a growing body of evidence indicating that Bcl-2
overexpression is implicated in cisplatin resistance in ovarian
cancer. However, the mechanisms responsible for this activity are
undefined (4).
Numerous studies have demonstrated that Bcl-2
functions in the mitochondria and it also has an established
anti-apoptotic role in the endoplasmic reticulum (ER). The
anti-apoptotic function of Bcl-2 is mediated by its effects on
intracellular Ca2+ homeostasis and dynamics (5). Ca2+ is an important second
messenger involved in regulating cell survival and apoptosis. ER,
the major intracellular Ca2+ storage organelle, is
involved in several biological processes. Various stimuli directly
target the ER to induce Ca2+ release into the cytosol
and mitochondria, thus inducing cytosolic and mitochondrial
Ca2+ overload and contributing to the induction of
apoptosis (6). Ca2+
release from the ER is mainly mediated by the inositol
1,4,5-trisphosphate receptor (IP3R), an IP3-gated Ca2+
channel located on the surface of the ER. Bcl-2 physically
interacts with IP3R to prevent pro-apoptotic Ca2+
transfer to the cytoplasm and mitochondria (7). Mitochondrial Ca2+ overload
is detrimental to its function by inducing mitochondrial
permeability transition pore (mPTP) opening and loss of the
mitochondrial membrane potential (Δψm), which triggers
mitochondrial-mediated apoptosis. Ca2+ entry into the
mitochondria is mainly mediated by voltage-dependent anion channel
1 (VDAC1), an outer mitochondrial membrane protein (8). Bcl-2 prevents mitochondrial
Ca2+ overload and cell death through binding to VDAC1
(9). However, it remains unclear
whether Bcl-2 contributes to cisplatin resistance in ovarian cancer
cells via inhibiting the ER Ca2+ release.
Ca2+ release from the ER to the
mitochondria regulates several mitochondrial processes (10). Notably, there is a tight interplay
between the ER and mitochondria in all eukaryotic cells. The
contact site between the ER and mitochondria, known as the
mitochondrial-associated membrane or MAM, is a signaling hub for
Ca2+ transfer between these organelles (11–13).
ER- and mitochondrial-associated proteins including IP3R, VDAC1 and
Grp75 are the basic components of the MAM. MAM provides a platform
to coordinate the release of Ca2+ from the ER and the
uptake of efficient mitochondrial Ca2+ (14). Arruda et al (15) reported that obesity leads to
increased mitochondrial Ca2+ levels via direct uptake
through the MAM junctions and not by the release of Ca2+
into the cytosol followed by mitochondrial uptake. Recent studies
have revealed that MAM can enhance apoptosis sensitivity by
increasing the transfer of Ca2+ into the mitochondria
(16). However, whether Bcl-2
promotes cisplatin resistance via modulating ER-mitochondrial
Ca2+ signaling remains unclear.
The aim of the present study was to determine
whether Bcl-2 reduces cisplatin cytotoxicity in SKOV3 cells by
blocking ER-mitochondrial Ca2+ signaling. Stable Bcl-2
overexpression in SKOV3 cells reduced the anticancer effects of
cisplatin both in vitro and in vivo, indicating a
novel therapeutic target for gene therapy in ovarian cancer.
Materials and methods
Antibodies (Abs) and drugs
Anti-caspase-3 (sc-7272; murine Ab; 1:200 dilution),
anti-caspase-4 (sc-56056; murine Ab; 1:200 dilution), anti-cleaved
caspase-4 (sc-22173-R; rabbit Ab; 1:200 dilution) and
anti-caspase-9 (sc-56073; murine Ab; 1:200 dilution) Abs were
purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Anti-cleaved caspase-3 (ab2302; rabbit Ab; 1:200 dilution),
anti-cleaved caspase-9 (ab2324; rabbit Ab; 1:400 dilution),
anti-PDI (ab2792; murine Ab; 1:1,000 dilution), anti-VDAC1
(ab14734; murine Ab; 1:1,000 dilution), anti-CHOP (ab11419; murine
Ab; 1:1,000 dilution), anti-IP3R (ab5804; rabbit Ab; 1:1,000
dilution) and 2-aminoethyl diphenylborinate (2-APB, ab120124) Abs
were purchased from Abcam Ltd. (Hong Kong, China). Anti-β-actin
(60008–1-Ig; murine Ab; 1:2,000 dilution), anti-Bax (50599–2-Ig;
rabbit Ab; 1:2,000 dilution), anti-Bcl-2 (12789–1-AP; rabbit Ab;
1:1,000 dilution), anti-cytochrome c (cyto c)
(10993–1-AP; rabbit Ab; 1:1,000 dilution), anti-Grp78/BIP
(11587–1-AP; rabbit Ab; 1:1,000 dilution), peroxidase-conjugated
AffiniPure goat anti-mouse IgG (H+L) (SA00001-1; goat Ab; 1:2,000
dilution) and peroxidase-conjugated AffiniPure goat anti-rabbit IgG
(H+L) (SA00001-2; goat Ab; 1:2,000 dilution) Abs were purchased
from ProteinTech Group, Inc. (Chicago, IL, USA). The anti-calpain-1
catalytic subunit (#31038-1; rabbit Ab; 1:4,000 dilution) Ab was
purchased from Signalway Antibody LLC (SAB; College Park, MD, USA).
Cisplatin was purchased from Sigma-Aldrich (St. Louis, MO, USA) and
dissolved in normal saline (NS) for in vitro use and animal
studies.
Cell culture
SKOV3 ovarian cancer cells were obtained from the
Chinese Academy of Medical Sciences (Beijing, China). Transfected
SKOV3 cells were maintained in Roswell Park Memorial Institute
(RPMI)-1640 culture (RPMI-1640; Gibco; Thermo Fisher Scientific,
Inc., Carlsbad, CA, USA) and supplemented with 10% (v/v) fetal calf
serum (FCS; Gibco; Thermo Fisher Scientific, Inc.) 100 mg/ml
streptomycin and 100 U/ml penicillin (each from Genview, Galveston,
TX, USA). The cells were incubated at 37°C in an atmosphere
containing 5% CO2.
Transfection
SKOV3 cells were seeded at 2×105
cells/well in a 24-well plate and grown until they reached 30–40%
confluency before transfection. The pcDNA3.1(+) or
pcDNA3.1(+)-Bcl-2 plasmids were directly transfected into the cells
using Lipofectamine™ 2000 Transfection Reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturers
protocol. Selection was performed 72 h later using media that
contained G418 (400 µg/ml). The culture continued for 14 days to
generate stable transfectants and G418-resistant clones were
isolated. The clones were further expanded and analyzed using
western blotting. The transfected cells were used for subsequent
experiments.
Cell viability assay
Cell viability was determined by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assays. The cells were plated at 1×104 cells/well in
96-well plates. The following day, cisplatin was added to the wells
and incubated for 24 h. Each treatment was repeated in three
independent tests. MTT (20 µl) was added to each well (MTT;
Sigma-Aldrich) and incubated for 4 h. Subsequently 150 µl dimethyl
sulphoxide (DMSO) was added to dissolve the formazan crystals.
