Introduction
Dormant solitary cells, or minimal dormant
metastases, have been recognized as the main cause of cancer
recurrence (1–3). Tumor dormancy is maintained in a
microenvironment unfavorable for tumor cell proliferation (3,4). When
the microenvironmental conditions shift to support tumor expansion,
dormant tumors may resume active growth and progression (3). Therefore, understanding the mechanisms
that regulate dormancy or the switch to a proliferative state is
crucial for identifying novel targets and designing interventions
to prevent disease recurrence.
Compared with highly invasive human cancer cells,
non-invasive human cancer cells constitutively have a less
prominent malignant phenotype, as the metastatic tumor cells
derived from non-invasive tumor cells may be more sensitive to the
restriction of a new microenvironment (5). Our previous study demonstrated that
tumor cells seeded at metastatic sites have a lower proliferative
potential, and remain quiescent over long periods of time (5). Following a period of dormancy in
metastatic sites, dormant tumor cells (DTCs) may ultimately transit
through an angiogenic switch and become clinically apparent
metastases (5,7), whose size or proliferative potential
is limited by the lack of a favorable tumor environment (6), suggesting that the microenvironment
serves a key role in tumor cell dormancy. However, whether tumor
dormancy is a consequence of deficiency or alteration of the proper
growth signals in the secondary target site require further
investigation.
Metastasis and recurrence is the leading cause of
death in patients suffering from cancer, and the lung is the one of
most common sites of metastasis (8). Alveolar epithelial cells II (AEC IIs)
serve a multifunctional role in the lung, including secretory,
synthetic and remodeling reservoirs for the lung epithelium to host
defense (9). The AEC IIs are the
progenitors of AEC Is, and the progenitor function of AEC IIs may
be activated when the lung epithelium encounters a variety of
stimulators, including acute lung injury or bacteria, among others,
to defend the alveolus from injury (10,11).
However, whether the tumor cells metastasizing to the lung are able
to stimulate AEC II transdifferentiation has not been examined in
detail.
Transforming growth factor β1 (TGF-β1) is a key
factor in the alteration of the tumor environment (14), that is secreted during the
transdifferentiation of AEC IIs to AEC Is (12,13);
these alterations in the tumor environment may be implicated in the
regulation of a variety of biological responses, including cell
proliferation and differentiation (14). The lung is one of most common sites
of metastasis (8). However, the
role of AEC II transdifferentiation during reactivation of DTCs has
not been fully elucidated.
In the present study, we investigated whether tumor
cells can stimulate the transdifferentiation of AEC IIs into AEC
Is, as well as whether AEC II transdifferentiation can induce
reactivation of DTCs in the lung. Our results revealed that tumor
cells may promote the transdifferentiation of AEC IIs into AEC Is.
Furthermore, the transdifferentiation of AEC IIs may, in turn,
promote the switch of metastasized DTCs to reactivate growth via
TGF-β1, secreted by AEC II transdifferentiation, leading to
increase the SNAI2 expression in DTCs. Targeting this process may
provide novel therapeutic strategies for the inhibition of the
dormant-to-proliferating metastatic switch.
Materials and methods
Ethics statement
All animal experiments were conducted according to
the relevant national and international guidelines, and were
approved by the Ethics Committee on Animal Experiments of the China
Three Gorges University and monitored by the Department of
Experimental Animals of the Yichang Central People's Hospital.
Cells and reagents
The human breast cancer cell lines MCF-7 and T-47D
were purchased from the Type Culture Collection of the Chinese
Academy of Sciences (Shanghai, China), and maintained in Dulbecco's
modified Eagle's medium (DMEM) with high glucose, 10% fetal bovine
serum (FBS) and antibiotics (Life Technologies, Carlsbad, CA, USA),
according to the guidelines. Lipopolysaccharide (LPS) was purchased
from Sigma-Aldrich (St. Louis, MO, USA). Growth factor-reduced 3D
Cultrex basement membrane extract (BME) was purchased from Trevigen
Inc. (Gaithersburg, MD, USA). Monensin was purchased from
Selleckchem.cn. Antibodies against surfactant protein C (SP-C) and
aquaporin 5 (AQP5), and all secondary antibodies, were purchased
from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and Cell
Signaling Technology Inc. (Beverly, MA, USA). The TGF-β receptor
kinase inhibitor LY2109761 was purchased from Selleckchem.cn.
Gene expression assessment by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells with
TRIzol® reagent (Invitrogen, Carlsbad, CA, USA). Total
RNA (100 ng) was used for reverse transcription using SuperScript
II RNase H reverse transcriptase (Invitrogen) in a volume of 25 µl.
Then, 1 µl of cDNA was amplified with SYBR-Green Universal PCR
Master Mix (Bio-Rad, Richmond, CA, USA) in duplicate.
Quantification of the expression of genes was performed using the
comparative CT method (Sequence Detector User Bulletin
2; Applied Biosystems, Carlsbad, CA, USA). The expression level of
each mRNA was normalized to GAPDH mRNA and expressed as n-fold
difference relative to the control (calibrator). The PCR was
conducted using the following parameters: 95°C for 5 min, and 40
cycles at 95°C for 10 sec, 60° for 20 sec and 72°C for 15 sec. The
relative quantity of mRNA was determined by RT-qPCR, as previously
described (15). The mRNA of GAPDH
was used as the internal control. The primer sequences were as
follows: SNAI2 sense, 5′-AGGAATCTGGCTGCTGTG-3′ and antisense,
5′-GGAGAAAATGCCTTTGGAC-3′; SP-C sense, 5′-GTCGTCGTGGTGATTGTAGGG-3′
and antisense, 5′-GAAGGTAGCGATGGTGTC TG-3′; AQP5 sense,
5′-CGTCAATGCGCTGAACAACAA-3′ and antisense, 5′-ACAGACAAGCCAATGGATAAG
G-3′; GAPDH sense, 5′-TCATTGACCTCAACTACATGGTTT-3′ and antisense,
5′-GAAGATGGTGATGGGATTTC-3′.
Western blot assay
Western blotting was performed as previously
described (5). Cell lysates (30 µg
of total protein) and prestained molecular weight markers were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) followed by the transfer onto
nitrocellulose membranes. The membranes were blocked in
Tris-buffered saline with 0.5% Triton X-100 (TBST) containing 5%
non-fat milk and probed with antibodies (SPC, sc-13979, 1:1,000;
AQP5, sc-514022, 1:1,000; SNAI2, #9585, 1:1,000; β-actin, sc-58673,
1:5,000) at 4°C with gentle shaking, overnight. After incubation
with the secondary antibody (sc-2357, 1:10,000; sc-516102,
1:10,000) conjugated with horseradish peroxidise at room
temperature with gentle shaking for 2 h, membranes were extensively
washed, and the immunoreactivity was visualized by enhanced
chemiluminescence according to the manufacturer's protocol (ECL
kit; Santa Cruz Biotechnology). Antibodies were purchased from
Santa Cruz Biotechnology and Cell Signaling Technology. The protein
expression data were analyzed using ImageJ software (NIH; National
Institutes of Health, Bethesda, MD, USA).
