Introduction
Chronic myeloid leukemia (CML) is a malignancy
characterized by a reciprocal translocation (9:22) resulting in the
Philadelphia chromosome, which triggers the formation of the fusion
oncoprotein Bcr-Abl. This is a constitutively active tyrosine
kinase with leukemogenic properties, required for the pathogenesis
of CML (1). The inhibition of
Bcr-Abl by imatinib and other tyrosine kinase inhibitors (TKIs) has
fundamentally improved the outcome for patients with CML in the
chronic phase (2,3). However, leukemic stem cells (LSCs)
seem to be capable of surviving even after continuous, long-term
TKI administration and the discontinuation of TKI in most cases
leads to disease recurrence. Thus, there is a need to improve
current CML therapy and to focus on therapeutic options that may
lead to eradication of LSCs, and total cure. Other signaling
pathways, in addition to that of Bcr-Abl, have been found to be
aberrantly expressed in CML cells and may thus be crucial for the
survival and maintenance of LSCs. Such pathways include
Wnt/β-catenin signaling, the tumor suppressor gene PML, Forkhead
box subgroup O transcription factors, sonic hedgehog signaling,
prosurvival protein Bcl2 and JAK/STAT signaling (4).
Formation of bioactive leukotrienes (LTs), which
have a recognized role in inflammatory processes (5), is another pathway suggested to be
involved in initiation and maintenance of LSCs (4). In mice, the arachidonate
5-lipoxygenase (5-LO) gene (Alox5), active in an early stage
of LT synthesis, has been reported to be upregulated in CML LSCs
rendering them resistant to TKI treatment. While the presence of
Alox5 is crucial for LSCs, it did not significantly affect
the functions of normal hematopoietic stem cells (6,7). In
the human, 5-LO converts arachidonic acid into the intermediate
LTA4, for further metabolism into LTB4 and the cysteinyl
(Cys) containing leukotrienes LTC4, LTD4 and
LTE4. The biological actions of CysLTs are mediated
through activation of CysLT1 and CysLT2 receptors (8). Overexpression of the cysteinyl LT1
receptor (CysLT1R) has been reported in renal cell carcinoma and
cancer of the bladder, prostate, testes, brain, breast and
colorectal region (9–13). High expression of CysLT1R in breast
and colorectal cancer is associated with poor prognosis (9,12).
Cysteinyl leukotriene signaling is involved in many tumorigenesis
cancer-related processes: proliferation, migration, invasion,
angiogenesis, genome instability, deregulation of cellular
energetics and apoptosis (11).
Montelukast is a selective oral CysLT1R antagonist,
approved in clinical practice for several years as a safe and
effective treatment for patients with asthma and allergic rhinitis
(14). In addition, a role in
apoptosis induction has recently been highlighted in several
malignancies. Montelukast inhibits the growth of urological,
colorectal and renal cell carcinoma by induction of apoptosis
(10,15–17).
Montelukast was found to induce reduced colony formation, apoptosis
and G1 arrest in colon cancer cells and decreased the growth of
mouse xenografts (18). In
neuroblastoma cell lines, montelukast induced caspase-dependent
apoptosis and cell cycle arrest in the subG1 phase (19). Montelukast also was found to inhibit
the cell migration and invasion of human glioblastoma cells by
suppressing MMP-2 and MMP-9 activities (20).
Our group has previously described that human CML
cells have a high capacity to synthesize bioactive LTs and that LTs
are capable of stimulating normal myeloid progenitor cell growth
(21,22). Recently, we also showed that
physiological concentrations of the CysLT1R antagonist montelukast,
alone or together with imatinib, induced a clear and dose-dependent
inhibition of the growth of human CML cells, while normal bone
marrow cells and fibroblasts were unaffected (23). Herein, we further analyzed the
mechanisms underlying the cytotoxic activity exerted by montelukast
on CML cells by assessing its relation to receptor expression and
induction of key proteins linked to apoptotic events.
One of the pathways that CysLT1 signaling modifies
is Wnt/β-catenin signaling. This pathway has been shown to be
important for development and survival of CML LSCs and potentially
represents a therapeutic target in CML treatment (24,25).
Herein, we showed that montelukast can indeed alter Wnt/β-catenin
signaling, indicating that inhibition of β-catenin could be a key
downstream mechanism for this compound in CML.
Materials and methods
Cell lines and inhibitors
CML cell lines (K562 and JURL-MK1) were obtained
from Leibniz-Institute DSMZ-German Collection of Microorganisms and
Cell Cultures (Braunschweig, Germany) and maintained in RPMI-1640
medium with 10% FBS, 2 mM L-glutamine (Invitrogen; Thermo Fisher
Scientific, Inc., Carlsbad, CA, USA) Pen Strep (Gibco; Thermo
Fisher Scientific, Inc.). The CysLT1R antagonist montelukast and
imatinib were purchased from Selleck Chemicals (Houston, TX,
USA).
Proliferation assay
Cell proliferation of JURL-MK1 cells was measured
using the MTT cell proliferation assay according to the
manufacturer's instructions as previously described (23). Cells were seeded in triplicate in
flat-bottomed 96-well plates at 30,000 cells/well and treated for
72 h in the presence of 1 µM montelukast and/or 0.1 imatinib
µM.
siRNA knockdown of CysLT1R
expression
K562 cells were transfected with 100 nM CysLT1R
siRNA (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 96
h using Dharmafect. Stealth RNAi negative control duplexes
(Invitrogen; Thermo Fisher Scientific, Inc.) were used as a
negative control for transfection. Ablation of CysLT1R by siRNA was
validated using western blotting as described below.
Non-transfected cells, cells transfected with CysLT1R siRNA or
Stealth RNAi negative control duplexes were transferred to a
96-well plate and treated with 2 and 3 µM montelukast or 1 µM
imatinib for 24 h and cell viability was tested with trypan blue
staining.
