Introduction
Colorectal cancer (CRC) is the third most common
type of cancer and the fourth leading cause of cancer-related
deaths worldwide (1). Globally,
~1.2 million patients are newly diagnosed with CRC each year, and
>600,000 succumb to this malignancy (2). At the time of their diagnosis, ~25% of
patients with CRC already have distal liver and lung metastases,
which often means that they have missed the opportunity for radical
surgery. In addition, 35–55% of patients with CRC with disease
progression may develop distal liver and lung metastases, resulting
in a marked decline in survival (3–5). It is
estimated that stage I patients with CRC have a 5-year survival
rate of 80–90%, while patients with advanced disease have a 5-year
survival rate of <10% (6). Early
diagnosis and early treatment are therefore of great significance
in improving the survival and prognosis of patients with CRC.
During the past several decades, there has been a
stable but slow improvement in the prognosis of CRC (1). However, this could be greatly improved
by precision medicine, which facilitates the improvement of disease
diagnosis and the development of novel treatments, and helps select
the optimal treatment strategy for CRC based on gene mutation
detection (7). Based on analyses of
cohorts comprised of twins from Sweden, Denmark and Finland,
heritable factors are thought to contribute ~35% to CRC (8). To date, a number of genes (including
APC, p53 and KRAS) have been identified to be
involved in the pathogenesis of CRC; however, there is still a
large number of unidentified genes associated with the pathogenesis
of CRC (9,10). Although several new hereditary CRC
susceptibility genes have been identified, including DNA repair
genes, DNA replication genes and genes related to the maintenance
of genome stability (11), there is
still no compelling evidence to support candidate genes for routine
genetic diagnosis (12). Since the
development of most CRC cases cannot be explained by known genes,
the identification of CRC susceptibility genes is a high priority
and is urgently needed.
As a comprehensive and coordinated effort to
accelerate our understanding of the molecular basis of cancer, The
Cancer Genome Atlas (TCGA) has generated comprehensive,
multi-dimensional maps of the key genomic changes in multiple
cancer types (13). In the present
study, we screened a gene that has been shown to be differentially
expressed in the development and progression of CRC,
Spermatid-associated (SPERT), based on the TCGA dataset.
Subsequently, we examined the effect of SPERT knockdown on
the proliferation and apoptosis of the human CRC cell line RKO, and
explored the mechanisms underlying these effects.
Materials and methods
Screening of target genes and
evaluation of correlations between gene expression and
clinicopathological characteristics
The TCGA dataset contains colon adenocarcinoma
(COAD) and rectum adenocarcinoma (READ) data. When we analyzed the
difference between cancer and peri-cancer expression for candidate
gene screening, we selected paired samples from RNA-sequencing
(RNA-Seq) and RNA-Seq Version 2 (RNA-SeqV2) to analyze the data (50
pairs). There were 41 pairs of colon cancer samples with
pathological information and 9 pairs of rectal cancer samples with
pathological information. For each gene symbol, the transcript with
the highest expression was used for analysis (original reads
>50), and transcripts were normalized with the Trimmed Mean of
M-values (TMM) method (14). The
following criteria were used for the screening of candidate genes:
i) genes that have already been reported to be involved in CRC were
excluded; ii) multi-transmembrane protein genes were excluded,
because multiple transmembrane proteins generally have large
molecular weights that are not easy to perform gene manipulation
(e.g., knocked down or overexpressed), and most transmembrane
proteins play the role of signal transmission and transduction;
iii) genes with undefined annotations (such as open reading frames)
were excluded; iv) genes with >100 publications in PubMed were
excluded intended to ensure the originality and innovation of the
genes we have screened. In order to render the analysis data more
convincing, the range of analysis was extended to 624 RNA-Seq
samples of colorectal cancer from the TCGA dataset after obtaining
the differentially expressed SPERT gene in pairs of samples,
to further analyze the difference of SPERT gene expression
in different pathological stages of colorectal cancer.
Construction of shRNA lentiviral
vectors
According to the RNA interference (RNAi) sequence,
multiple RNAi target sequences (19–21 nucleotides in length) were
designed, using the SPERT gene as a template. Following assessment
by the design software, the sequence 5′-ACAAGATCCTACAGGTCTT-3′ was
selected as an RNAi target (shSPERT): Forward primer,
5′-TACTTCTCCCCATCCGCCTCC-3′ and reverse primer,
5′-GCGACGTGGTCCTTCTTCACC-3′. The scramble sequence
5′-TTCTCCGAACGTGTCACGT-3′ served as an RNAi negative control
(shCtrl): Forward primer, 5′-CCATGATTCCTTCATATTTGC-3′ and reverse
primer, 5′-GTAATACGGTTATCCACGCG-3′. The shRNA lentiviral vector
targeting the SPERT gene (LV-SPERT-RNAi) and the control lentiviral
vector (LV-shRNA-NC) were constructed and packaged by Shanghai
Genechem Co., Ltd. (Shanghai, China). The lentiviral vector
contained a GFP open reading frame that could quickly and directly
display the efficiency of lentiviral infection, which helped us to
judge the expression of exogenous knockdown sequences in cultured
cells. In addition, there was also a FLAG-tag which could be
recognized by the Anti-Flag antibody, so it was convenient to
detect and identify the target protein containing FLAG by western
blotting.
Detection of SPERT expression in CRC
cell lines by RT-qPCR
The human CRC cell lines RKO, SW480 and HCT116 were
purchased from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China), and were screened to determine SPERT
expression. Total RNA was isolated from cells using
TRIzol® reagent (Shanghai Pufei Biotech Co., Ltd.,
Shanghai, China) and reverse-transcribed into cDNA. SPERT
gene expression (forward primer, 5′-TACTTCTCCCCATCCGCCTCC-3′ and
reverse primer, 5′-GCGACGTGGTCCTTCTTCACC-3′) was quantified in
these three cells lines using a Roche LightCycler® 480
Real-Time PCR platform (Roche Diagnostics, Indianapolis, IN, USA)
with 40 cycles at 95°C for 5 sec and at 60°C for 30 sec, while
GAPDH (forward primer, 5′-TGACTTCAACAGCGACACCCA-3′ and
reverse primer, 5′-CACCCTGTTGCTGTAGCCAAA-3′) served as the internal
control. The relative quantity was calculated using the
2−ΔΔCq method. Detection of SPERT expression was
repeated in triplicate in each cell line. Of the three cell lines,
the cell line expressing the highest level of SPERT was
selected for use in further experiments.
The same RT-qPCR method was used to quantify
SPERT mRNA levels after transfection of the chosen cell
line, RKO, with the shRNA lentiviral constructs (as described
below).
RKO cell culture and transfection
Human CRC RKO cells were incubated in RPMI-1640
medium (Beyotime Institute of Biotechnology, Shanghai, China)
supplemented with 10% fetal bovine serum (Ausbian, Adelaide,
Australia) at 37°C in an atmosphere of 5% CO2. Log-phase
cells were digested with trypsin (Sangon Biotech Co., Ltd.,
Shanghai, China), and prepared as cell suspensions with complete
DMEM (Corning Life Sciences, Manassas, VA, USA) at a density of
3×104-5×104 cells/ml. Cells were then seeded
into culture plates and grown until they reached 15–30% confluence.
Subsequently, the cells were transfected with shSPERT or shCtrl at
a multiplicity of infection (MOI) of 10, in the presence of
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Carlsbad, CA, USA). The medium was changed to
RPMI-1640 at 8–12 h post-transfection, and the target gene
expression was observed under an Olympus IX71 inverted fluorescence
microscope (Olympus, Tokyo, Japan) at 72 h post-transfection.
Western blot analysis
At 48 h post-transfection, total protein was
extracted using RIPA lysis buffer and quantified using a BCA
protein assay kit (both from Beyotime Institute of Biotechnology).
