Introduction
Glioblastomas are highly malignant brain tumors that
have a poor prognosis for patients despite advanced surgery and
subsequent aggressive treatment with combined radiotherapy and
chemotherapy (1). Over 90 years
ago, Bailey and Cushing established the term glioblastoma
multiforme (2) for the astrocytoma
World Health Organization (WHO) grade IV, paying tribute to the
pronounced heterogeneity of this type of tumor. This heterogeneity
has been observed and investigated in a variety of aspects of
glioblastoma biology, and attributed to the differential response
of patients to therapy (3,4). Cell cycle control and DNA repair
mechanisms have been identified as key aspects at the evolutionary
origin of the pronounced inter- and intratumoral genetic
heterogeneity of gliomas, amongst others (5,6).
However, a recent study indicated an important role of the
apolipoprotein B mRNA editing enzyme catalytic polypeptide-like
(APOBEC) enzyme family in genetic instability and heterogeneity, as
specific patterns of DNA mutagenesis have been observed in various
types of human tumors (7). The
evolutionarily highly conserved APOBEC enzyme family members have
cytidine desaminase activity, by which they exert
post-transcriptional editing of the mRNA by converting cytosine to
uracil; for example, in the mRNA of apolipoprotein B and
neurofibromin (8). Certain APOBEC
family members also support the innate immune system by editing the
viral genome, particularly the genome of retroviruses like the
human immunodeficiency virus HIV (9). In cancer, APOBEC family members are
key players in the induction of cancer evolution and heterogeneity
(10). In particular, APOBEC3B has
been identified as the major cause for typical mutagenesis
patterns, for example in breast (11), bladder, cervix, head and neck, and
lung cancer (12). Although a
recent study has addressed a putative APOBEC enzyme-caused
mutational signature in gliomas (13), to the best of our knowledge, there
is no information available on the APOBEC3B protein in human
gliomas. Thus, the present study investigated the expression and
functional role of APOBEC3B in human glioma tissue and cell
lines.
Materials and methods
Tissue and cell lines
Glioma samples of different malignancy grades were
obtained at the Department of Neurosurgery, University Medical
Center Schleswig-Holstein UKSH (Kiel, Germany) in accordance with
the Declaration of Helsinki (1975) with approval from the Ethics
Committee of the University of Kiel (Kiel, Germany) and after
written informed consent was obtained from the donors (file
reference, D536/15). Diagnosis was established by a pathologist and
malignancy grades were determined according to the WHO
classification system. Non-neoplastic brain tissue samples were
obtained from the Institute of Legal Medicine, University Kiel.
Glioma cell lines LN229, A172, T98G and U251MG were purchased from
the European Collection of Cell Cultures (Salisbury, UK) or the
American Type Culture Collection (Manassas, VA, USA) and cultured
in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine
serum (FBS) (Invitrogen; Thermo Fisher Scientific, Inc., Waltham,
MA, USA). Cells were routinely checked for Mycoplasma contamination
using bisbenzimide staining, and for identity/purity by short
tandem repeat profiling at the Department of Forensic Medicine,
University of Kiel where the Powerplex HS Genotyping kit (Promega
Corporation, Madison, WI, USA) and a 3500 Genetic analyzer (Thermo
Fisher Scientific, Inc.) were employed.
Cell stimulations and clustered
regularly interspaced short palindromic repeats (CRISPR)/CRISPR
associated protein 9 (Cas9) silencing
In order to analyze the regulation of APOBEC3B
expression, 5.0×104 LN229 cells/well were seeded in
six-well plates or 2.0×104 LN229 cells were seeded on
glass cover slips and grown for 24 h in DMEM with 10% FBS. Cells
were then treated with 100 µg/ml temozolomide or a corresponding
amount of the solvent DMSO for a total of 10 days, during which the
media were changed every 2–3 days, and the DMSO-treated cells were
detached and seeded again at the same densities on day 6. After 10
days, cells were harvested for mRNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR), or
the glass slides with cells were used for immunocytochemistry
(ICC).
For CRISPR/Cas9 silencing, 5.0×104 LN229
cells/well were seeded in six-well plates and transfected with 1 µg
of the APOBEC3B Double Nickase plasmid (cat. no. sc-401700-NIC) or
the corresponding Double Nickase Control plasmid (cat. no.
sc-437281) using the UltraCruz Transfection reagent (all from Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) for 24 h. Following the
manufacturer's protocol, selection was performed with puromycin
(Invitrogen; Thermo Fisher Scientific, Inc.), and clones were
selected and analyzed using RT-qPCR, ICC and western blot
analysis.
RT-qPCR
For mRNA expression analysis, cells were harvested
and tissues were homogenized using QIAzol Lysis reagent (Qiagen
GmbH, Hilden, Germany) and total RNA was isolated according to the
manufacturer's protocol. Genomic DNA was digested by RNase-free
DNase (1 U/µl; Promega Corporation) and cDNA was synthesized using
RevertAid™ H Minus M-MuLV Reverse Transcriptase (200 U/µl; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
RT-qPCR was performed as previously described (14) using the following TaqMan Primer
probes (Applied Biosystems; Thermo Fisher Scientific, Inc.): hGAPDH
(Hs99999905_m1) and hAPOBEC3B (Hs00358981_m1). Samples were
analyzed in duplicates and cycle threshold (Cq) values were
measured with the ABI PRISM 7500 Sequence Detection system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling
conditions were as follows: 2 min at 50°C and 10 min 95°C, followed
by 40 cycles of 15 sec at 95°C and 1 min at 60°C. Cq values were
used to calculate ∆Cq values [Cq(APOBEC3B) - Cq(GAPDH)]. Due to
logarithmic reaction process, a ∆Cq value of 3.33 corresponds to
one magnitude lower gene expression compared with that of GAPDH.
For stimuli-induced mRNA regulation, ∆∆Cq values were calculated as
follows: ∆∆Cq = 2 −
[∆Cq(stimulus-temozolomide) −
∆CT(control-DMSO)]. For statistical analysis,
undetectable samples were calculated as the anticipated maximum
cycle number-Cq (GAPDH).
Immunohistochemistry and ICC
Immunohistochemistry was performed as previously
described (15). Briefly, cryostat
sections from three different fresh-frozen glioblastoma samples
were fixed in ice-cold acetone/methanol (1:1) for 10 min at room
temperature and washed with Tris-buffered saline with 0.1% Tween
(TBS-T). The samples were then blocked for autofluorescence with 1%
Sudan black (in 70% ethanol) for 1 h at room temperature and for
unspecific binding with 0.5% glycine/0.5% bovine serum albumin
(SERVA Electrophoresis GmbH, Heidelberg, Germany) in TBS and
incubated with the primary antibodies overnight at 4°C.
