Introduction
Glioblastoma multiforme (GBM) is the most common and
aggressive type of brain malignancy, and is characterized by high
invasiveness, therapeutic resistance and recurrence (1,2). Due
to the difficulty of total surgical resection, temozolomide
(TMZ)-based chemotherapy has been the standard first-line adjuvant
treatment for patients with GBM, targeting residual tumor cells
with the aim of extending the progression-free and overall survival
times (3,4).
TMZ belongs to a group of alkylating agents that are
able to cross the blood-brain barrier (BBB) and reach therapeutic
concentrations in the activated form of compound
5–3-(methyl)-1-(triazen-1-yl) imidazole-4-carboxamide (MTIC), which
can cause DNA lesions mainly by methylating the
O6-guanine. The subsequent mismatch repair (MMR)
response in DNA replication, alerted by the misrepair of methylated
guanine and thymine (instead of cytidine), causes either
double-strand breaks or critical recombinogenic lesions. This
eventually induces apoptosis and inhibits proliferation of
malignant cells (5,6).
Despite fundamental clinical benefits,
chemoresistance is an inevitable limitation of TMZ administration.
Mechanistically, chemoresistance can be divided into two
categories: Intrinsic and extrinsic. The former can mostly be
attributed to inherent gene characteristics at the beginning of
therapy, such as lack of MGMT gene methylation. Extrinsic
chemoresistance, also referred to as acquired resistance, is
conferred by tumor cells that gradually develop genetic alterations
to antagonize TMZ toxicity. In many cases, the response to TMZ is
still poor even if MGMT promoter methylation is detected;
therefore, there is a greater interest in acquired chemoresistance,
which is more likely to be reversible and targetable (7). In order to uncover the mechanisms of
acquired chemoresistance, we first established a variant
glioblastoma cell line to simulate the refractory tumor bulk under
clinical treatment. Then, we performed a protein microarray, which
can provide more reliable evidence directly from interactions among
expressed proteins and activities, as compared with cDNA microarray
(8), in order to characterize
potential molecular mechanisms.
Characteristics of cell stemness are thought to be
associated with the tumorigenic capacity and migratory nature of
malignant glioblastoma cells, an area that is gaining more interest
in the field of cancer treatment. In accordance with results from a
number of laboratories indicating that GSCs have an innate ability
to resist the effects of therapeutic agents (9,10), the
pathway investigation in the present study uncovered stem-like
signaling in the TMZ-resistant U251 cell line, which highlighted
the central role of SRC in the networks. SRC belongs to a family of
non-receptor tyrosine kinases (SFKs), which contain a unique
N-terminal sequence, followed by four SRC homology (SH) domains and
a C-terminal negative regulatory sequence. Dephosphorylation at
Y530 in the C-terminus by protein tyrosine phosphatases (PTPs), and
the subsequent autophosphorylation at Y416 triggers the interaction
of SFKs with multiple downstream factors involved in cell adhesion,
migration, invasion, proliferation and angiogenesis. In addition,
several SFK members have been reported to be involved in cell
stemness and epithelial-mesenchymal transition (EMT) phenotypes of
multiple cancer types, including GBM (37). In the present study, we also
examined the role of SRC in TMZ-resistance. Collectively, the
present study presents a comprehensive insight into the defense
strategies of glioblastoma cells, which may provide potential
targets for improving TMZ-based chemotherapy.
Materials and methods
Experimental procedure
Cell culture and transfection
The human glioblastoma cell lines U251 and U373 were
purchased from the Chinese Academy of Sciences. The glioblastoma
cell lines were cultured in DMEM supplemented with 10% fetal bovine
serum, 1.5 mM glutamine, 100 IU/ml penicillin and 100 µg/ml
streptomycin. Cells were maintained at 37°C in a humidified 5%
CO2/95% air incubator, and harvested for passage with
trypsin (0.5 mg/ml) and EDTA (0.2 mg/ml) when they had reached
confluence.
For sphere culture and self-renewal assays, U251
cells were cultured in DMEM/F-12 supplemented with N2, B27
supplements (Thermo Fisher Scientific, Inc., Waltham, MA, USA), 20
ng/ml basic fibroblast growth factor (bFGF) and 20 ng/ml epidermal
growth factor (EGF; Sigma-Aldrich, St. Louis, MO, USA) in a
low-adhesive plate for 10 days to allow spheres to form. Cells were
plated at a density of ~1×104 live cells/dish, and the
medium was changed every 3–4 days. After obvious spheres were
observed (~2 weeks later), these spheres were disaggregated with
Accutase (Invitrogen; Thermo Fisher Scientific, Inc., Carlsbad, CA,
USA), followed by repetitive pipetting up and down, and then plated
for secondary culture and maintained in aforementioned GSC medium
for a further 10 days. When the majority of spheres reached ≥50 µm
in diameter, they were viewed under a phase-contrast microscope for
morphological comparison. The neurosphere diameters of each cell
line were measured for a total of 15 fields in triplicate
experiments.
The GV358-RNAi vector, which co-expressed the GFP
protein and specific short hairpin RNA (shRNA) against human SRC
(cat. no. 54150) or a non-targeting scramble shRNA (both from
Shanghai GeneChem Co., Ltd., Shanghai, China) as a control, were
commercially synthesized and constructed according to the
manufacturer's instructions. The GV358-RNAi vectors containing
SRC-specific and control shRNAs were transfected into 293FT cells
using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's instructions. Complete
lentiviral expression vectors were generated and denoted as Si-SRC
and Si-Scr lentivirus, respectively. The lentiviral vectors were
transfected into glioblastoma cells in the presence of 5 µg/ml
polybrene and cultured before they were subjected to cell viability
and sphere-formation assays.
Isolation of a TMZ-resistant cell
line
For the isolation of resistant cells, TMZ (Tasly
Pharmaceutical Co., Ltd., Tianjin, China) was dissolved in DMSO
(12.5 mg/ml stock solution) and diluted to the desired
concentrations. Parental U251 cells were consecutively re-exposed
to an incremental TMZ pulse from initiation of 1 µg/ml to reach the
desired concentration for a period of 6 months. Briefly, cells were
plated in 25 cm2 flasks in their usual medium and
allowed to attach overnight, following which the medium was
replaced with TMZ containing medium of double concentration
followed by TMZ-free fresh medium after 2 h, for an additional
10–12 days of regrowth. When there was no obvious cell loss
observed, cells were collected and subjected to an MTT assay.