Total protein was then separated by 10% SDS-PAGE, and transferred
to PVDF membranes (EMD Millipore, Bedford, MA, USA), which were
blocked and incubated at 4°C overnight. The blots were then
incubated with mouse anti-FLAG monoclonal antibodies (1:2,000
dilution; cat. no. F1804; Sigma-Aldrich Trading Co. Ltd., Shanghai,
China), washed three times (10 min each) with TBST, incubated with
the secondary antibody (goat anti-mouse IgG; dilution, 1:2,000;
cat. no. sc-2005; Santa Cruz Biotechnology, Co., Ltd., Shanghai,
China) for 1 h, and washed three times (10 min each) with TBST. The
membranes were viewed with an Odyssey fluorescence imaging system
(LI-COR Biosciences, Lincoln, NE, USA), and immunoreactive protein
bands were visualized with the Pierce™ ECL Western Blotting
Substrate (Pierce, Rockford, IL, USA). ImageJ (version 1.46
release; National Institutes of Health, Bethesda, MD, USA) software
was used for quantification of the western blots.
Celigo-based cell-counting and
viability assay
Log-phase cells were digested with trypsin,
resuspended in complete DMEM and counted. Cells were then seeded
into cell culture plates at a density of 2,000 cells/well in a
100-µl system, and three replicate wells were established for each
group. Cells were incubated at 37°C in an atmosphere of 5%
CO2. Commencing at day 2 post-seeding, the cells were
counted on a Celigo® Image Cytometer (Nexcelom
Bioscience, Lawrence, MA, USA) daily for 3–5 successive days. The
5-day cell growth curve was plotted.
MTT assay
Log-phase cells were digested with trypsin,
resuspended in complete DMEM and counted. Cells were then seeded
into cell culture plates, and three replicate wells were
established for each group. Cells were incubated at 37°C in an
atmosphere of 5% CO2. At day 2 post-seeding, 20 µl MTT
solution (5 mg/ml; GenView Corp., Houston, TX, USA) was added to
each well and incubated for 4 h, before the culture solution was
completely removed, leaving the formazan crystals on the bottom of
the well. Subsequently, 100 µl DMSO (Shanghai Shiyi Chemicals
Reagent Co., Ltd., Shanghai, China) was added to dissolve the
formazan crystals. Following vibration for 2–5 min, the optical
density (OD) was measured at 490 nm using a Tecan Infinite M200 Pro
Microplate Reader (Tecan Group, Ltd., Männedorf, Switzerland).
Caspase-Glo® 3/7 assay
Cells were seeded into 96-well plates, and incubated
at 37°C with 5% CO2 for 3–5 days. Following cell
counting, the cell density was adjusted to 1×104
cells/ml at room temperature. shSPERT- and shCtrl-transfected cells
were transferred to a new 96-well plate, with 100 µl medium/well.
Wells containing blank medium served as the negative controls.
Subsequently, 100 µl of Caspase-Glo reagent (Promega Corp.,
Madison, WI, USA) was added to each well, and the well content was
gently mixed with a plate shaker at 100–200 × g/min for 30 min.
Following incubation at 18–22°C for 0.5–3 h, the signal intensity
was measured.
Flow cytometry
Log-phase cells were harvested, digested with
trypsin, and resuspended in complete DMEM. The cell suspension and
supernatant were then transferred to a 5-ml centrifuge tube and
centrifuged at 300 × g/min for 5 min. Three replicate wells were
assigned for each group (≥5×105 cells). The supernatant
was then discarded, and the sediment was washed in 4°C precooled
D-Hanks' Balanced Salt Solution (pH 7.2–7.4). Subsequently, the
cells were washed with 1X binding buffer and centrifuged at 300 ×
g/min for 3 min. The supernatant was discarded, and the sediment
was resuspended in 200 µl of 1X binding buffer, and stained with 10
µl of Annexin V-APC (eBioscience, San Diego, CA, USA) at room
temperature in darkness for 10–15 min. To each tube, 500 µl of 1X
binding buffer was added, and the cells were then subjected to flow
cytometry. All measurements were repeated in triplicate.
Detection of SPERT-related biological
pathways
SPERT-related biological pathways were detected with
the PathScan® Signaling Antibody Array Kit (Cell
Signaling Technology, Inc., Danvers, MA, USA) following the
manufacturer's instructions. Briefly, the cells were lysed, and
incubated in 75 µl of 1X antibody mixture on a horizontal shaker
for 1 h. The antibody mixture was removed, and the cells were
washed four times with 1X wash buffer on a horizontal shaker (5 min
each time). Then, the cells were incubated in 75 µl of 1X
HRP-conjugated streptavidin on a horizontal shaker for 0.5 h.
Following removal of HRP-conjugated streptavidin, the cells were
washed four times with 1X wash buffer on a horizontal shaker (5 min
each time). Finally, the cell slides were immersed in 1X wash
buffer, visualized and analyzed. Cell slides were immersed in 1X
washing buffer. Exposure buffer was formulated with 9 ml
ddH20, 0.5 ml LumiGLO (Cell Signaling Technology,
Danvers, USA) and 0.5 ml peroxide, and the cell slides were
incubated with exposure buffer, then exposed within 1–2 sec using a
visualization system (Clinx ChemiScope 5300; Clinx Science
Instruments, Co., Ltd., Shanghai, China). The resulting images were
analyzed using the visualization system aforementioned, and raw
data were further analyzed manually.
Statistical analysis
All measurement data are presented as the mean ±
standard deviation (SD). Differences in the means between groups
were tested for statistical significance with the Student's t-test,
and comparison of proportions was conducted with a Chi-square test.
The association of SPERT expression with clinicopathological
characteristics was examined with a Mann-Whitney U test. All
statistical analyses were performed using SPSS statistical software
version 17.0 (SPSS, Inc., Chicago, IL, USA), and a P-value of
<0.05 was considered to indicate a statistically significant
difference.