Absorbance was assessed with a Vmax Microplate Reader (Molecular
Devices, LLC, Sunnyvale, CA, USA) at a wavelength of 570 nm.
Annexin V and cell death assay
The Muse™ Annexin V Dead Cell Assay kit (Ref. MCH
100105; Merck Millipore, Darmstadt, Germany) was used to monitor
cell death. Exponentially growing SKOV3/DDP cells were seeded into
6-well culture plates at a density of 2×105 cells/well.
After exposure to different experimental conditions for 24 h, the
cells were trypsinized and resuspended in RPMI-1640 with 10% FBS at
a concentration of 1×106 cells/ml. The cells were
incubated with Annexin V and Dead Cell Reagent in the dark at room
temperature for 20 min. Finally, the samples were assessed by flow
cytometry (Muse Cell Analyzer; Merck Millipore).
Calcium concentration analysis
The cytoplasmic Ca2+-sensitive
fluorescent dye Fluo-4/AM (Molecular Probes) and the mitochondrial
Ca2+-sensitive fluorescent dye Rhod-2/AM (AAT Bioquest,
Inc., Sunnyvale, CA, USA) were used to determine the
Ca2+ concentration according to the manufacturer's
instructions. Before exposure to different experimental conditions
for 24 h, the cells were incubated with Fluo-4/AM or Rhod-2/AM for
30 min at 37°C. The cell samples were then analyzed by confocal
laser microscopy. All experiments were performed in triplicate.
Mitochondrial membrane potential
(∆ψm)
Changes in the ∆ψm during the early stages of
apoptosis were assayed using the Muse MitoPotential Assay kit (Ref.
MCH 100110; Merck Millipore) in cells treated with cisplatin for 6
h. Briefly, the cells were harvested and the cell pellet was
suspended in assay buffer (1×105 cells/100 µl). The
MitoPotential dye working solution was added and the cell
suspension was incubated at 37°C for 20 min. After the addition of
Muse MitoPotential 7-AAD dye (propidium iodide) and incubation for
5 min, changes in the ∆ψm and in cellular plasma-membrane
permeabilization were assessed on the basis of the fluorescence
intensities of both dyes, which were analyzed by flow cytometry
(Muse Cell Analyzer; Merck Millipore).
Immunofluorescence staining and
confocal laser microscopy
The colocalization of IP3R and VDAC1 and the
expression of calpain-1 were examined by the indirect
immunofluorescence method. The cells were cultured on coverslips
overnight, then treated with the indicated drugs for 24 h and
rinsed with 0.1 M PBS three times. After incubation, the cells were
fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1%
Triton X-100 (Sigma-Aldrich) for 5 min, washed three times with
0.01 M phosphate-buffered saline (PBS) and then blocked for 30 min
in 5% (w/v) non-immune animal serum (goat; Beyotime Institute of
Biotechnology, Shanghai, China) PBS and incubated with primary
antibodies (IP3R, VDAC1 and PDI) overnight at 4°C. The following
day, the slides were incubated with the Alexa
Fluor-488/546-conjugated secondary antibody (1:400 dilution;
Invitrogen Life Technologies) for 1 h, and then stained with
Hoechst 33342 (2 µg/ml) for 2 min and washed three times with PBS.
After mounting, the cells were examined by Olympus FV1000 (Olympus,
Tokyo, Japan) confocal laser microscopy.
Protein preparation and western blot
analysis
The cells were treated with the indicated drugs for
24 h and various cells were harvested and lysed in lysis buffer (50
mM Tris-HCl, 1% NP40, 150 mM NaCl, 1 mM EDTA and 1 mM PMSF) for 30
min at 4°C. Total cell extracts were separated using 12% SDS/PAGE
gels and transferred to polyvinylidene fluoride (PVDF) membranes.
The membranes were blocked with 3% BSA and incubated with primary
antibodies diluted in blocking solution. The signals were
visualized using the chemiluminescent substrate method and the
SuperSignal West Pico kit (Pierce; Thermo Fisher Scientific).
β-actin was used as an internal control to normalize the loading
materials.
Transmission electron microscopy
The cells were treated with the indicated drugs for
24 h and were fixed with 2% paraformaldehyde and 2% glutaraldehyde
in 0.1 M phosphate buffer (pH 7.4) and then post fixed with 1%
OsO4 for 2 h. The cells were dehydrated with increasing
concentrations of alcohol (30, 50, 70, 90 and 100%), infiltrated
with LR white resin (62661; Sigma-Aldrich) twice for 1 h and
embedded in LR white resin. The solidified blocks were cut into
60-nm thicknesses and stained with uranyl acetate and lead citrate.
Samples were observed under a transmission electron microscope
(Hitachi H-7600; Hitachi High-Technologies Corp., Tokyo,
Japan).
Human tumor xenografts
BALB/c nude mice (4–6 weeks old) were purchased from
Beijing Vital River Laboratory Animal Technology Co., Ltd.
(Beijing, China). The animals were maintained in specific
pathogen-free conditions and in a controlled-light and humidity
environment. The animal experiments were conducted in accordance
with the National Institutes of Health Guide for the Care and Use
of Laboratory Animals. The transfected SKOV3 cells
(5×106) were subcutaneously injected into the right
flank of each mouse. Tumor volume (mm3) was assessed
every two days using a Vernier caliper and calculated as follows:
0.4 × (short length)2 × long length. Treatment was
initiated when the tumors reached a volume of 30 mm3 (on
the 10th day). The mice received NS intraperitoneally [(i.p.),
daily)] or 4 mg/kg cisplatin (i.p., every other day) for 8 days.
The mice were sacrificed and the experiment was terminated at the
end of the 18th day. The tumors were isolated, weighed and
imaged.
Immunohistochemistry
Immunohistochemical staining for cleaved caspase-3
was performed on 5-µm thick sections embedded in paraffin after
formalin fixation. The sections were de-paraffinized in xylene and
rehydrated in graded ethanol solutions (reducing concentration from
95 to 70%). The sections were incubated in
H2O2 solution (3% H2O2
in PBS buffer) for 30 min to block endogenous peroxidase activity.
Antigen retrieval was performed in retrieval buffer (pH 9.0, 20 mM
Tris, 0.05 mM EDTA, 0.05% Tween-20 buffer) in a 99°C bath for 20
min. The sections were subsequently incubated at 4°C overnight with
anti-cleaved caspase-3. After rinsing with PBS buffer, the
secondary antibody (MaxVision™ HRP-Polymer Anti-Rabbit IHC kit;
Maixin Bio, Beijing, China) was applied for 15 min at room
temperature (RT). The DAB (Maixin Bio) solution was used as the
chromogen. Finally, the sections were counterstained with
hematoxylin (Sigma-Aldrich) to identify the nuclei. The images were
captured and analyzed using a microscope (Leica DM4000 B; Leica
Microsystems GmbH, Wetzlar, Germany) with Image-Pro Plus 6.0
software (Media Cybernetics, Rockville, MD, USA).
Statistical analysis
Data are presented as the mean ± standard error
(SE). The significance of the difference in mean values within and
among multiple groups was examined with an ANOVA for repeated
measures followed by a Duncan's post hoc test. Student's t-test was
used to evaluate the significance of differences between two groups
of experiments (SigmaStat; SPSS, Inc., Chicago, IL, USA). A P-value
<0.05 was considered to indicate a statistically significant
difference.