Subsequently, the samples were washed with TBS-T and incubated with
the secondary antibodies for 1 h at 37°C. The samples then were
washed again with TBS-T, incubated with
4′,6-diamidino-2-phenylindole (DAPI; 1:30,000; Invitrogen; Thermo
Fisher Scientific, Inc.) for 30 min at room temperature to
counterstain the nuclei, washed with TBS-T, and embedded with
Shandon™ Immu-Mount™ (Thermo Fisher Scientific, Inc.). The primary
antibodies were as follows: Anti-human APOBEC3B (rabbit polyclonal;
1:100; cat. no. TA349029; OriGene Technologies, Inc., Rockville,
MD, USA); anti-human glial fibrillary acidic protein (GFAP; mouse
monoclonal; 1:100; cat. no. M0761; Dako; Agilent Technologies,
Inc., Santa Clara, CA, USA), anti-human made in borstel-1 (MIB-1;
mouse monoclonal; 1:100; cat. no. M7240; Dako; Agilent
Technologies, Inc.) and anti-human platelet endothelial cell
adhesion molecule (Pecam-1; goat polyclonal; 1:100; cat. no.
sc-1506; Santa Cruz Biotechnology, Inc.). The secondary antibodies
were as follows: Donkey anti-rabbit IgG Alexa Fluor 555 (cat. no.
A-31572); donkey anti-mouse IgG Alexa Fluor 488 (cat. no. A-21202);
and donkey anti-goat IgG Alexa Fluor 488 (cat. no. A-11055) (all
1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.).
For immunocytochemical analysis, cells were seeded
on glass cover slips (2.0×104 cells/cover slip) and
grown for 1 day (or for the subsequent treatment period). The cells
were fixed with ice-cold acetone-methanol for 10 min at room
temperature, blocked with 0.5% glycine/0.5% bovine serum albumin in
PBS for 1 h at room temperature and incubated with anti-human
APOBEC3B (rabbit polyclonal; 1:100) overnight at 4°C. The cells
were then washed with PBS + 0.1% Tween (PBS-T; 3X) and incubated
with donkey anti-rabbit IgG Alexa Fluor 555 (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C for 1 h. Subsequently, the cells
were washed with PBS-T (3X), the nuclei were counterstained with
DAPI for 30 min at room temperature, and the slips were embedded
using Immu-Mount.
The slides were viewed and imaged using a Zeiss
Axiovert 200 M fluorescence microscope (Zeiss AG, Oberkochem,
Germany) at ×400 and ×630 magnification. In order to compare
differences in the expression of APOBEC3B, equal exposure times
were used.
Western blot analysis
For western blot analysis, cells were lysed (50 mM
Tris, 100 mM NaCl, 5 mM EDTA, 1% Triton X-100, pH 7.8), and protein
content was measured using the Bradford protein assay. Protein
content was adjusted to 5 µg/20 µl with sample buffer (10%
glycerin, 0.4% SDS, 50 mM dithiothreitol in 75 mM Tris), and
samples were separated by SDS-PAGE (10% acrylamide gels, 5 µg/lane)
and blotted onto a polyvinylidene fluoride membrane. The membrane
was blocked with 5% skim milk powder in TBS-T for unspecific
binding for 1 h at room temperature and incubated with anti-human
APOBEC3B (rabbit polyclonal; 1:250) overnight at 4°C. Subsequently,
the membrane was washed with TBS-T (3X) and incubated with
horseradish peroxidase-conjugated secondary antibody overnight at
4°C (goat anti-rabbit; 1:30,000; cat. no. sc-2004; Santa Cruz
Biotechnology, Inc.). The membrane was then washed again with
TBS-T, followed by chemoluminescence signal detection (Immobilon;
Merck KGaA, Darmstadt, Germany) using a Peqlab Fusion camera
(Peqlab Biotechnologie GmbH, Erlangen, Germany). Blots were
reprobed following stripping (ReBlot Plus Strong Antibody Striping
solution; Merck KGaA) for GAPDH (primary antibody mouse anti-human;
1:250; cat. no. sc-47724; goat anti-mouse horseradish
peroxidase-conjugated secondary antibody; 1:30,000; cat. no.
sc-2005; from Santa Cruz Biotechnology, Inc.) in order to compare
loading using the same protocol aforementioned.
Proliferation assay
In order to compare the proliferation of LN229
APOBEC3B CRISPR/Cas9 knockdown clones with that of control clones,
2.5×104 cells/well were seeded in 6-well plates in
triplicates, and cultured for 24, 48, 72 and 96 h. Cells were then
washed with PBS, harvested, and analyzed using a
CyQUANT® cell proliferation assay (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. As an
internal standard, 2.5×104 cells (APOBEC3B and control
clones) were lysed and measured, and fluorescence signals were
normalized to these samples.
Caspase 3/7 assay
In order to analyze the influence of APOBEC3B on the
sensitivity of cells to temozolomide, the most common
chemotherapeutic agent used in glioblastoma therapy,
3.0×105 APOBEC3B clone cells or control clone cells were
seeded in T25 flasks, and cultured for 24 h. Subsequently, the
medium was replaced by fresh DMEM with 10% FBS and different
concentrations of temozolomide (or an equal volume of DMSO as a
solvent control), and the cells were treated for a further 48 h.
Caspase 3/7 activity was measured as previously described (16).
Statistical analysis
All values are presented as the mean ± standard
deviation. Expression values from tumor samples were compared with
non-neoplastic brain tissue samples using one-way analysis of
variance (ANOVA) with Dunnet's multiple comparison post hoc test.
To analyze induction of APOBEC3B expression, a paired Student's
t-test was used. For statistical analysis of biological effects of
APOBEC3B silencing (proliferation and caspase 3/7 assays) one-way
ANOVA with Bonferroni's multiple comparison post hoc test was used
to compare control clones with APOBEC3B knockdown clones at
different time points (proliferation) or upon different
temozolomide treatment doses (caspase 3/7 assay). P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression and drug-induced regulation
of APOBEC3B in gliomas
As the initial step, the expression and cellular
localization of APOBEC3B in solid human astrocytoma samples of
different malignancy grades, as well as in commercial glioblastoma
cell lines, were investigated. Additionally, the regulation of the
amounts of APOBEC3B mRNA and protein was analyzed in
temozolomide-treated glioblastoma cells.
APOBEC3B was expressed in all of the glioma samples
in considerable amounts (mean ΔCq value range, 8.0–9.0 for
astrocytoma grade I–IV; grade IV astrocytoma is also known as
glioblastoma). Compared with the normal brain control samples,
APOBEC3B was significantly overexpressed in glioblastoma tissue.