Analysis of apoptotic morphology and
cytochrome c staining
K562 cells were treated with 2 µM montelukast for 24
and 48 h. Imatinib (1 µM)-treated cells were used as a positive
control for induction of apoptotic morphology. Cells (20,000)were
resuspended in 100 µl PBS and cytospinned onto glass sides at 26 ×
g for 4 min. Glass slides were fixed in ice-cold methanol for 3 min
followed by ethanol gradient (100, 85 and 70%) for 2 min each and
kept at −20°C. Nuclear morphology of cells was examined by staining
with 4,6′diamidino-2-phenylindole dihydrochloride (DAPI) (Vector
Laboratories, Inc., Burlingame, CA, USA). Apoptosis was defined by
the presence of fragmented nuclei. In total, 200 cells were counted
and the percentage of apoptotic cells was presented.
In order to analyze the release of cytochrome
c from mitochondria, K562 cytospins were blocked with buffer
(3% BSA, 0.2% Triton X-100, 10 mM HEPES, pH 7.4) for 60 min and
stained with the primary antibody against cytochrome c (Cell
Signaling Technology, Danvers, MA, USA) for 16 h at 4°C. Cytospins
were incubated with Alexa 488 secondary antibody (Invitrogen;
Thermo Fisher Scientific) and counterstained with DAPI for nuclear
visualization. Fluorescence microscope Axio Imager.Z2 (Zeiss AG,
Oberkochen, Germany), equipped with a 100-W mercury lamp, a CCD
camera (C4742-95, Hamamatsu) was used.
Analysis of caspase activation
Active caspase-3 level (a marker of cells undergoing
apoptosis) was measured in K562 cells treated with 2 and 4 µM
montelukast for 24 h with PE Active Caspase-3 Apoptosis kit (BD
Biosciences, San Jose, CA, USA) according to manufacturer's
protocol by flow cytometry, using a CyAn ADP Analyzer (Beckman
Coulter, Inc., Brea, CA, USA). The results were analyzed with
FlowJo software, version 9.4.11 (Tree Star, Inc., Ashland, OR,
USA). In order to ascertain whether cell death induced by
montelukast is caspase-dependent, we treated cells with Z-VAD with
or without montelukast. Cells were pretreated with 50 µM Z-VAD for
30 min and then montelukast (2 µM) was added and treated for
additional 72 h. Cell viability was measured using the MTT cell
proliferation assay, as previously described (23).
Western blot analysis
To assess apoptosis induction, K562 and JURL-MK1
cells were treated with 2, 3 or 5 µM montelukast for 8 h.
Alterations in Wnt/β-catenin signaling were examined in K562 cells
treated with 30 nM montelukast for 15, 60, 90 and 180 min.
Whole-cell lysates were prepared in RIPA buffer (50 mM Tris-HCl, pH
7.4, 150 mM NaCl, 0.5% Igepal, 5 mM EDTA and 0.1% SDS) and western
blot analysis was performed as previously described (23). The following primary antibodies were
used: CysLT1R (120500; Cayman Chemicals, Ann Arbor, MI, USA); PARP1
(H250; sc-7150; Santa Cruz Biotechnology); Bax (2772), β-catenin
(9582), phospho-β-catenin (9561), βTrCP (4394; all from Cell
Signaling Technology); c-myc (ab56; Abcam, Cambridge, UK). Antibody
against β-tubulin (T7816; Sigma-Aldrich, Stockholm, Sweden) was
used as control of equal loading. IR-Dye-linked secondary
antibodies (926–32211 and 926–68070; LI-COR Biosciences, Lincoln,
NE, USA) were used to image bands on the Odyssey platform.
Ubiquitination of β-catenin
Whole-cell lysates of K562 cells treated with 30 nM
montelukast were obtained at 15, 60 and 90 min as described above.
Lysates were subjected to immunoprecipitation using magnetic
Dynabeads Protein A (10006D; Invitrogen; Thermo Fisher Scientific,
Inc.) and anti-β-catenin antibody (610154; BD Transduction
Laboratories, San Jose, CA, USA), according to the manufacturer's
instructions. Binding between β-catenin and β-TrCP was analyzed by
immunoblot analysis as described above.
Statistical analysis
All statistical analyses were performed using Prism
Software (GraphPad Software, Inc., La Jolla, CA, USA) and the
statistical significance of data was determined as *P<0.05;
**P<0.01; ***P<0.001 (as indicated in the figures). For
comparison between two groups, either a paired or unpaired t-test
(Student's t-test) was used. When three or more groups were
compared, ANOVA followed by Tukey HSD post-hoc test was used. All
values are expressed as the mean ± standard error of the mean
(SEM).