Results
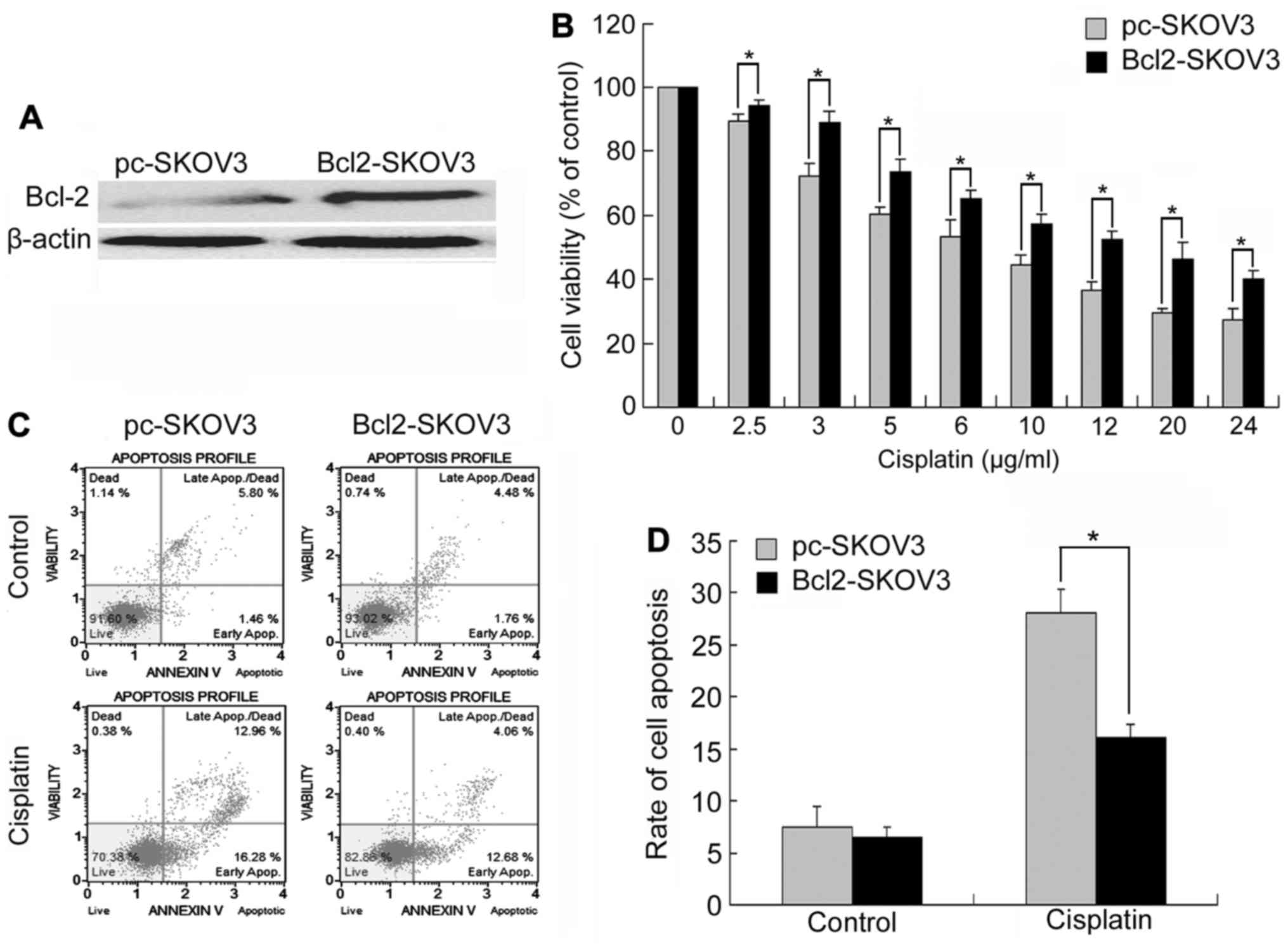
Bcl-2 overexpression reduces
cisplatin-induced growth inhibition and apoptosis in SKOV3
cells
To study the effect of Bcl-2 on cisplatin-induced
growth inhibition and apoptosis, we constructed a SKOV3 cell line
stably overexpressing Bcl-2 by pcDNA3.1(+)-Bcl-2 plasmids. Western
blot analysis verified that SKOV3 cells transfected with
pcDNA3.1(+)-Bcl-2 (Bcl-2-SKOV3 cells) had higher levels of Bcl-2
compared with those transfected with pcDNA3.1(+) (pc-SKOV3 cells)
(Fig. 1A). We treated Bcl-2-SKOV3
and pc-SKOV3 cells with 0–24 µg/ml cisplatin for 24 h and then
assessed cell viability using MTT assays. Although cisplatin
inhibited the growth and viability of both cell lines in a
dose-dependent manner, Bcl2-SKOV3 cells were more resistant to
cisplatin than pc-SKOV3 cells (Fig.
1B).
Subsequently, we treated both cell lines with 6
µg/ml cisplatin for 24 h, and then assessed the levels of apoptosis
by flow cytometry. Cisplatin induced a higher apoptosis rate in the
pc-SKOV3 cells than in the Bcl2-SKOV3 cells (Fig. 1C and D). In conclusion, Bcl-2
inhibited cisplatin-induced apoptosis in SKOV3 cells.
