Introduction
Breast cancers are the most malignant and leading
cause of cancer-related deaths in women worldwide (1). Among all breast cancers,
triple-negative breast cancers (TNBCs) are the most aggressive,
metastatic, and difficult to treat due to the lack of estrogen
receptors (ER), progesterone receptors (PR), and human epidermal
growth factor receptor 2 (Her2) (2). Accordingly, TNBCs exhibit a poorer
clinical outcome compared to other breast cancer subtypes (3). Moreover, the molecular heterogeneity
of TNBC increases the difficulty in improving survival rates.
Despite the development of hormonal and HER2-targeted therapies,
patients with TNBC rarely respond to these therapeutic options
(4,5). Thus, conventional chemotherapy such as
the use of taxanes and anthracyclines still remains the mainstay
for the treatment of TNBC patients (6,7). The
poor prognosis of TNBC patients along with the limited
effectiveness of therapeutic options necessitates the development
of alternative therapeutic approaches for the treatment of
TNBCs.
Heat shock protein 90 (Hsp90) is a molecular
chaperone that regulates the folding, stability, and function of
its substrate proteins, referred to as ‘client’ proteins (8,9). These
clients include epidermal growth factor receptor (EGFR), human
epidermal growth factor receptor 2 (Her2), anaplastic lymphoma
kinase (Alk), mesenchymal-epidermal transition factor (Met),
protein kinase B (Akt), cyclin-dependent kinase 4 (Cdk4), hypoxia
inducible factor 1 (HIF-1α), and matrix metalloproteinase 9 (MMP9).
Consequently, inhibition of the Hsp90 protein induces the
degradation of these oncogenic proteins via the
ubiquitin-proteasome pathway (10,11)
and results in a simultaneous disruption of multiple signaling
pathways in the proliferation and survival of cancers (12,13).
Therefore, Hsp90 has emerged as a promising therapeutic target for
the treatment of TNBCs (9).
Geldanamycin, a benzoquione ansamycin antibiotic was
first identified as a natural product Hsp90 inhibitor in 1994
(14). Since then, numerous
geldanamycin analogues such as 17-AAG, 17-DMAG, and IPI-504 were
entered into clinical trials (15).
Despite their clinical effects, geldanamycin analogues revealed
several drawbacks in their clinical application such as formulation
and toxicity issues (16). Hence,
significant efforts have been directed towards improving drug-like
properties and toxicities, resulting in diverse synthetic small
molecule inhibitors. These Hsp90 inhibitors are divided into three
major classes according to their core structures, purine,
resorcinol, and benzamide. The purine class is designed by
mimicking the ATP structure and includes PU-H71 (17), BIIB02 (18), and CUDC-305 (19) and the resorcinol class includes
AT13387 (20), STA-9090 (21), and NVP-AUY922 (22). The benzamide class is another
important class of Hsp90 inhibitors, including TAS-116 (23), XL888 (24), and SNX-5422 (25). Although a number of Hsp90 inhibitors
have been entered into clinical trials, none of the Hsp90
inhibitors are clinically approved yet.
In our ongoing effort to develop potent Hsp90
inhibitors, we previously revealed that a small molecule,
6,7-dihydrothieno[3,2-c] pyridin-5(4H)-yl amide (DPide, 1) displays
anticancer effects on cancer cells and this observation prompted us
to direct our efforts toward evaluating the biological activity of
DPide (1) and its analogue
Oxo-DPide (2) against MDA-MB-231
TNBC cell line (26–28).
Materials and methods
Chemistry
All reagents and solvents were purchased from
Sigma-Aldrich (Milwaukee, WI, USA) and Alfa Aesar (Haverhill, MA,
USA), and used without further purification. All experiments
dealing with moisture-sensitive compounds were carried out under an
argon atmosphere. Concentration or solvent removal under reduced
pressure was carried out using a rotary evaporator. Analytical thin
layer chromatography was performed on precoated silica gel
F254 TLC plates (Merck, Darmstadt, Germany) with
visualization under UV light. Column chromatography was conducted
under medium pressure on silica (Merck Silica Gel 40–63 µm) or
performed using a Biotage SP1 flash purification system with
prepacked silica gel cartrides (Biotage AB, Uppsala, Sweden). NMR
analyses were carried out using a JNM-ECZ500R spectrometer (500
MHz) manufactured by JEOL, Ltd. (Tokyo, Japan). Chemical shifts
were reported in parts per million (δ). The deuterium lock signal
of the sample solvent was used as a reference, and coupling
constants (J) were given in hertz (Hz). The splitting
pattern abbreviations were as follows: s, singlet; d, doublet; t,
triplet; q, quartet; dd, doublet of doublets; m, multiplet. The
purities of all final compounds were confirmed to be higher than
95% by analytical HPLC performed with a dual pump Shimadzu LC-6AD
system equipped with a VP-ODS C18 column (4.6×250 mm, 5 µm;
Shimadzu Corporation, Kyoto, Japan).