Isolation and culture of AEC IIs
AEC IIs were isolated from pathogen-free Kunming
(KM) mice (20-25 g) by an improved method (16). The animal experimental protocols
were approved by the Ethics Committee of China Three Gorges
University. Mice were acclimated to the laboratory conditions, with
free access to food and water in a facility with controlled
temperature (22°C) on a 12/12-h light/dark cycle, for 1 week prior
to the experiments. In brief, adult mouse lungs were lavaged with
PBS, and then digested with elastase (1 mg/ml). The cell mixture
was filtered through a 200- and a 100-µm nylon mesh (twice through
each), plated on mouse IgG-coated plates and incubated at 37°C for
3-4 h to remove macrophages and other pulmonary cells containing an
Fc-fragment. The purity of AEC IIs was >96±1.8%, as determined
by modified Papanicolaou staining, and the viability was
>98±2.1%. The detailed protocols were applied as previously
described (16).
Transdifferentiation of AEC IIs
AEC IIs were seeded onto 6-well plastic culture
dishes at a density of 1.5×106 cells/dish, and cultured
in media with 10% FBS with or without breast tumor cells
(5×103 cells/dish), with the cells contacting each other
(co-cultured contact) or divided with a Transwell assay
(co-cultured no contact). The media were changed after the first 24
h, and on alternate days thereafter. The level of TGF-β1 and other
cell factors secreted into the media were determined using ELISA
kits from R&D Systems (Minneapolis, MN, USA). AEC Is are the
components of the alveolar epithelium most susceptible to injury.
Following lung injury, AEC Is are destroyed and AEC IIs proliferate
and differentiate into AEC Is to repair the epithelium (17). Therefore, this in vitro
transdifferentiation model may be closely associated with the
process occurring in vivo during lung injury and repair. It
should also be noted that isolating AEC IIs from the lung and
culturing them may mimic lung disturbance and repair.
Preparation of super-TDA
The transdifferentiation of AEC IIs to AEC I-like
cells was evident from day 3 onwards and complete by day 5, as
shown by the present study, as well as previous data (16,29).
AEC IIs were seeded onto 6-well plastic culture plates at a density
of 1.5×106 cells/dish and cultured in media with 10% FBS
for 4 days, after which time the medium was replaced with fresh
medium (1 ml) without FBS and the cells were incubated for a
further 24 h, followed by harvesting of the supernatants from the
cultures. The supernatants were then passed through a 0.22-µm
filter and stored at −80°C until further use.
An in vitro 3D system inducing tumor
dormancy
The cell culture system was pre-coated with an
adequate amount of Cultrex growth factor-reduced BME (protein
concentration, 14-15 mg/ml; thickness, 1-2 mm) in a humidified
incubator with 5% CO2 at 37°C for 30 min; the culture
plates were then rinsed 3 times with 10 ml PBS (pH 7.4) for further
use. The tumor cells were suspended in a mixture of 2% FBS and 2%
Cultrex® DMEM mixture (CFD). The cells were cultured on
the pre-coated slides and re-fed every 2 days (29), and the proliferative ability of the
cells was then determined via an MTT assay and flow cytometry.
MTT proliferation assay
Plates with 96-well were coated with 50-100 µl BME.
Non-invasive MCF-7 or T-47D cells (1.5×103 to
2.0×103/well) were re-suspended in 200 µl CFD and grown
on the BME pre-coated plates. The Cell Titer 96 Aqueous One
Solution cell proliferation assay kit (Promega Corporation,
Madison, WI, USA) was used to measure cell proliferation according
to the manufacturer's instructions.
Cell cycle analysis by flow
cytometry
The tumor cells (2.5×105/well) were
cultured under 3D conditions in the presence of Super-TDA (50%) for
5 days; the tumor cells were then analyzed using propidium iodide
(PI; Molecular Probes, Invitrogen) to distinguish non-viable cells.
DNA synthesis or the total DNA content was measured by flow
cytometry using a FACSCalibur flow cytometer (BD Accuri™ C6).
Cell transfection
For the downregulation of SNAI2, MCF-7 cells were
transduced with SNAI2 shRNA lentiviral particles, or control shRNA
lentiviral particles (Santa Cruz Biotechnology) according to the
manufacturer's protocol. Following selection with puromycin, the
cells were used for further experiments.
Statistical analysis
Data were pooled from 3 independent experiments with
a total of 6 samples in each group. Results are expressed as the
mean ± SD and interpreted by one-way ANOVA. Differences were
considered to be statistically significant when P<0.05.
Results
Tumor cells promote
transdifferentiation of AEC IIs into AEC Is in vitro
The isolated AEC IIs were seeded on plastic dishes
and cultured for several days. The transdifferentiation of AEC IIs
to AECI-like cells was evident from day 3 onwards and was completed
by day 5, as previously shown (16). When the isolated AEC IIs
(1.5×106/well) were co-cultured in the presence of tumor
cells (MCF-7 or T-47D, 5×103/well), morphometric
observation of isolated AEC IIs discerned characteristics almost
typical of AEC Is at 3 days. To further characterize the cell
phenotype, qPCR and western blotting with specific labels generated
to the AEC IIs phenotypic marker SP-C (18) and to the AEC Is phenotypic marker
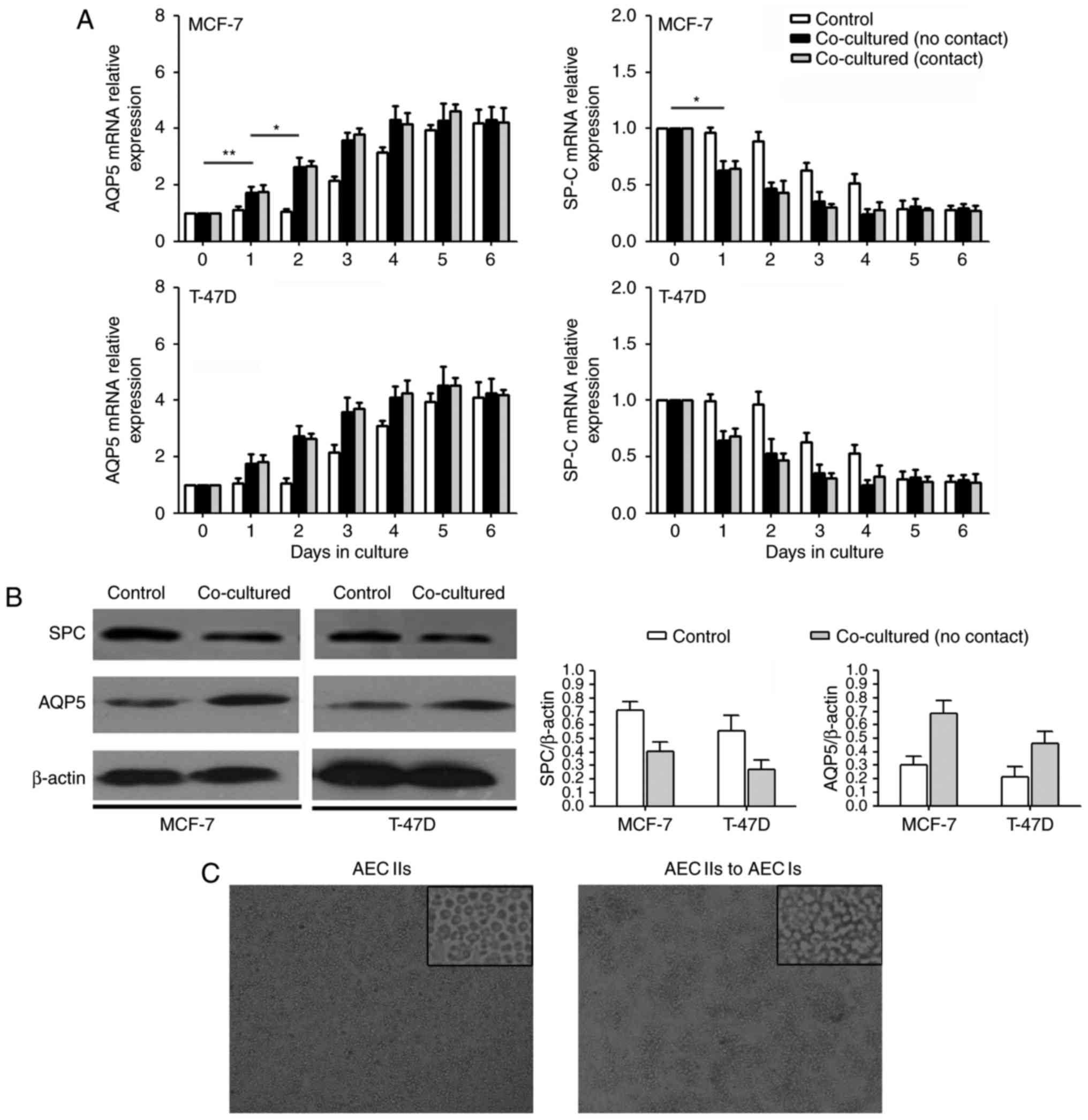
AQP5 (18) were used. Our results
revealed that the AEC IIs exhibited a significant decrease in SP-C
mRNA expression (Fig. 1A) and a
significant increase in AQP5 mRNA expression (Fig. 1B) when co-cultured with tumor cells
(MCF-7 and T-47D) for 3 day. Furthermore, western blot analysis
revealed that SP-C protein expression decreased and AQP5 protein
expression increased in AEC IIs over time (Fig. 1B). In addition, the changes during
AEC II transdifferentiation did not differ between the co-culture
contact and co-culture no contact group. The transition images of
AEC IIs to AEC Is is shown in Fig.