Results
CysLT1R expression and importance for
montelukast cytotoxicity
We previously showed clear expression of CysLT1R in
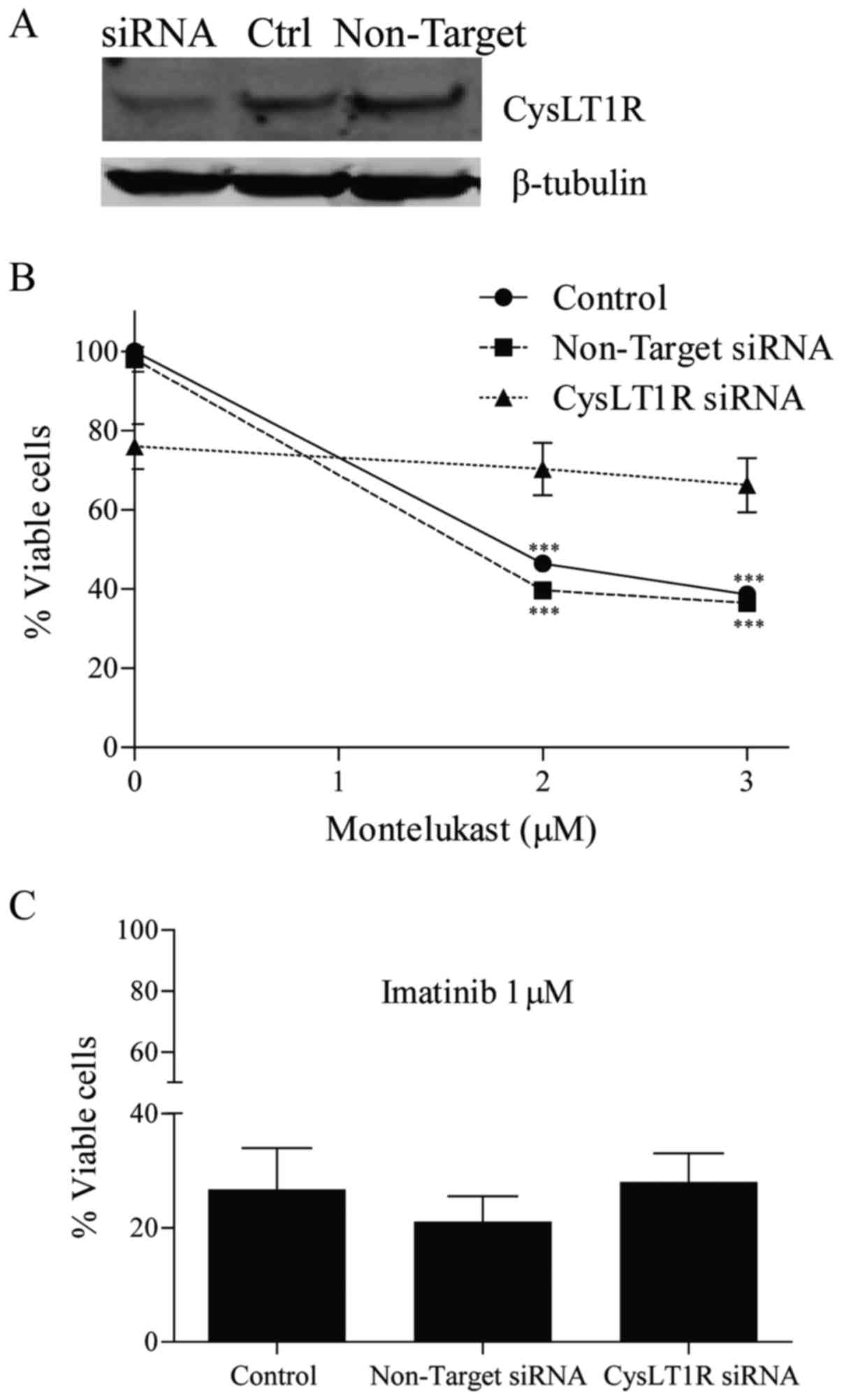
K562, KU812 and KCL22 CML cells (23). To show the importance of CysLT1R for
the growth of CML cells, the receptor was knocked down using siRNA
in K562 cells. After 96 h, this knockdown reduced the receptor
expression by approximately 80%, while control cells showed
maintainence of receptor expression (Fig. 1A). Depletion of CysLT1R resulted in
a 25% decrease in K562 cell growth, as compared to the control
cells and to cells exposed to non-target siRNA (P=0.05) (Fig. 1B). Next, untransfected cells and
cells transfected with CysLT1R siRNA or non-target siRNA were
treated with montelukast or imatinib for 24 h. The growth of cells
with knocked-down CysLT1 receptor was unaffected by 2 µM
montelukast, while this receptor antagonist significantly reduced
the viability of both cell lines exposed to non-target siRNA (by
50%) and control cells without siRNA (by 54%) (Fig. 1B). Using 3 µM montelukast, 62% of
control cells and 63% of cells with non-target siRNA died, but only
14% of cells exposed to CysLT1R siRNA. Imatinib-induced inhibition
of cell growth was similar in all three cell subpopulations
(Fig. 1C).