Results
Associations of SPERT expression with
clinicopathological characteristics
All data were obtained from highly reliable genetic
disease databases, and the gene list was finally obtained following
random condensation (Table I). In
fact, we screened ~6,000 disease-related genes in different tumor
types, but to ensure the originality and innovation of the
candidate genes, we selected the gene SPERT through the four
screening conditions aforementioned. We first analyzed
high-throughput RNA-sequencing data of the colon adenocarcinoma
(COAD) and rectal adenocarcinoma (READ) cohorts of TCGA, and found
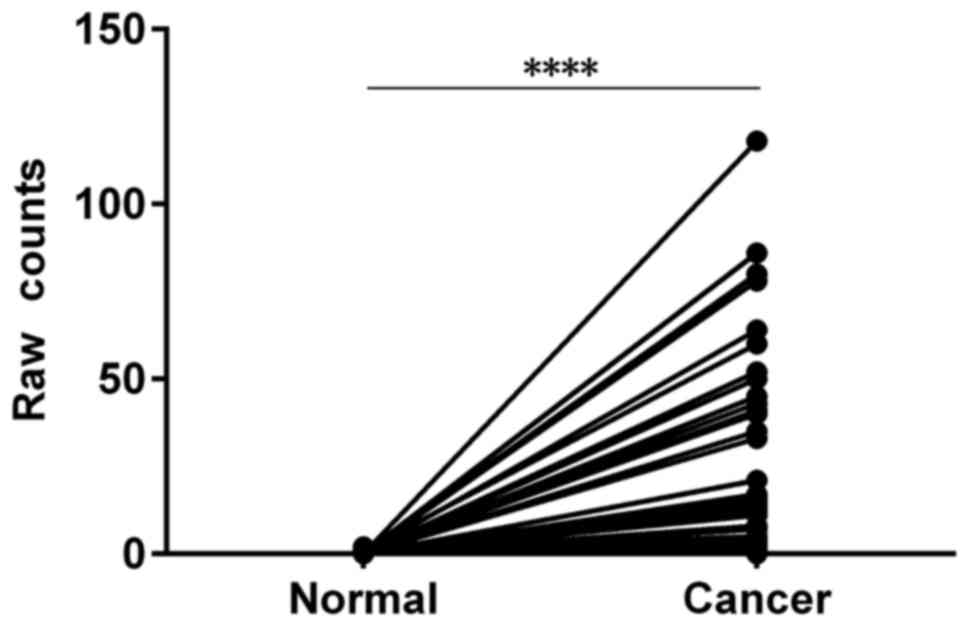
that SPERT expression was significantly increased in CRC
tissues compared with peri-cancer tissues (P<0.0001) (Table II and Fig. 1). Then, we analyzed the relationship
between SPERT expression and various clinicopathological
parameters of patients with CRC. The Mann-Whitney U test revealed
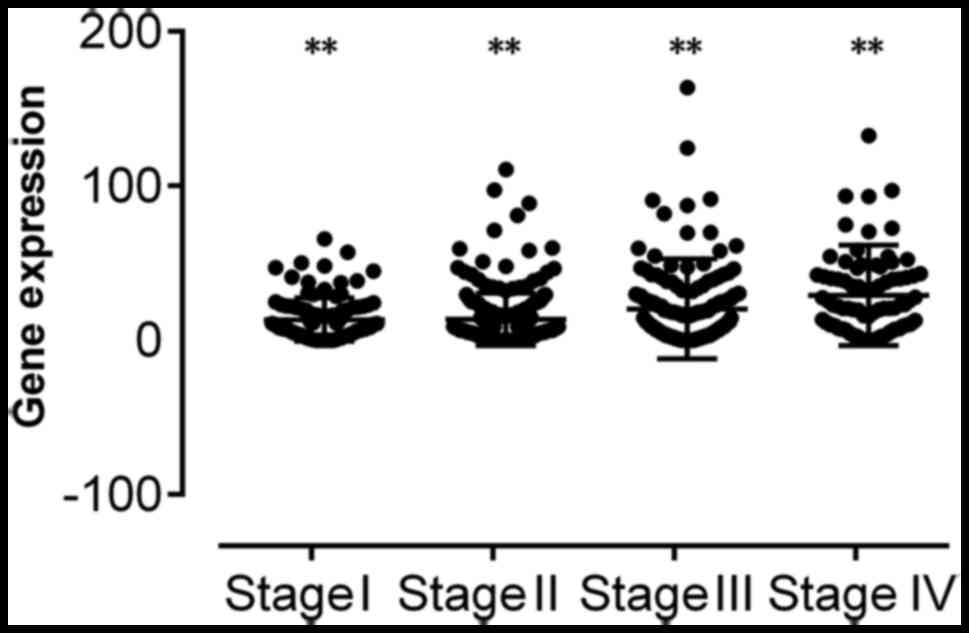
that SPERT expression was associated with N, M and
pathological stages in patients with CRC (all P<0.01) (Table III and Fig. 2).
 | Table I.Gene list for analysis based on The
Cancer Genome Atlas dataset. |
Table I.
Gene list for analysis based on The
Cancer Genome Atlas dataset.
| Gene ID | Gene name | No. of
transcripts | Publications in
PubMed | Novoseek disease
relationships for the gene | MalaCards disease
relationships for the gene |
|---|
| 220082 | SPERT | 3 | 13 | 0 | 0 |
| Table II.SPERT expression in colorectal
cancer and peri-cancer specimens in The Cancer Genome Atlas
dataset. |
Table II.
SPERT expression in colorectal
cancer and peri-cancer specimens in The Cancer Genome Atlas
dataset.
|
|
|
|
| No. of samples |
|---|
|
|
|
|
|
|
|---|
| ID | Gene symbol | FC | P-value | Total | With unchanged
SPERT expression | With upregulated
SPERT expression | With downregulated
SPERT expression |
|---|
| 220082 | SPERT | 100.975 | 2.94E-56 | 50 | 5 | 45 | 0 |
| Table III.Associations of SPERT
expression with clinicopathological features in human colorectal
cancer patients. |
Table III.
Associations of SPERT
expression with clinicopathological features in human colorectal
cancer patients.
|
| SPERT expression,
n |
|
|
|---|
|
|
|
|
|
|---|
| Clinicopathological
features | Low | High | Total, n | P-value |
|---|
| N |
| N0 | 197 | 155 | 352 | <0.001 |
|
N1/2 | 110 | 155 | 265 |
|
| Total | 307 | 310 | 617 |
|
| M |
| M0 | 243 | 215 | 458 | <0.001 |
| M1 | 27 | 61 | 88 |
|
| Total | 270 | 276 | 546 |
|
| Pathological
stage |
| Stage
I | 56 | 49 | 105 | 0.001 |
| Stage
II | 131 | 98 | 229 |
|
| Stage
III | 87 | 92 | 179 |
|
| Stage
IV | 28 | 62 | 90 |
|
| Total | 302 | 301 | 603 |
|
SPERT expression in various human CRC
cell lines
In order to fully demonstrate the effect of
SPERT on CRC cells, we determined the cell line with the
highest content of SPERT and selected this for use in
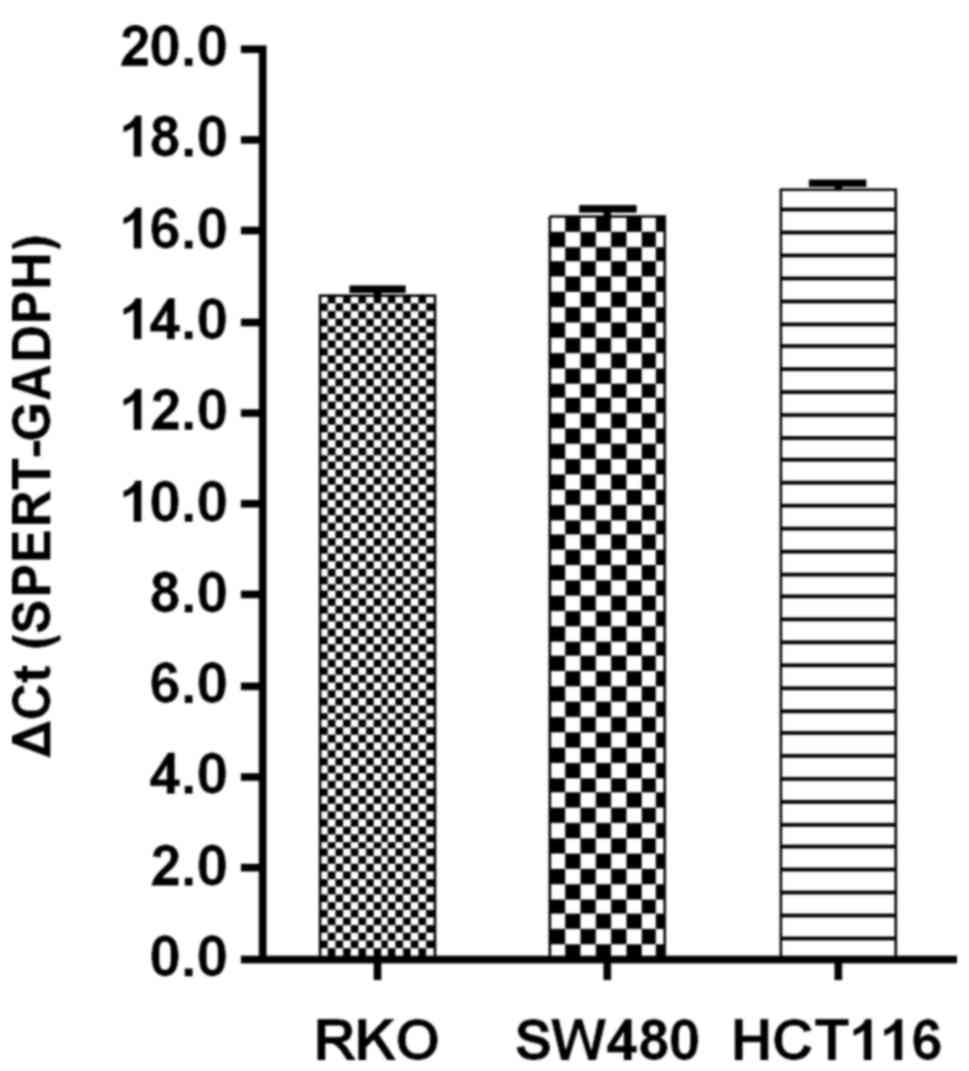
subsequent experiments. RT-qPCR was used to quantify SPERT
expression in the different cell lines (RKO, SW480 and HCT116
cells), and the results revealed that SPERT was most
prominent in RKO cells, as revealed in Fig. 3. Therefore, RKO cells were selected
for the subsequent experiments.