TMZ sensitivity and cell proliferation
assays
Cell growth was determined by an MTT colorimetric
assay. Cells were plated onto 96-well plates (5×105
cells/well) in culture medium overnight to allow adherence. The
medium was then replaced with fresh medium containing increasing
concentrations of TMZ (0–128 µg/ml). After 120 h, the medium was
replaced with MTT for 4 h in the dark at 37°C, and then shaken in
DMSO for 15 min to fully dissolve the formazan crystals. Absorbance
(A) of cells was measured at 590 nm with a microplate reader (Tecan
Group, Ltd., Männedorf, Switzerland). A total of 3 independent
experiments were performed in quadruplicate wells (plus control).
The inhibition ratio was calculated as follows: P = (1 - A in
treated cells/A in control cells) × 100%. The 50% inhibitory
concentration (IC50) of all experimental groups was
obtained.
For the cell proliferation experiment, MTT was added
in each well absent of TMZ at the beginning of cell plating (0 h),
and at 24, 48, 72, 96 and 120 h afterwards. The absorbance of each
well was measured according to the aforementioned procedure, and
the relative number of viable cells, which represented the
proliferation rate, was calculated as: Absorbance value at each
time/absorbance value at 0 h.
Cell cycle and apoptosis assay
Subconfluent cell cultures were incubated with 48
µg/ml TMZ for 72 h, then trypsinized, washed, collected and
centrifuged (1,000 rpm) for 5 min, fixed in 1.5 ml 75% ice-cold
ethanol and stored at 4°C overnight. After resuspension in cold
PBS, the cell suspension was incubated in 0.2 mg/ml propidium
iodide (PI) containing 0.1% Triton X-100 and RNase A (1 mg/ml;
Sigma-Aldrich) in the dark for 30 min at 4°C. Cell cycle
distribution was determined using fluorescence-activated cell
sorting (FACS) analysis (FACSCalibur™; BD Biosciences, Franklin
Lakes, NJ, USA), with excitation at 488 nm and emission at 630 nm.
For the apoptosis experiment, the cell suspension was evaluated
using a PI/Annexin apoptosis detection kit, according to the
aforementioned procedure.
Protein screening and bioinformatics
analysis
Grouping
In the present study, TMZ-induced U251 cells were
denoted as the treatment group while parental U251 cells were
denoted as the control group.
Construction of the protein chips
Proteins from U251 and U251R cells were extracted
and assessed to meet the concentration criterion. SET100 Protein
Microarrays (Full Moon BioSystems, Inc., Sunnyvale, CA, USA),
containing 1,358 antibody probes, were constructed for the two
groups according to the manufacturer's instructions. Protein-mixed
microarrays were then subjected to signal scanning (GenePix 4000B,
Axon Instruments; Molecular Devices, LLC, Sunnyvale, CA, USA).
Protein investigation
Protein signals were acquired using Genepix Pro 6.0
(Axon Instruments; Molecular Devices, LLC) repeatedly with GAPDH as
the internal control. Relative protein expression was calculated as
follows: Average/GAPDH. Acquired TMZ resistance-related
differentially expressed proteins were defined by the threshold as
±1.5-fold change.
Gene Ontology (GO) and pathway
analysis
The significant GOs of the genes corresponding to
differentially expressed proteins were analyzed using the GO
database (http://www.geneontology.org) and
online software DAVID (http://david.abcc.ncifcrf.gov) for gene annotation and
hierarchical categorization. All signaling pathways were analyzed
using data from KEGG (http://www.genome.jp/kegg) and NCBI (http://www.ncbi.nlm.nih.gov) databases. P-values for
the differentially expressed genes in all GO categories and
pathways were calculated using the two-sided Fisher's exact test to
obtain the enrichment value as -log10 (P-value).
TMZ resistance-related signal
network
The Human Protein Reference Database was used to
search for differentially-expressed proteins detected via
microarray. A TMZ resistance-related signal transduction network
was constructed using Cytoscape software (http://www.cytoscape.org). The significance of
molecules in the network was represented by the area of each node
and determined by the value of betweenness centrality, which was
calculated according to the number of effective interactions
through a particular molecular node. The greater the betweenness
value, the more control it possesses in interactive pathways.
Upregulated molecules were marked red; downregulated were marked
blue; and pink was used to mark the stem and EMT-like
molecules.
Protein extraction and western
blotting
Proteins (50 µg) were extracted from subconfluent
cultures and loaded onto polyacrylamide-SDS gels, separated by
electrophoresis, and transferred to nitrocellulose membranes. After
blocking with 5% non-fat milk in PBST for 1 h at room temperature,
the membranes were blotted with primary antibodies overnight at
4°C, followed by incubation with peroxidase-conjugated secondary
antibodies for 50 min at room temperature. Bands on the membranes
were visualized using enhanced chemiluminescence (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The primary antibodies,
including those against SRC (1:1,000; cat. no. ab47405), YES
(1:1,000; cat. no. ab109265), LYN (1:1,000; cat. no. ab1890) and
GAPDH (1:1,000; cat. no. ab9485) were all purchased from Abcam
(Cambridge, MA, USA). GAPDH was used as the internal control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNAs were extracted from two experimental cell
lines, using the RNeasy Mini kit (Qiagen, Venlo, The Netherlands).
First-strand cDNA was reverse transcribed from 1 µg total RNA using
the SuperScript First-Strand cDNA system and amplified by Platinum
SYBR Green qPCR SuperMix-UDG (both from Invitrogen; Thermo Fisher
Scientific, Inc.). For each PCR reaction, a Master mix was prepared
that included Platinum SYBR-Green qPCR SuperMix-UDG, forward
primer, reverse primer, and 10 ng template cDNA. GAPDH was used as
the internal control. The thermocycling conditions were as follows:
95°C for 2 min for the first step; 40 cycles of 95°C for 10 sec,
55°C for 15 sec and 72°C for 10 sec for the second step; 95°C for 1
min, 60°C for 30 sec and 95°C for 30 sec for the last step. Data
were analyzed from three independent experiments and presented as
the mean ± SD. The fold-change quantification of target genes was
calculated with the 2−∆∆Ct method. Primer pairs used to
detect the mRNA levels of genes were as follows: YES forward,
5′-GCCTGTCAGTACAAGTGTGAG-3′ and reverse,
5′-AAAGGCGTTACCCCTGAGGAT-3′; SRC forward, 5′-TGGCAAGATCACCAGACGG-3′
and reverse, 5′-GGCACCTTTCGTGGTCTCAC-3′; LYN forward,
5′-TTCTGGTCTCCGAGTCACTCA-3′ and reverse, 5′-GCCGTCCACTTAATAGGGAA
CT-3′; GAPDH forward, 5′-CATGAGAAGTATGACAACAGCCT-3′ and reverse,
5′-AGTCCTTCCACGATACCAAAGT-3′.