1C. Therefore, our results demonstrated that the tumor cells
promoted AEC II transdifferentiation into AEC Is in
vitro.
Establishment of solitary DTC model in
vitro
Cell dormancy may be defined as a non-proliferative
state or an arrested stage in the cell cycle that results in a
prolonged G0 phase. Due to their small size and non-invasive
nature, these DTCs remain asymptomatic and, in most cases,
undetected. Unfortunately, these DTCs are resistant to conventional
therapies targeting actively dividing cells, which likely accounts
for disease recurrence following apparent successful treatment of
the primary tumors (19,20). However, the reasons for the
activation of DTCs are not clear, mainly due to lack of a DTC
model.
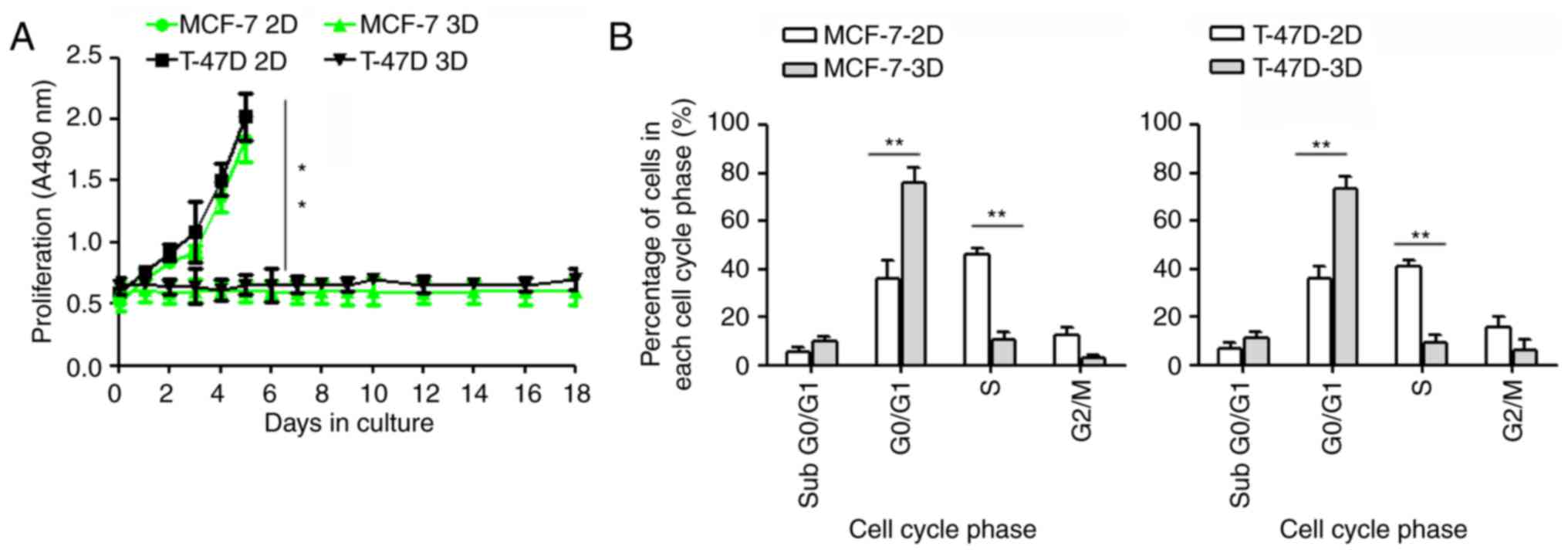
The 3D BME culture system was used as a model for
DTCs, as described above. MCF-7 cells exhibit dormant behavior when
cultured in the 3D BME system, consistent with their behavior at
distant sites in vivo (6,21). In
the present study, we successfully established a solitary DTC model
for MCF-7 and T-47D cells. We found that both MCF-7 and T-47D cells
did not proliferate, and remained quiescent throughout the entire
experimental 18-day culture period when cultured in 3D BME
(P≤0.01). In addition, both breast cancer cell lines proliferated
readily when cultured on a 2D plastic substrate (Fig. 2A).
We further investigated the cell cycle of dormant
MCF-7 and T-47D cells using flow cytometry. Our results revealed
that a high percentage of MCF-7 and T-47D cells remained in the
G0/G1 phase (P≤0.01), with a smaller S and G2/M cell population
(P≤0.01), when cultured under 3D conditions for 5 days, suggesting
that the cells remained at an arrested stage of a prolonged G0
phase (Fig. 2B).
In brief, MCF-7 and T-47D cells were quiescent when
cultured in the 3D BME system (3D).