SPERT expression following shRNA
transfection
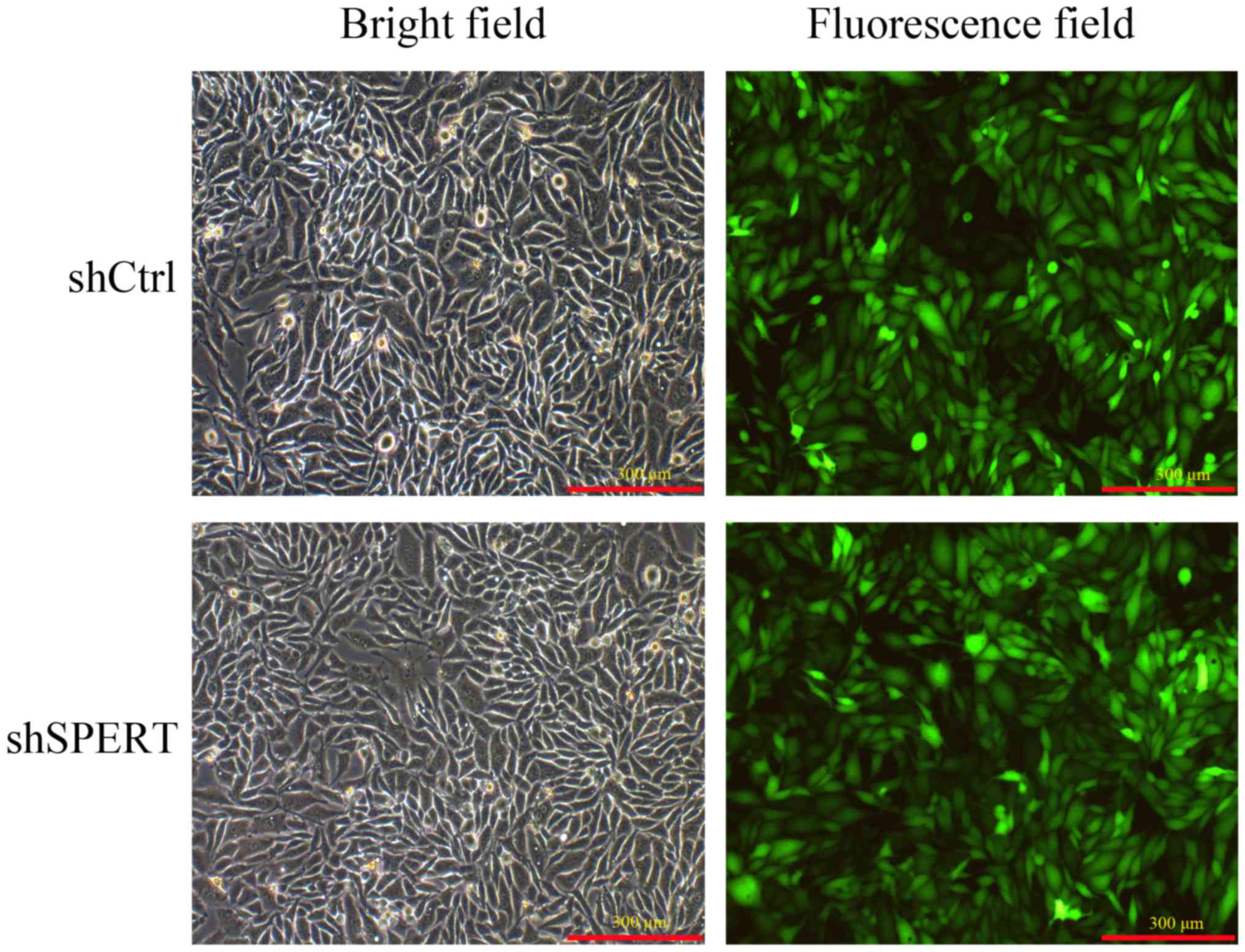
Fluorescence microscopy revealed that >80% of RKO
cells were successfully infected with LV-SPERT-RNAi or LV-shRNA-NC
at 72 h post-transfection, indicating a high success rate of
lentiviral vector infection (Fig.
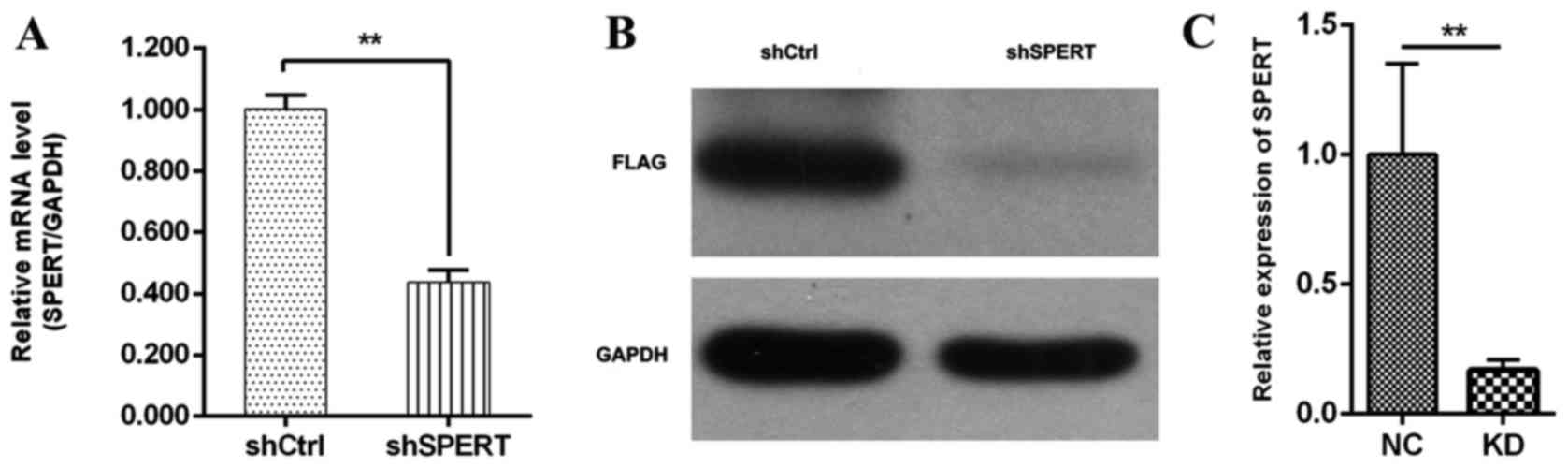
4). RT-qPCR detected decreased SPERT mRNA expression in
shSPERT-transfected RKO cells than in shCtrl-transfected cells
(0.437±0.040 vs. 1.001±0.046, respectively, P<0.01), and the
transfection efficiency was 90.3% (Fig.