The mean ΔCq values were 11.2 for the normal brain samples, and
8.4, 9.1, 8.2 and 8.0 for grade I–IV astrocytoma samples,
respectively (Fig. 1, left). A ∆Cq
value of 3.33 corresponds to one magnitude lower gene expression
compared with that of the reference marker GAPDH. In the
glioblastoma samples, APOBEC3B was expressed in GFAP-positive
astrocytic tumor regions and was detected near to Pecam-1-positive
tumor vessels, but was not co-stainable with MIB-1 (antigen Ki-67),
which is a marker of active proliferating cells (Fig. 1, right).
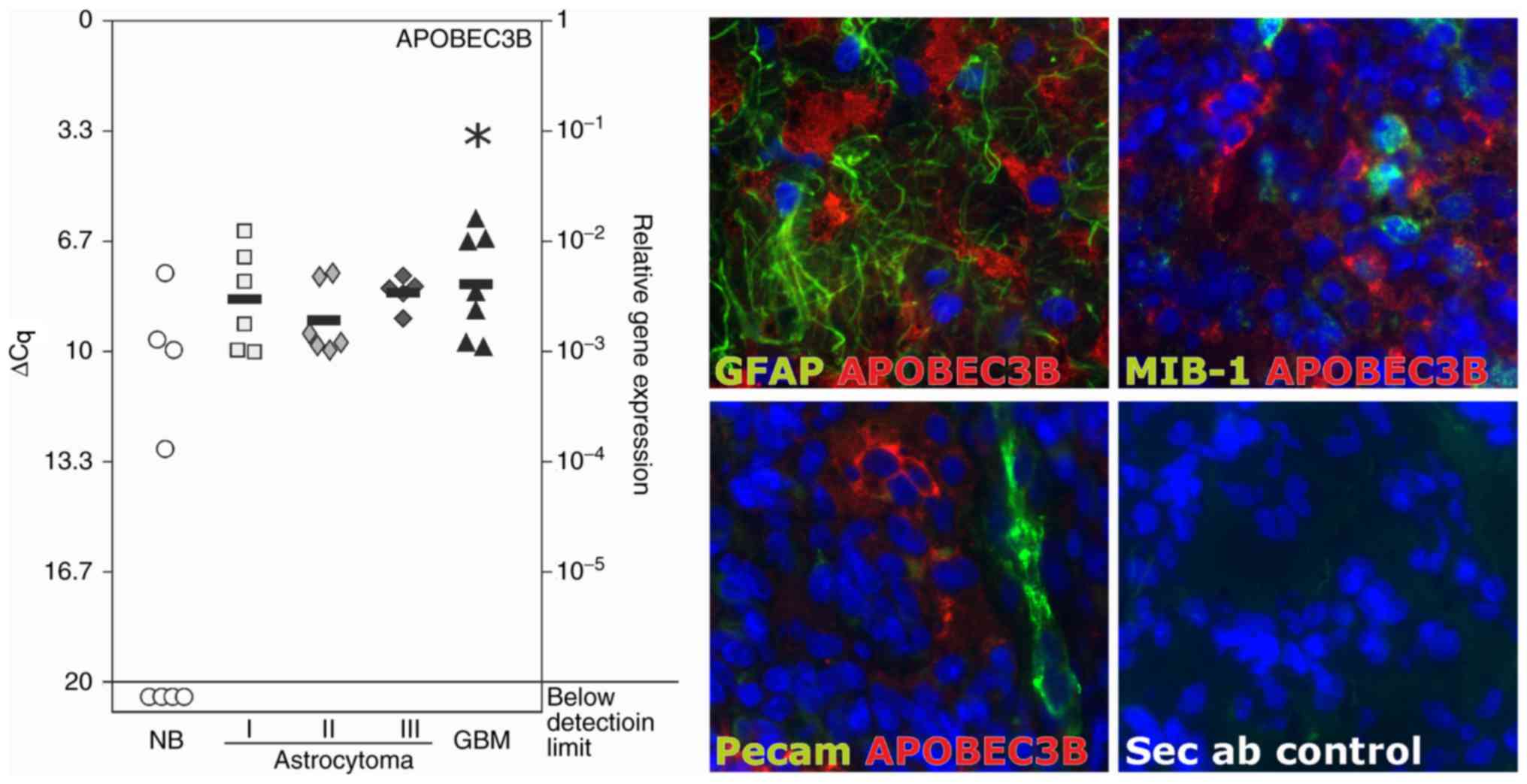 | Figure 1.Expression of APOBEC3B in solid human
gliomas. Left: Transcription of APOBEC3B was analyzed in eight NB,
six grade I astrocytoma, six grade II astrocytoma, five grade III
astrocytoma and seven grade IV astrocytoma/GBM using RT-qPCR. In
all of the investigated glioma samples, APOBEC3B mRNA was clearly
detectable. In the grade IV tumors, the levels of mRNA expression
were significantly higher compared with those in the non-neoplastic
brain tissues. Black bars indicate mean values and a lower ΔCq
value of 3.33 corresponds to a magnitude higher expression. In a
number of the non-neoplastic brain tissue samples, APOBEC3B could
not be detected (below detection limit); thus, a mean bar could not
be included. Right: Expression and localization of APOBEC3B were
analyzed using immunohistochemistry in the human glioblastoma
samples. APOBEC3B (red) was detected on the protein level in
GFAP-positive tumor regions (green), frequently in small cell
clusters and in proximity to blood vessels (stained by Pecam-1;
green), and co-staining with MIB-1, a marker of proliferating
cells, was not evident. For secondary antibody controls, primary
antibodies were respectively omitted. Representative examples of
three individual glioblastoma samples are shown at ×630
magnification. *P<0.05, one-way analysis of variance followed by
Dunnet's multiple comparison test. APOBEC3B, apolipoprotein B mRNA
editing enzyme catalytic polypeptide-like protein 3B; Cq, cycle of
threshold; GBM, glioblastoma multiforme; GFAP, glial acidic
fibrillary protein; MIB-1, made in Borstel-1; NB, normal brain;
Pecam-1, platelet endothelial cell adhesion molecule; Sec ab,
secondary antibody control. |
With the exception of the U251MG cell line, APOBEC3B
was evidently detectable in the glioblastoma cell lines
investigated (A172, LN229, T98G and U251MG) at the transcriptional
level, as determined using RT-qPCR (Fig. 2, left). The mean ΔCq values were as
follows: 8.4±0.7 (A172), 6.7±0.2 (LN229), 6.8±1.1 (T98G) and
18.5±3.0 (U251MG) (n=4 independent experiments). Furthermore,
APOBEC3B expression was evaluated at the translational level using
ICC in the LN229, T98G and U251MG cell lines (n=2 independent
experiment; Fig. 2, right). When
stimulating LN229 glioblastoma cells with temozolomide (100 µg/ml
for 10 days), the standard chemotherapeutic drug used in
glioblastoma treatment, APOBEC3B mRNA expression was increased up
to 9-fold (range, 3.5 to 9.2-fold; mean, 6.1±1.9-fold; Fig. 3, left; n=8 independent experiments;
P<0.05) of that in the DMSO control group. In accordance with
this, APOBEC3B protein was detectable in markedly higher amounts in
the temozolomide-treated LN229 cells compared with those of the
DMSO control-treated cells (Fig. 3,
right; n=2 independent experiments).