Statistical analysis
Data analysis was performed using GraphPad Prism 7
software (GraphPad Software, Inc., La Jolla, CA, USA). The α level
for type I error was set at 0.05 for rejecting the null hypothesis.
Descriptive data are presented as the mean ± standard deviation
(SD). The survival rate at each TMZ concentration, cell cycle
distribution and apoptosis rate for the two glioblastoma cell lines
were analyzed with the Student's t-test. P-value <0.05 was
considered to indicate a statistically significant difference.
Results
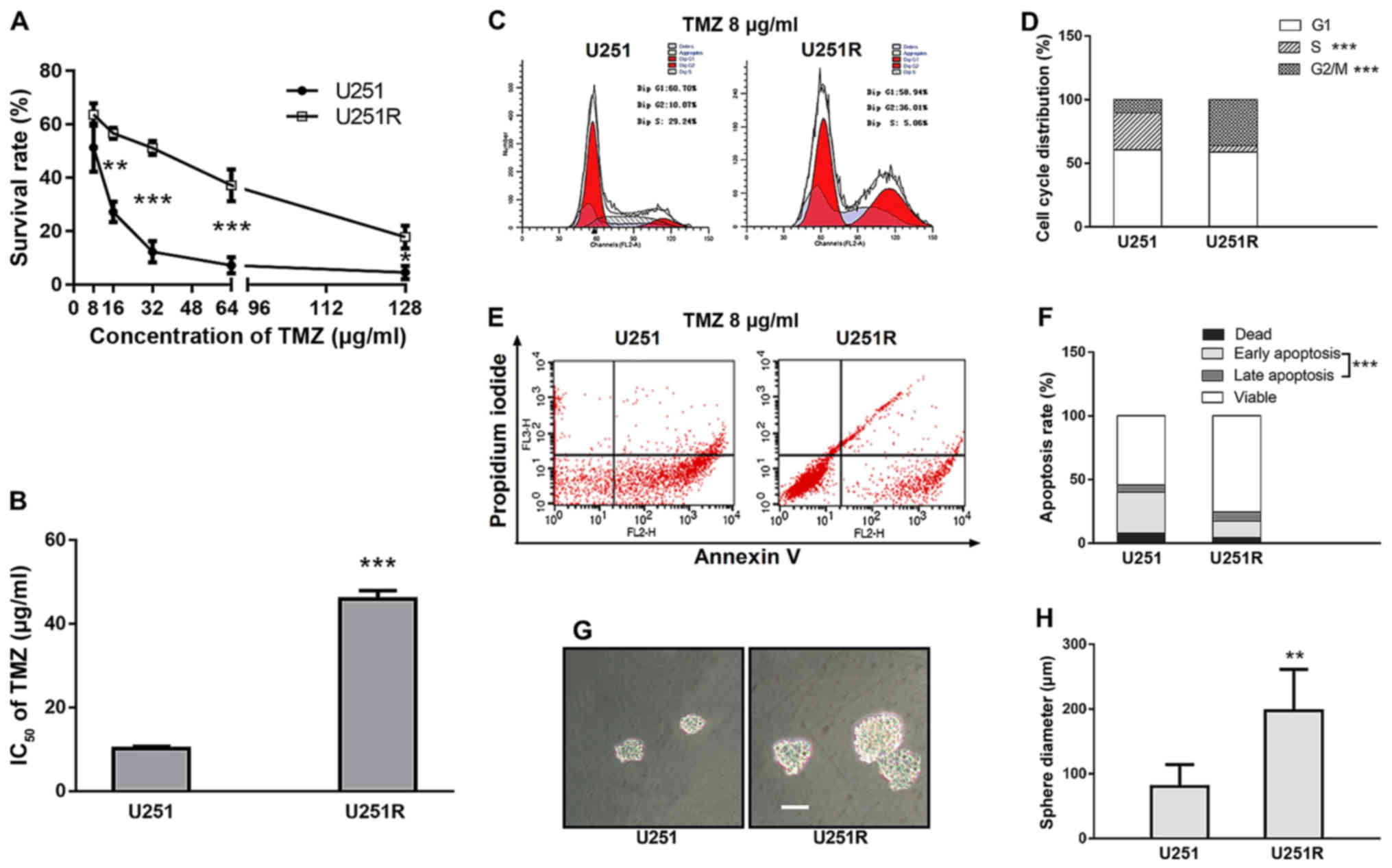
Acquisition of TMZ resistance in the
U251 glioblastoma cell line
After initial killing of a large majority of cells
at each TMZ concentration period, the remaining cells survived and
retained a normal growing state. Finally, a stable resistant
phenotype was observed. MTT results revealed that both parental
U251 cells and TMZ-induced U251 cells exhibited a prominent and
concentration-dependent decrease in the viable cell number.
Notably, a 4-fold increase in the IC50 value was
observed in the TMZ-induced U251 cell line, compared with that in
the parental cells (45.19 and 10.89 µg/ml, respectively) (Fig. 1A and B). After 10 passages in
TMZ-free medium, and storage for 1 year at −80°C after the last
exposure to TMZ, the resistance characteristics remained constant.
The subline isolated from the U251 cells was referred to as
U251R.
TMZ induction causes G2/M arrest,
duplication inhibition, apoptosis attenuation and increases
self-renewal ability in chemoresistant U251 cells
Cell cycle distribution was determined by FACS
analysis. With 8 µg/ml TMZ exposure, U251R cells exhibited a higher
proportion of G2/M phase arrest and a lower proportion of S phase
compared with the parental U251 cells (36.22 vs. 10.36%, P<0.001
and 4.97 vs. 29.13%, P<0.001, respectively) (Fig. 1C and D), whereas the percentage of
G0/G1 phase remained unaltered between the two cell cultures.
Assuming that prolonged G2/M distribution was likely to be linked
with cell survival, we further assessed cell apoptosis by Annexin
V/PI staining. As expected, apoptosis was significantly attenuated
in TMZ-resistant U251R cells compared with that in the control
group (19.95 vs. 37.83%, P<0.001) (Fig. 1E and F), which supports the link
between clonogenic survival and G2/M arrest. Another assumption was
that reduced S-phase proportion indicated a slower growth rate.
Indeed, there was a trend of reduced proliferation in the U251R
cell line compared with that in the U251 cell line, although the
difference was not significant (P-value of the group according to
time interaction was 0.089, data not shown).