The supernatant of
transdifferentiation of AEC IIs to AEC Is (Super-TDA) may promote
DTC growth by altering the cell cycle
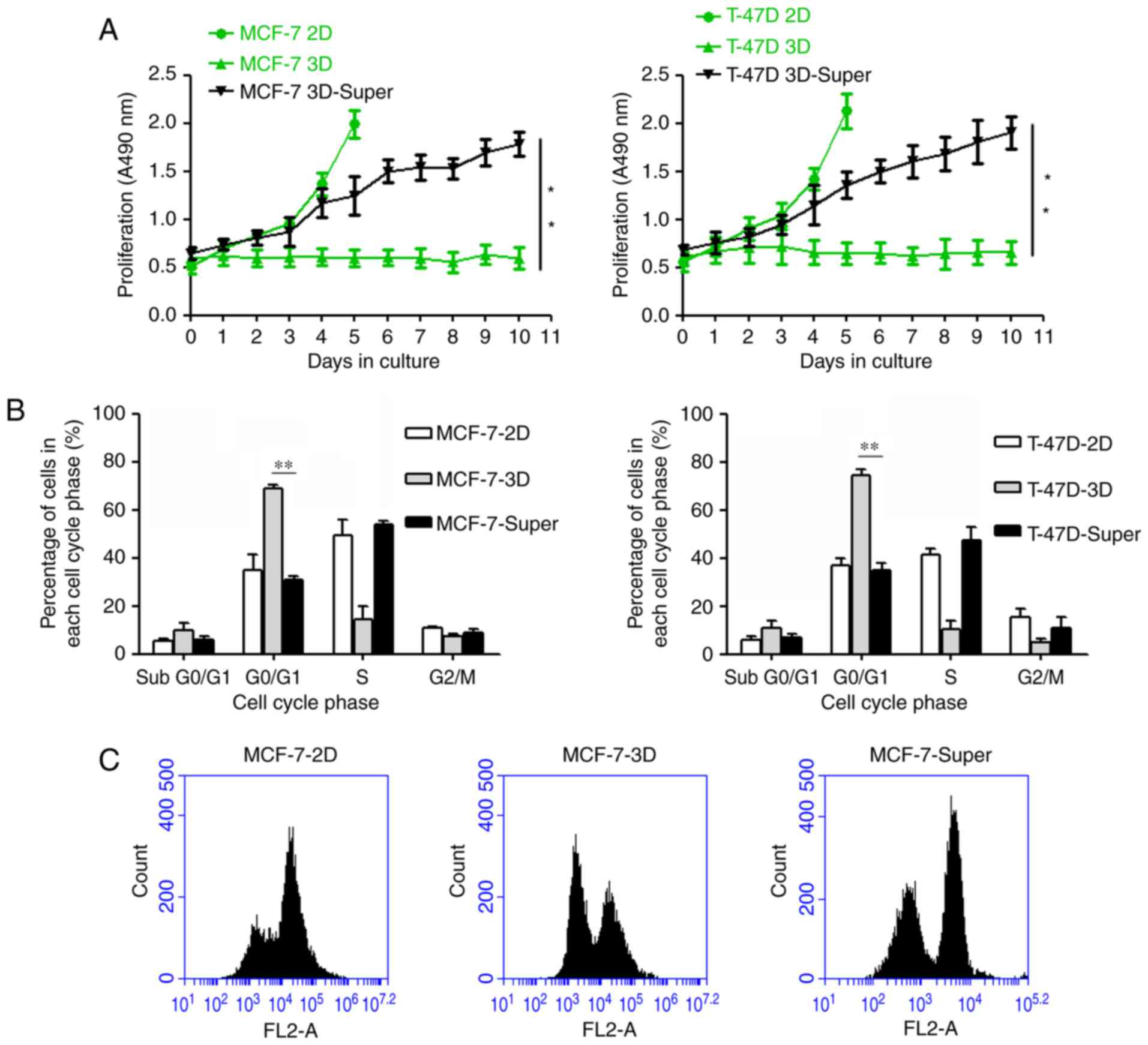
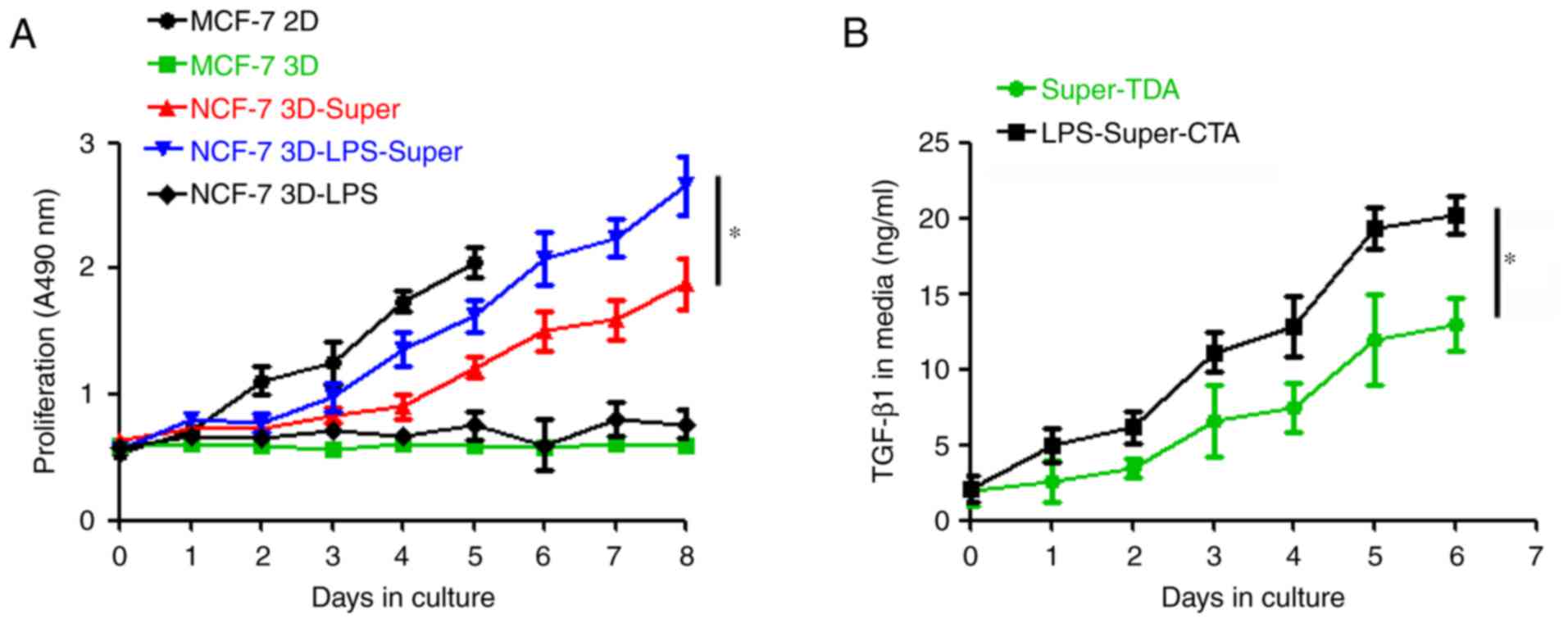
To determine whether Super-TDA induces a
dormant-to-proliferating switch of the DTCs, DTCs were cultured
with the Super-TDA (50%), and we observed that the DTCs were
reactivated and proliferated stably (Fig. 3A). We further investigated whether
the Super-TDA affected the cell cycle of dormant MCF-7 and T-47D
cells using flow cytometry. Our results revealed that a high
percentage of MCF-7 and T-47D cells remained in G0/G1 when cultured
under 3D conditions for 5 days (Fig.
3B). However, the established-DTCs exhibited a significantly
higher S and G2/M cell population when co-cultured with Super-TDA
(50%) for 5 days, compared with parental cells (P≤0.01). In
conclusion, Super-TDA promoted the dormant-to-proliferating switch
by altering the cell cycle.