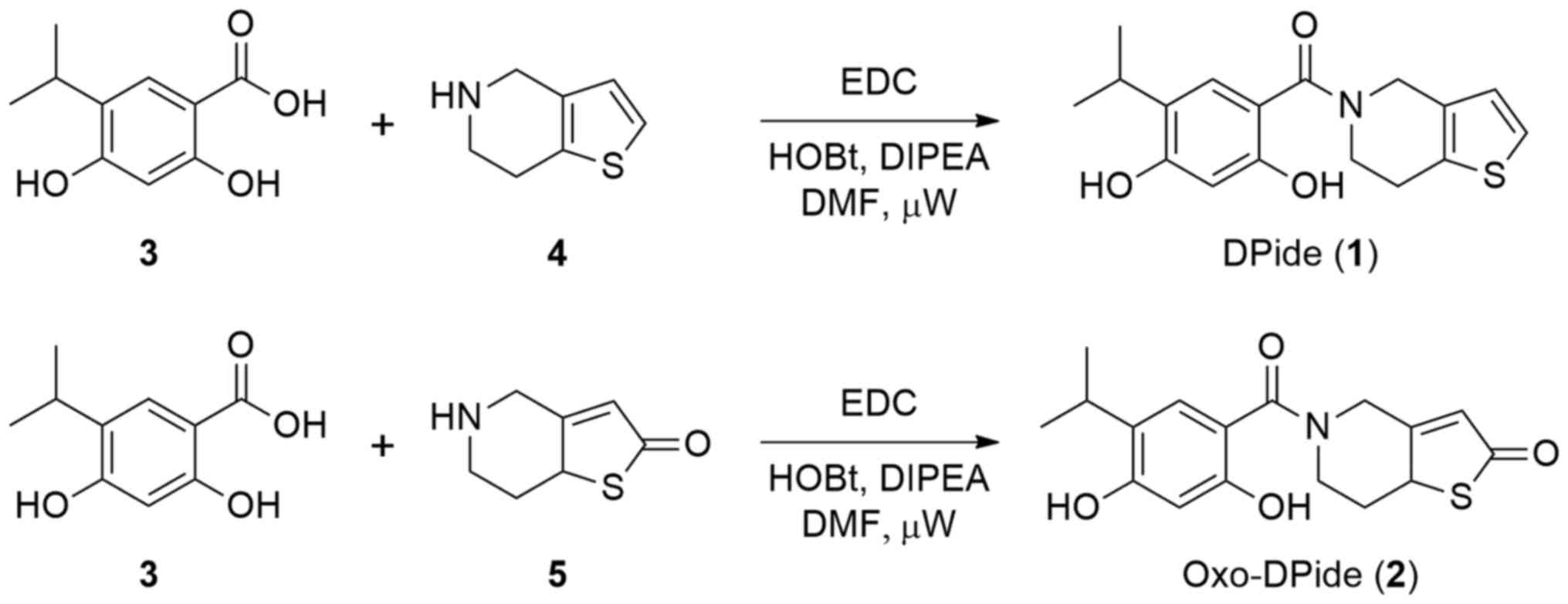
Synthesis of
5-(2,4-Dihydroxy-5-isopropylbenzoyl)-5,6,7,7a-tetrahydrothieno[3,2-c]pyridin-2(4H)-one
(Oxo-Dpide, 2)
A mixture of compound 2 (0.20 g, 1.02 mmol),
5,6,7,7a-tetrahydrothieno[3,2-c]pyridin-2(4H)-one hydrochloride
(0.29 ml, 1.53 mmol), 1-ethyl-3-(3-dimethylaminopropyl)
carbodiimide (0.39 g, 2.04 mmol), and
N,N-diisopropylethylamine (0.36 ml, 2.04 mmol) in 4 ml of
DMF was stirred under microwave irradiation at 80°C and 30 bar for
3 h. The mixture was concentrated under reduced pressure and
purified by MPLC to afford Oxo-DPide (2) in 10% yield. Rf=0.19
(5:5 ethyl acetate: hexane). 1H NMR (500 MHz,
CD3OD) δ 7.02 (s, 1H), 6.37 (s, 1H), 5.32 (d,
J=14.5 Hz, 1H), 4.49–4.46 (m, 1H), 3.79 (d, J=14.5
Hz, 1H), 3.49 (s, 1H), 3.25 (t, J=14.5 Hz, 1H), 3.18–3.12
(m, 1H), 2.53–2.49 (m, 1H), 1.91–1.85 (m, 1H), 1.19 (d,
J=6.5 Hz, 6H). ESI MS (m/e) 334.12
[M+1]+.
Cell culture & materials
H1975 cells (from the ATCC, Manassas, VA, USA)
(NSCLC) were grown in RPMI-1640 with L-glutamine supplemented with
streptomycin (500 mg/ml), penicillin (100 U/ml), and 10% fetal
bovine serum (FBS). Triple-negative breast cancer cells MDA-MB-231
(from the ATCC) were grown in DMEM high glucose, supplemented with
streptomycin (500 mg/ml), penicillin (100 U/ml), and 10% fetal
bovine serum (FBS). Cells were grown to confluence at 37°C in a
humidified atmosphere with 5% CO2. DPide was prepared
following a previously reported procedure (26). For in vitro studies, DPide
and geldanamycin (Alexis Biochemicals; Enzo Life Sciences, Inc.,
Farmingdale, NY, USA) were dissolved in DMSO. Antibodies for EGFR
(1:1,000; rabbit anti-human mAb; cat. no. 4267) Her2 (1:1,000;
rabbit anti-human mAb; cat. no. 2165), Met (1:1,000; rabbit
anti-human mAb; cat no. 8198), Akt (1:1,000; rabbit anti-human pAb;
cat. no. 9272) c-Raf (1:1,000; rabbit anti-human pAb; cat. no.
9422), Cdk4 (1:1,000; mouse anti-human mAb; cat. no. 2906), Hsp90
(1:1,000; rabbit anti-human pAb; cat. no. 4874), Hsp70 (1:1,000;
rabbit anti-human pAb; cat. no. 4872), PARP (1:1,000; rabbit
anti-human pAb; cat. no. 9542) and β-actin (1:1,000; rabbit
anti-human mAb; cat. no. 4970) were purchased from Cell Signaling
Technology (Beverly, MA, USA). Anti-mouse (1:5,000; goat anti-mouse
pAb; cat. no. sc-2005) and anti-rabbit antibodies (1:5,000; goat
anti-rabbit pAb; cat. no. sc-2004) were purchased from Santa Cruz
Biotechnology (Dallas, TX, USA).
Cell proliferation assay
H1975 cells (2×103 cells/well) were
seeded in a 96-well plate. The medium volume was brought to 100 µl,
and the cells were allowed to attach for 14 h. The cells were then
incubated with DPide or Oxo-DPide (0.01, 0.1, 0.05, 0.1, 0.5, 1, 5,
10, 30, 50, 70 and 100 µM) at 37°C with 5% CO2 for 3
days. MDA-MB-231 cells (2×103 cells/well) were seeded in
a 96-well plate, the medium volume was brought to 100 µl, and the
cells were allowed to attach for 14 h. The cells were then
incubated with DPide (0.01, 0.1, 1 and 5 µM) at 37°C with 5%
CO2 for 1, 2, and 3 days. CellTiter 96 Aqueous One
Solution reagent (Promega Corp., Madison, WI, USA) was added into
each well following the manufacturer's instructions. The absorbance
at 490 nm was read on the Tecan Infinite F200 Pro plate reader, and
values were expressed as the percentage of the absorbance from
cells incubated in DMSO alone.
Western blotting
After being treated with DPide (0.05, 0.1, 0.5, 1,
and 2 µM) or GA (1 µM) for 24 h, MDA-MB-231 cells were harvested
and lysed in ice-cold lysis buffer (25 mM Tris-HCl pH 7.6, 150 mM
NaCl, 1% NP40, 1% sodium deoxycholate, 0.1% SDS, 1 mM
phenylmethylsulfonyl fluoride). The 30 µg of lysate per lane was
separated by SDS-PAGE and transferred to a PVDF membrane (Bio-Rad
Laboratories, Hercules, CA, USA). After being blocked with 5% skim
milk in TBS with 0.1% Tween-20 (TBS-T), the membrane was incubated
with the corresponding antibodies in TBS-T at 4°C overnight. The
proteins were visualized using an enhanced chemiluminescence (ECL)
reagent according to the manufacturer's instructions (GE
Healthcare, Pittsburgh, PΑ, USA).
Reverse-transcriptase PCR
(RT-PCR)
RT-PCR was performed using the RT-PCR kit (Bio-Rad
Laboratories) following the manufacturer's protocol. Briefly, total
RNA was extracted from cultured cells using TRIzol reagent (Thermo
Fisher Scientific, Hampton, NH, USA), reverse transcribed, and then
subjected to PCR. The following primers were used for the
amplification of human Met, Akt, Cdk4, Hsp90 and β-actin: Met,
5′-AAGAGGGCATTTTGGTTGTG-3′ (forward) and 3′-GATGATTCCCTCGGTCAGAA-5′
(reverse); Akt, 5′-TTTTATTTCTCGGGTGCAT-3′ (forward) and
3′-CATTTCCCTACGTGAATCGG-5′ (reverse); Cdk4, 5′-CCGAAGTTCTTCTGCAGTCC
(forward) and 3′-AGGCAGAGATTCGCTTGTGT (reverse); Hsp90,
5′-GAGCAGTACGCTTGGGAGTC (forward) and 3′-CCAGATGGGCTTTGTTTTGT
(reverse); β-actin, 5′-AGAAAATCTGGCACCACACC-3′ (forward) and
3′-CCATCTCTTGCTCGAAGTCC-5′ (reverse).