In order to characterize the potential effect of
TMZ-induction on the self-renewal activity of tumor cells, we
performed a sphere-formation assay with both U251 and U251R cell
lines. TMZ-resistant U251R cells grew with more notable generation
of spheres in serum-free cultures compared with U251 cells. The
average diameter of the neurospheres formed from U251R cells was
198 µm, whereas that of U251 cells was 80 µm (Fig. 1G and H). This observation is
notable, since recent research indicated that cancer stem cells
(CSCs) are characterized by therapeutic resistance (25). The results led us to further inspect
the stemness-related pathways of TMZ-resistant cells.
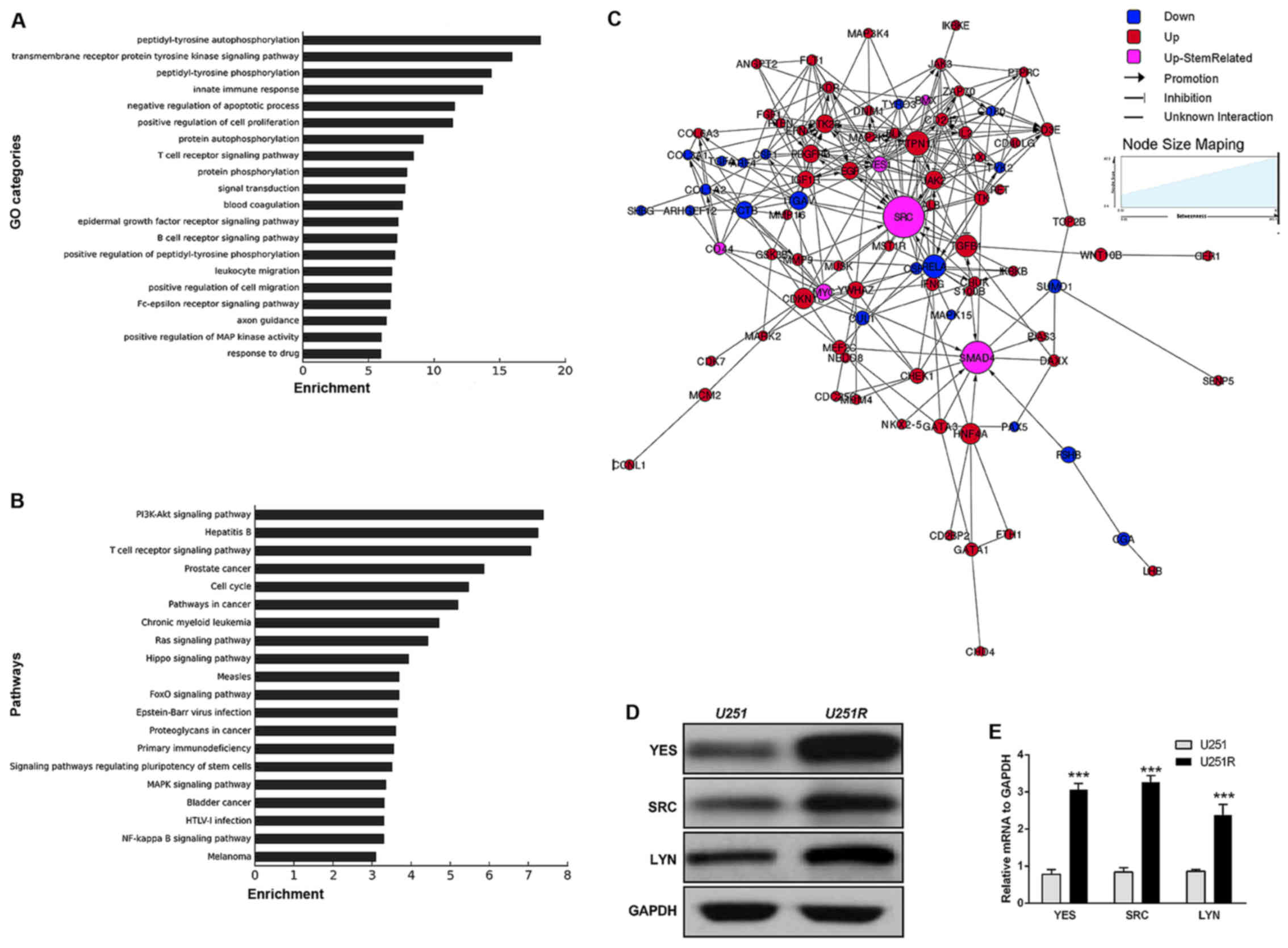
Acquired TMZ resistance-related
differentially expressed proteins
Using a relative expression level of 0.5-fold change
as the threshold, proteins in the U251 and U251R cell lines were
compared. As a result, more than 200 significant differentially
expressed proteins were obtained (data not shown), including
Ferritin, JAK2, MYC, MDM4, PARP, SOX2 and SRC. Of these, 160
proteins were upregulated and 40 proteins were downregulated.
GOs of the differentially expressed proteins were
statistically analyzed. According to the enrichment list, the top
20 GO categories in which differentially expressed genes were
identified included: Transmembrane receptor protein tyrosine kinase
function, negative regulation of cell apoptosis, positive
regulation of cell proliferation, protein phosphorylation and drug
response process (Fig. 2A).
Similarly, pathways of the genes corresponding to differentially
expressed proteins were also statistically analyzed. The results
indicated that up and downregulated genes predominantly
participated in 20 significant pathways, including the PI3K-Akt,
Ras, MAPK and FoxO signaling pathways, as well as some
stemness-related signaling pathways (Fig. 2B).
Acquired TMZ resistance-related
molecular networks
According to the expression dataset acquired from
the protein microarray assay, TMZ resistance-related molecules and
the pathways in which they participated were investigated using the
Human Protein Reference Database. On this basis, a signal
transfection network was constructed (Fig. 2C). The proteins marked in pink were
recognized as stemness- and EMT-related. They were at relatively
significant positions, and interacted with downstream and upstream
molecules participating in cell differentiation, proliferation,
invasion and migration pathways. This molecular profiling, together
with the results of the sphere-formation assay, indicated a
phenotype transition from differentiation to stemness in
TMZ-resistant cells.