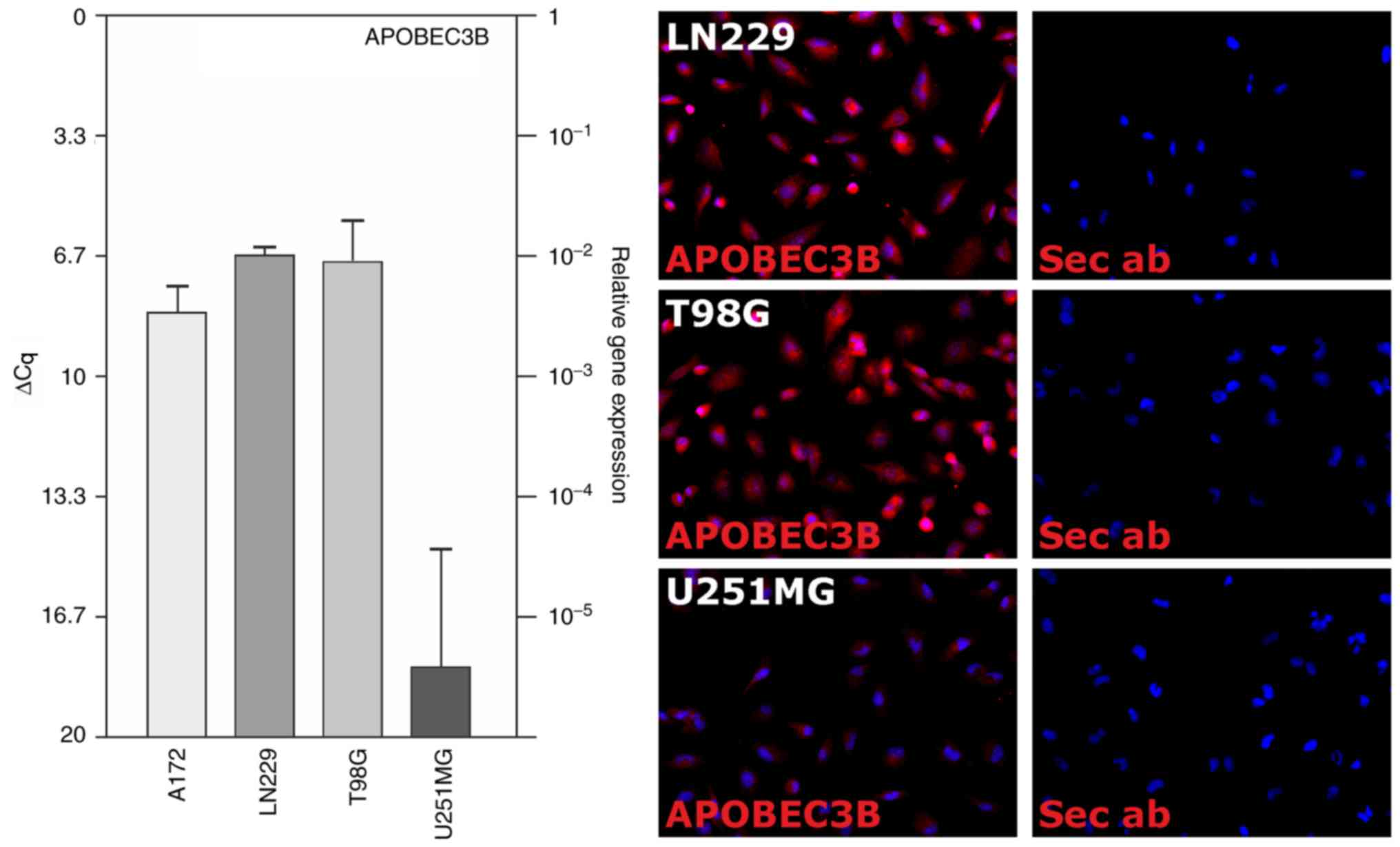 | Figure 2.Expression of APOBEC3B in human
glioblastoma cell lines. APOBEC3B expression was analyzed on the
mRNA (RT-qPCR, left) and protein (ICC, right) levels in human
glioblastoma cell lines. Clearly detectable levels of mRNA
expression were observed in the A172, LN229 and T98G cells, while
APOBEC3B was hardly detected in the U251MG cells. At the protein
level, APOBEC3B expression was exemplarily analyzed in the LN229,
T98G and U251MG cells. Data are presented as the mean ΔCq values ±
standard deviation from n=4 individual RNA preparations and
exemplary results from n=2 individual immunostainings at ×400
magnification. APOBEC3B, apolipoprotein B mRNA editing enzyme
catalytic polypeptide-like protein 3B; Cq, cycle of threshold; Sec
ab, secondary antibody control; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; ICC,
immunocytochemistry. |
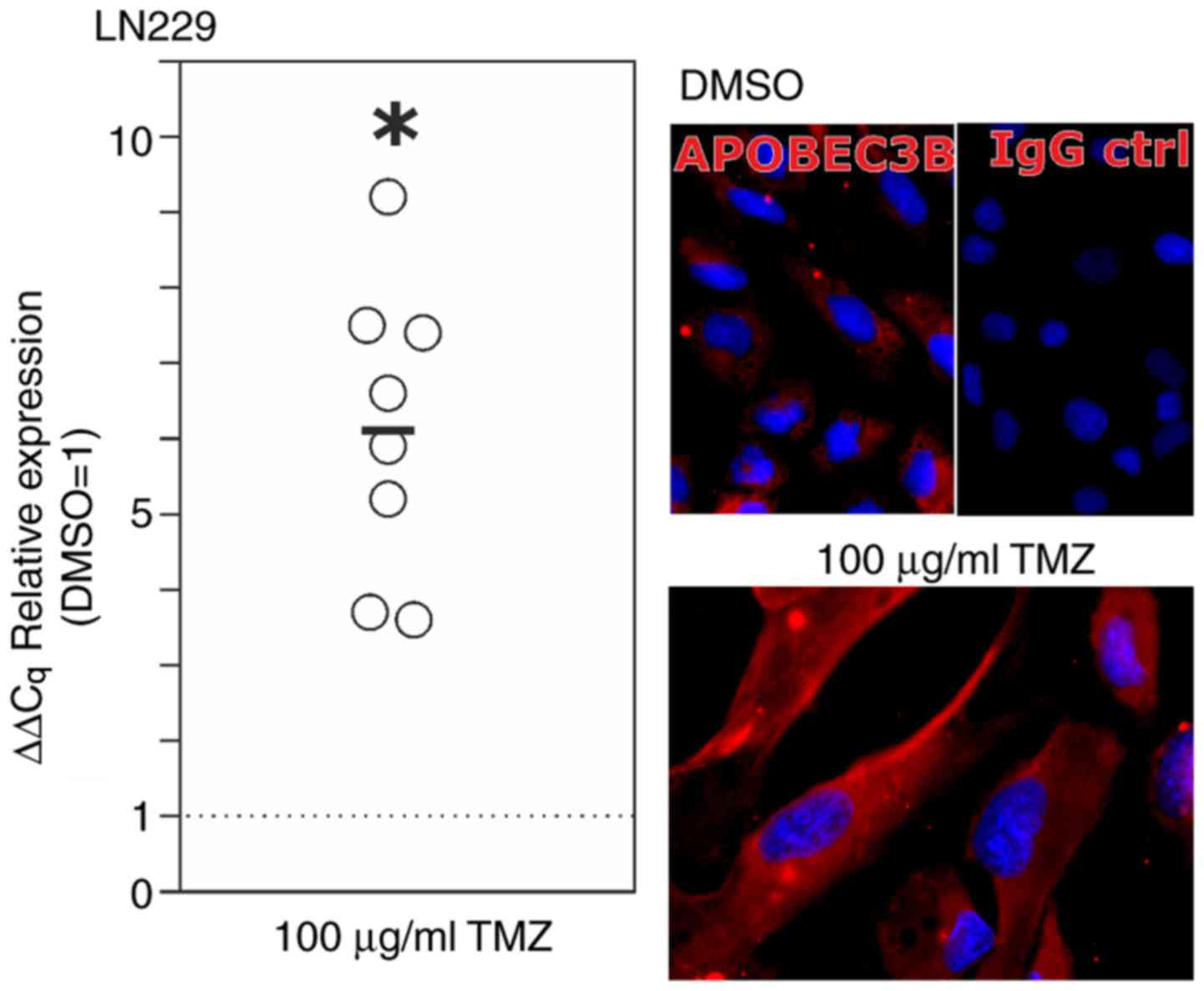 | Figure 3.Regulation of APOBEC3B upon TMZ
treatment in vitro. LN229 glioma cells were treated for 10
days with 100 µg/ml TMZ or equal volumes of the solvent DMSO as a
control. Expression analysis using RT-qPCR (left, circles indicate
independent experiments, black bar indicates mean value from n=8
independent experiments, *P<0.05; Student's two-tailed t-test)
and ICC (representative examples of n=2 independent stimulations)
demonstrated a significant induction (mean, 6.1-fold) of APOBEC3B
upon TMZ treatment. For the microscopic images, the same
magnifications (×630) and exposure times were used. APOBEC3B,
apolipoprotein B mRNA editing enzyme catalytic polypeptide-like