Colony formation assay
MDA-MB-231 cells (1×105 cells/well) were
seeded in a 6-well plate and treated with DPide (0.1 and 0.5 µM) at
37°C with 5% CO2 for 21 days. After treatment, the
medium removed and stained with 0.1% of crystal violet in 10%
ethanol at room temperature for 30 min. The image was acquired with
an inverted phase contrast microscope (Olympus, Japan) using a 100X
objective.
Wound healing assay
MDA-MB-231 cells (2×105 cells/well) were
seeded in a 6-well plate, and the cells were allowed to attach for
24 h. The 80% confluent cells were wounded with a linear scratch
using a disposable 200-µl micropipette tip. The cells were washed
with medium to remove cell debris and covered with serum free
medium containing DPide (0.1 and 0.5 µm or GA (1 µm). After being
incubated for 24 h, the migrated cells were determined under an
inverted phase contrast microscope (Olympus, Tokyo, Japan) using a
100X objective.
Gelatin zymography assay
MDA-MB-231 (5×105 cells/well) cells were
seeded in a 6-well culture plate and treated with DPide (0.05, 0.5,
and 1 µm or GA (1 µm) for 24 h. Supernatants were collected and
loaded on a 10% SDS-PAGE gel with gelatin (0.2 mg/ml). Following
electrophoresis, the gels were washed for 40 min in 2.5% Triton
X-100. After washing with deionized water, the gels were incubated
for an additional for 72 h at 37°C in incubation buffer (50 mM
Tris, 0.15 M NaCl, 10 mM CaCl2, 0.05% sodium azide, pH 7.8). The
gel was stained with staining solution (1% Commassie Brilliant Blue
G-250, 5% MeOH, 10% acetic acid). The number of pixels per band was
used to determine the enzyme activity in each group. The gelatin
zymography experiment was repeated three times with independent
samples.
Fluorescence polarization (FP)
assay
All fluorescence polarization experiments were
conducted in 96-well, black, round-bottom plates using a microplate
reader. For FP assay experiments, to each well, HFB buffer (20 mM
HEPES pH 7.3, 50 mM KCl, 5 mM MgCl2, 20 mM
Na2MoO4, 0.01% NP-40), 30 nM recombinant
Hsp90α full length protein, 5 nM fluorescein isothiocyanate labeled
geldanamycin (GA-FITC) inhibitor, 0.1 mg/ml bovine globulin (BGG),
2 mM 1,4-dithiothreitol (DTT) and inhibitors (0.001, 0.01, 0.1,
0.5, 1, 5, 10, 50, and 100 µM) were added. All wells had a final
volume of 100 µl in the HFB buffer. The plates were incubated at
4°C for 14 h. The polarization values in millipolarization units
(mP) were assessed at an excitation wavelength at 495 nm and an
emission wavelength at 530 nm. All experimental data were analyzed
using Prism software (Graphpad Software, San Diego, CA, USA).
Molecular modeling
In silico docking of Oxi-DPide (1) or Oxi-DPide (2) with the 3D coordinates of the X-ray
crystal structures of the N-terminal domain of human Hsp90α
(PDB code: 2XJX) was accomplished using the AutoDock program
downloaded from the Molecular Graphics Laboratory of the Scripps
Research Institute (La Jolla, CA, USA). In the docking experiments
carried out, Gasteiger charges were placed on the X-ray structures
of the N-terminal domain of Hsp90 along with Oxi-DPide
(1) or Oxi-DPide (2) using tools from the AutoDock suite. A
grid box centered on the N-terminal Hsp90 domain with
definitions of 60×60×60 points and 0.375 Å spacing was chosen for
ligand docking experiments. The docking parameters consisted of
setting the population size to 150, the number of generations to
27,000, and the number of evaluations to 25,000,000, while the
number of docking runs was set to 100 with a cutoff of 1 Å for the
root-mean-square tolerance for the grouping of each docking run.
The docking model of human Hsp90α with Oxi-DPide (1) or Oxi-DPide (2) are depicted in Fig. 1 and the images were generated using
PyMol1.3 (DeLano Scientific LLC, Palo Alto, CA, USA).
Statistical analysis
Statistical analysis was performed with the
Student's t-test (two-tailed) using Prism GraphPad software.
Differences with *P<0.05, **P<0.01, ***P<0.001 were
considered statistically significant. Data were represented as the
mean ± SD.