TMZ induction upregulates SFK family
kinases in chemoresistant U251 cells at both the protein and mRNA
levels
It was previously reported that glioma stem cells
(GSCs) isolated from GBM patient tissues are more resistant than
normal GBM cells to TMZ and radiotherapy (25). Consistent with this, the results of
our sphere formation assay strengthened the
stemness-chemoresistance link in GBM cells. Further inspection of
the molecular signatures revealed a variety of signaling pathways
that were related to the stem-like phenotype. Among them, a group
of SRC family kinases (SFKs) attracted particular attention. It has
previously been reported that SFKs can mediate stem-like signaling
and EMT in breast cancer, and in head and neck cancer (37,38).
Thus, to demonstrate that SFKs are indeed upregulated in the
chemoresistant U251R cell line, we conducted an immunoblotting
experiment, the results of which were in accordance with those of
the microarray experiment, revealing upregulation of SRC, YES and
LYN (Fig. 2D). Furthermore, RT-qPCR
analysis confirmed that the mRNA levels of these proteins in
TMZ-resistant U251 cells were notably higher compared with those in
the TMZ-sensitive cells (relative expression 3.68 for YES, 2.33 for
LYN and 2.89 for SRC) (Fig.
2E).
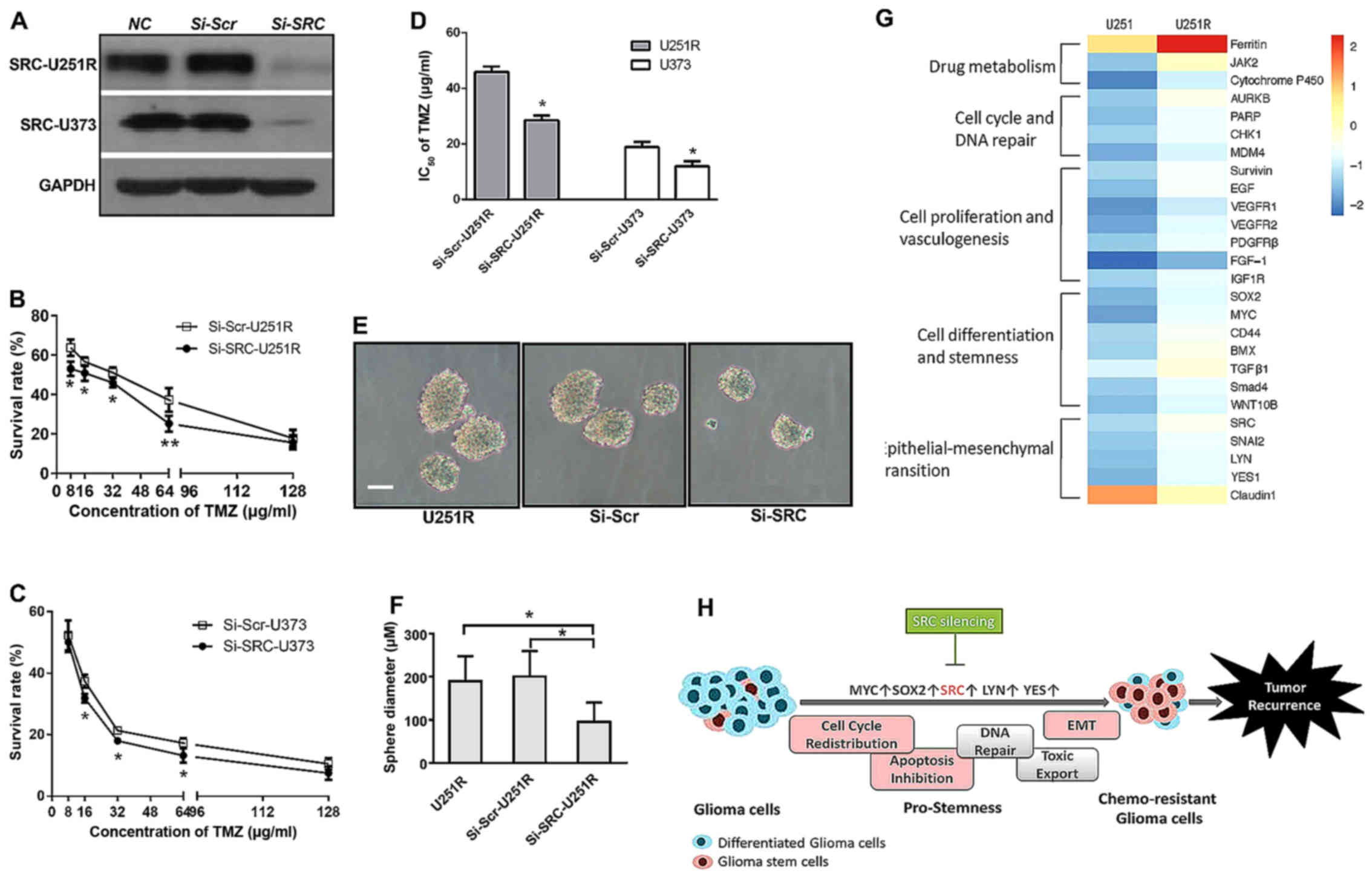
shRNA-mediated SRC knockdown reduces
self-renewal activity and enhances TMZ sensitivity
It was previously found that inhibitors of several
SFK proteins, including an ATP-competitive dual SFK/ABL inhibitor
dasatinib, suppressed the migration of GSCs in vitro
(37). Consistent with this, we
revealed that the characteristics of TMZ-resistant U251R cells were
similar to those of GSCs, and a series of SFK members were
significantly involved in the signaling to confer TMZ resistance.
We then surmised that inhibition of SFKs would affect the TMZ
sensitivity of U251R cells, which was hypothetically related to
cell stemness. In particular, SRC was not only significantly
upregulated, but also located in the central hub of signaling
networks. Therefore, we selectively performed transient siRNA
transfection against human SRC and successfully inhibited SRC
expression in the U251R and U373 cell lines (Fig. 3A). Then, we performed a viability
assay and the results revealed that lentivirus-induced SRC
inhibition partially reversed the increased survival curve of
resistant U251R (Fig. 3B), and
decreased the survival rate of the U373 cells (Fig. 3C). Accordingly, the IC50
values calculated based on the survival curve were clearly reduced
in both cell lines (45.91 vs. 28.58 µg/ml in U251R, and 18.89 vs.
11.98 µg/ml in U373) (Fig. 3D).
Next, we repeated the sphere-formation assay to observe the effect
of SRC-knockdown on cell self-renewal. The increase in sphere
diameter in U251R cells was abolished after Si-SRC transfection.
The average diameter was 186 µm in U251R, 200 µm in scramble
siRNA-transfected U251R, and 102 µm in SRC siRNA-transfected U251R
cells (Fig. 3E and F). These
collective results indicated that enhanced sensitivity to TMZ in
U251R and U373 cells is associated with an SRC-mediated stem-like
phenotype.