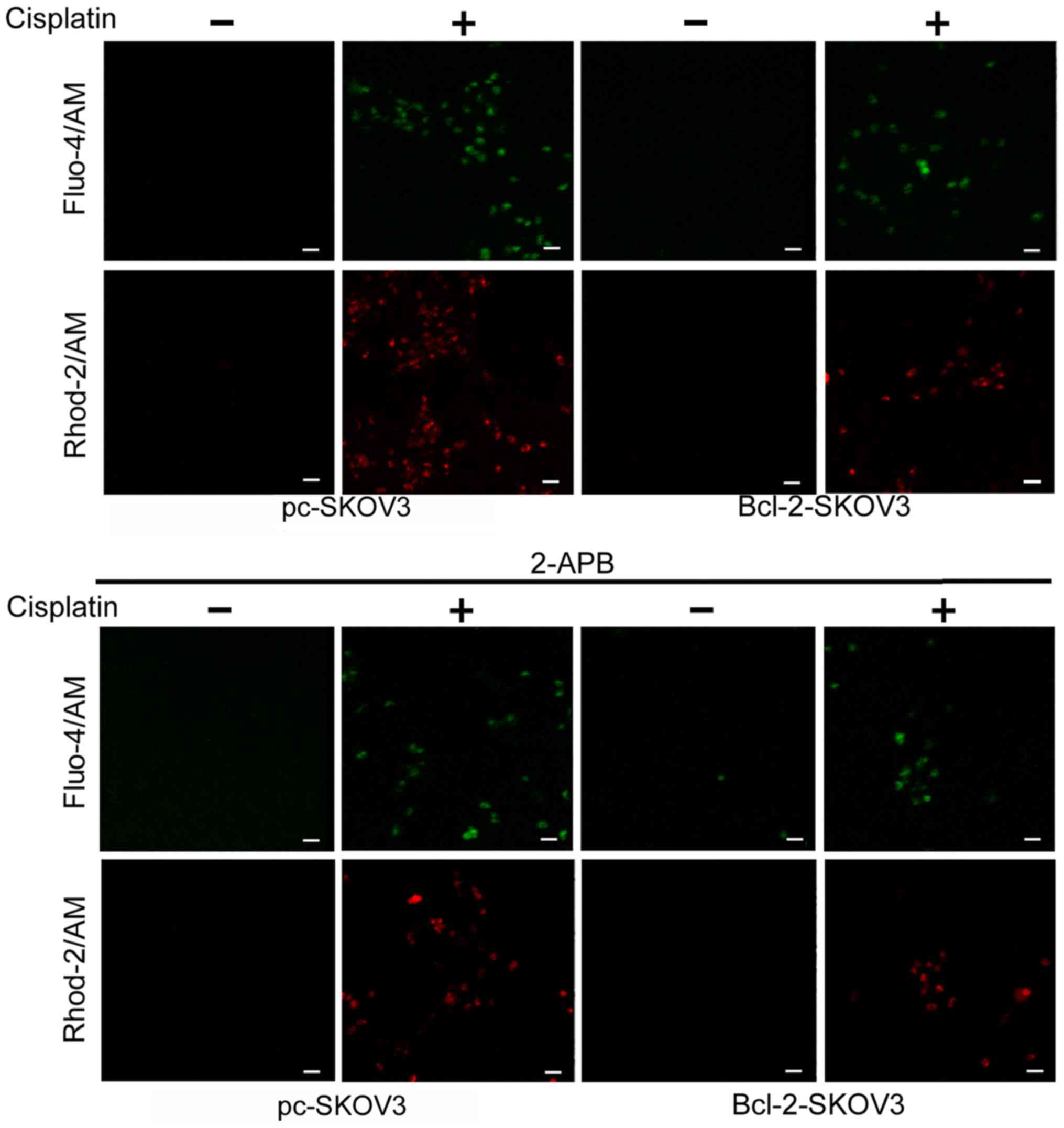
Bcl-2 overexpression reduces
cisplatin-induced Ca2+ release from the ER to the
cytoplasm and mitochondria
Previous studies indicate that Bcl-2 modulates
cytosolic and mitochondrial Ca2+ levels in cancer cells
(17). We assessed the relative
cytosolic and mitochondrial Ca2+ levels in Bcl-2-SKOV3
and pc-SKOV3 cells treated with 6 µg/ml cisplatin for 24 h using
the Fluo-4/AM and Rhod-2/AM indicators, respectively. Cisplatin
treatment induced higher cytosolic and mitochondrial
Ca2+ levels in pc-SKOV3 cells than in Bcl-2-SKOV3 cells
(Fig. 2, upper panel). In order to
demonstrate that cisplatin-induced cytosolic and mitochondrial
Ca2+ accumulation derives from the ER, the IP3R chelator
2-APB was administered to both cell types in the presence and
absence of cisplatin. Confocal microscopy revealed that 2-APB
inhibited cisplatin-induced cytosolic and mitochondrial
Ca2+ accumulation in both cell types (Fig. 2, lower panel). These findings
demonstrated that Bcl-2 overexpression reduced cisplatin-induced
Ca2+ release from the ER to the cytoplasm and
mitochondria.
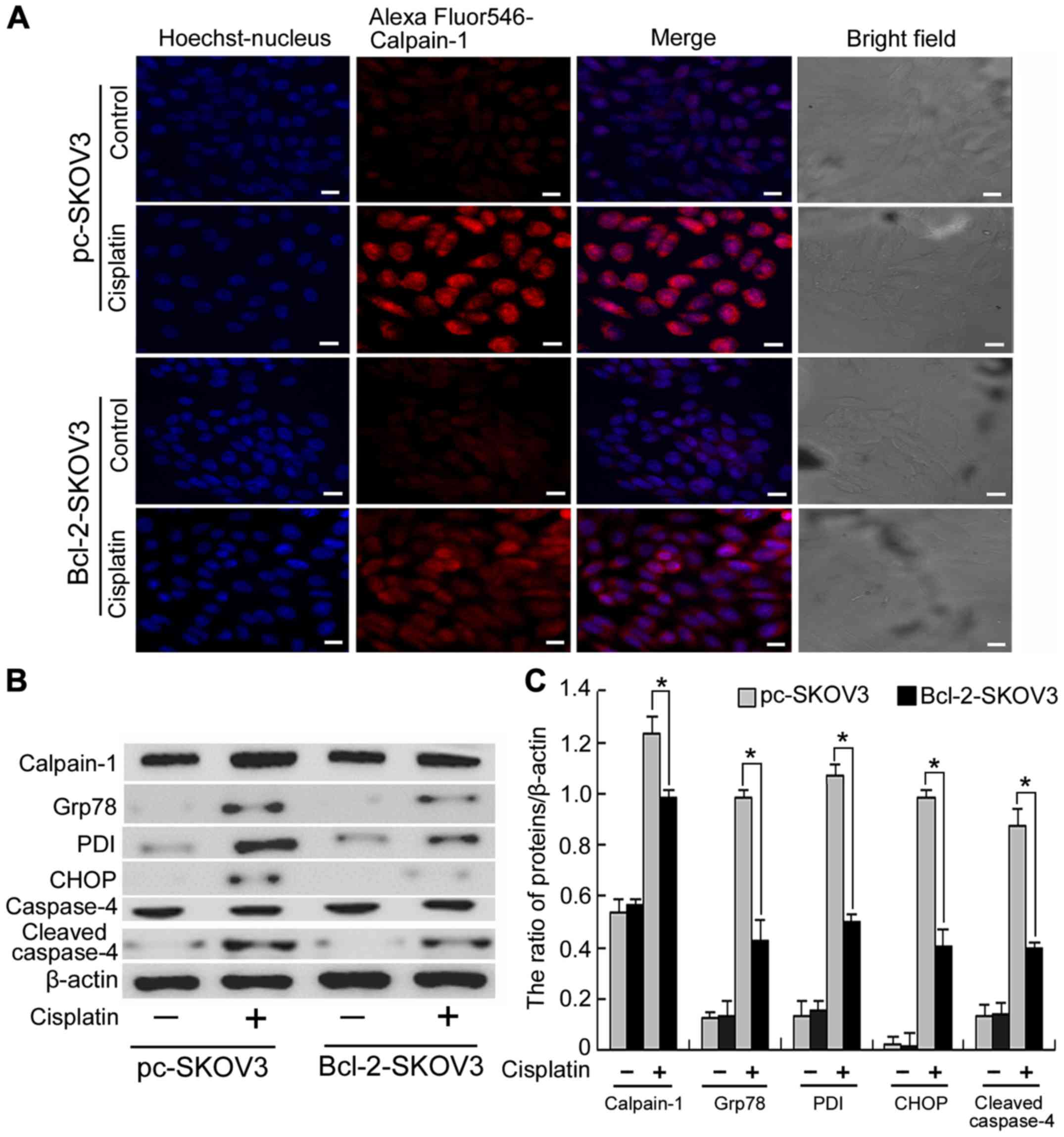
Bcl-2 overexpression inhibits the
activation of ER stress-mediated apoptosis by cisplatin in SKOV3
cells
Calpain-1, a Ca2+-activated protease, is
involved in many cellular events and is activated by cytosolic
Ca2+ accumulation (18).