Results
Molecular docking and drug design
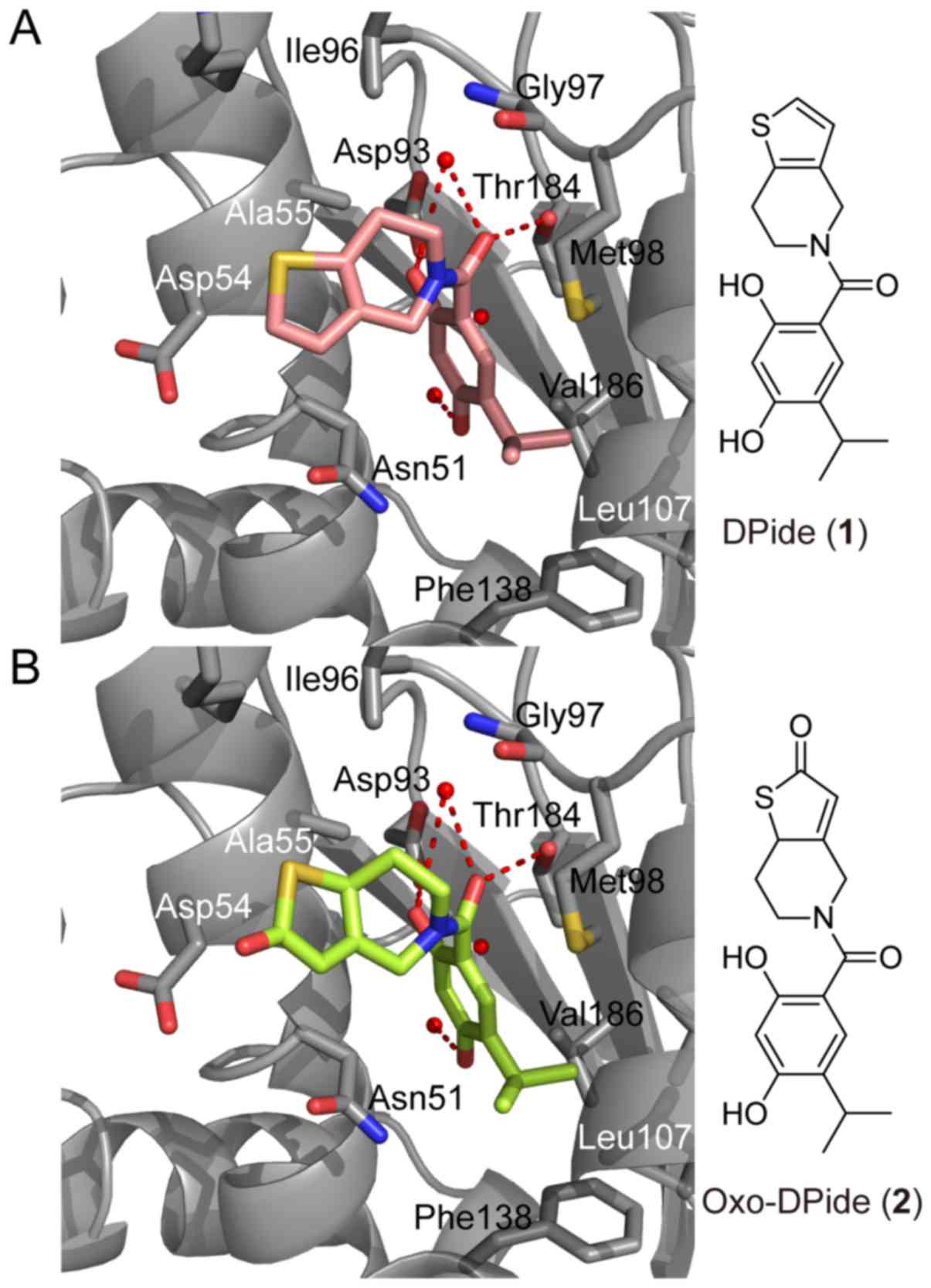
Our previous study indicated that DPide (1) strongly bound to the ATP-binding pocket
in the N-terminal domain of Hsp90α (Fig. 1A) (26). The resorcinol ring and the carbonyl
group of DPide (1) formed the
hydrogen bonding interactions with Asp93, Thr184, and conserved
water molecules, while the isopropyl group of DPide (1) was imbedded into the hydrophobic cavity
formed by Leu107, Phe138, and Val186. The 6,7-dihydrothieno[3,2-c]
pyridine-5(4H)-yl moiety of DPide (1) was nicely positioned in the entry
region composed of Asp54, Ala55, and Ile96. The docking pose of
DPide (1) suggested that installing
a carbonyl group on DPide (1) may
form an additional hydrogen bonding interaction with the carboxylic
acid side chain of Asp54. Therefore, we rationally designed
Oxo-DPide (2) having an extra
carbonyl group adjacent to the sulfur atom and performed the
docking simulation shown in Fig.
1B. To experimentally examine whether Oxo-DPide (2) can form an additional hydrogen bonding
interaction with the carboxylic acid side chain of Asp54, we first
commenced the synthesis of DPide (1) and Oxo-DPide (2).
Chemistry
DPide (1) was
synthesized following a previously reported procedure (Fig. 2) (26). Briefly, the amide coupling reaction
of carboxylic acid 3 with 4,5,6,7-tetrahydrothieno[3,2-c]pyridine
(4) using EDC afforded DPide
(1) in 52% yield. Similarly,
Oxo-DPide (2) was obtained by the
reaction of carboxylic acid 3 with commercially available compound
5 using EDC, HOBt, and DIPEA in DMF under microwave irradiation at
80°C for 3 h in 10% yield.
Comparative Hsp90 binding affinity and
anti-proliferative activity of DPide and Oxo-DPide
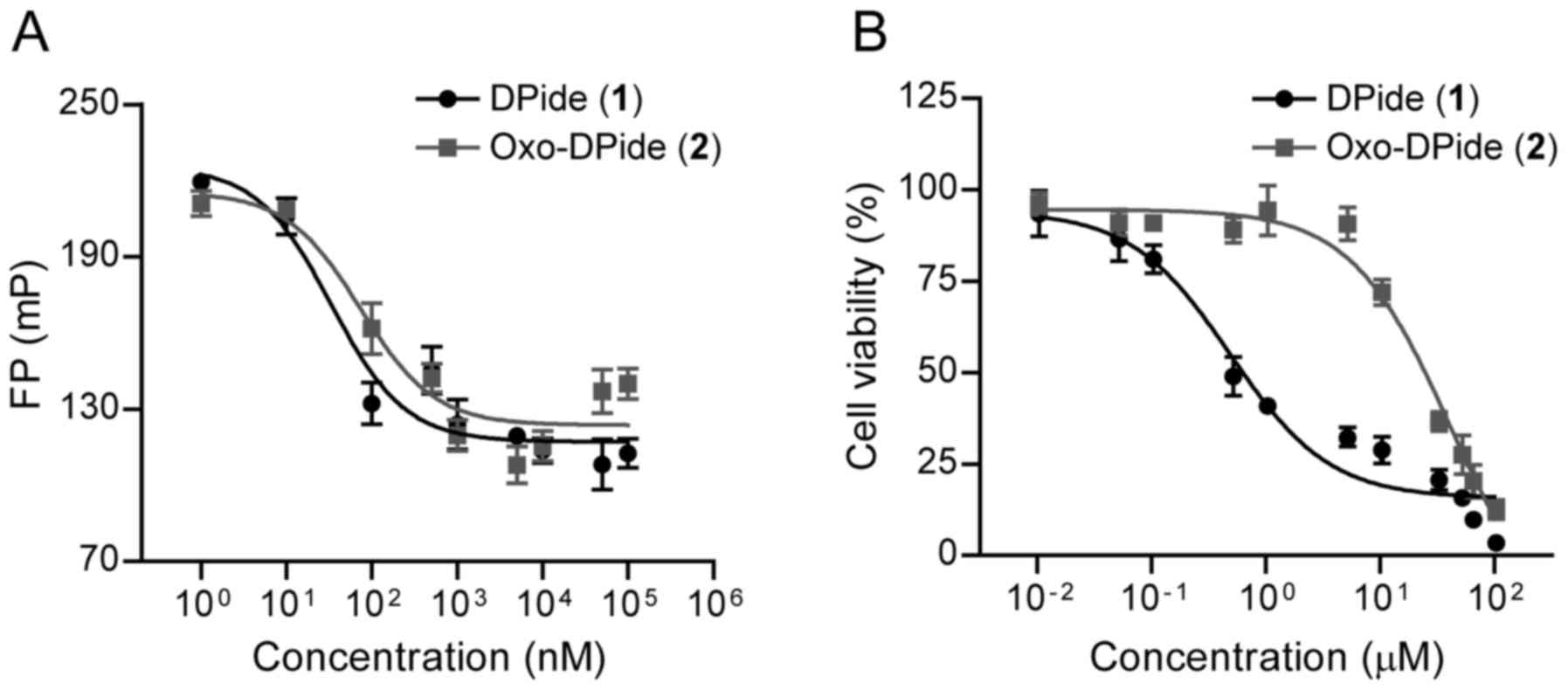
With DPide and Oxo-DPide in hand, we first assessed
the direct binding affinity of DPide and Oxo-DPide to human Hsp90α
with a fluorescence piolarization (FP) assay in which FITC-labeled
geldanamycin was used as a fluorescence probe. As shown in Fig. 3A and Table I, DPide directly bound to the
ATP-binding pocket of Hsp90α with an IC50 value of 33.3
nM, which was more potent than Oxo-DPide (IC50=73.7 nM).