Discussion
The underlying mechanism for acquired TMZ resistance
in glioblastoma is considered to be complex (10). In addition to the well-studied
mechanisms of adenosine triphosphate binding cassette (ABC)
superfamily-associated drug transport to avoid the penetration of
chemotherapeutics into the brain (11–13),
glioblastoma cells may also initiate DNA damage repair. This could
include enhanced base excision repair (BER), nucleotide excision
repair (NER), impaired MMR and the more specific
single/double-stand break repair pathways to restore DNA integrity
(6,14–16).
In addition, two equally important cell fate-decisive processes,
apoptosis and autophagy, are heavily involved in chemoresistance.
The former is usually determined by the balance of Bcl-2 family
members, comprised of pro-apoptotic and anti-apoptotic factors
(13,17,18),
while the latter depends on the stage and extent of lysosomal
degradation of chemotherapeutic toxicants (19,20).
Moreover, drug metabolism activity may serve as another
reinforcement to protect cancer cells from drug-induced
cytotoxicity, which is represented by increased expression of
glutathione S-transferase (GST) in glioblastoma cells (21). Finally, two cell
differentiation-related phenotypes, EMT and cancer stemness (e.g.,
glioma stem cells), have been shown to give tumors quiescent
properties, in order to evade the therapeutic window of most
alkylating agents (9,22).
In the present study, we managed to successfully
isolate U251R cells with a four-fold IC50 value,
compared with the parental U251 cell line, by incremental TMZ
induction, which imitated clinical TMZ administration. FACS
analysis revealed an increased rate of G2/M arrest and a decreased
S-phase fragment, as well as a reduced apoptotic rate in U251R
cells compared with the parental U251 cells. These results
indicated that the acquisition of TMZ chemoresistance could be
attributed to cell cycle alterations and apoptosis suppression.
G2/M arrest, a distinct phenomenon following TMZ treatment, is
widely accepted to be critical for TMZ toxicity caused by
O6-G lesions and the subsequent deleterious MMR response
(19). It is argued that the DNA
repair response is tightly linked with the G2 checkpoint and G2/M
arrest, with the notion that glioblastoma cells can initiate DNA
repair pathways when arrested by TMZ treatment so as to prevent
cells from apoptotic death (23,24).
The current findings of both G2/M arrest and apoptosis inhibition
support the view that G2/M arrest induced by TMZ is not just a
destructive but also a reconstructive process for glioma cells.
Another effect of TMZ on glioblastoma cells was the reduced
duplication phase. This has previously been reported in six
glioblastoma cell lines treated with 100 µM TMZ for 72 h (19). Similarly, our FACS study found that
a reduced rate of S-phase cells was accompanied by slower
proliferation in the U251R cell line (data not shown). Previously,
Ye et al reported that GSCs are slow-cycling and
tumor-initiating cells. Additionally, Chen et al indicated
that CD133+ head-neck cancer stem cells expressed low
levels of epithelial differentiation maker CK18 and had a reduced
apoptosis rate (25,26). Since DNA repair, apoptosis
suppression and slower growth are all characteristics of cell
stemness, we performed a sphere-formation assay in the present
study, and found that TMZ-treated U251R cells exhibited greater
self-renewal capacity compared with TMZ-sensitive cells.
Considering previous findings with our current results, we suggest
that U251R cells may acquire stem-like attributes, which confer
their chemoresistance during TMZ treatment.
Next, we performed a high-throughput comparative
protein microarray in order to further investigate the mechanisms
for acquired chemoresistance. We first noticed that MGMT, MMR
members (MSH2/3/6) and ABC family proteins, which participate in
DNA repair and drug detoxification, were not significantly altered
between U251 and U251R cell lines. Although the methylation status
of the MGMT promoter could not be determined by the antibody-based
screening, the results implied that the mechanisms of
chemoresistant glioma cells are versatile. In addition, 200
differentially expressed molecules, participating in a variety of
biological processes and signaling pathways, were identified.
Ferritin is an intracellular iron storage protein
that localizes to the cell nuclei to protect DNA from oxidative
damage. It has been reported that ferritin blockade can increase
tumor sensitivity to chemotoxins in glioblastoma cells (27). As the most significant upregulated
protein in the TMZ-resistant cell line, ferritin is likely to play
a critical role in the regulation of TMZ chemoresistance. It is
well known that cytochrome P450s (CYPs) are crucial to drug
metabolism in addition to GST. Increasing expression of cytochrome
P450 in U251R indicated that cytochrome P450 is likely to
participate in the process of detoxification by accelerating TMZ
metabolism (28). JAK2 and AURKB
are two other upregulated molecules able to catalyze
phosphorylation of a spectrum of protein substrates. A recent study
found that JAK2-mediated Y41 phosphorylation and AURKB-mediated S10
phosphorylation on histone H3 were associated with
radio-chemoresistance in patients with GBM. Kinase inhibitors
targeting AURKB enhanced both radiation- and TMZ-sensitivity
(29,30). In addition, AURKB is essential for
G2 checkpoint-related G2/M transition. Therefore, from our cell
cycle analyses, it is reasonable to surmise that there is a close
relationship between G2/M arrest and the upregulation of AURKB
expression. We noticed the upregulated expression of various DNA
repair- and apoptosis-associated proteins, including PARP, CHK1,
MDM4 and Survivin (31–34), which was concordant with the
observed G2/M arrest and apoptosis inhibition in U251R cells.
Furthermore, transmembrane receptor tyrosine kinase (i.e., growth
factor receptor) has previously been correlated with glioblastoma
therapeutic resistance through MDR1 (a major membrane protein
transporter) (35,36). According to the protein-chip
results, several members of growth factor receptor signaling
pathways, including EGF, VEGFR1, VEGFR2, PDGFRβ, FGF-1 and IGF-1R,
were upregulated in the U251R cell line.
Most notably, a group of stemness- and EMT-related
proteins, including MYC, SRC, SOX2, CD44, BMX, TGFβ1, SMAD4, SNAI2,
LYN and YES, were overexpressed in U251R cells, whereas epithelial
proteins, such as Claudin1 and E-cadherin, were downregulated. In
the past, Auger et al identified a stem cell-like genotype
in SNB-19 TMZ-resistant variants, while Ye et al observed
slow-cycling and strong radiation-resistance in patient
tumor-derived GSC cultures (25,37).