5A). Consistently, western blotting detected a reduction of
82.25±0.25% in SPERT expression in the shSPERT-transfected RKO
cells than in the shCtrl-transfected cells (P<0.01) (Fig. 5B and C). These results confirmed
that RNAi could effectively reduce the endogenous expression of the
target gene.
Effect of SPERT knockdown on RKO cell
growth and proliferation
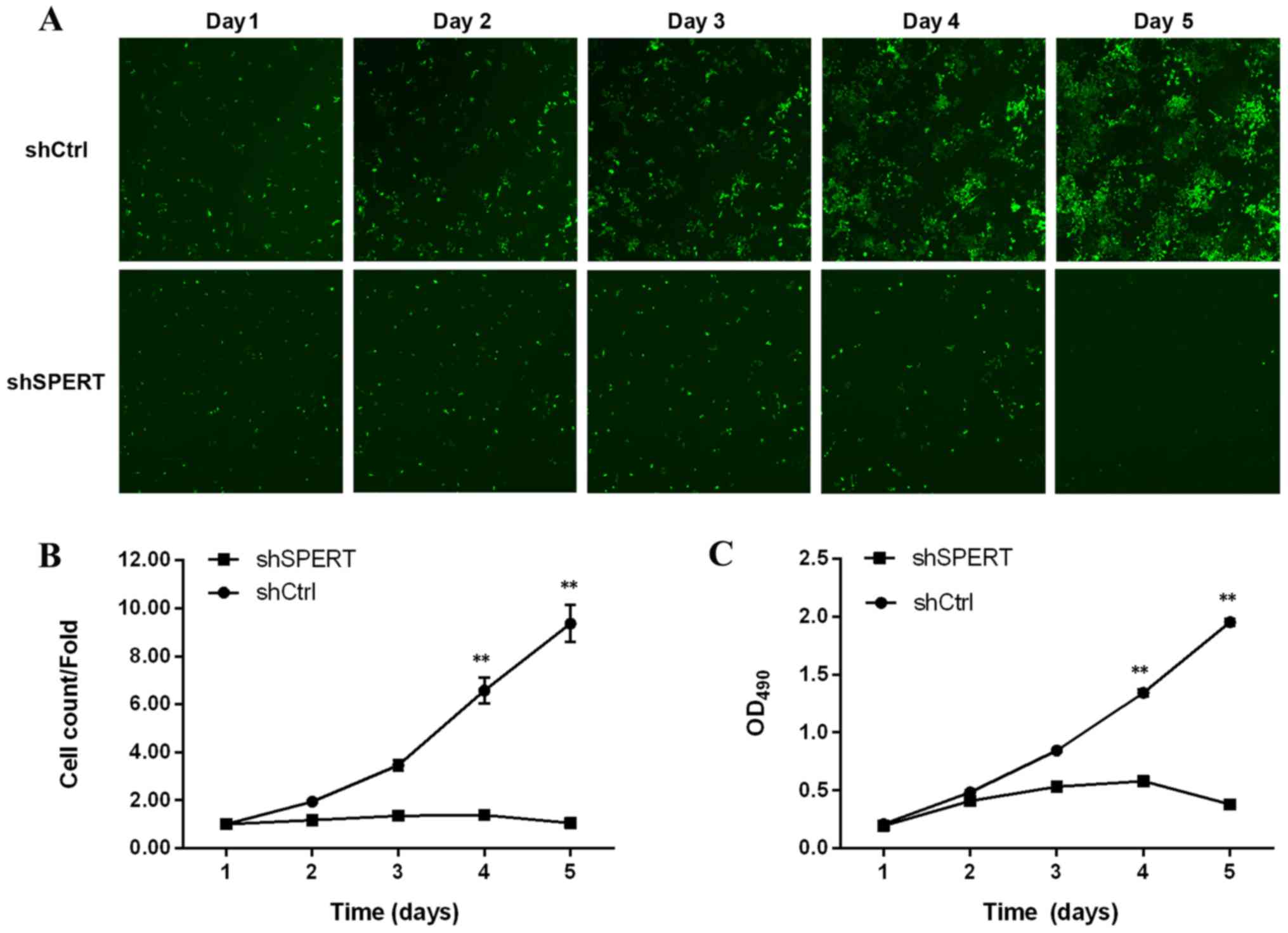
shSPERT-transfected RKO cells exhibited a decline in
number on days 1–5 post-transfection, while the number of
shCtrl-transfected cells increased, indicating that shRNA-induced
knockdown of SPERT inhibited RKO cell proliferation
(Fig. 6A and B). The MTT assay
revealed a clear decrease in the proliferation of
shSPERT-transfected RKO cells relative to shCtrl-transfected RKO
cells (P<0.01), demonstrating that shRNA-induced knockdown of
SPERT suppressed RKO cell proliferation (Fig. 6C).
Effect of SPERT knockdown on RKO cell
apoptosis
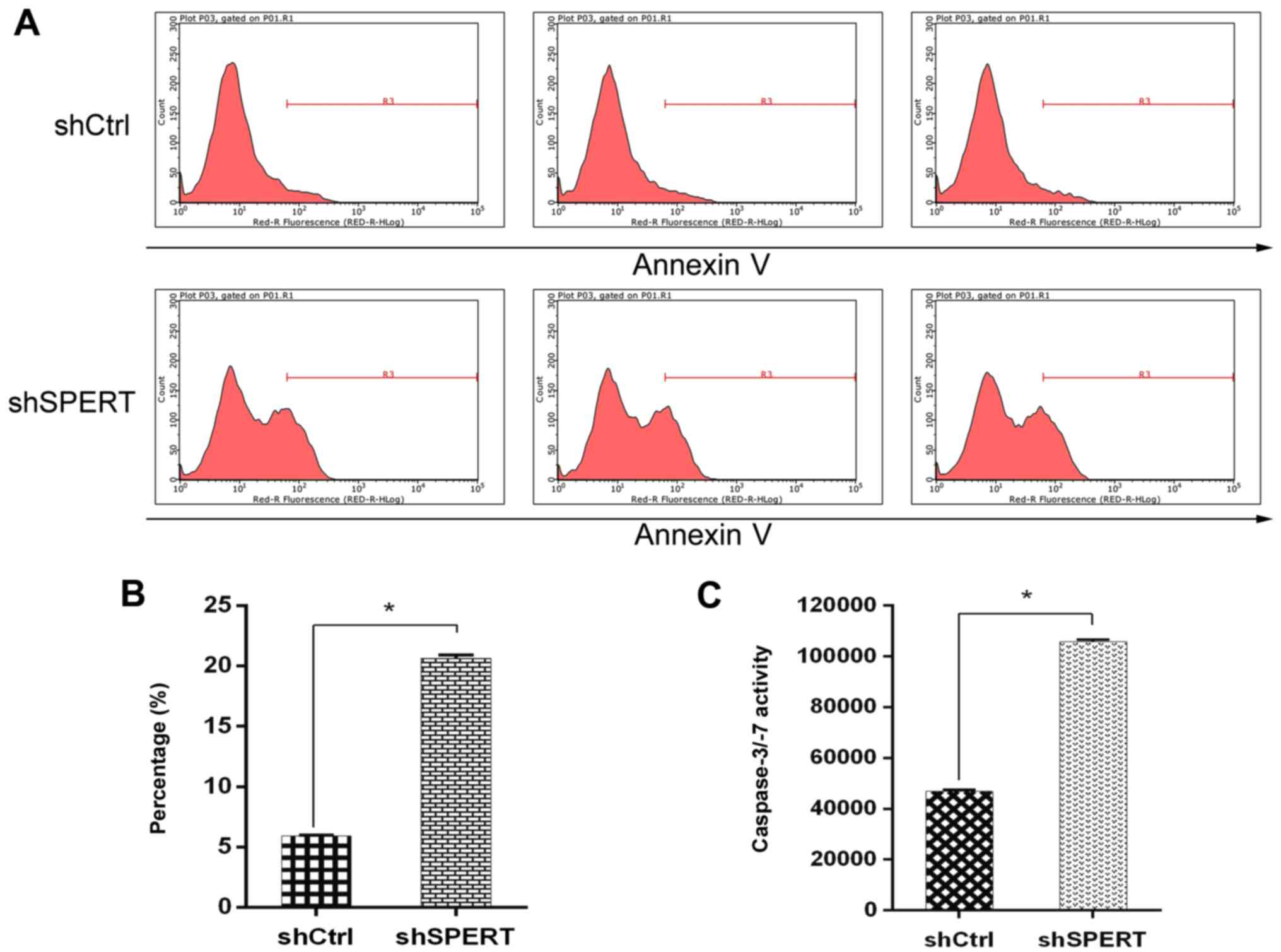
The Caspase-Glo 3/7 assay detected an increase in
the caspase-3/7 activity and the number of apoptotic cells in the
shSPERT-transfected RKO cells than in the shCtrl-transfected cells
(P<0.05) (Fig. 7C).