As previously demonstrated Bcl-2 inhibits cisplatin-induced
cytosolic Ca2+ accumulation. Hence, we investigated
whether Bcl-2 inhibits cisplatin-induced calpain-1 expression in
SKOV3 cells. Confocal microscopy revealed that cisplatin induced
higher levels of calpain-1 expression in pc-SKOV3 cells than in
Bcl-2-SKOV3 cells (Fig. 3A),
indicating that Bcl-2 inhibits cisplatin-induced calpain-1
expression. Calpain-1 induces ER stress by promoting the unfolded
protein response. Subsequently, we assessed the expression levels
of calpain-1 and the ER stress markers PDI and Grp78 in pc-SKOV3
and Bcl-2-SKOV3 cells treated with cisplatin. Western blot analysis
revealed that cisplatin induced higher levels of all three proteins
in pc-SKOV3 cells than in Bcl2-SKOV3 cells (Fig. 3B and C). In addition, CHOP and
cleaved caspase-4 are required for the ER stress-mediated
apoptosis. Cisplatin induced higher CHOP and cleaved caspase-4
levels in pc-SKOV3 cells than in Bcl-2-SKOV3 cells (Fig. 3B and C). These data indicated that
Bcl-2 inhibited induction of calpain-1 expression and activation of
ER stress-mediated apoptosis by cisplatin.
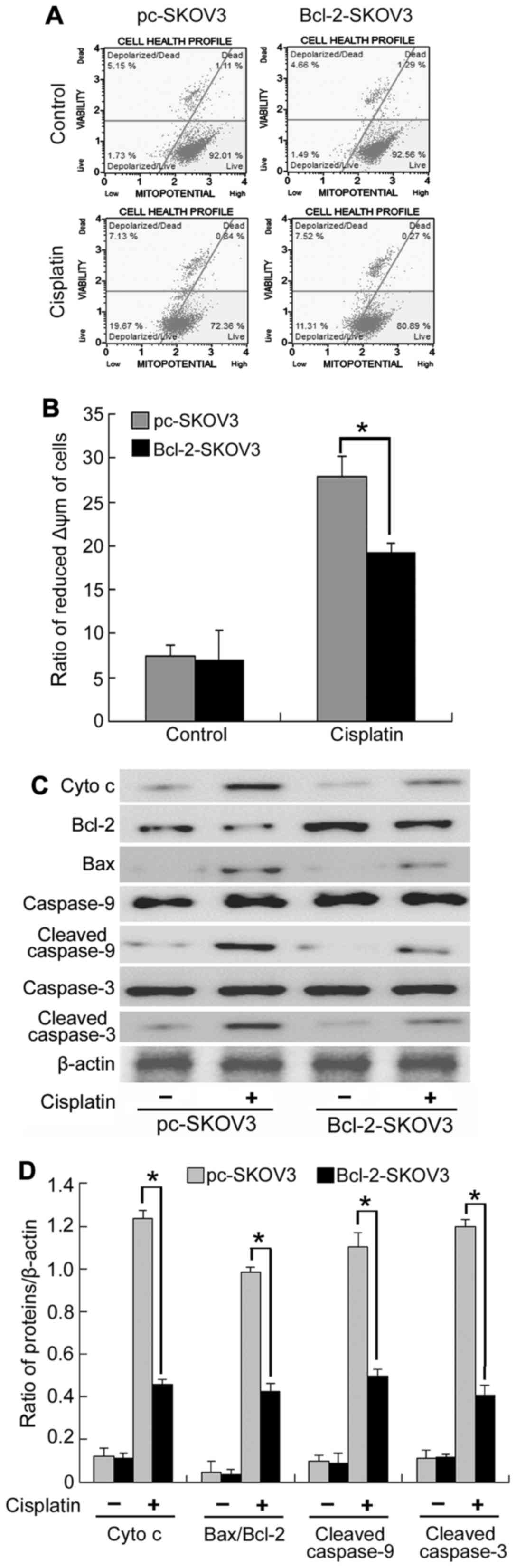
Bcl-2 overexpression inhibits the
activation of the mitochondrial apoptosis pathway by cisplatin in
SKOV3 cells
As mitochondrial Ca2+ overload is the
major cause of decreased Δψm, we assessed Δψm in Bcl-2-SKOV3 and
pc-SKOV3 cells treated with cisplatin using flow cytometry. The
results indicated that Bcl-2 inhibited the cisplatin-induced
decrease in Δψm in SKOV3 cells (Fig.
4A). Decreased Δψm can lead to mitochondrial swelling and
mitochondrial membrane rupture, which increases cytochrome c
efflux from the mitochondria, thus, activating the mitochondrial
apoptosis pathway. Subsequently, we assessed mitochondrial
apoptosis by analyzing the expression of cytochrome c,
Bcl-2, Bax, cleaved caspase-9 and cleaved caspase-3. Western blot
analysis indicated that cisplatin induced a higher expression of
cytochrome c, Bax/Bcl-2 ratio, cleaved caspase-9 and cleaved
caspase-3 in pc-SKOV3 cells than in Bcl-2-SKOV3 cells (Fig. 4B and C), indicating that Bcl-2
inhibits the activation of the mitochondrial apoptosis pathway by
cisplatin. Collectively, these results demonstrated that Bcl-2
inhibited the induction of the mitochondrial apoptosis pathway by
cisplatin via altering mitochondrial Ca2+ levels in
SKOV3 cells.