TGF-β1 secreted during the
transdifferentiation of AEC IIs is one of the major stimulants
involved in inducing DTC reproliferation
TGF-β1 is a key factor altering the tumor
environment, and is implicated in the regulation of a variety of
biological responses (14),
including proliferation and differentiation of tumor cells. To
further identify factors involved in inducing the growth of DTCs in
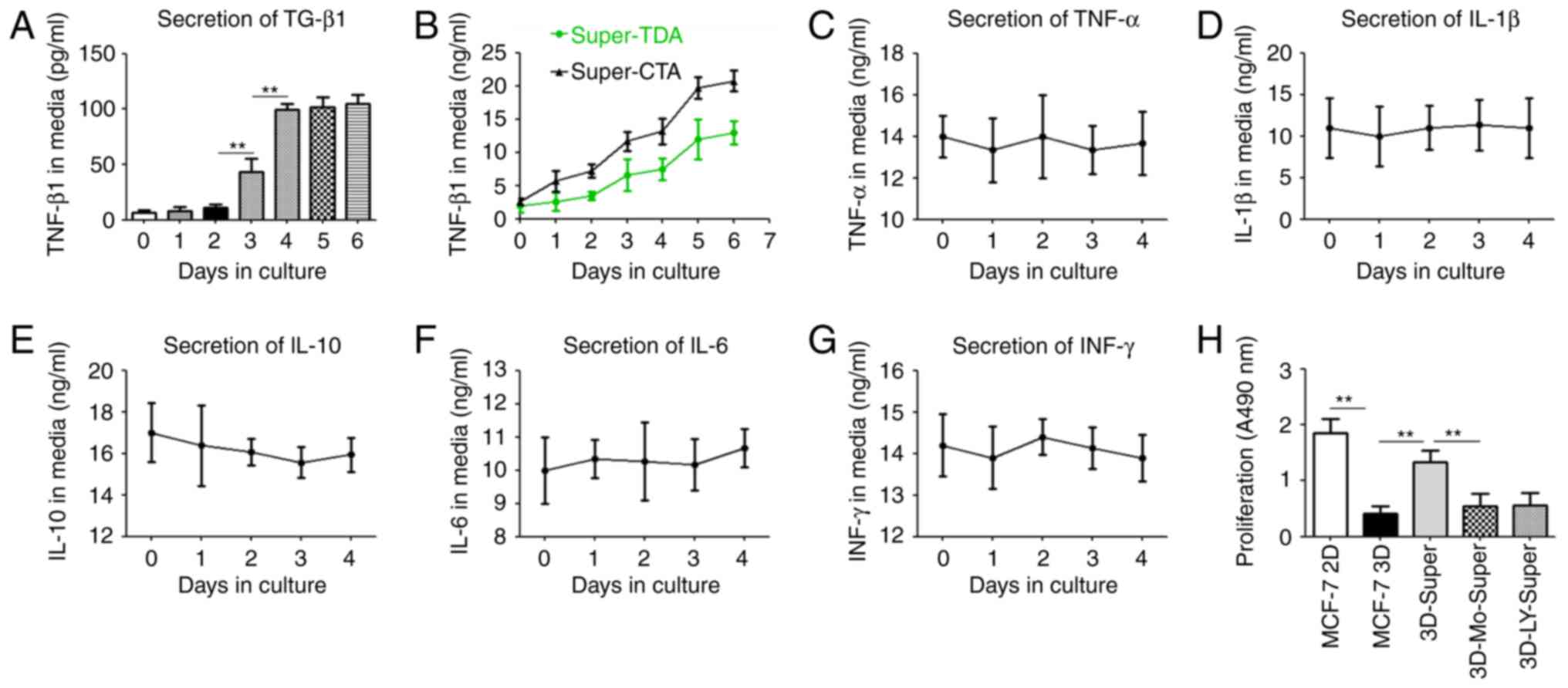
Super-TDA, TGF-β1 expression was evaluated by sandwich ELISA
(Fig. 4). Our data revealed that
the level of secreted-TGF-β1 was markedly increased with the
progression of the transdifferentiation of AEC IIs to AEC Is,
particularly on day 5, and the amount of TGF-β1 were nearly 12
times that in the control group (P≤0.01) (Fig. 4A). Furthermore, we observed that the
amount of secreted TGF-β1 increased markedly and more quickly when
AEC IIs were co-cultured with tumor cells, suggesting that tumor
cells enhanced the production and secretion of TGF-β1 from AEC IIs
during transdifferentiation (Fig.
4B). However, The secretion levels of other cell factors, such
as TNF-α, IL-1β1, IL-10, IL-6 and INF-γ, remained unchanged.
(P>0.05) (Fig. 4B-G).
Furthermore, the addition of the TGF-β receptor kinase inhibitor
LY2109761 or monensin inhibited the DTCs induced by Super-TDA
(Fig. 4H), indicating that the
autocrine effect of TGF-β1 through transdifferentiation of AEC IIs
is one of the major stimulators inducing DTC growth.
LPS stimulates the growth of DTCs by
promoting the secretion of TGF-β1 in the Super-TDA
TLR4 expressed on tumor cells has been found to
contribute to tumor progression by promoting tumor cell
proliferation, resistance to apoptosis and evasion from immune
attack (22,23). Therefore, we hypothesized that LPS
may also participate in the activation of DTCs, and we used LPS to
simulate inflammatory damage. We then incubated the DTCs with
Super-TDA pretreated with or without LPS for several days, as shown
in Fig. 5A. We found that although
LPS could not induce DTC reproliferation directly, the reactivation
and proliferation period of DTCs was obviously shortened to 2 days
when the DTCs were co-cultured with Super-TDA pretreated with LPS.
Of note, the proliferative potential of the reactivated DTCs was
obviously increased compared with that of the control groups,
suggesting that LPS facilitates DTC growth. We also found that LPS
promoted the release of TGF-β1 from AEC IIs undergoing
transdifferentiation (Fig. 5B),
indicating that LPS may participate in the growth of DTCs by
increasing the TGF-β1 secretion from Super-TDA.