Additionally, EMT is commonly regarded to be closely associated
with cell stemness, since EMT transition could lead to misplaced
stemness properties, while cancer stem cells are capable of
aberrantly communicating with peri-tumor niches for migration like
mesenchymal cells (26).
Collectively, with the observed stem-like behavior of U251R cells,
we propose the hypothesis that glioma cells developing stemness
convert to chemoresistant glioma cells (Fig. 3H).
SFKs form a family of non-receptor tyrosine kinases
that is intensively involved in glioma tumorigenesis, invasion and
migration. There are nine members of the SFK family, of which c-Src
(also referred to as SRC), Fyn, Lyn, Yes and Lck were shown to be
highly expressed in GBM samples (38,39).
Our protein-chip based observations indicated significant
upregulation of and a central role for SRC in the networks
regulating TMZ-resistance. Accordingly, SRC silencing was able to
reverse the acquired survival advantage of U251R cells, while
sensitizing U373 cells to TMZ treatment. Empirically, SRC has been
linked to cancer cell stemness. Boivin et al and Chen et
al proposed that CD133 is a novel binding partner of SRC. SRC
inhibitor PP2 reduced the expression of SOX2-interacting stemness
marker Oct4 and could reverse the EMT pattern in head-neck cancer
stem cells (26,40). Consistent with this, we observed
that SRC silencing in U251R cells inhibited their self-renewal, a
prominent biological characteristic of stem-like cells. Moreover,
Li et al demonstrated that SRC/Akt/mTOR signaling cascades
are important for the maintenance of leukemic stemness (41). In fact, our previous study reported
a suppressive effect of Akt2 knockdown on TMZ sensitivity both
in vitro and in vivo (13). Therefore, the regulatory axis of
SRC/AKT2 remains promising, although the current findings may not
establish a direct connection between SRC-mediated stemness
interruption and TMZ chemoresistance.
There were several limitations of the present study,
which may weaken the clinical relevance of the results. Firstly,
the variant U251R cell line was not derived from patients with GBM.
Secondly, radiation treatment was not administered along with TMZ
to induce the therapy resistance in U251 cells. However, we
depicted a network with a wide range of molecules and signaling
pathways conferring the acquired TMZ resistance, and proposed a
promising therapeutic modality targeting the pro-stemness process
that is regulated by SRC.
Acknowledgements
The authors thank Professor Yicheng Lu, as a forever
mentor, for his outstanding academic guidance.
References
|
1
|
Bleeker FE, Molenaar RJ and Leenstra S:
Recent advances in the molecular understanding of glioblastoma. J
Neurooncol. 108:11–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Van Meir EG, Hadjipanayis CG, Norden AD,
Shu HK, Wen PY and Olson JJ: Exciting new advances in
neuro-oncology: The avenue to a cure for malignant glioma. CA
Cancer J Clin. 60:166–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Newlands ES, O'Reilly SM, Glaser MG, Bower
M, Evans H, Brock C, Brampton MH, Colquhoun I, Lewis P,
Rice-Edwards JM, et al: The Charing Cross Hospital experience with
temozolomide in patients with gliomas. Eur J Cancer. 32A:2236–2241.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: European Organisation for Research and Treatment of
Cancer Brain Tumor and Radiotherapy Groups; National Cancer
Institute of Canada Clinical Trials Group: Radiotherapy plus
concomitant and adjuvant temozolomide for glioblastoma. N Engl J
Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang J, Stevens MF, Laughton CA,
Madhusudan S and Bradshaw TD: Acquired resistance to temozolomide
in glioma cell lines: Molecular mechanisms and potential
translational applications. Oncology. 78:103–114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
D'Atri S, Tentori L, Lacal PM, Graziani G,
Pagani E, Benincasa E, Zambruno G, Bonmassar E and Jiricny J:
Involvement of the mismatch repair system in temozolomide-induced
apoptosis. Mol Pharmacol. 54:334–341. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu C and Shervington A: Chemoresistance in
gliomas. Mol Cell Biochem. 312:71–80. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hall DA, Ptacek J and Snyder M: Protein
microarray technology. Mech Ageing Dev. 128:161–167. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sørensen MD, Fosmark S, Hellwege S, Beier
D, Kristensen BW and Beier CP: Chemoresistance and chemotherapy
targeting stem-like cells in malignant glioma. Adv Exp Med Biol.
853:111–138. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Izumiya M, Kabashima A, Higuchi H,
Igarashi T, Sakai G, Iizuka H, Nakamura S, Adachi M, Hamamoto Y,
Funakoshi S, et al: Chemoresistance is associated with cancer stem
cell-like properties and epithelial-to-mesenchymal transition in
pancreatic cancer cells. Anticancer Res. 32:3847–3853.
2012.PubMed/NCBI
|
|
11
|
von Manstein V, Yang CM, Richter D, Delis
N, Vafaizadeh V and Groner B: Resistance of cancer cells to
targeted therapies through the activation of compensating signaling
loops. Curr Signal Transduct Ther. 8:193–202. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Spiegl-Kreinecker S, Buchroithner J,
Elbling L, Steiner E, Wurm G, Bodenteich A, Fischer J, Micksche M
and Berger W: Expression and functional activity of the
ABC-transporter proteins P-glycoprotein and multidrug-resistance
protein 1 in human brain tumor cells and astrocytes. J Neurooncol.
57:27–36. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cui Y, Lin J, Zuo J, Zhang L, Dong Y, Hu
G, Luo C, Chen J and Lu Y: AKT2-knockdown suppressed viability with
enhanced apoptosis, and attenuated chemoresistance to temozolomide
of human glioblastoma cells in vitro and in vivo. Onco Targets
Ther. 8:1681–1690. 2015.PubMed/NCBI
|
|
14
|
Zhang J, Stevens MF and Bradshaw TD:
Temozolomide: Mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bobola MS, Kolstoe DD, Blank A,
Chamberlain MC and Silber JR: Repair of 3-methyladenine and abasic
sites by base excision repair mediates glioblastoma resistance to
temozolomide. Front Oncol. 2:1762012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fu D, Calvo JA and Samson LD: Balancing
repair and tolerance of DNA damage caused by alkylating agents. Nat
Rev Cancer. 12:104–120. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Happold C, Roth P, Wick W, Schmidt N,
Florea AM, Silginer M, Reifenberger G and Weller M: Distinct
molecular mechanisms of acquired resistance to temozolomide in
glioblastoma cells. J Neurochem. 122:444–455. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wertz IE, Kusam S, Lam C, Okamoto T,
Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al:
Sensitivity to antitubulin chemotherapeutics is regulated by MCL1
and FBW7. Nature. 471:110–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin C-J, Lee C-C, Shih YL, Lin CH, Wang
SH, Chen TH and Shih CM: Inhibition of mitochondria- and
endoplasmic reticulum stress-mediated autophagy augments
temozolomide-induced apoptosis in glioma cells. PLoS One.