protein 3B; Cq, cycle of threshold; DMSO, dimethylsulfoxide; IgG
ctrl, IgG control; TMZ, temozolomide; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; ICC,
immunocytochemistry. |
Biological functions of APOBEC3B in
glioblastoma
In order to investigate the biological functions of
APOBEC3B in glioblastoma, stable APOBEC3B CRISPR/Cas9 knockdown
LN229 clones were established. The knockdown efficiency was
confirmed using RT-qPCR, western blot analysis and ICC. All methods
demonstrated clearly diminished levels of APOBEC3B expression at
the mRNA and protein levels. At the mRNA level (n=3) APOBEC3B
expression was reduced to 1.7±0.8% in comparison with the
corresponding control CRISPR/Cas9 clones (data not shown). At the
protein level, evident reduced signals were observed in the western
blot experiments (Fig. 4A) and no
APOBEC3B signals in the APOBEC3B CRISPR/Cas9 knockdown LN229 clones
were identified by ICC (equal exposure times, Fig. 4A).
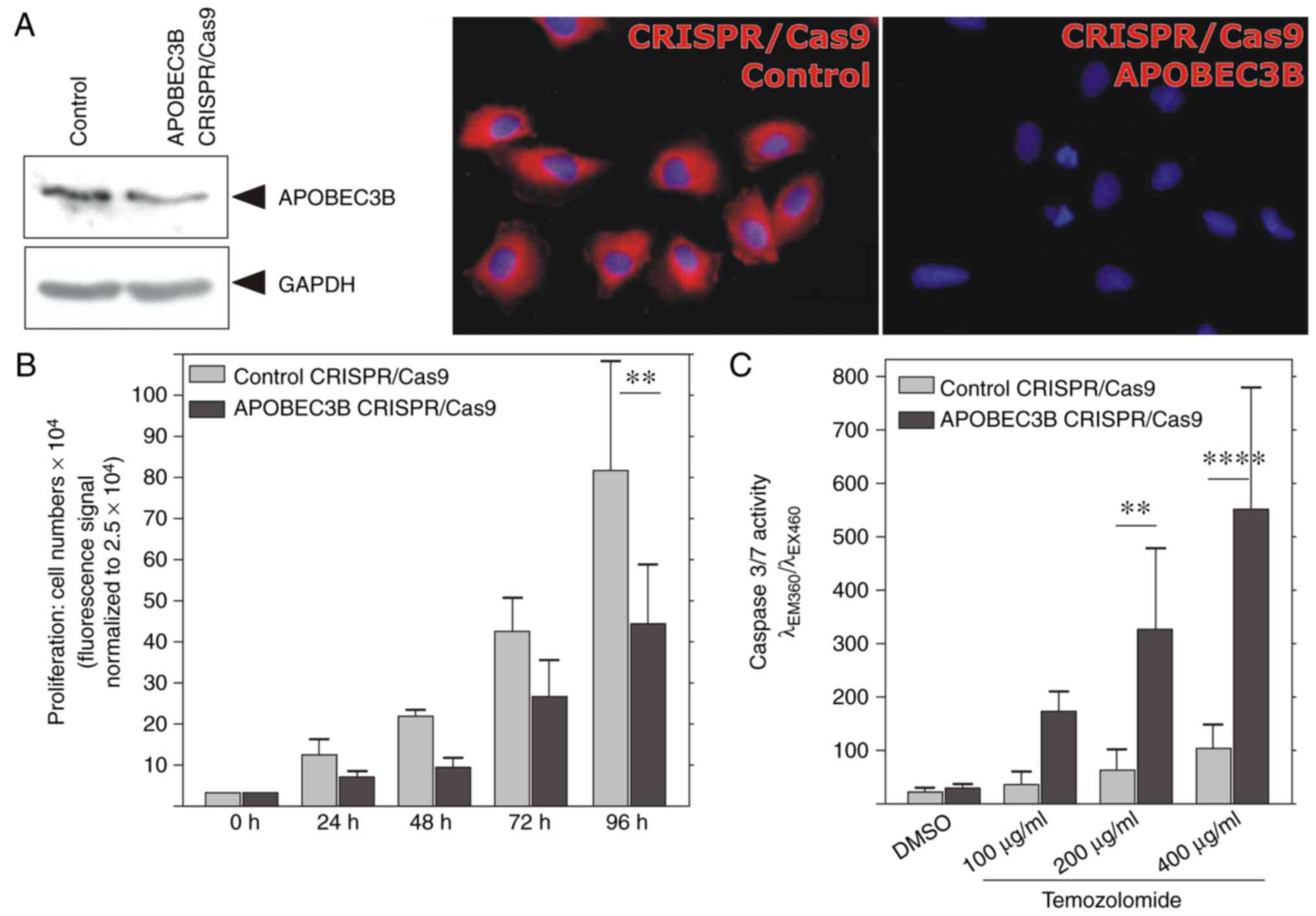 | Figure 4.Knockdown of APOBEC3B reduces
proliferation and enhances sensitivity of cells to TMZ. (A) In
LN229-derived CRISPR/Cas9 knockdown clones of APOBEC3B, expression
levels of APOBEC3B were markedly reduced compared with the control
clones, as shown by western blotting (detection of GAPDH served as
the loading control) and ICC. For the microscopic images, the same
magnifications (×400) and exposure times were used. (B)
LN229-derived CRISPR/Cas9 knockdown clones of APOBEC3B proliferated
less compared with the corresponding control clones in a time
period of 48–96 h, as detected by measurement of the DNA content.