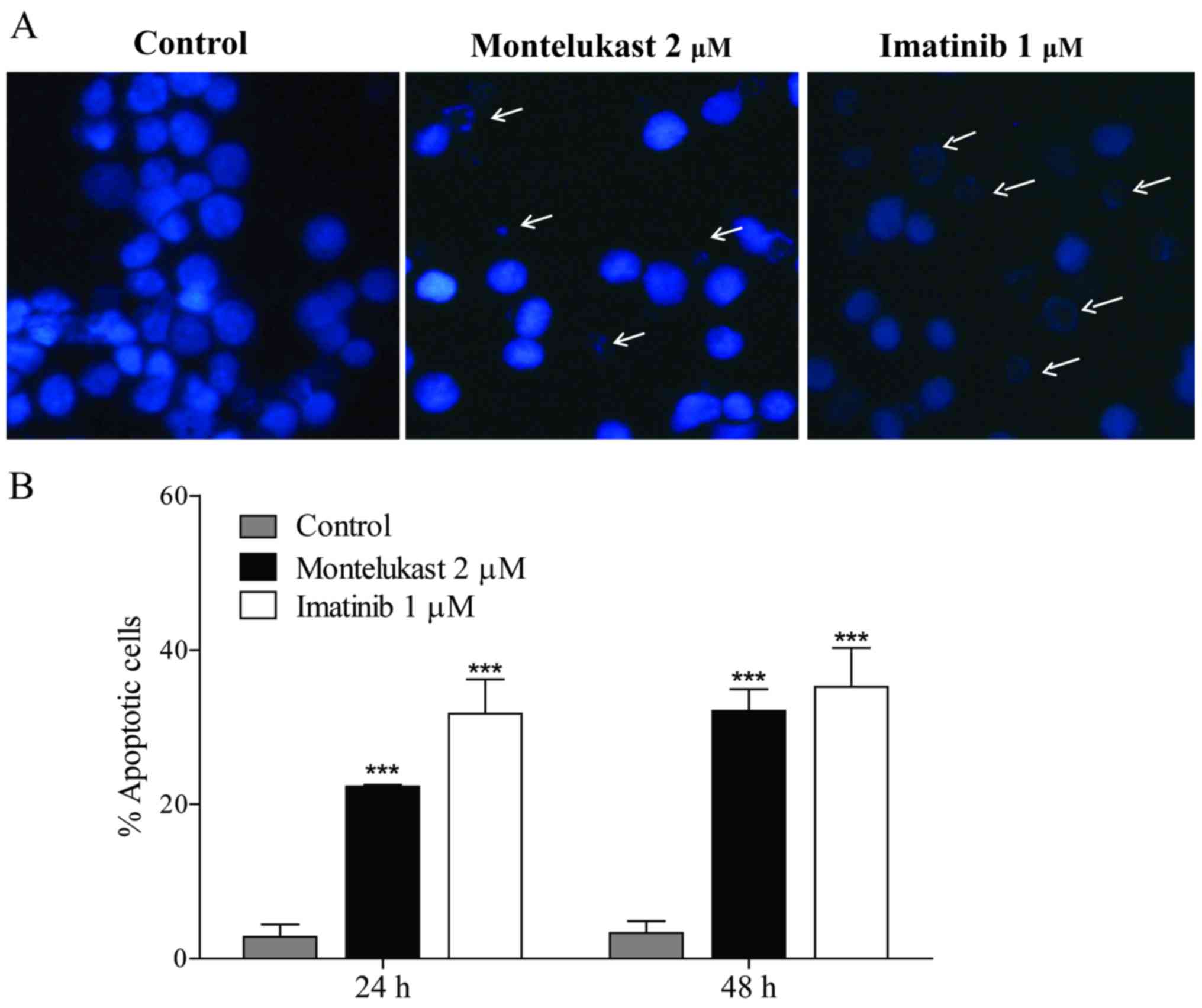
Montelukast induces the apoptosis of
K562 and JURL-MK-1 cells
To understand the mechanism by which montelukast
induces growth prevention in CML, cell morphology was examined in
the K562 cells. Montelukast induced apoptotic morphology in 22% of
K562 cells after 24 h and in 32% after 48 h of treatment. Imatinib
was used as a positive control and induced apoptotic morphology in
32% and 35% cells after 24 and 48 h, respectively (Fig. 2).
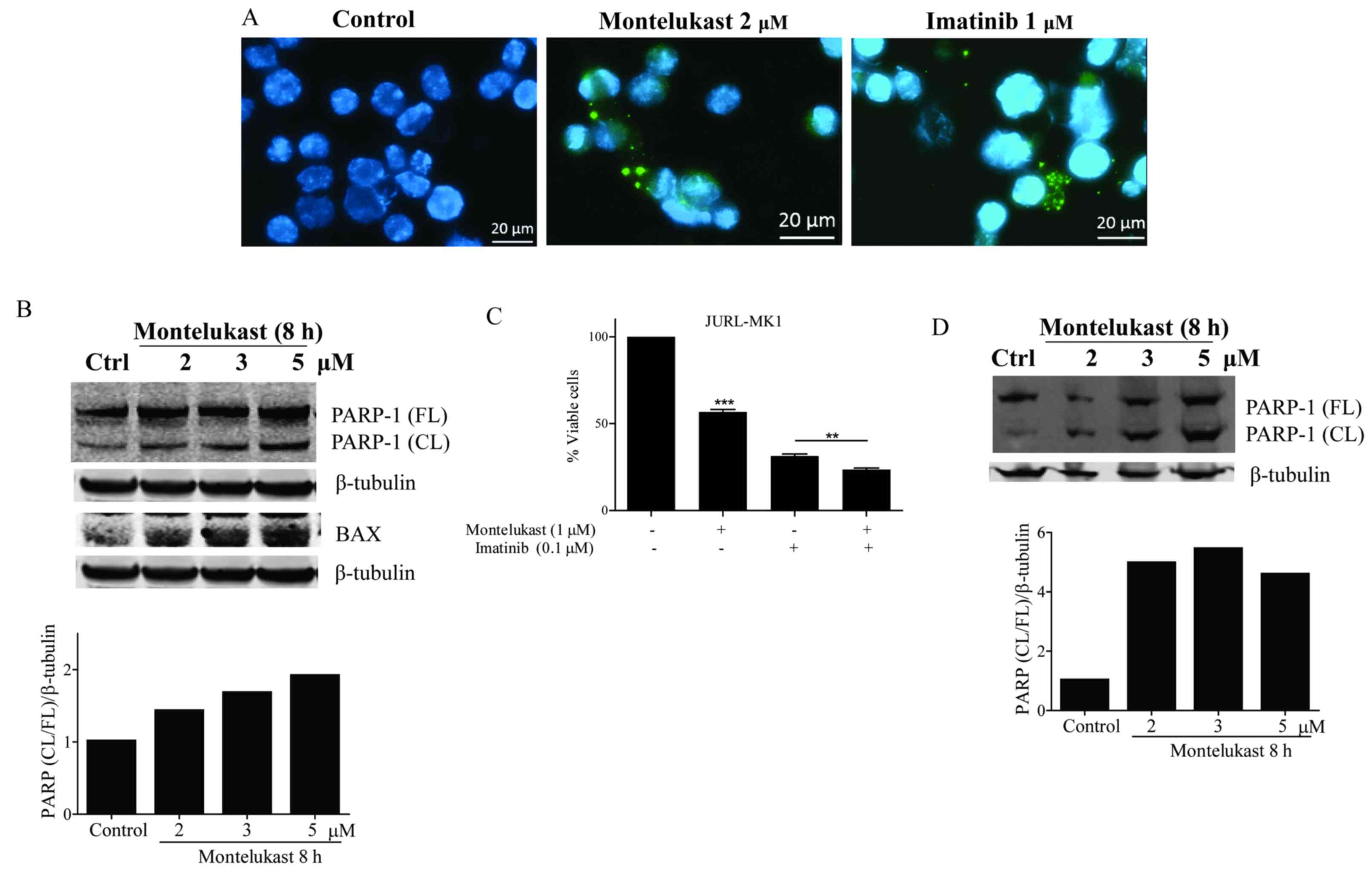
Next, cytochrome c release from
mitochondria was examined
Treatment with montelukast, as well as with
imatinib, caused cytosol localization of cytochrome c
(Fig. 3A). Release of cytochrome
c leads to activation of caspases and cleavage of the
caspase-3 substrate PARP-1. Montelukast treatment resulted in
PARP-1 protein cleavage in the K562 cells. Ratio of cleaved vs.