Additionally, flow cytometry detected a higher apoptotic rate in
the shSPERT-transfected RKO cells than in the shCtrl-transfected
cells (20.65±0.26 vs. 5.93±0.06%, respectively, P<0.05)
(Fig. 7A and B). These findings
demonstrated that shRNA-induced knockdown of SPERT promoted
RKO cell apoptosis.
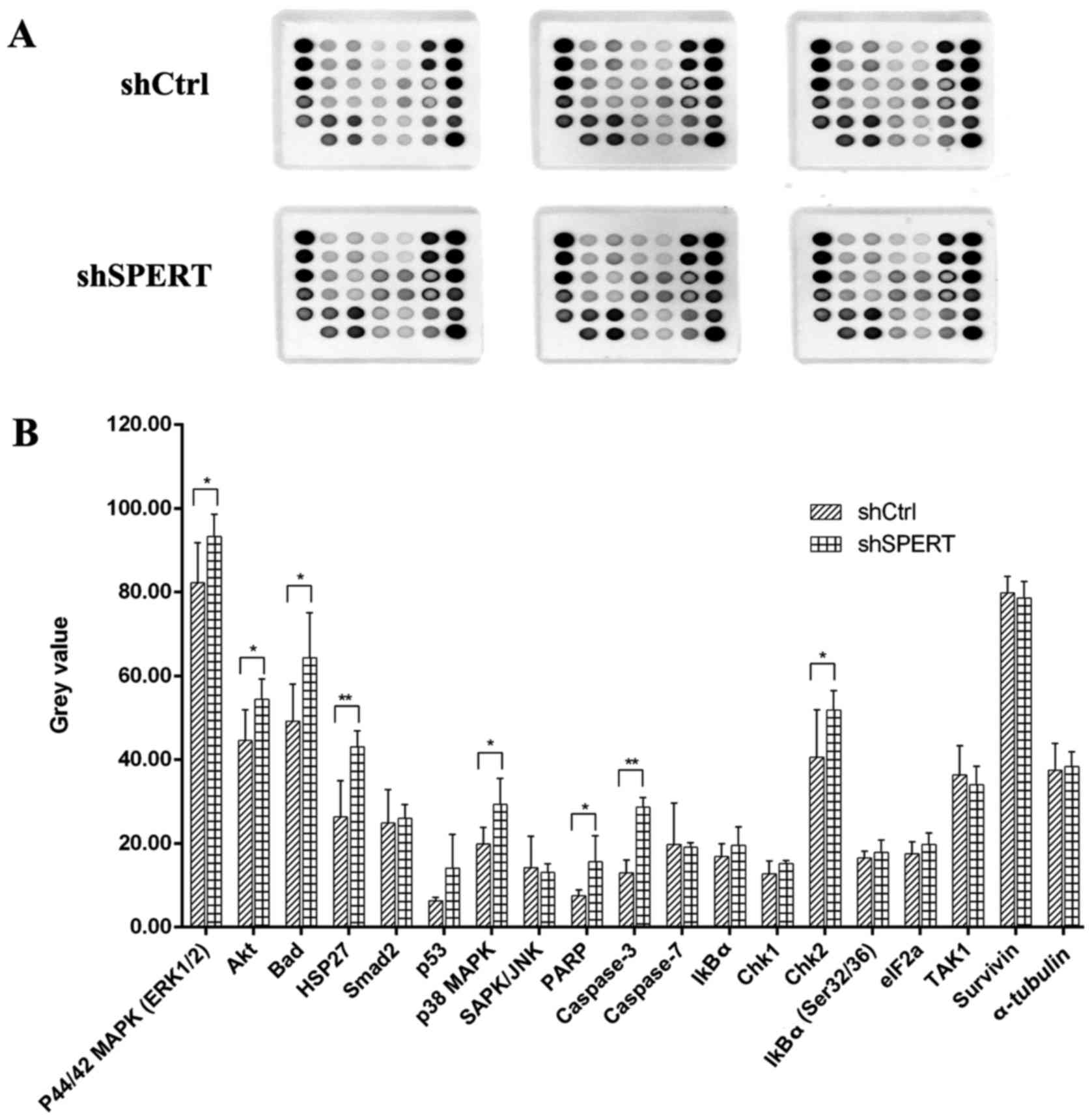
SPERT-related biological pathways
To evaluate the SPERT-associated biological
pathways, we used the PathScan® Signaling Antibody Array
Kit from Cell Signaling Technology. The PathScan Stress and
Apoptosis Signaling Antibody Array Kit (Chemiluminescent Readout)
uses glass slides as the planar surface and is based upon the
sandwich immunoassay principle. The array kit allows for the
simultaneous detection of 19 signaling molecules that are involved
in the regulation of the stress response and apoptosis.
Target-specific capture antibodies are spotted in duplicate on
nitrocellulose-coated glass slides. Each kit contains two slides
allowing for the interrogation of 32 different samples and the
generation of 608 data points in a single experiment. As shown in
Fig. 8, the levels of
phosphorylated p44/42 MAPK (ERK1/2), Akt, Bad, HSP27, p38 MARK and
Chk2, as well as PARP and caspase-3 expression were elevated in
shSPERT-transfected RKO cells compared with shCtrl-transfected
cells (P<0.05, P<0.01), indicating that shRNA-induced
knockdown of SPERT may suppress RKO cell proliferation and
promote cell apoptosis via these proteins.
Discussion
CRC is one of the most common gastrointestinal
cancers (1). Currently, surgery
remains the primary treatment for CRC (15), and neoadjuvant and adjuvant
radiochemotherapy have been shown to be effective in preventing
post-surgical recurrence and improving the survival rate in
patients with CRC (16,17). In addition, the introduction of
targeted therapy has been shown to increase the median survival
time from 3.6–6 to 24–28 months in patients with metastatic CRC
(18–20).
CRC is a genetically heterogeneous disease (21). To date, the roles of APC, p53 and
KRAS in the pathogenesis of CRC have been demonstrated; however,
the contribution of other genes to the pathogenesis of CRC remains
unclear (9,10). Systems biology studies have shown
gene instability, microsatellite instability and methylation
abnormalities in CRC (22).
However, TCGA datasets provide valuable bases for clinical
diagnosis, treatment and precision medicine for CRC (23–25).