**P<0.01, one-way analysis of variance followed by Bonferroni's
multiple comparison test. (C) When exposed to different
concentrations of TMZ for 48 h, LN229 APOBEC3B-knockdown cells
exhibited significantly higher levels of caspase 3/7 activity
compared with those of the corresponding control clones upon 200
and 400 µg/ml TMZ treatment. **P<0.01 and ****P<0.0001,
one-way analysis of variance followed by Bonferroni's multiple
comparison test. The levels of basic caspase 3/7 activity in
DMSO-treated cells were not significantly altered in the APOBEC3B
CRISPR/Cas9 clones, compared with those of the control clones. Data
are presented as the mean values ± standard deviation of n=3
independent experiments. APOBEC3B, apolipoprotein B mRNA editing
enzyme catalytic polypeptide-like protein 3B; GAPDH, glyceraldehyde
3-phosphate dehydrogenase; TMZ, temozolomide; CRISPR, clustered
regularly interspaced short palindromic repeats; Cas9, CRISPR
associated protein 9. |
Investigation of the proliferative potential of
APOBEC3B CRISPR/Cas9 knockdown LN229 clones for ≤96 h compared with
that of their control-transfected counterparts revealed that LN229
clones with diminished APOBEC3B expression were characterized by a
reduction in proliferation levels (Fig.
4B; n=3 independent experiments; P<0.01 at 96 h).
Furthermore, as demonstrated by caspase 3/7 activity, APOBEC3B
CRISPR/Cas9 knockdown LN229 clones exhibited significantly higher
levels of sensitivity to temozolomide treatment compared with those
of the controls (Fig. 4C).
Increasing concentrations of temozolomide (100–400 mg/ml; 48 h)
yielded increasing levels of caspase 3/7 activity in the control
cells; however, in the APOBEC3B knockdown clones, the
concentration-dependent induction of the caspase 3/7 activity was
significantly more prominent (200 µg/ml TMZ, P<0.01; 400 µg/ml
TMZ, P<0.0001). The caspase 3/7 activity in DMSO-treated samples
was not significantly different between the APOBEC3B and control
CRISPR/Cas9 clones. Thus, APOBEC3B mediates proliferative and
anti-apoptotic effects in human glioblastoma cells.
In summary, the APOBEC3B enzyme was evidently
detectable in human glioma samples of different malignancy grades,
and was observable in astrocytic tumor regions and nearby tumor
vessels. Furthermore, the APOBEC3B enzyme was inducible upon
temozolomide treatment, and mediated proliferative and
anti-apoptotic effects in human glioblastoma cells.
Discussion
A previous study has demonstrated that members of
the APOBEC family may cause a characteristic pattern of mutations
in a several types of cancer (7).
Apart from their physiological role in mRNA editing, this enzyme
family has important functions in the innate immune response to
retroviruses and retrotransponsons, and may also contribute to
tumor heterogeneity and progression via its DNA-desaminase activity
(9,10). In the present study, it was
demonstrated that APOBEC3B, a member of the APOBEC family, was
overexpressed in glioblastoma (WHO grade IV), and was identified in
astrocytic tumor regions and nearby tumor vessels. To date,
APOBEC3B has only been detected in human leptomeninges and
meningioma, to the best of our knowledge (17). However, Talagas et al
(18) reported that certain
oligodendroglial intracranial tumors that do not exhibit 1p/19q
deletions are characterized by various other mutations, including a
homozygous deletion at 22q13.1 (APOBEC3B gene). To the best of our
knowledge, there is no data regarding on the role of APOBEC3B in
astroglial tumors to date. Focusing on another APOBEC family member
in astroglial tumors, a recent study indicated an important role
for APOBEC3G in mesenchymal glioma, a highly malignant brain tumor
type (19). APOBEC3G knockdown
attenuated the proliferation and invasion of glioblastoma cell
lines by influencing the transforming growth factor
(TGF)-β-signaling pathway via smad2, which resulted in decreased
expression levels of proteins, including thrombospondin-1 and
matrix-metalloproteinase-2 (19).
However, the exact mechanism by which APOBEC3G regulates the TGF-β
pathway remains unclear. Notably, Wang et al (20) revealed that APOBEC3G is upregulated
in human astrocytoma (U87MG) cells by stimulation with different
interferons, interleukin-1 and tumor necrosis factor. In contrast,
other cytokines, including interleukin-4 or −6 and particularly
TGF-β did not induce APOBEC3G expression. In the present study,
expression of APOBEC3B was induced in cultured glioblastoma cells
by temozolomide, the most commonly used chemotherapeutic in glioma
therapy. Particularly in light of the proliferative and
anti-apoptotic effects of APOBEC3B demonstrated in the present
study, this induction may also influence the progression of
glioma.