We next explored the anti-proliferative effect of DPide and
Oxo-DPide on H1975 and MDA-MB-231 cancer cell lines. Consistent
with the result from the FP assay, DPide displayed very potent
anti-proliferative activity against H1975 and MDA-MB-231 cells with
GI50 values of 0.478 and 1.67 µM respectively, while
Oxo-DPide exerted less efficient potency against H1975 and
MDA-MB-231 cells with GI50 values of 20.7 and 35.8 µM
(Fig. 3B and Table I). Due to the higher potency of
DPide than Oxo-DPide towards Hsp90α as well as the two cancer cell
lines, we decided to further investigate the biological activity of
DPide but not Oxo-DPide against TNBCs.
 | Table I.Inhibitory activity against Hsp90α
and H1975 cancer cells. |
Table I.
Inhibitory activity against Hsp90α
and H1975 cancer cells.
| Compound | Hsp90α
(FP)a
(IC50; nM) | H1975b (GI50; µM) |
MDA-MB-231b (GI50; µM) |
|---|
| DPide (1) | 33.3 | 0.478 | 1.67 |
| Oxo-DPide (2) | 73.7 | 20.7 | 35.8 |
DPide significantly inhibits the
proliferation of MDA-MB-231 cells
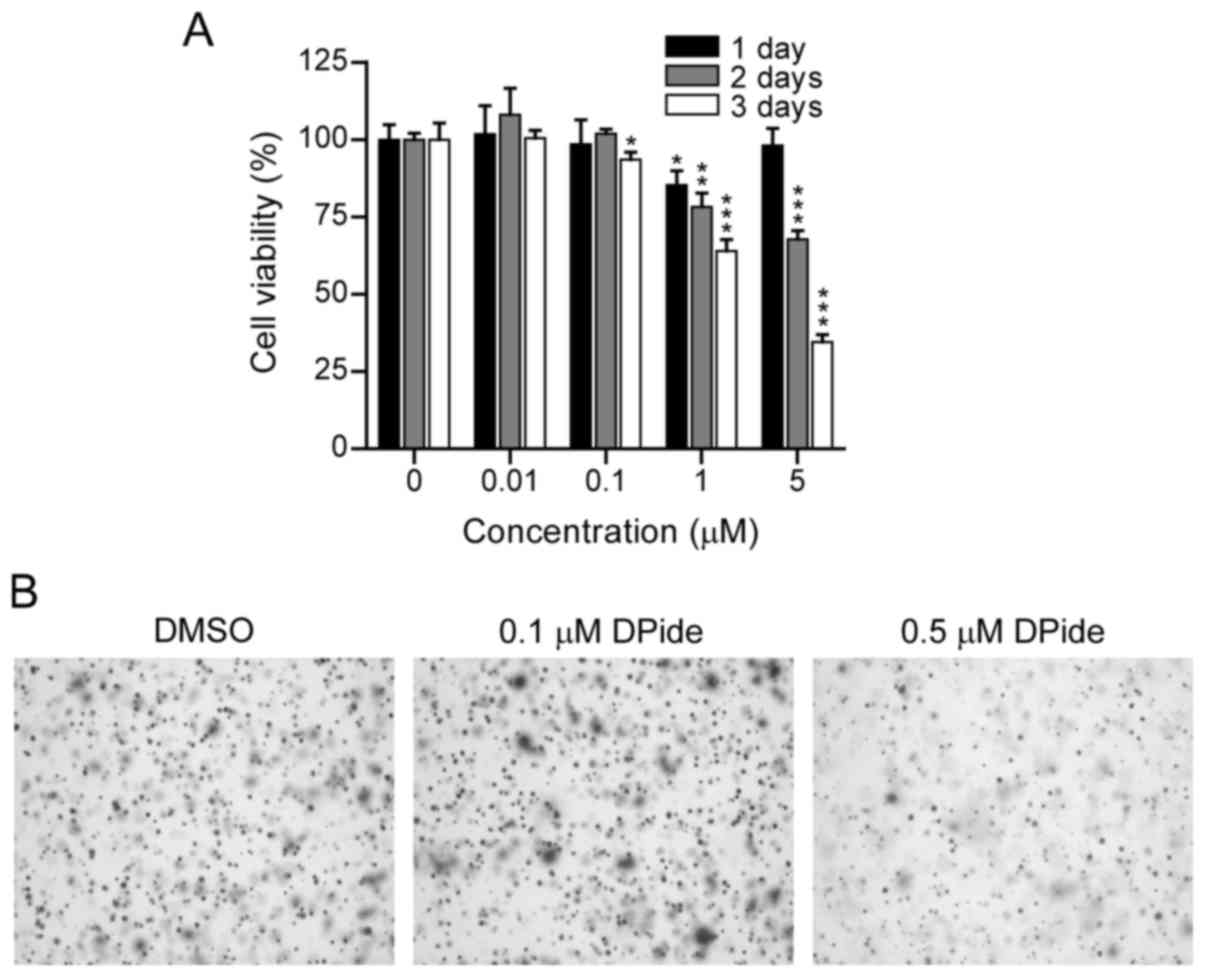
To examine the anticancer activity of DPide against
MDA-MB-231 cells, we first evaluated the dose- and time-dependent
effect of DPide on the growth of MDA-MB-231 cells. MDA-MB-231 cells
were treated with DPide at various concentrations (0.01, 0.1, 1,
and 5 µM) for 1, 2, and 3 days and the cell viability was
determined using an MTS assay (Fig.
4A). The cell viability assay revealed that DPide afforded very
potent cellular efficacy against MDA-MB-231 cells in a dose- and
time-dependent manner. Notably, the exposure of cells with DPide
(0.01, 0.1, 1, and 5 µM) for 1 day displayed only a minimal effect
on cell viability.
Anchorage-independent cell growth is a hallmark of
oncogenic transformation and considered as a key metastatic
potential for malignant cancer cells to proliferate away from their
site of origin. Accordingly, we investigated the effect of DPide on
the anchorage-independent cell growth and the soft-agar colony
formation assay revealed that the treatment of cells with 0.5 µM
DPide inhibited the anchorage-independent growth of MDA-MB-231
cells, indicating that the metastatic potential of MDA-MB-231 cells
could be suppressed by DPide (Fig.
4B).