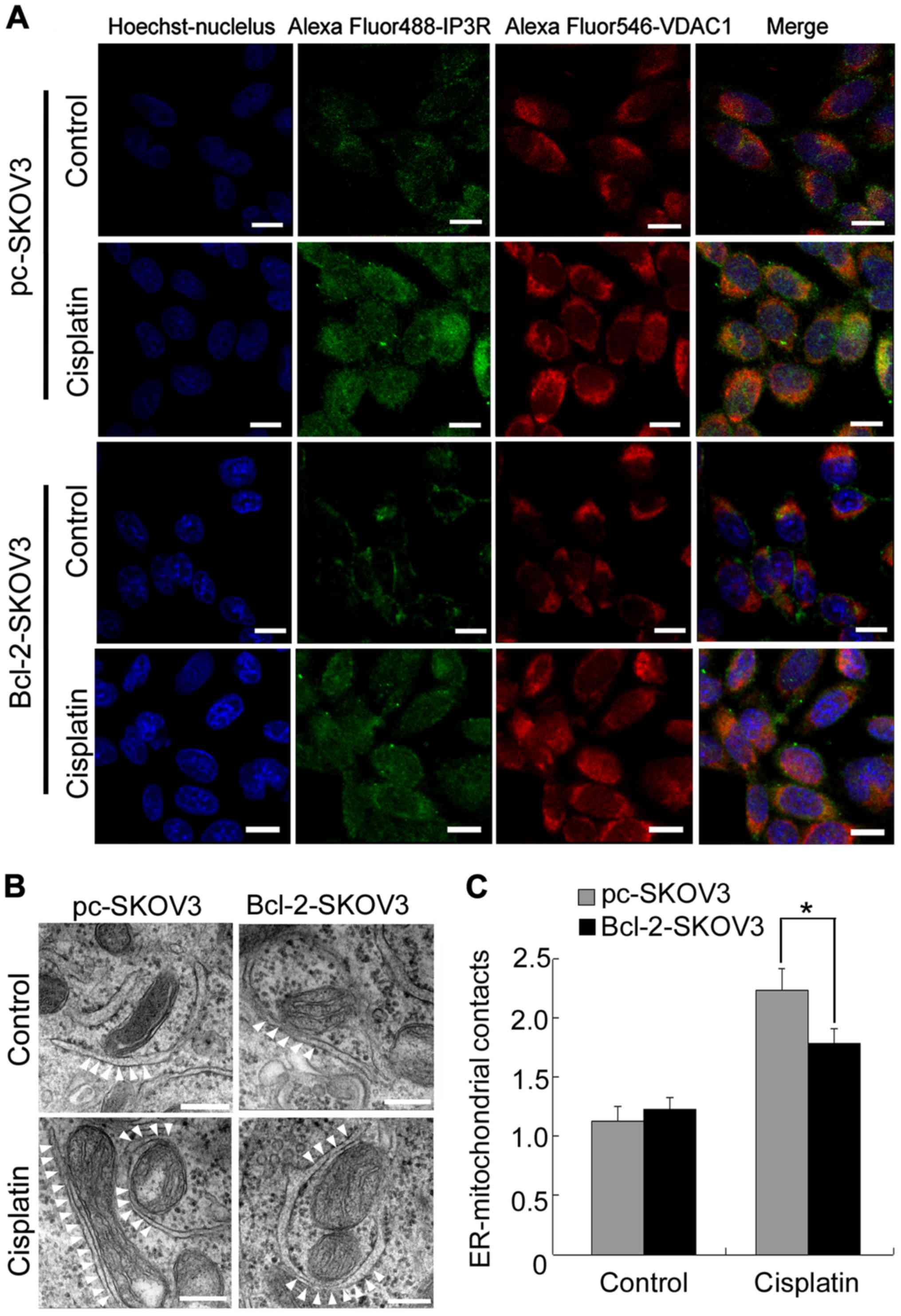
Bcl-2 overexpression blocks
cisplatin-induced ER-mitochondrial interactions in SKOV3 cells
Close proximity of the organelles facilitates direct
Ca2+ transfer from the ER to mitochondria (16). Therefore, we investigated whether
Bcl-2 inhibited cisplatin-induced mitochondrial Ca2+
accumulation by decreasing ER-mitochondrial interactions in SKOV3
cells. Confocal microscopy revealed that cisplatin induced
IP3R-VDAC1 colocalization to a greater extent in pc-SKOV3 cells
than in Bcl-2-SKOV3 cells (Fig.
5A). Subsequently, we used electron microscopy to examine
ER-mitochondrial interactions in pc-SKOV3 and Bcl-2-SKOV3 cells
after cisplatin treatment. Significantly more contact points
between the two organelles were detected in pc-SKOV3 cells than in
Bcl-2-SKOV3 cells at 24 h after cisplatin treatment (Fig. 5B and C). These results revealed that
Bcl-2 reduced the number of cisplatin-induced ER-mitochondrial
interactions in SKOV3 cells.
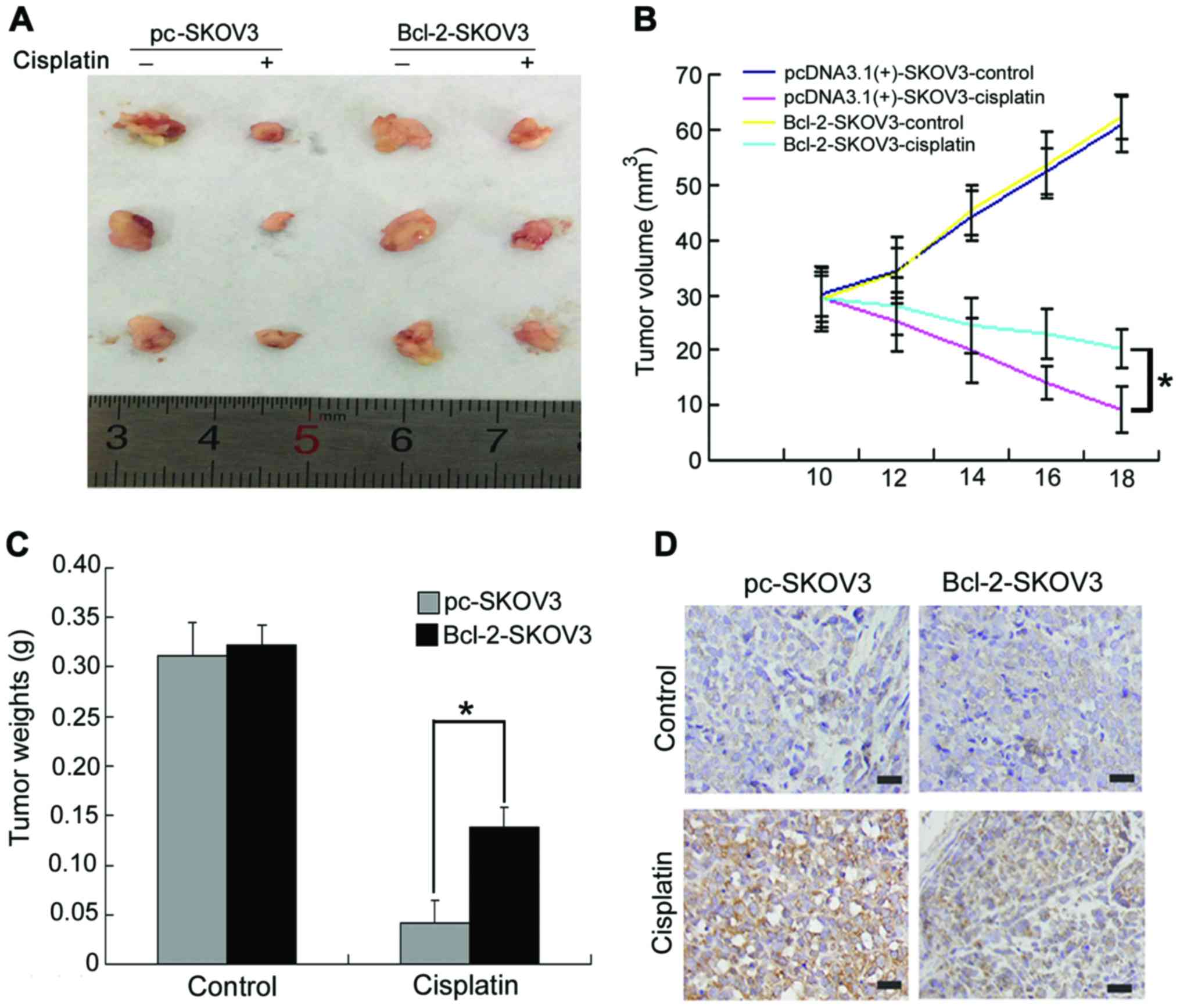
Bcl-2 attenuates the in vivo antitumor
activity of cisplatin in SKOV3 human ovarian cancer xenografts
To further validate the effect of Bcl-2
overexpression on cisplatin-induced SKOV3 cell apoptosis in
vivo, we established pc-SKOV3 and Bcl-2-SKOV3 xenograft models.