APOBEC3B is known to be involved in several types of
cancer (12,21–27)
and the results of the present study suggest that APOBEC3B may
serve a more prominent role in brain cancer progression than
previously assumed. Taking into account that Periyasamy et
al (28) demonstrated that p53
regulates the expression of APOBEC3B in order to limit its
potential mutagenic activity, the results of the present study are
interesting. Glioblastoma are well known to be characterized by
multiple mutations or allelic loss of the p53 tumor suppressor gene
(3), and the A172, LN229, U251MG
and T98G glioblastoma cell lines used in the present study exhibit
missense or small frameshift mutations of the p53 gene [A172: p53
gene position 242, wt TGC, mut TTC (29); LN229: p53 gene position 164, wt AAG,
mut GAG; U251MG: p53 gene position 273, wt CGT, mut CAT; and T98G:
p53 gene position 237, wt ATG, mut ATA (30)]. As mutations or loss of the p53 gene
result in elevated levels of APOBEC3B expression followed by
increased levels of desaminase activity in cancer cells (28), the detectable amounts of APOBEC3B in
glioblastoma cells may also be a consequence of p53 mutations.
However, this paradigm does not apply absolutely as the U251MG
cells were characterized by low levels of APOBEC3B expression (mean
ΔCq, 18.5) in the present study, and this cell line also contains
p53 point mutations. APOBEC3B expression has been demonstrated to
be regulated by other mechanisms, for example by Myb-related
protein B (encoded by MYB proto-oncogene like 2), which binds to
the APOBEC3B promoter causing its transactivation (31), or by the classical nuclear (NF)-κB
signaling pathway (32). Three
NF-κB binding sites have been identified in the APOBEC3B promoter
(32). In accordance with this,
Leonard et al (33)
demonstrated that protein kinase C activation causes the
recruitment of RELB proto-oncogene NF-κB subunit (the gene encoding
the transcription factor RelB), but not RELA proto-oncogene NF-κB
subunit (the gene encoding the transcription factor p65), to the
APOBEC3B promoter, indicating the involvement of the non-canonical
NF-κB signaling.
With regards to its functional role, the results of
the present study indicate that APOBEC3B mediates proliferative and
anti-apoptotic effects in human glioblastoma cells, and, therefore,
may promote glioma progression. However, a previous study reported
that high levels of APOBEC3B expression causes hypersensitivity to
small-molecule inhibitors that target the DNA damage response,
suggesting that APOBEC3B overexpression also imparts targetable
vulnerabilities upon cells (34).
In summary, APOBEC3B appears to serve a role in
intracranial brain tumors, particularly in glioblastoma,
contributing to glioma growth and heterogeneity by promoting the
establishment of a mutational signature, and supporting glioma
progression. APOBEC3B may therefore be a potential target for
future therapeutic strategies against glioma.
Acknowledgements
The authors would like to thank Ms. Judith Becker,
Ms. Martina Burmester and Ms. Sonja Dahle (all from the Institute
of Anatomy, University Kiel, Germany) for expert technical
assistance.
Funding
The present study was supported by a grant from the
Familie Mehdorn Foundation and by the Deutsche
Forschungsgemeinschaft (grant no. RTG2154; projects 7 and 8).
Availability of data and materials
The raw datasets analyzed in this study are
available from the corresponding author upon request.
Authors' contributions
RL, MS, JHF, CS and KH were involved in the
conception and design of the study. CS performed the experiments
and contributed to the data analysis, RL and MS provided essential
materials. JHF and KH planned the experimental studies, analyzed
the data and wrote the manuscript. All authors have critically
revised and approved the manuscript.
Ethics approval and consent to
participate
Glioma samples of different malignancy grades were
obtained at the Department of Neurosurgery, University Medical
Center Schleswig-Holstein UKSH (Kiel, Germany) in accordance with
the Declaration of Helsinki (1975) with the approval of the Ethics
Committee of the University of Kiel (Kiel, Germany) and after
written informed consent was obtained from the donors (file
reference, D536/15).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
APOBEC
|
apolipoprotein B mRNA editing enzyme,
catalytic polypeptide-like
|
|
Cq
|
cycle of threshold
|
|
GBM
|
glioblastoma multiforme
|
|
GFAP
|
glial fibrillary acidic protein
|
|
HIV
|
human immunodeficiency virus
|
|
ICC
|
immunocytochemistry
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
References
|
1
|
Kreth FW, Thon N, Simon M, Westphal M,
Schackert G, Nikkhah G, Hentschel B, Reifenberger G, Pietsch T,
Weller M, et al: German Glioma Network: Gross total but not
incomplete resection of glioblastoma prolongs survival in the era
of radiochemotherapy. Ann Oncol. 24:3117–3123. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bailey P and Cushing H: A classification
of the tumours of the glioma group on a histogenetic basis: With a
correlated study of prognosisMedium 8vo. J.B. Lippincott Company;
Philadelphia, London, and Montreal, with 108 illustrations: pp.
1751926, https://doi.org/10.1002/bjs.1800145540
|
|
3
|
Brennan CW, Verhaak RG, McKenna A, Campos
B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ,
Berman SH, et al: The somatic genomic landscape of glioblastoma.
Cell. 155:462–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hegi ME, Diserens AC, Godard S, Dietrich
PY, Regli L, Ostermann S, Otten P, Van Melle G, de Tribolet N and
Stupp R: Clinical trial substantiates the predictive value of
O-6-methylguanine DNA methyltransferase promoter treated
with temozolamide. Clin Cancer Res. 10:1871–1874. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dirks PB and Rutka JT: Current concepts in
neuro-oncology: The cell cycle - A review. Neurosurgery.
40:1000–1013. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stark AM, Doukas A, Hugo HH, Hedderich J,
Hattermann K, Mehdorn Maximilian H and Held-Feindt J: Expression of
DNA mismatch repair proteins MLH1, MSH2, and MSH6 in recurrent
glioblastoma. Neurol Res. 37:95–105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roberts SA, Lawrence MS, Klimczak LJ,
Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL,
Saksena G, et al: An APOBEC cytidine deaminase mutagenesis pattern
is widespread in human cancers. Nat Genet. 45:970–976. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wedekind JE, Dance GS, Sowden MP and Smith
HC: Messenger RNA editing in mammals: New members of the APOBEC
family seeking roles in the family business. Trends Genet.
19:207–216. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sheehy AM, Gaddis NC, Choi JD and Malim
MH: Isolation of a human gene that inhibits HIV-1 infection and is
suppressed by the viral Vif protein. Nature. 418:646–650. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Swanton C, McGranahan N, Starrett GJ and
Harris RS: APOBEC enzymes: Mutagenic fuel for cancer evolution and
heterogeneity. Cancer Disc. 5:704–712. 2015. View Article : Google Scholar
|
|
11
|
Burns MB, Lackey L, Carpenter MA, Rathore
A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N,
Nikas JB, et al: APOBEC3B is an enzymatic source of mutation in
breast cancer. Nature. 494:366–370. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Burns MB, Temiz NA and Harris RS: Evidence
for APOBEC3B mutagenesis in multiple human cancers. Nature Genet.