7:e387062012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kogias E, Osterberg N, Baumer B, Psarras
N, Koentges C, Papazoglou A, Saavedra JE, Keefer LK and Weyerbrock
A: Growth-inhibitory and chemosensitizing effects of the
glutathione-S-transferase-π-activated nitric oxide donor PABA/NO in
malignant gliomas. Int J Cancer. 130:1184–1194. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu G, Yuan X, Zeng Z, Tunici P, Ng H,
Abdulkadir IR, Lu L, Irvin D, Black KL and Yu JS: Analysis of gene
expression and chemoresistance of CD133+ cancer stem
cells in glioblastoma. Mol Cancer. 5:672006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takuro H, Kazuhide A and Yuichi H: The Cdk
inhibitor flavopiridol enhances temozolomide-induced cytotoxicity
in human glioma cells. J Neurooncol. 115:169–178. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hirose Y, Katayama M, Mirzoeva OK, Berger
MS and Pieper RO: Akt activation suppresses Chk2-mediated,
methylating agent-induced G2 arrest and protects from
temozolomide-induced mitotic catastrophe and cellular senescence.
Cancer Res. 65:4861–4869. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ye F, Zhang Y, Liu Y, Yamada K, Tso JL,
Menjivar JC, Tian JY, Yong WH, Schaue D, Mischel PS, et al:
Protective properties of radio-chemoresistant glioblastoma stem
cell clones are associated with metabolic adaptation to reduced
glucose dependence. PLoS One. 8:e803972013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen Y-S, Wu M-J, Huang C-Y, Lin SC,
Chuang TH, Yu CC and Lo JF: CD133/Src axis mediates tumor
initiating property and epithelial-mesenchymal transition of head
and neck cancer. PLoS One. 6:e280532011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu X, Madhankumar AB, Slagle-Webb B,
Sheehan JM, Surguladze N and Connor JR: Heavy chain ferritin siRNA
delivered by cationic liposomes increases sensitivity of cancer
cells to chemotherapeutic agents. Cancer Res. 71:2240–2249. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Isvoran A, Louet M, Vladoiu DL, Craciun D,
Loriot MA, Villoutreix BO and Miteva MA: Pharmacogenomics of the
cytochrome P450 2C family: Impacts of amino acid variations on drug
metabolism. Drug Discov Today. 22:366–376. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pacaud R, Cheray M, Nadaradjane A,
Vallette FM and Cartron PF: Histone H3 phosphorylation in GBM: A
new rational to guide the use of kinase inhibitors in anti-GBM
therapy. Theranostics. 5:12–22. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Borges KS, Castro-Gamero AM, Moreno DA, da
Silva Silveira V, Brassesco MS, de Paula Queiroz RG, de Oliveira
HF, Carlotti CG Jr, Scrideli CA and Tone LG: Inhibition of Aurora
kinases enhances chemosensitivity to temozolomide and causes
radiosensitization in glioblastoma cells. J Cancer Res Clin Oncol.
138:405–414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tentori L, Ricci-Vitiani L, Muzi A,
Ciccarone F, Pelacchi F, Calabrese R, Runci D, Pallini R, Caiafa P
and Graziani G: Pharmacological inhibition of poly(ADP-ribose)
polymerase-1 modulates resistance of human glioblastoma stem cells
to temozolomide. BMC Cancer. 14:1512014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tang TK, Chiu SC, Lin CW, Su MJ and Liao
MH: Induction of survivin inhibition, G2/M cell cycle arrest and
autophagic on cell death in human malignant glioblastoma cells.
Chin J Physiol. 58:95–103. 2015.PubMed/NCBI
|
|
33
|
Zhao Z, Liu Y, He H, Chen X, Chen J and Lu
YC: Candidate genes influencing sensitivity and resistance of human
glioblastoma to Semustine. Brain Res Bull. 86:189–194. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Eich M, Roos WP, Nikolova T and Kaina B:
Contribution of ATM and ATR to the resistance of glioblastoma and
malignant melanoma cells to the methylating anticancer drug
temozolomide. Mol Cancer Ther. 12:2529–2540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Munoz JL, Rodriguez-Cruz V, Greco SJ,
Nagula V, Scotto KW and Rameshwar P: Temozolomide induces the
production of epidermal growth factor to regulate MDR1 expression
in glioblastoma cells. Mol Cancer Ther. 13:2399–2411. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Popescu AM, Alexandru O, Brindusa C,
Purcaru SO, Tache DE, Tataranu LG, Taisescu C and Dricu A:
Targeting the VEGF and PDGF signaling pathway in glioblastoma
treatment. Int J Clin Exp Pathol. 8:7825–7837. 2015.PubMed/NCBI
|
|
37
|
Auger N, Thillet J, Wanherdrick K, Idbaih
A, Legrier ME, Dutrillaux B, Sanson M and Poupon MF: Genetic
alterations associated with acquired temozolomide resistance in
SNB-19, a human glioma cell line. Mol Cancer Ther. 5:2182–2192.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han X, Zhang W, Yang X, Wheeler CG,
Langford CP, Wu L, Filippova N, Friedman GK, Ding Q,
Fathallah-Shaykh HM, et al: The role of Src family kinases in
growth and migration of glioma stem cells. Int J Oncol. 45:302–310.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Choi YL, Bocanegra M, Kwon MJ, Shin YK,
Nam SJ, Yang JH, Kao J, Godwin AK and Pollack JR: LYN is a mediator
of epithelial-mesenchymal transition and a target of dasatinib in
breast cancer. Cancer Res. 70:2296–2306. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Boivin D, Labbé D, Fontaine N, Lamy S,
Beaulieu E, Gingras D and Béliveau R: The stem cell marker CD133
(prominin-1) is phosphorylated on cytoplasmic tyrosine-828 and
tyrosine-852 by Src and Fyn tyrosine kinases. Biochemistry.
48:3998–4007. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li XY, Jiang LJ, Chen L, Ding ML, Guo HZ,
Zhang W, Zhang HX, Ma XD, Liu XZ, Xi XD, et al: RIG-I modulates
Src-mediated AKT activation to restrain leukemic stemness. Mol
Cell. 53:407–419. 2014. View Article : Google Scholar : PubMed/NCBI
|