DPide inhibits the chaperone function
of Hsp90 and downregulated Hsp90 client proteins via the
ubiquitin-proteasomal pathway
To uncover the underlying mechanism of DPide, we
next analyzed the effect of DPide on the expression of Hsp90 client
proteins in MDA-MB-231 cells. Hsp90 is well-known for maintaining
the stability of EGFR, Her2, Met, Akt, c-Raf, and Cdk4. Thus, the
inhibition of Hsp90 results in the degradation of these client
proteins via the ubiquitin-proteasome pathway. As shown Fig. 5A, the exposure of cells to DPide
induced a significant reduction of EGFR, Her2, Met, Akt, c-Raf, and
Cdk4 in a dose-dependent manner, consequently leading to the
cleavage of PARP. In contrast, DPide increased the cellular level
of Hsp70 and Hsp90 proteins, upregulation of which is a cellular
hallmark of Hsp90 inhibition. The expression of non-Hsp90-dependent
protein, β-actin, remained unchanged as expected. To further
clarify the mechanism of DPide in detail, we next assessed the mRNA
levels of Met, Akt, Cdk4, and Hsp90 by semi-quantitative RT-PCR
(Fig. 5B). As expected, DPide did
not significantly alter the mRNA levels of Met, Akt, and Cdk4,
indicating that the downregulation of those protein levels occurred
via proteasomal degradation but not transcriptional regulation. In
contrast, DPide treatment increased the mRNA level of Hsp90 in a
dose-dependent manner, in that inhibition of Hsp90 chaperone
function by DPide induced proteotoxic stress, activated HSF1, and
transcriptionally expressed chaperone proteins such as Hsp90,
Hsp70, Hsp47, and Hsp27 by feedback mechanism (29). Collectively, these results
demonstrated that DPide inhibited the chaperone function of Hsp90
and downregulated Hsp90 client proteins via the
ubiquitin-proteasomal pathway.
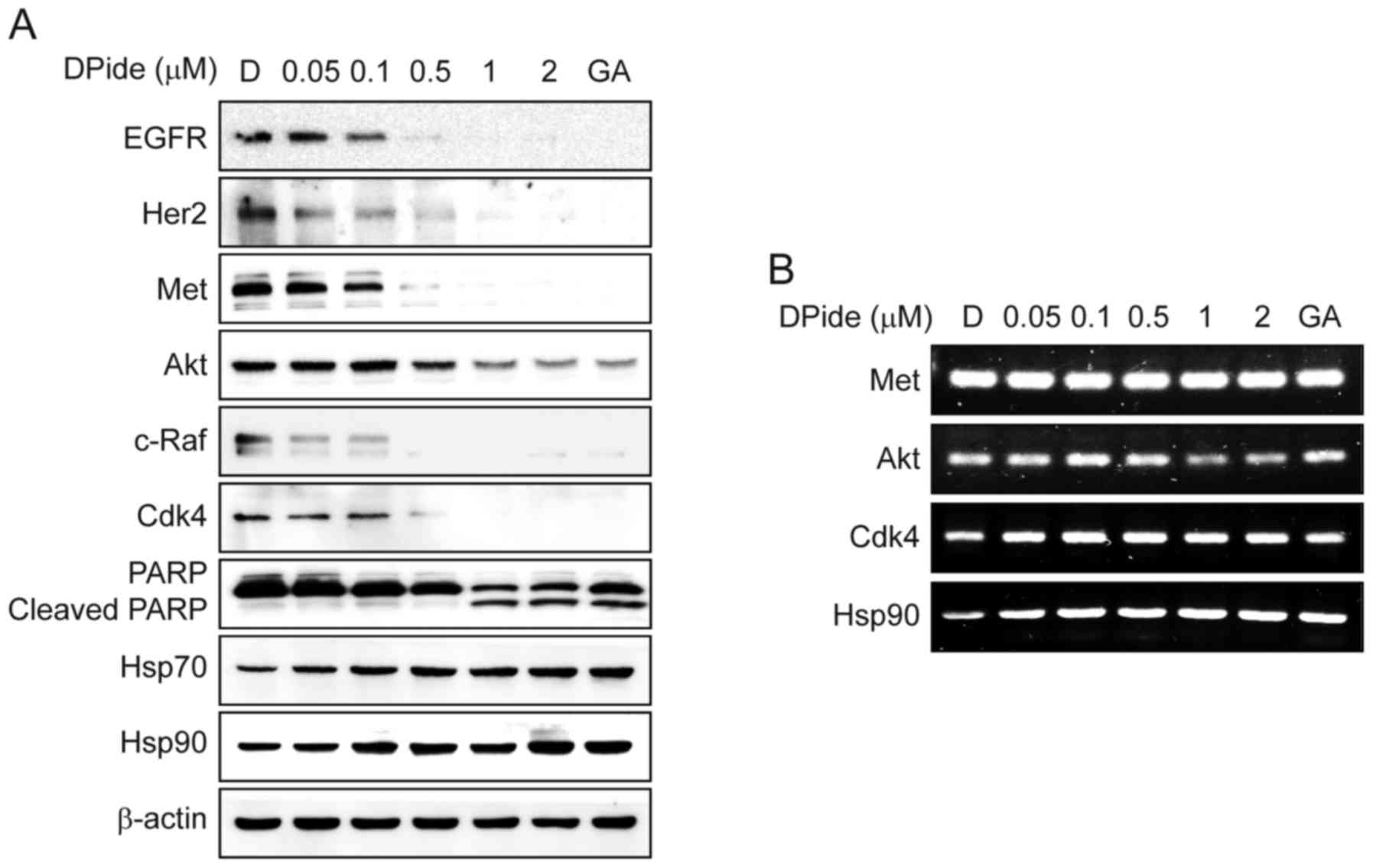 | Figure 5.DPide inhibits the chaperone function
of Hsp90 and induces the degradation of client proteins. (A)
MDA-MB-231 cells were treated with the indicated concentrations of
DPide for 24 h and the expression of EGFR, Her2, Met, Akt, c-Raf,
Cdk4, PARP, Cleaved PARP, Hsp70, Hsp90, and β-actin was analyzed
using western blotting. Geldanamycin (GA, 1 µM) and DMSO (D, 0.5%)
were employed as a positive and a negative control, respectively.
(B) Effect of DPide on the transcriptional levels of Met, Akt,
Cdk4, and Hsp90 were assessed by semi-quantitative RT-PCR.