Mice bearing xenograft tumors were treated with NS or cisplatin for
8 days, as described in the Materials and Methods section.
Cisplatin inhibited the in vivo growth of pc-SKOV3 xenograft
tumors more efficiently than that of Bcl-2-SKOV3 cells (Fig. 6A and B). In addition, cisplatin
treatment led to a greater reduction in the weight of pc-SKOV3
xenograft tumors compared with Bcl-2-SKOV3 xenograft tumors
(Fig. 6C). Regarding the effects of
BCL-2 overexpression on cisplatin-induced apoptosis, a greater
number of cleaved caspase-3-positive cells was observed in pc-SKOV3
than in Bcl-2-SKOV3 xenograft tumors after cisplatin treatment
(Fig. 6D). These data indicated
that Bcl-2 attenuated the in vivo antitumor activity of
cisplatin in ovarian xenograft tumors.
Discussion
Ovarian carcinoma is a common gynecological
malignancy with an increasing incidence worldwide (2,18).
Although cisplatin is one of the most widely used chemotherapeutic
drugs used to treat ovarian cancer, the development of
chemoresistance in ovarian cancer patients is a major problem
(19). The resistance of ovarian
cancer cells to cisplatin is partially dependent on Bcl-2, a
prosurvival factor (20). Nishioka
et al (21) reported that
nicotine increases the resistance of H5800 lung cancer cells to
cisplatin by Bcl-2 stabilization via preventing its degradation.
Consistent with this findings, we demonstrated that Bcl-2 inhibited
cisplatin-induced apoptosis in SKOV3 cells both in vitro and
in vivo (Figs. 1 and
6). Bcl-2 overexpression suppressed
the proapoptotic response to ER Ca2+ release and
inhibited cancer cell sensitivity to various stimuli (22). In addition, the present study
revealed that Bcl-2 attenuated the cisplatin-induced release of ER
Ca2+ into the cytosol and mitochondria (Fig. 2).
Intracellular Ca2+ is an important second
messenger with a pivotal role in signal transduction pathways that
regulate a wide variety of cellular processes, including gene
expression, protein synthesis and apoptosis. Cellular
Ca2+ homeostasis is crucially important for the proper
function of normal and cancer cells. Elevated intracellular
Ca2+ levels are responsible for inducing or modulating
the apoptotic response (23).
Cytosolic Ca2+ elevation activates calpain-1, a
Ca2+-dependent protease. Calpain-1 overexpression is
reported to promote caspase-4 activation and increase the level of
ER stress-mediated apoptosis (24).
Wang et al (25) reported
that the MDL28170 (a calpain inhibitor) attenuated ER
stress-mediated apoptosis by inhibiting CHOP and caspase-12.
Similarly, in the present study, we found that Bcl-2 attenuated the
induction of calpain-1 expression and activation of ER
stress-mediated apoptosis by cisplatin (Fig. 3).
Mitochondrial Ca2+ uptake is essential
for regulating aerobic metabolism, ATP production and cell
survival. However, mitochondrial Ca2+ overload can lead
to mitochondrial swelling and a decrease in Δψm, which in turn,
induces the release of mitochondrial apoptotic factors (such as
cytochrome c) into the cytosol thus activating the
mitochondrial apoptosis pathway (26,27).
Hu et al (28) reported that
apigenin and 5-fu co-treatment increased mitochondrial membrane
depolarization, thus inducing mitochondrial apoptosis via
decreasing the Bcl-2 expression. Our data revealed that Bcl-2
attenuated cisplatin-induced decrease in Δψm and cisplatin-induced
elevated Bax/Bcl-2 ratio, cytochrome c, cleaved caspase-9
and cleaved caspase-3 expression in SKOV3 cells (Fig. 4). These results indicated that Bcl-2
attenuated cisplatin-induced activation of the mitochondrial
apoptosis pathway by inhibiting ER Ca2+ release into the
mitochondria.
Subsequently, we investigated the mechanism
responsible for the Bcl-2 inhibition of cisplatin-induced ER
Ca2+ release into the mitochondria. Recent studies
revealed that MAM is crucial for the correct communication,
including the efficient transmission of physiological and
pathological Ca2+ signals, between the ER and
mitochondria (29). Mitochondrial
Ca2+ uptake mainly takes place through the MAM. Under
normal physiological conditions, there is little contact between
the ER and mitochondria and low physiological Ca2+
release maintains mitochondria function and cell survival. However,
an increase in the number of contacts between these two organelles
can lead to Ca2+ release and thus mitochondrial
Ca2+ overload-induced apoptosis (30–32).
Notably, FATE1, a component of MAM, is reported to antagonize
mitochondrial Ca2+ overload and chemotherapy-induced
apoptosis by decreasing the number of ER-mitochondrial contacts,
suggesting that MAM is responsible for mitochondrial
Ca2+ overload-induced apoptosis (33). Our results revealed that Bcl-2
inhibited cisplatin-induced ER-mitochondrial interaction in SKOV3
cells (Fig. 5), indicating that
Bcl-2 inhibits cisplatin-induced ER Ca2+ release into
mitochondria by reducing the number of ER-mitochondrial
interactions.
In conclusion, we demonstrated that Bcl-2 attenuated
cisplatin-induced Ca2+ release from the ER into the
cytosol and mitochondria, thus inhibiting cisplatin-induced ER
stress-mediated apoptosis and activation of the mitochondrial
apoptosis pathway. Furthermore, we revealed that decreased
ER-mitochondrial crosstalk is responsible for Bcl-2 attenuation of
cisplatin-induced mitochondrial Ca2+ accumulation in
SKOV3 cells. Thus, Bcl-2 may be a novel marker of cisplatin
resistance and thus, a potential therapeutic target for ovarian
cancer chemotherapy.
Acknowledgements
The present study was supported by the National
Nature and Science Foundation of China (NSFC 81372793) and the
Department of Education of Jilin Province Project (no. 2016237).
The authors would like to thank Director Dominic James from Liwen
Bianji (Edanz Group China) for the language editing of this
study.
References
|
1
|
Muralidhar GG and Barbolina MV: The
miR-200 family: Versatile players in epithelial ovarian cancer. Int
J Mol Sci. 16:16833–16847. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sapiezynski J, Taratula O,
Rodriguez-Rodriguez L and Minko T: Precision targeted therapy of
ovarian cancer. J Control Release. 243:250–268. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guo P, Xiong X, Zhang S and Peng D:
miR-100 resensitizes resistant epithelial ovarian cancer to
cisplatin. Oncol Rep. 36:3552–3558. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dai Y, Jin S, Li X and Wang D: The
involvement of Bcl-2 family proteins in AKT-regulated cell survival
in cisplatin resistant epithelial ovarian cancer. Oncotarget.