In this study, the colon adenocarcinoma (COAD) and
rectum adenocarcinoma (READ) datasets in the TCGA were used, and
RNA-Seq and RNA-SeqV2 were employed to analyze gene expression in
paired COAD and READ samples. SPERT was selected from the
screening. There was a significant difference in SPERT
expression between cancer and peri-cancer specimens in patients
with CRC, and SPERT gene expression was significantly
associated with lymph node metastasis, distal metastasis and
pathological stages. Our data indicated that SPERT was
involved in the progression of CRC, and may serve as an indicator
for clinicopathological staging in patients with CRC.
In the present study, knockdown of SPERT was
found to suppress human CRC RKO cell proliferation and promote cell
apoptosis. It is therefore hypothesized that SPERT
overexpression may be involved in the development, progression and
metastasis of CRC. SPERT, which is located on human
chromosome 13q14.13, encodes the SPERT protein (also known as CBY2
and Nurit), which contains 338 amino acids and has a molecular
weight of 51,570 Da. SPERT belongs to the Chibby protein family and
has a quaternary structure of homodimers. It is a highly conserved
gene in mammals, it is expressed in humans, Rhesus monkeys, mice
and rats, but it is not expressed in Drosophila melanogaster
or Caenorhabditis elegans (26). However, the function of the SPERT
protein has remained unknown until now. It is reported that the
SPERT protein contains a leucine zipper motif and two coiled-coil
regions, and the leucine zipper motif is involved in
homodimerization or oligomerization, which is associated with the
regulation of oncogene expression (27). It is known that SPERT proteins may
form dimers in mice, and the C-terminal coiled-coil domain mediates
protein dimerization, which may link with Nek1 kinase (26,28).
Nek1, a member of the NIMA-related kinase family, is aberrantly
expressed in multiple cancer types, and aberrant Nek1 expression
was reported to cause abnormal regulation of the entire cell cycle
and abnormal cell proliferation, thereby resulting in cancer
development and progression (29,30).
Further studies to examine the interplay between the SPERT protein
and Nek1 appear justified.
To date, the exact role of SPERT in CRC remains
unclear. In this study, the PathScan® Signaling Antibody
Array Kit was employed to detect changes in key signaling molecules
involved in stress and apoptosis signaling pathways. Our findings
revealed upregulation of phosphorylated p44/42 MAPK (ERK1/2), Akt,
Bad, HSP27, p38 MARK and Chk2, in addition to elevated PARP and
Caspase-3 expression in shSPERT-transfected RKO cells, indicating
that these proteins are involved in the progression of CRC, in
which MAPK and PI3K/Akt signaling plays a predominant role. Bad is
the downstream target of the MAPK and PI3K/Akt signaling pathways.
MAPK regulates the function of the proapoptotic protein Bad through
the phosphorylation of serine 112, while PI3K/Akt regulates its
function by phosphorylation of serine 136. The dissociation of
phosphorylated Bad from anti-apoptotic factor Bcl-2 leads to an
increase in the activity of free Bcl-2, which inhibits apoptosis
(31). Caspase-3 and HSP27 are the
downstream target proteins of the p38 MAPK/HSP27 signaling pathway,
and the most important substrate of caspase-3 is the
poly(ADP-ribose) polymerase PARP. PARP cleavage is closely related
to cell apoptosis and is one of the markers of cell apoptosis and
caspase activation (32). MAP
kinase cascades mediate Hsp27 phosphorylation, MAPKAPK2 and
MAPKAPK3 regulate the function of the heat shock protein
27 (HSP27) by phosphorylating it on distinct sites, Ser-15,
Ser-78 and Ser-82. Downstream, HSP27 also blocks the activation of
caspase-9 and subsequent activation of caspase-3, thereby
inhibiting the remaining of the proteolytic caspase cascade
(33,34).
It has been revealed that both the MAPK and PI3K/Akt
pathways, which are associated with cancer initiation and
progression, serve critical roles in CRC progression and greatly
contribute to intestinal epithelial cell differentiation (35–39).
Ras mutations have been detected in 35–45% of patients with
CRC (40–43), while 9–11% of patients with CRC
harbor BRAF mutations (44–46).
KRAS and BRAF have been demonstrated to contribute to
CRC progression (43,45). ERK1 and ERK2, which are located
downstream of the Ras oncogene protein, bind to cell-surface
receptor tyrosine kinases and G-protein-coupled receptors,
resulting in Ras and BRAF activation (47) and subsequent MEK1/MEK2 and ERK1/ERK2
phosphorylation (48). The
Ras/Raf/MEK/ERK cascade has been revealed to be involved in the
mediation of cell growth signals, cell survival and cancer invasion
(49), and is strongly associated
with the differentiation, pathological stage and prognosis of
cancers, which may be used to guide the selection of targeted drugs
for use in clinical cancer therapies (50). Our findings revealed that knockdown
of SPERT significantly increased the levels of
phosphorylated p44/42 MAPK (ERK1/2) and p38 MAPK; this may explain
why SPERT knockdown suppressed RKO cell proliferation and
promoted cell apoptosis. Further studies are required to
investigate the detailed mechanisms underlying the SPERT-mediated
effects on the MAPK and PI3K/Akt signaling pathways.
In summary, the results of the present study
demonstrated that SPERT expression was significantly
upregulated in CRC specimens, and knockdown of SPERT
suppressed CRC cell growth and promoted apoptosis via the MAPK and
PI3K/Akt signaling pathways. SPERT may serve as an oncogene
for CRC, and may be a promising biomarker for predicting the poor
prognosis of CRC. In addition, SPERT may be a potential
target for the treatment of CRC.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by a grant
from the National Natural Science Foundation of China (no.
81272465).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SZC and LZZ conceived and designed the study. LZZ
performed the experiments and wrote the paper. SZC reviewed and
edited the manuscript. All authors read and approved the manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
GBD 2015 Mortality and Causes of Death
Collaborators: Global, regional, and national life expectancy,
all-cause mortality, and cause-specific mortality for 249 causes of
death, 1980–2015: A systematic analysis for the Global Burden of
disease study 2015. Lancet. 388:1459–1544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kelly C and Cassidy J: Chemotherapy in
metastatic colorectal cancer. Surg Oncol. 16:65–70. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bouche O, Conroy T, Michel P, Penna C and
Tournigand C: Metastatic colorectal cancer. Gastroenterol Clin
Biol. 30:2S30–2S42. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yau T, Chan P, Ching Chan Y, Wong BC,
Liang R and Epstein RJ: Current management of metastatic colorectal
cancer-the evolving impact of targeted drug therapies. Aliment
Pharmacol Ther. 27:997–1005. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilkes G and Hartshorn K: Clinical update:
Colon, rectal, and anal cancers. Semin Oncol Nurs. 28:e1–e22. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tran NH, Cavalcante LL, Lubner SJ,
Mulkerin DL, LoConte NK, Clipson L, Matkowskyj KA and Deming DA:
Precision medicine in colorectal cancer: The molecular profile
alters treatment strategies. Ther Adv Med Oncol. 7:252–262. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lichtenstein P, Holm NV, Verkasalo PK,
Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A and Hemminki
K: Environmental and heritable factors in the causation of
cancer-analyses of cohorts of twins from Sweden, Denmark, and
Finland. N Engl J Med. 343:78–85. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wood LD, Parsons DW, Jones S, Lin J,
Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al: The
genomic landscapes of human breast and colorectal cancers. Science.
318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sjöblom T, Jones S, Wood LD, Parsons DW,
Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al:
The consensus coding sequences of human breast and colorectal
cancers. Science. 314:268–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nassiri M, Kooshyar MM, Roudbar Z, Mahdavi
M and Doosti M: Genes and SNPs associated with non-hereditary and
hereditary colorectal cancer. Asian Pac J Cancer Prev.