MDA-MB-231 cells were treated with indicated concentrations of
DPide for 24 h. Total RNA was isolated from the cells,
reverse-transcribed to cDNA, and then assessed by semi-quantitative
RT-PCR. Geldanamycin (GA, 1 µM) and DMSO (D, 0.5%) were employed as
a positive and a negative control, respectively. |
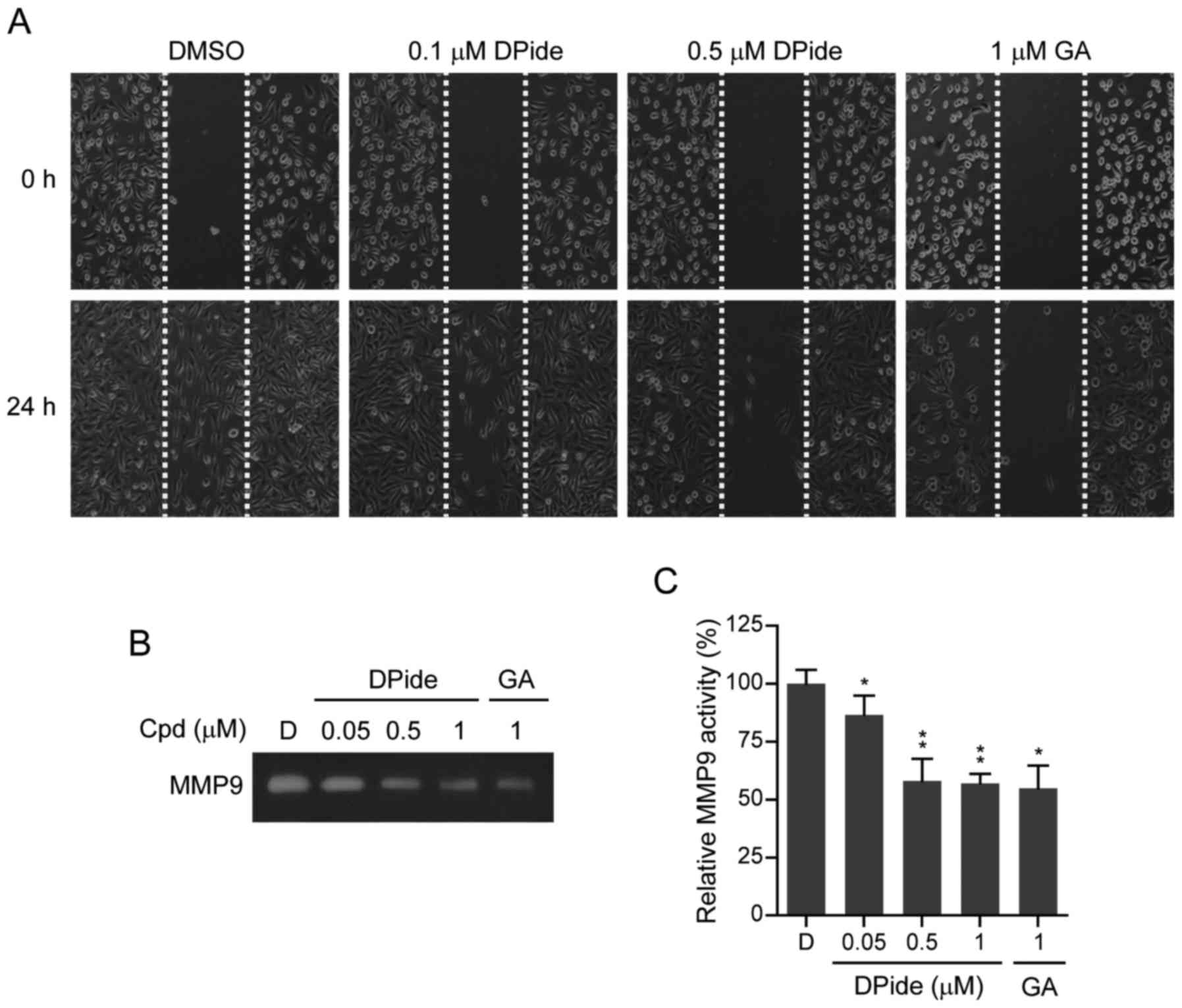
DPide inhibits the migration and MMP9
activity of MDA-MB-231 cells
MDA-MB-231 cells have a mesenchymal phenotype and
are highly aggressive metastatic breast cancer cells. An increase
of mobility has been associated with the metastatic potential of
cancer cells. To explore the anti-metastatic activity of DPide, we
next examined the effect of DPide on the mobility of MDA-MB-231
cells. Wounds were first formed by scratching the cell monolayer
with a pipette tip and wound closure of MDA-MB-231 cells in the
presence or absence of DPide was assessed by counting the number of
cells that had infiltrated the wounded area after 24 h. As shown in
Fig. 6A, treatment of cells with
0.1 and 0.5 µM of DPide for 24 h significantly inhibited the
migration of MDA-MB-231 cells compared to the DMSO control. Thus,
the in vitro wound-healing migration assay revealed that
DPide inhibited the migration of MDA-MB-231 cells.
Matrix metalloproteinases (MMPs) are a group of
zinc-dependent proteases that are involved in the degradation and
remodelling of the extracellular matrix (ECM). By degrading the
matrix, MMPs provide physical space within the matrix for tumor
growth, invasion, and metastasis. It has been reported that MMP9 is
secreted by various types of malignant cancer cells and contributes
to cancer metastasis by breaking down various EMC molecules, which
allows metastatic cancer cells to be more invasive (30). Therefore, we next investigated the
effect of DPide on MMP9 activity by performing gelatin zymography.
As shown in Fig. 6B and C, DPide
significantly suppressed MMP9 activity in MDA-MB-231 cells in a
dose-dependent manner. Treatment of cells with 0.05, 0.5, and 1 µM
of DPide decreased MMP9 activity by 14, 42, and 43%, compared to
DMSO control, respectively. Collectively the results clearly
indicated that DPide inhibited the migration of MDA-MB-231 cells
and suppressed MMP9 activity of MDA-MB-231 cells.
Discussion
In the present study, the design, synthesis, and
biological evaluation of Hsp90 inhibitors DPide and Oxo-DPide were
described. Our results demonstrated that DPide more strongly bound
to the N-terminal domain of Hsp90α (IC50=33.3 nM)
than Oxo-DPide (IC50=73.7 nM) in the FP assay. Moreover,
the FP enzyme affinity of DPide agreeably translated into
satisfactory cellular activity with GI50 values of 0.478
and 1.67 µM against H1975 and MDA-MB-231 cells, respectively. In
contrast, Oxo-DPide inhibited the proliferation of H1975 and
MDA-MB-231 cells with GI50 values of 20.7 and 35.8 µM,
respectively. The treatment of MDA-MB-231 cells with DPide led to
the degradation of EGFR, Her2, Met, Akt, c-Raf, and Cdk4 and the
consequent cleavage of PARP in a dose-dependent manner. In
contrast, the treatment of MDA-MB-231 cells with DPide upregulated
the protein level of Hsp70 and Hsp90. RT-PCR experiments revealed
that the exposure of MDA-MB-231 cells with DPide did not alter the
mRNA levels of Met, Akt, and Cdk4, suggesting that the
downregulation of Hsp90 client proteins was associated with
proteasomal degradation but not transcriptional downregulation.
DPide efficiently suppressed the migration and MMP9 activity of
MDA-MB-231 cells in a dose-dependent manner. Overall, these
findings strongly support that a synthetic small molecule, DPide
offers an effective treatment strategy against TNBCs.
Acknowledgements
The present study was supported by the Bisa Research
Grant of Keimyung University in 2015.
Competing interests
The authors declare thay they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Anders C and Carey LA: Understanding and
treating triple-negative breast cancer. Oncology (Williston Park).
22:1233–1243. 2008.PubMed/NCBI
|
|
3
|
Yadav BS, Sharma SC, Chanana P and Jhamb
S: Systemic treatment strategies for triple-negative breast cancer.