full length PARP-1 increased in a dose dependent manner (Fig. 3B). Furthermore, we observed a
dose-dependent increase in proapoptotic Bax protein signal in K562
cells upon montelukast treatment (Fig.
3B). The growth of JURL-MK1 cells was significantly inhibited
by montelukast, and when combined with imatinib the receptor
antagonist induced further growth inhibition (Fig. 3C). PARP-1 protein was also cleaved
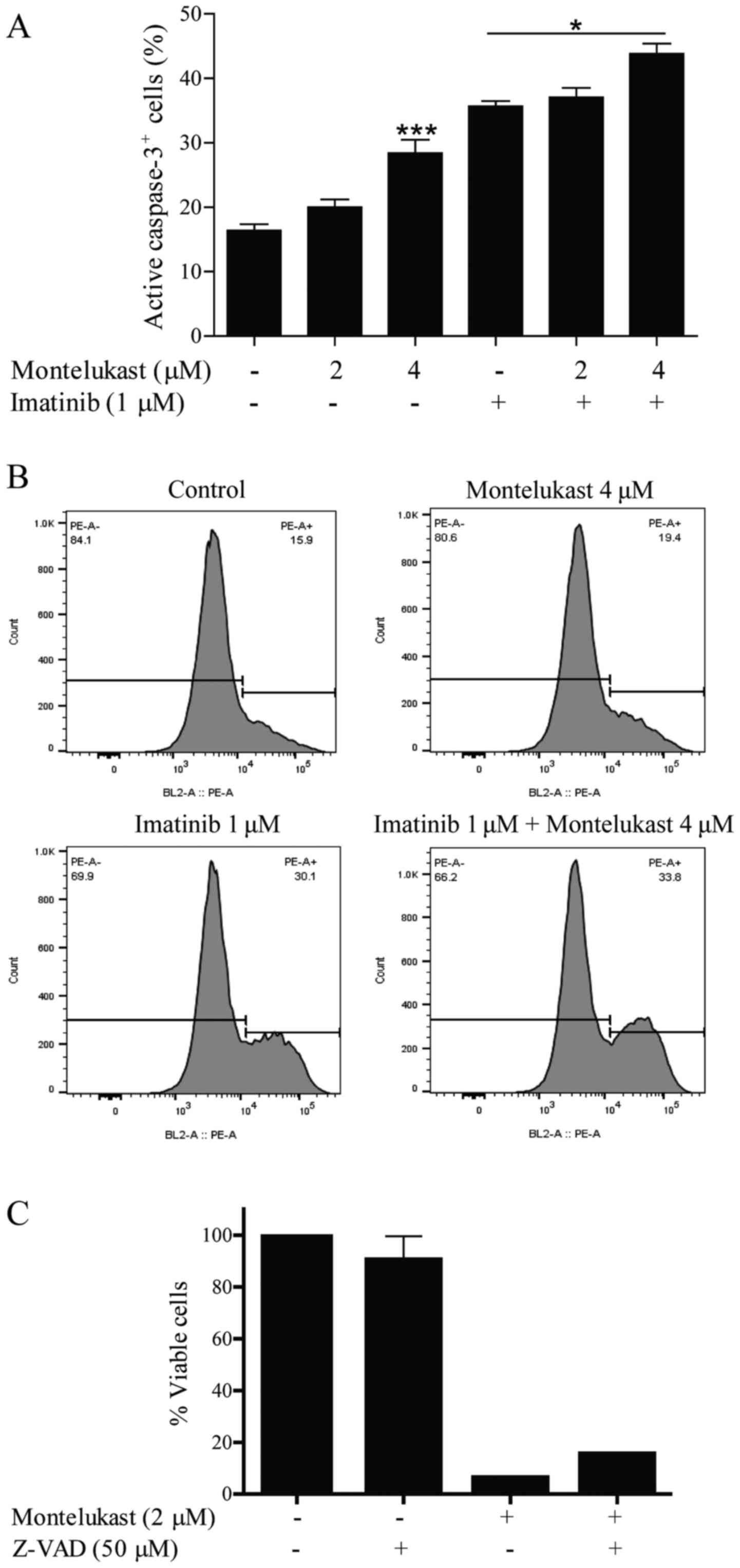
in JURL-MK-1 cells treated with montelukast (Fig. 3D). Montelukast (4 µM) also induced
significant caspase-3 activation (Fig.
4A and B). Imatinib, which was used as a positive control for
apoptosis induction, activated caspase-3 but when used in
combination with 4 µM montelukast, caspase-3 was further activated.