45:977–983. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Broad Institute TCGA Genome Data Analysis
Center: Analysis of mutagenesis by APOBEC cytidine deaminase
(P-MACD). Broad Institute of MIT and Harvard. 2015.doi:
10.7908/C12V2FBX.
|
|
14
|
Held-Feindt J, Hattermann K, Müerköster
SS, Wedderkopp H, Knerlich-Lukoschus F, Ungefroren H, Mehdorn HM
and Mentlein R: CX3CR1 promotes recruitment of human
glioma-infiltrating microglia/macrophages (GIMs). Exp Cell Res.
316:1553–1566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hattermann K, Li G, Hugo HH, Mentlein R,
Mehdorn HM and Held-Feindt J: Expression of the chemokines CXCL12
and CX3CL1 and their receptors in human nerve sheath tumors. Histol
Histopathol. 28:1337–1349. 2013.PubMed/NCBI
|
|
16
|
Hattermann K, Holzenburg E, Hans F, Lucius
R, Held-Feindt J and Mentlein R: Effects of the chemokine CXCL12
and combined internalization of its receptors CXCR4 and CXCR7 in
human MCF-7 breast cancer cells. Cell Tiss Res. 357:253–266. 2014.
View Article : Google Scholar
|
|
17
|
Johnson MD, Reeder JE and O'Connell M:
APOBEC3B expression in human leptomeninges and meningiomas. Oncol
Lett. 12:5344–5348. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Talagas M, Marcorelles P, Uguen A, Redon
S, Quintin-Roué I, Costa S, Férec C, Morel F, Hieu PD and De
Braekeleer M: Identification of a novel population in high-grade
oligodendroglial tumors not deleted on 1p/19q using array CGH. J
Neurooncol. 109:405–413. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Wu S, Zheng S, Wang S, Wali A,
Ezhilarasan R, Sulman EP, Koul D and Yung Alfred WK: APOBEC3G acts
as a therapeutic target in mesenchymal gliomas by sensitizing cells
to radiation-induced cell death. Oncotarget. 8:54285–54296.
2017.PubMed/NCBI
|
|
20
|
Wang YJ, Wang X, Zhang H, Zhou L, Liu S,
Kolson DL, Song L, Ye L and Ho WZ: Expression and regulation of
antiviral protein APOBEC3G in human neuronal cells. J Neuroimmunol.
206:14–21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fanourakis G, Tosios K, Papanikolaou N,
Chatzistamou I, Xydous M, Tseleni-Balafouta S, Sklavounou A,
Voutsinas GE and Vastardis H: Evidence for APOBEC3B mRNA and
protein expression in oral squamous cell carcinomas. Exp Mol
Pathol. 101:314–319. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jin Z, Han YX and Han XR: The role of
APOBEC3B in chrondosarcoma. Oncol Rep. 32:1867–1872. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leonard B, Hart SN, Burns MB, Carpenter
MA, Temiz NA, Rathore A, Vogel RI, Nikas JB, Law EK, Brown WL, et
al: APOBEC3B upregulation and genomic mutation patterns in serous
ovarian carcinoma. Cancer Res. 73:7222–7231. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Olson ME, Harris RS and Harki DA: APOBEC
enzymes as targets for virus and cancer therapy. Cell Chem Biol.
25:36–49. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sasaki H, Suzuki A, Tatematsu T, Shitara
M, Hikosaka Y, Okuda K, Moriyama S, Yano M and Fujii Y:
APOBEC3B gene overexpression in non-small-cell lung cancer.
Biomed Rep. 2:392–395. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Warren CJ, Westrich JA, Doorslaer KV and
Pyeon D: Roles of APOBEC3A and APOBEC3B in human papillomavirus
infection and disease progression. Viruses. 9:pii: E233. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu PF, Chen YS, Kuo TY, Lin HH, Liu CW and
Chang LC: APOBEC3B: A potential factor suppressing growth of human
hepatocellular carcinoma cells. Anticancer Res. 35:1521–1527.
2015.PubMed/NCBI
|
|
28
|
Periyasamy M, Singh AK, Gemma C, Kranjec
C, Farzan R, Leach DA, Navaratnam N, Pálinkás HL, Vértessy BG,
Fenton TR, et al: p53 controls expression of the DNA deaminase
APOBEC3B to limit its potential mutagenic activity in cancer cells.
Nucleic Acids Res. 45:11056–11069. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gomez-Manzano C, Fueyo J, Kyritsis AP,
Steck PA, Roth JA, Mcdonnell TJ, Steck KD, Levin VA and Yung WK:
Adenovirus-mediated transfer of the p53 gene produces rapid
and generalized death of human glioma cells via apoptosis. Cancer
Res. 56:694–699. 1996.PubMed/NCBI
|
|
30
|
Van Meir EG, Kikuchi T, Tada M, Li H,
Diserens AC, Wojcik BE, Huang HJ, Friedmann T, de Tribolet N and
Cavenee WK: Analysis of the p53 gene and its expression in
human glioblastoma cells. Cancer Res. 54:649–652. 1994.PubMed/NCBI
|
|
31
|
Chou WC, Chen WT, Hsiung CN, Hu LY, Yu CJ,
Hsu HM and Shen CJ: B-Myb induces APOBEC3B expression
leading to somatic mutation in multiple cancers. Sci Rep.
7:440892017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maruyama W, Shirakawa K, Matsui H,
Matsumoto T, Yamazaki H, Sarca AD, Kazuma Y, Kobayashi M, Shindo K
and Takaori-Kondo A: Classical NF-κB pathway is responsible for
APOBEC3B expression in cancer cells. Biochem Biophys Res Comm.
478:1466–1471. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Leonard B, McCann JL, Starrett GJ,
Kosyakovsky L, Luengas EM, Molan AM, Burns MB, McDougle RM, Parker
PJ, Brown WL, et al: The PKC/NF-κB signaling pathway induces
APOBEC3B expression in multiple human cancers. Cancer Res.
75:4538–4547. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nikkilä J, Kumar R, Campbell J, Brandsma
I, Pemberton HN, Wallberg F, Nagy K, Scheer I, Vertessy BG,
Serebrenik AA, et al: Elevated APOBEC3B expression drives a
kataegic-like mutation signature and replication stress-related
therapeutic vulnerabilities in p53-defective cells. Br J Cancer.
117:113–123. 2017. View Article : Google Scholar : PubMed/NCBI
|