8:1354–1368. 2017.PubMed/NCBI
|
|
5
|
Hirata H, Lopes GS, Jurkiewicz A,
Garcez-do-Carmo L and Smaili SS: Bcl-2 modulates endoplasmic
reticulum and mitochondrial calcium stores in PC12 cells. Neurochem
Res. 37:238–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pan Z and Gollahon L: Paclitaxel
attenuates Bcl-2 resistance to apoptosis in breast cancer cells
through an endoplasmic reticulum-mediated calcium release in a
dosage dependent manner. Biochem Biophys Res Commun. 432:431–437.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cui C, Merritt R, Fu L and Pan Z:
Targeting calcium signaling in cancer therapy. Acta Pharm Sin B.
7:3–17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stary CM, Sun X, Ouyang Y, Li L and
Giffard RG: miR-29a differentially regulates cell survival in
astrocytes from cornu ammonis 1 and dentate gyrus by targeting
VDAC1. Mitochondrion. 30:248–254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arbel N and Shoshan-Barmatz V:
Voltage-dependent anion channel 1-based peptides interact with
Bcl-2 to prevent antiapoptotic activity. J Biol Chem.
285:6053–6062. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kilpatrick BS, Yates E, Grimm C, Schapira
AH and Patel S: Endo-lysosomal TRP mucolipin-1 channels trigger
global ER Ca2+ release and Ca2+ influx. J
Cell Sci. 129:3859–3867. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Theurey P, Tubbs E, Vial G, Jacquemetton
J, Bendridi N, Chauvin MA, Alam MR, Le Romancer M, Vidal H and
Rieusset J: Mitochondria-associated endoplasmic reticulum membranes
allow adaptation of mitochondrial metabolism to glucose
availability in the liver. J Mol Cell Biol. 8:129–143. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Area-Gomez E: Assessing the function of
mitochondria-associated ER membranes. Methods Enzymol. 547:181–197.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giorgi C, Wieckowski MR, Pandolfi PP and
Pinton P: Mitochondria associated membranes (MAMs) as critical hubs
for apoptosis. Commun Integr Biol. 4:334–335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qi H and Shuai J: Alzheimers disease via
enhanced calcium signaling caused by the decrease of endoplasmic
reticulum-mitochondrial distance. Med Hypotheses. 89:28–31. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arruda AP, Pers BM, Parlakgül G, Güney E,
Inouye K and Hotamisligil GS: Chronic enrichment of hepatic
endoplasmic reticulum-mitochondria contact leads to mitochondrial
dysfunction in obesity. Nat Med. 20:1427–1435. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krols M, Bultynck G and Janssens S:
ER-Mitochondria contact sites: A new regulator of cellular calcium
flux comes into play. J Cell Biol. 214:367–370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xie Q, Su J, Jiao B, Shen L, Ma L, Qu X,
Yu C, Jiang X, Xu Y and Sun L: ABT737 reverses cisplatin resistance
by regulating ER-mitochondria Ca2+ signal transduction
in human ovarian cancer cells. Int J Oncol. 49:2507–2519. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Albu DF, Albu CC, Văduva CC, Niculescu M
and Edu A: Diagnosis problems in a case of ovarian tumor - case
presentation. Rom J Morphol Embryol. 57:1437–1442. 2016.PubMed/NCBI
|
|
19
|
Zhu X, Ji M, Han Y, Guo Y, Zhu W, Gao F,
Yang X and Zhang C: PGRMC1-dependent autophagy by hyperoside
induces apoptosis and sensitizes ovarian cancer cells to cisplatin
treatment. Int J Oncol. 50:835–846. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tung MC, Lin PL, Cheng YW, Wu DW, Yeh SD,
Chen CY and Lee H: Reduction of microRNA-184 by E6 oncoprotein
confers cisplatin resistance in lung cancer via increasing Bcl-2.
Oncotarget. 7:32362–32374. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nishioka T, Luo LY, Shen L, He H,
Mariyannis A, Dai W and Chen C: Nicotine increases the resistance
of lung cancer cells to cisplatin through enhancing Bcl-2
stability. Br J Cancer. 110:1785–1792. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Williams A, Hayashi T, Wolozny D, Yin B,
Su TC, Betenbaugh MJ and Su TP: The non-apoptotic action of Bcl-xL:
Regulating Ca2+ signaling and bioenergetics at the
ER-mitochondrion interface. J Bioenerg Biomembr. 48:211–225. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen L, Wen N, Xia M, Zhang YU, Liu W, Xu
YE and Sun L: Calcium efflux from the endoplasmic reticulum
regulates cisplatin-induced apoptosis in human cervical cancer HeLa
cells. Oncol Lett. 11:2411–2419. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ma SH, Zhuang QX, Shen WX, Peng YP and Qiu
YH: Interleukin-6 reduces NMDAR-mediated cytosolic Ca2+
overload and neuronal death via JAK/CaN signaling. Cell Calcium.
58:286–295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang C, Shi D, Song X, Chen Y, Wang L and
Zhang X: Calpain inhibitor attenuates ER stress-induced apoptosis
in injured spinal cord after bone mesenchymal stem cells
transplantation. Neurochem Int. 97:15–25. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Demaurex N and Rosselin M: Redox control
of mitochondrial calcium uptake. Mol Cell. 65:961–962. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pendin D, Greotti E and Pozzan T: The
elusive importance of being a mitochondrial Ca2+
uniporter. Cell Calcium. 55:139–145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hu XY, Liang JY, Guo XJ, Liu L and Guo YB:
5-Fluorouracil combined with apigenin enhances anticancer activity
through mitochondrial membrane potential (ΔΨm)-mediated apoptosis
in hepatocellular carcinoma. Clin Exp Pharmacol Physiol.
42:146–153. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Galmes R, Houcine A, van Vliet AR,
Agostinis P, Jackson CL and Giordano F: ORP5/ORP8 localize to
endoplasmic reticulum-mitochondria contacts and are involved in
mitochondrial function. EMBO Rep. 17:800–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Joshi AU, Kornfeld OS and Mochly-Rosen D:
The entangled ER-mitochondrial axis as a potential therapeutic
strategy in neurodegeneration: A tangled duo unchained. Cell
Calcium. 60:218–234. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grimm S: The ER-mitochondria interface:
The social network of cell death. Biochim Biophys Acta.
1823:327–334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Raturi A and Simmen T: Where the
endoplasmic reticulum and the mitochondrion tie the knot: The
mitochondria-associated membrane (MAM). Biochim Biophys Acta.
1833:213–224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Doghman-Bouguerra M, Granatiero V, Sbiera
S, Sbiera I, Lacas-Gervais S, Brau F, Fassnacht M, Rizzuto R and
Lalli E: FATE1 antagonizes calcium- and drug-induced apoptosis by
uncoupling ER and mitochondria. EMBO Rep. 17:1264–1280. 2016.
View Article : Google Scholar : PubMed/NCBI
|