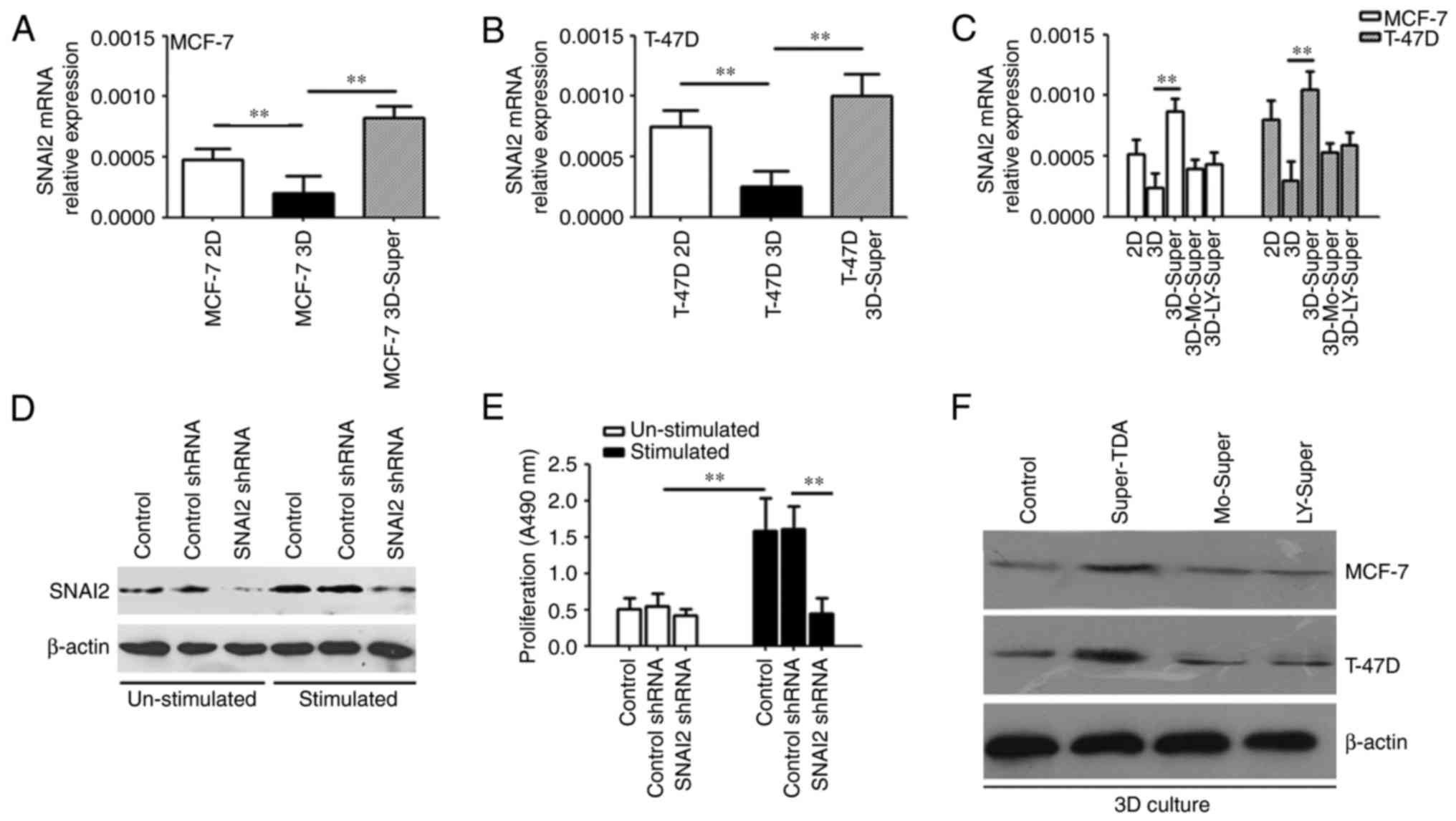
The transdifferentiation of AEC IIs
promotes the upregulation of SNAI2 expression in the reactivated
DTCs
SNAI2 serves an important role in the proliferation,
metastasis and angiogenesis of tumors (24). SNAI2 enhances the kinase activity of
cyclin D1/CDK4/CDK6, a central mediator in the transition from the
G1 to the S phase, which further promotes the switch of tumor cell
proliferation (25), indicating
that SNAI2 may participate in the dormant-to-proliferating switch
of DTCs.
To confirm the role of SNAI2 in the
dormant-to-proliferating switch of DTCs, we analyzed the dormant
and reactivated tumor cells by qPCR and western blotting. As shown
in Fig. 6A and B, Super-TDA
promoted DTC upregulation of SNAI2 expression, and the upregulation
of SNAI2 was inhibited by monensin or the TGF-β receptor kinase
inhibitor LY2109761 (Fig. 6C and
F). We then used SNAI2 shRNA to inhibit the Super-TDA-induced
increased expression of SNAI2 (Fig.
6D), which significantly hindered the re-proliferation of DTCs
induced by Super-TDA (Fig. 6E),
indicating that the upregulation of SNAI2 expression is required
for the dormant-to-proliferating switch of DTCs.
Discussion
Metastatic recurrence of carcinomas frequently
occurs after a long latency period following removal of the primary
tumor and administration of adjuvant therapy. Accumulating evidence
suggests that tumor cells that have seeded to metastatic sites are
resistant to conventional therapies, and remain dormant over long
periods of time in target organs (26). A likely explanation of this
phenomenon is that the presence of DTCs in secondary sites is
maintained if the microenvironment is unfavorable for tumor cell
proliferation (3,4). However, it remains unclear whether
tumor cells become dormant as a consequence of intrinsic defects,
or in response to inhibitory signals that they encounter in foreign
microenvironments.
Normal tissue homeostasis is maintained by signals
from cell-cell communication, cell-cell adhesion and cell-matrix
interactions, as well as more systemic mechanisms (e.g. hormones,
cytokines) of growth and differentiation control (27). When the tumor microenvironment is
disturbed, it may stimulate DTCs to resume active growth and
progression; otherwise, they would remain dormant in the secondary
target site (3,28). The lung is one of the most common
sites of metastasis (8). Our
previous and other studies demonstrated that the metastatic tumor
cells derived from non-invasive tumor cells exhibit lower
expression of the proliferation marker Ki-67 (5). It is well-known that the new
microenvironment may restrict the proliferation of disseminated
tumor cells (38). Different from highly invasive tumor cells, the
metastatic tumor cells derived from non-invasive tumor cells may be
more sensitive to the restrictions of the new microenvironment.
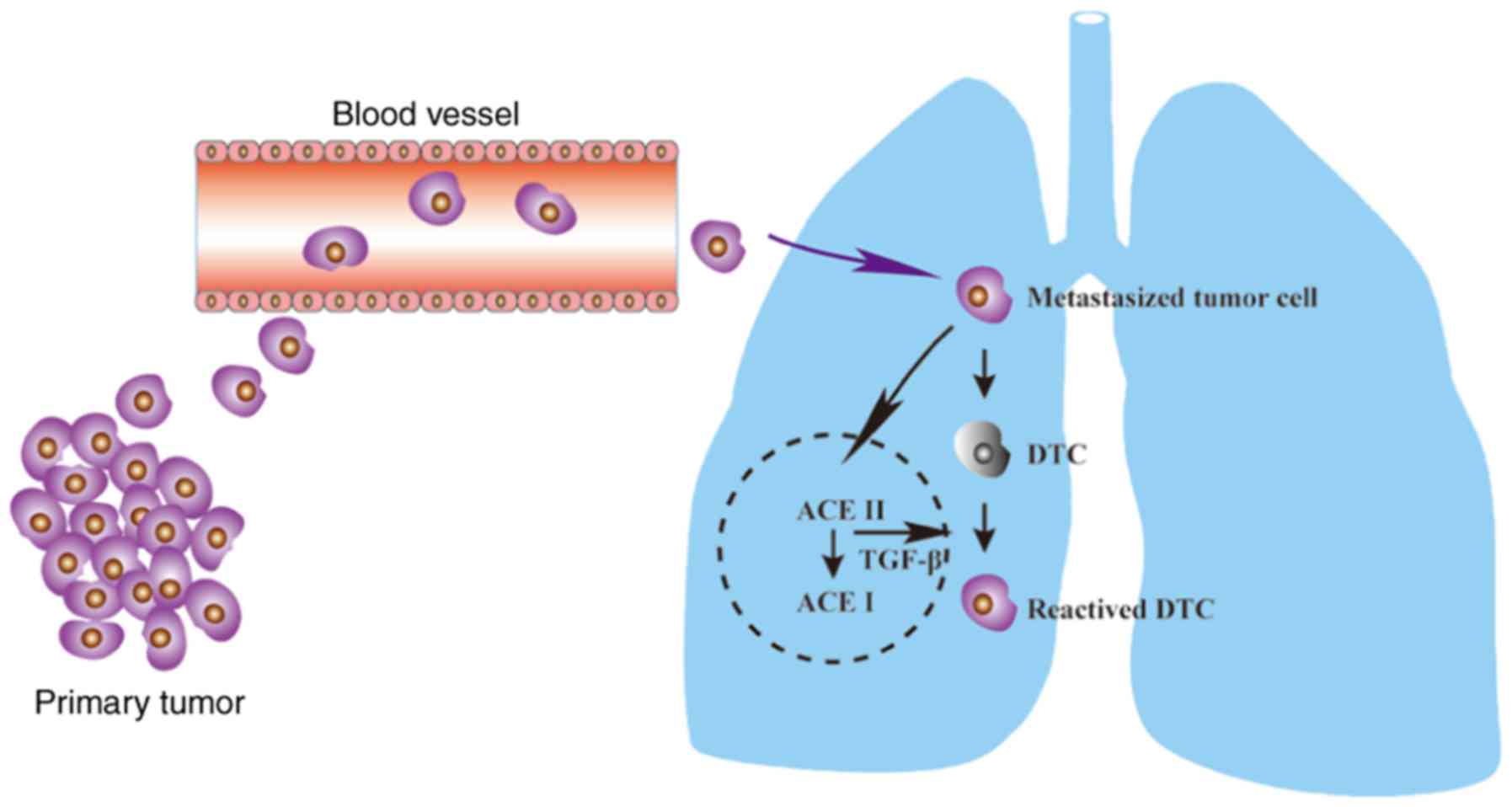
In the present study, we demonstrated that tumor
cells may promote the transdifferentiation of AEC IIs. Of note, the
transdifferentiation of AEC IIs may in turn induce a DTC switch to
reactivated growth by enhancing TGF-β1/SNAI2 signaling (Fig. 7). Targeting this process may provide
novel therapeutic strategies for inhibition of the
dormant-to-proliferating metastatic cell switch.