Moreover, the caspase inhibitor Z-VAD was able to partially
(although not statistically significant according to ANOVA test)
inhibit montelukast-induced cell death (Fig. 4C).
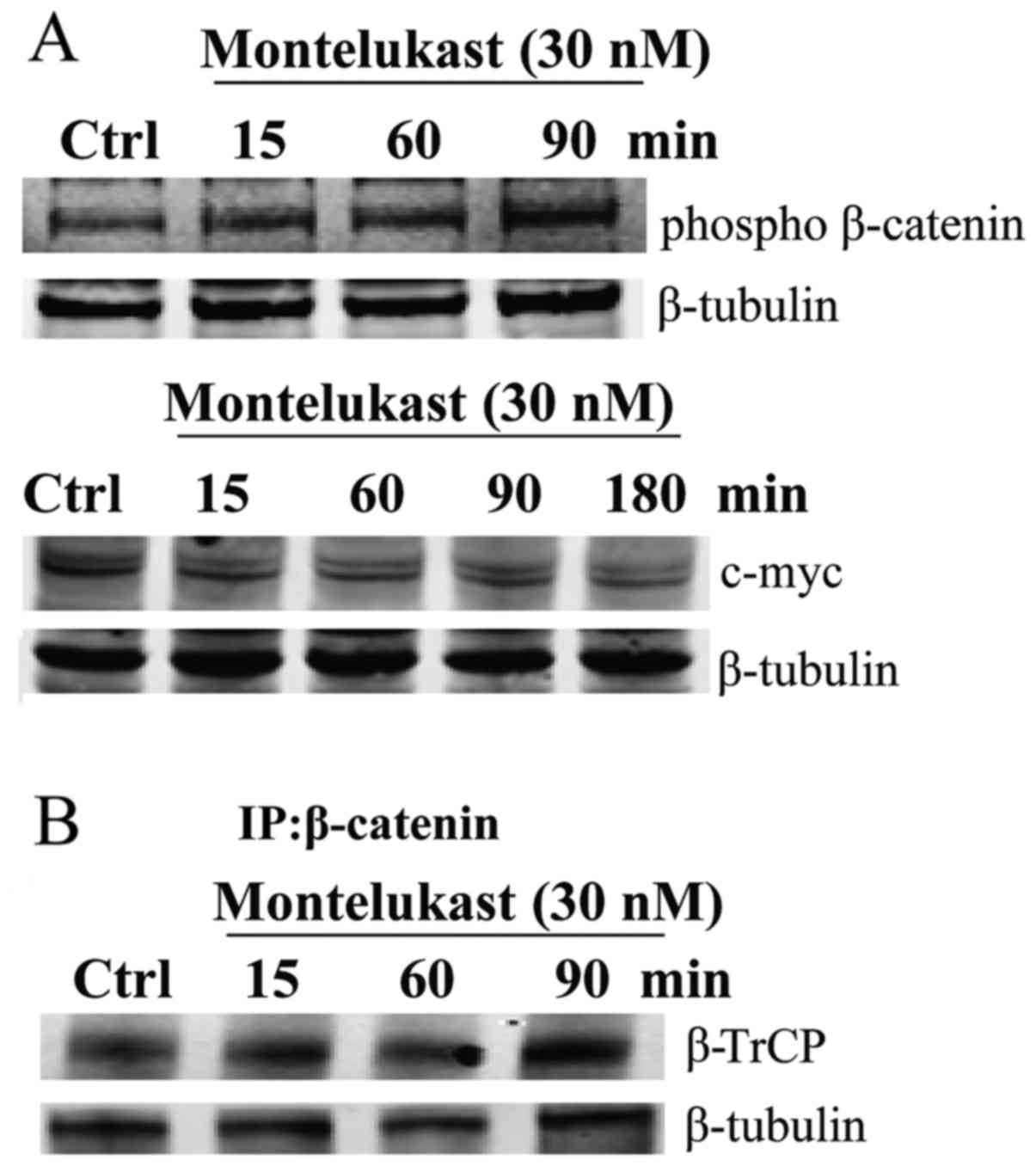
Montelukast induces alterations of
Wnt/β-catenin signaling
In the absence of Wnt signaling, β-catenin is
phosphorylated and polyubiquitinated by β-TrCP, leading to
degradation by the proteasome. Montelukast treatment induced
phosphorylation of β-catenin in a time-dependent manner, as well as
decreased expression of downstream target c-myc (Fig. 5A). Immunoprecipitation with
β-catenin revealed that montelukast enhanced the interaction
between β-catenin and ubiquitin ligase β-TrCP (Fig. 5B).
Discussion
The cysteinyl LT1 receptor (CysLT1R) has been
reported to be overexpressed in several carcinomas, a finding often
linked to an inferior clinical prognosis (9,12). The
action of CysLT1R can be blocked through antagonist binding to the
receptor (10,18).
In the present study, we examined whether the
expression of CysLT1R is crucial for the toxic effect exerted by
the CysLT1R antagonist montelukast. Previously, we demonstrated
that this antagonist has a clear inhibitory effect on the growth of
the CML cell lines K562, KU812 and KCL22, with IC50
concentrations ranging between 1 and 2 µM. CysLT1R was expressed in
all three cell lines (23). This
indicates that an adequate presence of the receptor might be a
prerequisite for the toxic, or cell inhibitory effect of the
antagonist. In order to prove our hypothesis, we knocked down the
CysLT1R receptor in K562 cells utilizing siRNA. When suppressing
the receptor montelukast induced only minor effects on the survival
of these CML cells, excluding off-target effects of the antagonist.
In contrast, imatinib showed a consistent growth inhibitory effect
on the CML cells, regardless of their CysLT1R receptor status.