World J Clin Oncol. 5:125–133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gajria D and Chandarlapaty S:
HER2-amplified breast cancer: Mechanisms of trastuzumab resistance
and novel targeted therapies. Expert Rev Anticancer Ther.
11:263–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garcia-Becerra R, Santos N, Diaz L and
Camacho J: Mechanisms of resistance to endocrine therapy in breast
cancer: Focus on signaling pathways, miRNAs and genetically based
resistance. Int J Mol Sci. 14:108–145. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gradishar WJ, Anderson BO, Balassanian R,
Blair SL, Burstein HJ, Cyr A, Elias AD, Farrar WB, Forero A,
Giordano SH, et al: NCCN guidelines insights breast cancer, version
1.2016. J Natl Compr Canc Netw. 13:1475–1485. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schiff R, Massarweh S, Shou J and Osborne
CK: Breast cancer endocrine resistance: How growth factor signaling
and estrogen receptor coregulators modulate response. Clin Cancer
Res. 9:447S–454S. 2003.PubMed/NCBI
|
|
8
|
Sankhala KK, Mita MM, Mita AC and Takimoto
CH: Heat shock proteins: A potential anticancer target. Curr Drug
Targets. 12:2001–2008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Whitesell L and Lindquist SL: HSP90 and
the chaperoning of cancer. Nat Rev Cancer. 5:761–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
da Silva VC and Ramos CH: The network
interaction of the human cytosolic 90 kDa heat shock protein Hsp90:
A target for cancer therapeutics. J Proteomics. 75:2790–2802. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang H and Burrows F: Targeting multiple
signal transduction pathways through inhibition of Hsp90. J Mol Med
(Berl). 82:488–499. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chiosis G, Vilenchik M, Kim J and Solit D:
Hsp90: The vulnerable chaperone. Drug Discov Today. 9:881–888.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mosser DD and Morimoto RI: Molecular
chaperones and the stress of oncogenesis. Oncogene. 23:2907–2918.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Whitesell L, Mimnaugh EG, De Costa B,
Myers CE and Neckers LM: Inhibition of heat shock protein
HSP90-pp60v-src heteroprotein complex formation by benzoquinone
ansamycins: Essential role for stress proteins in oncogenic
transformation. Proc Natl Acad Sci USA. 91:pp. 8324–8328. 1994;
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Trepel J, Mollapour M, Giaccone G and
Neckers L: Targeting the dynamic HSP90 complex in cancer. Nat Rev
Cancer. 10:537–549. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Supko JG, Hickman RL, Grever MR and
Malspeis L: Preclinical pharmacologic evaluation of geldanamycin as
an antitumor agent. Cancer Chemother Pharmacol. 36:305–315. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Caldas-Lopes E, Cerchietti L, Ahn JH,
Clement CC, Robles AI, Rodina A, Moulick K, Taldone T, Gozman A,
Guo Y, et al: Hsp90 inhibitor PU-H71, a multimodal inhibitor of
malignancy, induces complete responses in triple-negative breast
cancer models. Proc Natl Acad Sci USA. 106:pp. 8368–8373. 2009;
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lundgren K, Zhang H, Brekken J, Huser N,
Powell RE, Timple N, Busch DJ, Neely L, Sensintaffar JL, Yang YC,
et al: BIIB021, an orally available, fully synthetic small-molecule
inhibitor of the heat shock protein Hsp90. Mol Cancer Ther.
8:921–929. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bao R, Lai CJ, Wang DG, Qu H, Yin L,
Zifcak B, Tao X, Wang J, Atoyan R, Samson M, et al: Targeting heat
shock protein 90 with CUDC-305 overcomes erlotinib resistance in
non-small cell lung cancer. Mol Cancer Ther. 8:3296–3306. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Woodhead AJ, Angove H, Carr MG, Chessari
G, Congreve M, Coyle JE, Cosme J, Graham B, Day PJ, Downham R, et
al: Discovery of
(2,4-dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydrois
oindol-2-yl]methanone (AT13387), a novel inhibitor of the molecular
chaperone Hsp90 by fragment based drug design. J Med Chem.
53:5956–5969. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Trepel JB, Neckers LM and Giaccone
G: STA-9090, a small-molecule Hsp90 inhibitor for the potential
treatment of cancer. Curr Opin Investig Drugs. 11:1466–1476.
2010.PubMed/NCBI
|
|
22
|
Eccles SA, Massey A, Raynaud FI, Sharp SY,
Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall
F, et al: NVP-AUY922: A novel heat shock protein 90 inhibitor
active against xenograft tumor growth, angiogenesis, and
metastasis. Cancer Res. 68:2850–2860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ohkubo S, Kodama Y, Muraoka H,
Hitotsumachi H, Yoshimura C, Kitade M, Hashimoto A, Ito K, Gomori
A, Takahashi K, et al: TAS-116, a highly selective inhibitor of
heat shock protein 90α and β, demonstrates potent antitumor
activity and minimal ocular toxicity in preclinical models. Mol
Cancer Ther. 14:14–22. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bussenius J, Blazey CM, Aay N, Anand NK,
Arcalas A, Baik T, Bowles OJ, Buhr CA, Costanzo S, Curtis JK, et
al: Discovery of XL888: A novel tropane-derived small molecule
inhibitor of HSP90. Bioorg Med Chem Lett. 22:5396–5404. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Infante JR, Weiss GJ, Jones S, Tibes R,
Bauer TM, Bendell JC, Hinson JM Jr, Von Hoff DD, Burris HA III,
Orlemans EO and Ramanathan RK: Phase I dose-escalation studies of
SNX-5422, an orally bioavailable heat shock protein 90 inhibitor,
in patients with refractory solid tumours. Eur J Cancer.
50:2897–2904. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jeong JH, Oh YJ, Lho Y, Park SY, Liu KH,
Ha E and Seo YH: Targeting the entry region of Hsp90's ATP binding
pocket with a novel 6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl amide.
Eur J Med Chem. 124:1069–1080. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jeong CH, Park HB, Jang WJ, Jung SH and
Seo YH: Discovery of hybrid Hsp90 inhibitors and their
anti-neoplastic effects against gefitinib-resistant non-small cell
lung cancer (NSCLC). Bioorg Med Chem Lett. 24:224–227. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jeong JH, Oh YJ, Kwon TK and Seo YH:
Chalcone-templated Hsp90 inhibitors and their effects on gefitinib
resistance in non-small cell lung cancer (NSCLC). Arch Pharm Res.
40:96–105. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dai C and Sampson SB: HSF1: Guardian of
proteostasis in cancer. Trends Cell Biol. 26:17–28. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dechow TN, Pedranzini L, Leitch A, Leslie
K, Gerald WL, Linkov I and Bromberg JF: Requirement of matrix
metalloproteinase-9 for the transformation of human mammary
epithelial cells by Stat3-C. Proc Natl Acad Sci USA. 101:pp.
10602–10607. 2004; View Article : Google Scholar : PubMed/NCBI
|