AEC IIs have a multifunctional role in the lung,
including secretory, synthetic and remodeling reservoirs for the
lung epithelium to host defense (9). The progenitor function of AEC IIs is
activated when the lung epithelium encounters a variety of
conditions, including acute lung injury and inflammation, among
others (10). AEC II proliferation
and hyperplasia, followed by transdifferentiation, is a hallmark of
alveolar epithelial injury, and defends the alveolus from injury
(10). Apart from alveolar injury,
the present study confirmed that the tumor cells also stimulated
the transdifferentiation of AEC IIs into AEC Is and TGF-β1
secretion, suggesting that the metastasized tumor cells may
participate in the induction of the transdifferentiation of AEC IIs
to AEC Is in vivo, and AEC II transdifferentiation may be a
common occurrence during lung metastasis.
In addition, AEC II transdifferentiation may in turn
induce the DTC dormant-to-proliferating switch, which may explain
why the lung is one of most common metastatic and recurrence sites
(9); in addition, the metastasized
tumor cells may also affect the microenvironment of the metastatic
sites, which may be correlated with the dormant or proliferative
behavior of tumor cells at the metastatic sites in vivo.
TGF-β1, produced by AECs, is secreted during the
transdifferentiation of AEC IIs to AEC Is, and the autocrine
production of TGF-β1 has been shown to promote the
transdifferentiation of primary rat AEC IIs to AEC Is in
vitro (11,12). An autocrine loop of TGF-β1 in
mammary epithelium of pregnant mice has been shown to aid the
differentiation process (29). The
level of autocrine TGF-β1 was found to be markedly increased when
the AEC IIs were undergoing transdifferentiation phase, as shown by
ours and other data (13,29). Notably, the amount of TGF-β1 during
transdifferentiation of AEC IIs was 12-times higher than the basic
secretion level in the present study, suggesting that the amount of
TGF-β1 secreted by AEC IIs during transdifferentiation may be
sufficient to alter the tumor microenvironment and cause a variety
of biological responses, including cell proliferation (14). In addition, both monensin and
LY2109761 were able to block the dormant-to-proliferating switch
for DTCs induced by Super-TDA, further indicating that the TGF-β1
from Super-TDA may be the main stimulator of DTC reactivation and
proliferation in the lung.
The progenitor function of AEC IIs is activated when
the lung epithelium encounters a variety of conditions, including
acute lung injury and inflammation (10). TLR4 ligands (LPS) may be present
in vivo due to surgery, cell damage, or the presence of
bacteria in the tumor (30–32). TLR4 expressed on tumor cells has
been found to contribute to tumor progression by promoting tumor
cell proliferation, resistance to apoptosis and tumor evasion from
immune surveillance (22,23). Our results confirmed that LPS could
not induce DTC reactivation directly, but rather by enhancing the
secretion of TGF-β1 from AEC IIs undergoing transdifferentiation,
suggesting that inflammatory damage of the lung may participate in
enhancing the reactivation process of DTCs. This may explain the
fact that, when the lung epithelium encounters a variety of
conditions, including acute lung injury, the progenitor function of
AEC IIs is activated (10),
promoting self-repair as well as DTC reactivation at the same
time.
Although the expression of SNAI2 is very low in
DTCs, the proliferative potential of DTCs was reactivated along
with the increased expression of SNAI2 induced by Super-TDA. In
addition, the upregulation of SNAI2 expression was inhibited by
pretreatment with monensin and LY2109761 during the
transdifferentiation of AEC IIs. The increased expression of SNAI2
is a major risk factors for the tumor recurrence and metastasis
(33,34). SNAI2 may enhance the kinase activity
of cyclin D1/CDK4/CDK6, a central mediator in the transition from
the G1 to the S phase, which further promotes the proliferation
switch of tumor cells (25). Thus,
SNAI2 may serve a pivotal role in the dormant-to-proliferating
switch of DTCs.
In summary, tumor cell metastasis to the lung may
promote the transdifferentiation of AEC IIs to AEC Is in
vitro. In addition, Super-TDA may in turn induce the
reactivation of DTCs by enhancing TGF-β1/SNAI2 signaling.
Recurrence and metastasis are responsible for the majority of
cancer deaths. Given that the transdifferentiation of AEC IIs and
the enhanced expression of TGF-β1/SNAI2 are required for inducing
the reactivation and proliferation of DTCs, targeting one of these
stimuli or signaling pathways may be a viable approach to a
comprehensive strategy for cancer treatment.
Acknowledgements
We would like to thank Professor Changyi Xiao, Dr
Xiaokun Tu and Wei Wang for technical assistance in our
experiments. We acknowledge research funding from the National
Nature Science Foundation of China (grant nos. 81403163 and
81402404) and Yi Chang Scientific and Technological Bureau (grant
nos. A14301-04 and A14301-10).
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Super-TDA
|
supernatant of the
transdifferentiation of AEC IIs to AEC Is
|
|
Super-CTA
|
supernatant of co-cultured tumor cells
and AEC IIs by Transwell assay
|
|
AEC IIs
|
type II alveolar epithelial cells
|
|
3D
|
three dimensional
|
|
DTC
|
dormant tumor cell
|
|
BME
|
Cultrex growth factor-reduced basement
membrane extract
|
|
LPS
|
lipopolysaccharide
|
References
|
1
|
Goss PE and Chambers AF: Does tumour
dormancy offer a therapeutic target? Nat Rev Cancer. 10:871–877.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Páez D, Labonte MJ, Bohanes P, Zhang W,
Benhanim L, Ning Y, Wakatsuki T, Loupakis F and Lenz HJ: Cancer
dormancy: A model of early dissemination and late cancerrecurrence.