Moreover, we also showed that the presence of CysLT1R appears to be
of importance for the overall survival of K562 cells, since cells
with siRNA CysLT1R had a reduced viability by approximately 25%. It
is important to note that the knockout was not complete;
approximately 20% of the receptor was still detectable after
knockdown. This may provide an explanation why 75% of the K562
cells remained alive, in spite of (relative) CysLT1R absence.
Several studies performed in different malignancies
have shown that montelukast is able to induce cell apoptosis
(10,15–19).
In order to further understand the mechanisms behind
montelukast-induced cytotoxicity in CML we examined the morphology
of K562 cells. Montelukast caused prominent apoptotic morphology of
the cell nuclei, similar to that induced by imatinib, which we used
as a positive control. It is known that the proapoptotic Bax
protein is overexpressed and the mitochondrial membrane
permeabilized at an early stage of apoptosis, leading to release of
cytochrome c and activation of caspases (26). We noted that montelukast, in a
dose-dependent manner, induced an overexpression of Bax. We further
observed cytochrome c release from cells treated with
montelukast, as well as from cells treated with imatinib.
Montelukast also caused PARP-1 cleavage and caspase-3 activation in
a dose-dependent manner. Importantly, when montelukast was added to
imatinib, a significant increase in caspase activation was
observed, compared to when imatinib was added alone. These results
are in line with our previous observation, that addition of
montelukast to imatinib induced additional inhibitory growth
effects on CML cell lines and on primary CML patient cells
(23). In addition to K562 we also
assessed the CML cell line JURL-MK1 and again observed cleavage of
the PARP-1 protein upon montelukast treatment. This indicates that
apoptosis induction is not specific only for the K562 cell line but
it is activated in other CML cell lines as well in response to
montelukast treatment.
Various downstream signaling pathways are activated
by CysLT1R in other malignancies (11,27).
Ligand binding to CysLT1R induces β-catenin nuclear translocation,
translocation and activation of NF-κB, activation of transcription
factor cAMP response element-binding protein (CREB), activation of
the Ras/Raf/MEK/ERK pathway leading to proliferation, migration and
survival. To better understand the mechanism of action of
montelukast we aimed to ascertain whether montelukast alters these
pathways in the CML cell line K562. Canonical Wnt pathway's
downstream effector β-catenin is required for the progression of
CML and maintenance of LSCs (25,28,29).
In active Wnt/β-catenin signaling β-catenin is translocated into
the nucleus where it activates the transcription of c-myc,
survivin, and cyclin D1 resulting in proliferation of cells
(29). Therefore genetic and
pharmacologic inhibition of β-catenin can eradicate
imatinib-resistant CML LSCs (25).
If Wnt/β-catenin signaling is negatively regulated, cytoplasmic
β-catenin is phosphorylated and marked with ubiquitin mediated by
β-transducin-repeat-containing protein (β-TrCP) for
proteasome-dependent degradation (29). Montelukast induced phosphorylation
of β-catenin, decreased expression of c-myc and enhanced
interaction of β-catenin and β-TrCP. Although the full mechanisms
remain to be elucidated, these observations suggest that
montelukast is able to inhibit Wnt/β-catenin signaling. We observed
no alterations in NF-κB, CREB or Ras/Raf/MEK/ERK pathway as a
result of montelukast treatment (data not shown).
Taken together, our results indicate that addition
of montelukast to imatinib may be a promising, novel clinical
treatment option in CML, perhaps particularly beneficial for
individual CML patients presenting with CysLT overexpression.
Acknowledgements
The authors would like to thank Dr Metka Novak for
the valuable input regarding the cytochrome c staining
experiment.
Funding
The study was financially supported by Emil
Andersson Fund for Medical Research, Medical Research Funds of Umeå
University and Västernorrland County Council, the Cancer Research
Foundations of Radiumhemmet, the National Board of Health and
Welfare, Karolinska Institutet's foundations and funds.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
AZ and EYK contributed equally to conception and
design, acquisition, analysis and interpretation of data, writing
and revising the manuscript. DS performed selected analyses and
participated in data interpretation. LS contributed to the design,
writing and critical revision of the manuscript. AN and JW were
involved in the conception of the study. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Heisterkamp N, Stam K, Groffen J, de Klein
A and Grosveld G: Structural organization of the bcr gene and its
role in the Ph' translocation. Nature. 315:758–761. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hoglund M, Sandin F, Hellström K, Björeman
M, Björkholm M, Brune M, Dreimane A, Ekblom M, Lehmann S, Ljungman
P, et al: Tyrosine kinase inhibitor usage, treatment outcome, and
prognostic scores in CML: Report from the population-based Swedish
CML registry. Blood. 122:pp. 1284–1292. 2013, View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gambacorti-Passerini C, Antolini L, Mahon
FX, Guilhot F, Deininger M, Fava C, Nagler A, Della Casa CM, Morra
E, Abruzzese E, et al: Multicenter independent assessment of
outcomes in chronic myeloid leukemia patients treated with
imatinib. J Natl Cancer Inst. 103:553–561. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Morotti A, Panuzzo C, Fava C and Saglio G:
Kinase-inhibitor-insensitive cancer stem cells in chronic myeloid
leukemia. Expert Opin Biol Ther. 14:287–299. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Funk CD: Prostaglandins and leukotrienes:
Advances in eicosanoid biology. Science. 294:1871–1875. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen Y, Hu Y, Zhang H, Peng C and Li S:
Loss of the Alox5 gene impairs leukemia stem cells and
prevents chronic myeloid leukemia. Nat Genet. 41:783–792. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Y, Li D and Li S: The Alox5
gene is a novel therapeutic target in cancer stem cells of chronic
myeloid leukemia. Cell Cycle. 8:3488–3492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Peters-Golden M, Gleason MM and Togias A:
Cysteinyl leukotrienes: Multi-functional mediators in allergic
rhinitis. Clin Exp Allergy. 36:689–703. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ohd JF, Nielsen CK, Campbell J, Landberg
G, Löfberg H and Sjölander A: Expression of the leukotriene D4
receptor CysLT1, COX-2, and other cell survival factors in
colorectal adenocarcinomas. Gastroenterology. 124:57–70. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Matsuyama M and Yoshimura R:
Cysteinyl-leukotriene1 receptor is a potent target for the
prevention and treatment of human urological cancer. Mol Med Rep.