14:5609–5614. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hampel H: Genetic testing for hereditary
colorectal cancer. Surg Oncol Clin N Am. 18:687–703. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cancer Genome Atlas Research Network;
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA,
Ellrott K, Shmulevich I, Sander C and Stuart JM: The cancer genome
atlas Pan-cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin Y, Golovnina K, Chen ZX, Lee HN,
Negron YL, Sultana H, Oliver B and Harbison ST: Comparison of
normalization and differential expression analyses using RNA-Seq
data from 726 individual Drosophila melanogaster. BMC
Genomics. 17:282016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Coffey JC and Dockery P: Colorectal
cancer: Surgery for colorectal cancer-standardization required. Nat
Rev Gastroenterol Hepatol. 13:256–257. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sauer R, Fietkau R, Wittekind C, Rödel C,
Martus P, Hohenberger W, Tschmelitsch J, Sabitzer H, Karstens JH,
Becker H, et al: Adjuvant vs. neoadjuvant radiochemotherapy for
locally advanced rectal cancer: The German trial CAO/ARO/AIO-94.
Colorectal Dis. 5:406–415. 2003.
|
|
17
|
Benson AB Rd: Should we consider adjuvant
therapy for rectal cancer after neoadjuvant chemoradiotherapy? Clin
Adv Hematol Oncol. 14:778–781. 2016.PubMed/NCBI
|
|
18
|
Arnold D and Seufferlein T: Targeted
treatments in colorectal cancer: State of the art and future
perspectives. Gut. 59:838–858. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seeber A and Gastl G: Targeted therapy of
colorectal cancer. Oncol Res Treat. 39:796–802. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pai SG and Fuloria J: Novel therapeutic
agents in the treatment of metastatic colorectal cancer. World J
Gastrointest Oncol. 8:99–104. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu YW, Zhang HF, Liang R, Xie ZR, Luo HY,
Zeng YJ, Xu Y, Wang LM, Kong XY and Wang KH: Colorectal cancer
genetic heterogeneity delineated by multi-region sequencing. PLoS
One. 11:e01526732016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sonachalam M, Shen J, Huang H and Wu X:
Systems biology approach to identify gene network signatures for
colorectal cancer. Front Genet. 3:802012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee H, Palm J, Grimes SM and Ji HP: The
cancer genome atlas clinical explorer: A web and mobile interface
for identifying clinical-genomic driver associations. Genome Med.
7:1122015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Z, Jensen MA and Zenklusen JC: A
practical guide to the cancer genome atlas (TCGA). Methods Mol
Biol. 1418:111–141. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hartman M, Loy EY, Ku CS and Chia KS:
Molecular epidemiology and its current clinical use in cancer
management. Lancet Oncol. 11:383–390. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Feige E, Chen A and Motro B: Nurit, a
novel leucine-zipper protein, expressed uniquely in the spermatid
flower-like structure. Mech Dev. 117:369–377. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alber T: Structure of the leucine zipper.
Curr Opin Genet Dev. 2:205–210. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Takemaru K, Yamaguchi S, Lee YS, Zhang Y,
Carthew RW and Moon RT: Chibby, a nuclear beta-catenin-associated
antagonist of the Wnt/Wingless pathway. Nature. 422:905–909. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Letwin K, Mizzen L, Motro B, Ben-David Y,
Bernstein A and Pawson T: A mammalian dual specificity protein
kinase, Nek1, is related to the NIMA cell cycle regulator and
highly expressed in meiotic germ cells. EMBO J. 11:3521–3531.
1992.PubMed/NCBI
|
|
30
|
Upadhya P, Birkenmeier EH, Birkenmeier CS
and Barker JE: Mutations in a NIMA-related kinase gene,
Nek1, cause pleiotropic effects including a progressive
polycystic kidney disease in mice. Proc Natl Acad Sci USA.
97:217–221. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
She QB, Solit DB, Ye Q, O'Reilly KE, Lobo
J and Rosen N: The BAD protein integrates survival signaling by
EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor
cells. Cancer Cell. 8:287–297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ivana Scovassi A and Diederich M:
Modulation of poly(ADP-ribosylation) in apoptotic cells. Biochem
Pharmacol. 68:1041–1047. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bitar KN: HSP27 phosphorylation and
interaction with actin-myosin in smooth muscle contraction. Am J
Physiol Gastrointest Liver Physiol. 282:G894–G903. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bruey JM, Paul C, Fromentin A, Hilpert S,
Arrigo AP, Solary E and Garrido C: Differential regulation of HSP27
oligomerization in tumor cells grown in vitro and in vivo.
Oncogene. 19:4855–4863. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fang JY and Richardson BC: The MAPK
signalling pathways and colorectal cancer. Lancet Oncol. 6:322–327.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Miki H, Yamada H and Mitamura K:
Involvement of p38 MAP kinase in apoptotic and proliferative
alteration in human colorectal cancers. Anticancer Res.
19:5283–5291. 1999.PubMed/NCBI
|
|
37
|
Abubaker J, Bavi P, Al-Harbi S, Ibrahim M,
Siraj AK, Al-Sanea N, Abduljabbar A, Ashari LH, Alhomoud S,
Al-Dayel F, et al: Clinicopathological analysis of colorectal
cancers with PIK3CA mutations in Middle Eastern population.
Oncogene. 27:3539–3545. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ollikainen M, Gylling A, Puputti M,
Nupponen NN, Abdel-Rahman WM, Butzow R and Peltomäki P: Patterns of
PIK3CA alterations in familial colorectal and endometrial
carcinoma. Int J Cancer. 121:915–920. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Taupin D and Podolsky DK:
Mitogen-activated protein kinase activation regulates intestinal
epithelial differentiation. Gastroenterology. 116:1072–1080. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
De Roock W, Claes B, Bernasconi D, De
Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V,
Papamichael D, Laurent-Puig P, et al: Effects of KRAS, BRAF,
NRAS, and PIK3CA mutations on the efficacy of cetuximab
plus chemotherapy in chemotherapy-refractory metastatic colorectal
cancer: A retrospective consortium analysis. Lancet Oncol.
11:753–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Herreros-Villanueva M, Chen CC, Yuan SS,
Liu TC and Er TK: KRAS mutations: Analytical considerations.
Clin Chim Acta. 431:211–220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Andreyev HJ, Norman AR, Cunningham D,
Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N,
et al: Kirsten ras mutations in patients with colorectal cancer:
The ‘RASCAL II’ study. Br J Cancer. 85:692–696. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Andreyev HJ, Norman AR, Cunningham D,
Oates JR and Clarke PA: Kirsten ras mutations in patients with
colorectal cancer: The multicenter ‘RASCAL’ study. J Natl Cancer
Inst. 90:675–684. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Deng G, Bell I, Crawley S, Gum J, Terdiman
JP, Allen BA, Truta B, Sleisenger MH and Kim YS: BRAF
mutation is frequently present in sporadic colorectal cancer with
methylated hMLH1, but not in hereditary nonpolyposis colorectal
cancer. Clin Cancer Res. 10:191–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rajagopalan H, Bardelli A, Lengauer C,
Kinzler KW, Vogelstein B and Velculescu VE: Tumorigenesis:
RAF/RAS oncogenes and mismatch-repair status. Nature.
418:9342002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Roskoski R Jr: ERK1/2 MAP kinases:
Structure, function, and regulation. Pharmacol Res. 66:105–143.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shama J, Garcia-Medina R, Pouysségur J and
Vial E: Major contribution of MEK1 to the activation of ERK1/ERK2
and to the growth of LS174T colon carcinoma cells. Biochem Biophys
Res Commun. 372:845–849. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hilger RA, Scheulen ME and Strumberg D:
The Ras-Raf-MEK-ERK pathway in the treatment of cancer. Onkologie.
25:511–518. 2002.PubMed/NCBI
|