Clin Cancer Res. 18:645–653. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zappalà G, McDonald PG and Cole SW: Tumor
dormancy and the neuroendocrine system: An undisclosed connection?
Cancer Metastasis Rev. 32:189–200. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Alizadeh AM, Shiri S and Farsinejad S:
Metastasis review: From bench to bedside. Tumour Biol.
35:8483–8523. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou YH, Liao SJ, Li D, Luo J, Wei JJ, Yan
B, Sun R, Shu Y, Wang Q, Zhang GM and Feng ZH: TLR4
ligand/H2O2 enhances TGF-β1 signaling to
induce metastatic potential of non-invasive breast cancer cells by
activating non-Smad pathways. PLoS One. 8:e659062013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barkan D, Kleinman H, Simmons JL, Asmussen
H, Kamaraju AK, Hoenorhoff MJ, Liu ZY, Costes SV, Cho EH, Lockett
S, et al: Inhibition of metastatic outgrowth from single dormant
tumor cells by targeting the cytoskeleton. Cancer Res.
68:6241–6250. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aguirre-Ghiso JA: Models, mechanisms and
clinical evidence for cancer dormancy. Nat Rev Cancer. 7:834–846.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu
W, Giri DD, Viale A, Olshen AB, Gerald WL and Massagué J: Genes
that mediate breast cancer metastasis to lung. Nature. 436:518–524.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warburton D and Bellusci S: The molecular
genetics of lung morphogenesis and injury repair. Paediatr Respir
Rev. 5 Suppl A:S283–S287. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fehrenbach H: Alveolar epithelial type II
cell: Defender of the alveolus revisited. Respir Res. 2:33–46.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu W, Xu B, Zhao Y, Yang N, Liu C, Wen G
and Zhang B: Wnt5a reverses the inhibitory effect of hyperoxia on
transdifferentiation of alveolar epithelial type II cells to type I
cells. J Physiol Biochem. 71:823–838. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barkauskas CE, Cronce MJ, Rackley CR,
Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW and Hogan BL:
Type 2 alveolar cells are stem cells in adult lung. J Clin Invest.
123:3025–3036. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao L, Yee M and O'Reilly MA:
Transdifferentiation of alveolar epithelial type II to type I cells
is controlled by opposing TGF-β and BMP signaling. Am J Physiol
Lung Cell Mol Physiol. 305:L409–L418. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Catalano V, Turdo A, Di Franco S, Dieli F,
Todaro M and Stassi G: Tumor and its microenvironment: A
synergistic interplay. Semin Cancer Biol. 23:522–532. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liao SJ, Zhou YH, Yuan Y, Li D, Wu FH,
Wang Q, Zhu JH, Yan B, Wei JJ, Zhang GM and Feng ZH: Triggering of
Toll-like receptor 4 on metastatic breast cancer cells promotes
αvβ3-mediated adhesion and invasive migration. Breast Cancer Res
Treat. 133:853–863. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan YL, Zhou YH and Ye H: An improved
method for the isolation and primary culture of mouse AEC II.
Hainan Med J. 27:3280–3282. 2016.
|
|
17
|
Zhang L, Zhao S, Yuan L, Wu H, Jiang H and
Luo G: Hyperoxia-mediated LC3B activation contributes to the
impaired transdifferentiation of type II alveolar epithelial cells
(AECIIs) to type I cells (AECIs). Clin Exp Pharmacol Physiol.
43:834–843. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Isakson BE, Lubman RL, Seedorf GJ and
Boitano S: Modulation of pulmonary alveolar type II cell phenotype
and communication by extracellular matrix and KGF. Am J Physiol
Cell Physiol. 281:C1291–C1299. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Almog N: Molecular mechanisms underlying
tumor dormancy. Cancer Lett. 294:139–146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goss P, Allan AL, Rodenhiser DI, Foster PJ
and Chambers AF: New clinical and experimental approaches for
studying tumor dormancy: Does tumor dormancy offer a therapeutic
target? APMIS. 116:552–568. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weidenfeld K, Schif-Zuck S, Abu-Tayeh H,
Kang K, Kessler O, Weissmann M, Neufeld G and Barkan D: Dormant
tumor cells expressing LOXL2 acquire a stem-like phenotype
mediating their transition to proliferative growth. Oncotarget.
7:71362–71377. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang B, Zhao J, Li H, He KL, Chen Y, Chen
SH, Mayer L, Unkeless JC and Xiong H: Toll-like receptors on tumor
cells facilitate evasion of immune surveillance. Cancer Res.
65:5009–5014. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang EL, Qian ZR, Nakasono M, Tanahashi T,
Yoshimoto K, Bando Y, Kudo E, Shimada M and Sano T: High expression
of Toll-like receptor 4/myeloid differentiation factor 88 signals
correlates with poor prognosis in colorectal cancer. Br J Cancer.
102:908–915. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shih JY and Yang PC: The EMT regulator
SNAI2 and lung carcinogenesis. Carcinogenesis. 32:1299–1304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Casimiro MC, Velasco-Velázquez M,
Aguirre-Alvarado C and Pestell RG: Overview of cyclins D1 function
in cancer and the CDK inhibitor landscape: Past and present. Expert
Opin Investig Drugs. 23:295–304. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao H, Chakraborty G, Lee-Lim AP, Mo Q,
Decker M, Vonica A, Shen R, Brogi E, Brivanlou AH and Giancotti FG:
The BMP inhibitor Coco reactivates breast cancer cells at lung
metastatic sites. Cell. 150:764–779. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Glick AB and Yuspa SH: Tissue homeostasis
and the control of the neoplastic phenotype in epithelial cancers.
Semin Cancer Biol. 15:75–83. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Friberg S and Nyström A: Cancer
metastases: Early dissemination and late recurrences. Cancer Growth
Metastasis. 8:43–49. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhaskaran M, Kolliputi N, Wang Y, Gou D,
Chintagari NR and Liu L: Trans-differentiation of alveolar
epithelial type II cells to type I cells involves autocrine
signaling by transforming growth factor beta 1 through the Smad
pathway. J Biol Chem. 282:3968–3976. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pidgeon GP, Harmey JH, Kay E, Da Costa M,
Redmond HP and Bouchier-Hayes DJ: The role of
endotoxin/lipopolysaccharide in surgically induced tumour growth in
a murine model of metastatic disease. Br J Cancer. 81:1311–1317.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tsan MF and Gao B: Endogenous ligands of
Toll-like receptors. J Leukoc Biol. 76:514–519. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Loze MT, et al: DAMP-mediated
autophagy contributes to drug resistance. Autophagy. 7:112–114.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheng WY, Kandel JJ, Yamashiro DJ, Canoll
P and Anastassiou D: A multi-cancer mesenchymal transition gene
expression signature is associated with prolonged time to
recurrence in glioblastoma. PLoS One. 7:e347052012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hasan M, Sharma R and Saraya A: Slug is a
predictor of poor prognosis in esophageal squamous cell carcinoma
patients. PLoS One. 8:e828462013. View Article : Google Scholar : PubMed/NCBI
|