3:245–251. 2010.PubMed/NCBI
|
|
11
|
Burke L, Butler CT, Murphy A, Moran B,
Gallagher WM, O'Sullivan J and Kennedy BN: Evaluation of cysteinyl
leukotriene signaling as a therapeutic target for colorectal
cancer. Front Cell Dev Biol. 4:1032016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Magnusson C, Liu J, Ehrnström R, Manjer J,
Jirström K, Andersson T and Sjölander A: Cysteinyl leukotriene
receptor expression pattern affects migration of breast cancer
cells and survival of breast cancer patients. Int J Cancer.
129:9–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang WP, Hu H, Zhang L, Ding W, Yao HT,
Chen KD, Sheng WW, Chen Z and Wei EQ: Expression of cysteinyl
leukotriene receptor 1 in human traumatic brain injury and brain
tumors. Neurosci Lett. 363:247–251. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Virchow JC and Bachert C: Efficacy and
safety of montelukast in adults with asthma and allergic rhinitis.
Respir Med. 100:1952–1959. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matsuyama M, Funao K, Hayama T, Tanaka T,
Kawahito Y, Sano H, Takemoto Y, Nakatani T and Yoshimura R:
Relationship between cysteinyl-leukotriene-1 receptor and human
transitional cell carcinoma in bladder. Urology. 73:916–921. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matsuyama M, Hayama T, Funao K, Kawahito
Y, Sano H, Takemoto Y, Nakatani T and Yoshimura R: Overexpression
of cysteinyl LT1 receptor in prostate cancer and CysLT1R antagonist
inhibits prostate cancer cell growth through apoptosis. Oncol Rep.
18:99–104. 2007.PubMed/NCBI
|
|
17
|
Funao K, Matsuyama M, Naganuma T, Kawahito
Y, Sano H, Nakatani T and Yoshimura R: The cysteinylLT1 receptor in
human renal cell carcinoma. Mol Med Rep. 1:185–189. 2008.PubMed/NCBI
|
|
18
|
Savari S, Liu M, Zhang Y, Sime W and
Sjölander A: CysLT(1)R antagonists inhibit tumor growth in a
xenograft model of colon cancer. PLoS One. 8:e734662013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sveinbjörnsson B, Rasmuson A, Baryawno N,
Wan M, Pettersen I, Ponthan F, Orrego A, Haeggström JZ, Johnsen JI
and Kogner P: Expression of enzymes and receptors of the
leukotriene pathway in human neuroblastoma promotes tumor survival
and provides a target for therapy. FASEB J. 22:3525–3536. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Piromkraipak P, Sangpairoj K, Tirakotai W,
Chaithirayanon K, Unchern S, Supavilai P, Power C and Vivithanaporn
P: Cysteinyl leukotriene receptor antagonists inhibit migration,
invasion, and expression of MMP-2/9 in human glioblastoma. Cell Mol
Neurobiol. 38:559–573. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stenke L, Mansour M, Reizenstein P and
Lindgren JA: Stimulation of human myelopoiesis by leukotrienes B4
and C4: interactions with granulocyte-macrophage colony-stimulating
factor. Blood. 81:352–356. 1993.PubMed/NCBI
|
|
22
|
Stenke L, Samuelsson J, Palmblad J,
Dabrowski L, Reizenstein P and Lindgren JA: Elevated white blood
cell synthesis of leukotriene C4 in chronic myelogenous leukaemia
but not in polycythaemia vera. Br J Haematol. 74:257–263. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yektaei-Karin E, Zovko A, Nilsson A,
Näsman-Glaser B, Kanter L, Rådmark O, Wallvik J, Ekblom M, Dolinska
M, Qian H, et al: Modulation of leukotriene signaling inhibiting
cell growth in chronic myeloid leukemia. Leuk Lymphoma.
58:1903–1913. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao C, Blum J, Chen A, Kwon HY, Jung SH,
Cook JM, Lagoo A and Reya T: Loss of beta-catenin impairs the
renewal of normal and CML stem cells in vivo. Cancer Cell.
12:528–541. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Heidel FH, Bullinger L, Feng Z, Wang Z,
Neff TA, Stein L, Kalaitzidis D, Lane SW and Armstrong SA: Genetic
and pharmacologic inhibition of β-catenin targets
imatinib-resistant leukemia stem cells in CML. Cell Stem Cell.
10:412–424. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reed JC: Proapoptotic multidomain
Bcl-2/Bax-family proteins: Mechanisms, physiological roles, and
therapeutic opportunities. Cell Death Differ. 13:1378–1386. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Savari S, Vinnakota K, Zhang Y and
Sjölander A: Cysteinyl leukotrienes and their receptors: Bridging
inflammation and colorectal cancer. World J Gastroenterol.
20:968–977. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou H, Mak PY, Mu H, Mak DH, Zeng Z,
Cortes J, Liu Q, Andreeff M and Carter BZ: Combined inhibition of
β-catenin and Bcr-Abl synergistically targets tyrosine kinase
inhibitor-resistant blast crisis chronic myeloid leukemia blasts
and progenitors in vitro and in vivo. Leukemia. 31:2065–2074. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang X and Hao J: Development of
anticancer agents targeting the Wnt/β-catenin signaling. Am J
Cancer Res. 5:2344–2360. 2015.PubMed/NCBI
|