Introduction
Acute leukemia is the most common form of childhood
cancer, accounting for ~30% of pediatric cancer cases (1,2).
Despite the considerable progress that has been made in its
treatment, this disease remains a leading cause of pediatric
cancer-associated mortality, and its prognosis is unfavorable for
children with relapsed or refractory disease. The principal cause
of treatment failure is chemotherapy resistance (2,3).
An emerging concept suggests that leukemia cells and
their interactions with the bone marrow (BM) microenvironment are
the primary causes of acute leukemia relapse, due to the survival
of residual cells following chemotherapy (4–6). A
novel mechanism for the protection exerted by BM mesenchymal
stromal cells (MSCs) on acute lymphoblastic leukemia (ALL) cells
against chemotherapy has been identified. Pivotal to this mechanism
is a multiprotein complex expressed on the plasma membrane of
leukemic cells, consisting of potassium voltage-gated channel
subfamily H member 2 (hERG1) channels, the β1 subunit of integrin
receptors and the stromal cell-derived factor 12 (CXCL12) receptor,
C-X-C chemokine receptor type 4 (CXCR4) (hERG1/β1/CXCR4) (7). These data gave support to the
previously demonstrated functional link between hERG1 K+
channels and CXCL12 in acute leukemic cell migration (8). Overall, by controlling leukemia cell
survival and motility, the hERG1/β1/CXCR4 complex has emerged as a
target of choice for anti-chemoresistance strategies. Indeed,
blocking hERG1 with classical hERG1-specific blockers overcomes
MSC-induced chemoresistance, in vitro and in vivo, in
ALL mouse models (7).
hERG1 is a voltage-dependent potassium channel, the
functional relevance of which in human leukemia has been repeatedly
proven (7,9–11).
Furthermore, the alternative transcript of the hERG1 gene,
hERG1B, is an independent prognostic factor of a high risk
of relapse in pediatric T-ALL (12). Although a number of hERG1-specific
blockers exist on the market, a number of them produce adverse
cardiac side effects (13,14). Besides developing hERG1B-specific,
non-cardiotoxic blockers (15), an
alternative strategy to target the hERG1/β1/CXCR4 complex in
leukemia cells may involve targeting CXCR4 and its ligand
CXCL12.
The chemokine receptor CXCR4 is overexpressed in
leukemia and is associated with poor outcomes in ALL and acute
myeloid leukemia (AML) (6,16). The ligand CXCL12 is constitutively
produced by MSCs, particularly under hypoxic conditions (17), and contributes to the migration and
survival of leukemic blast cells through the activation of
phosphatidylinositol 3-kinase/RAC-α serine/threonine-protein kinase
(Akt) and mitogen-activated protein kinase (MAPK) pathways
(5). Notably, chemotherapeutic
treatments have been demonstrated to upregulate CXCR4 expression.
Such upregulation represents a mechanism of acquired resistance in
pediatric AML (18). A number of
tools have been developed to block CXCR4/CXCL12 interactions, and
they are currently under different stages of development (5). Research focused on targeting chemokine
receptors has also identified promising molecules, which have
subsequently been unsuccessful in clinical trials. Certain of these
molecules suffered from low oral bioavailability, e.g. the C-C
chemokine receptor type 5 (CCR5) inhibitor TAK-779 (19), while others gave rise to severe side
effects, e.g. Aplaviroc, also inhibiting CCR5 and causing
hepatotoxicity (20). On the other
hand, chemokines, commonly considered ‘undruggable’ due to their
small size and shallow surfaces, have re-emerged as drug
development targets through novel biochemical approaches (21).
Novel tools targeting CXCL12 have recently been
developed by means of combining paramagnetic fragment-based nuclear
magnetic resonance (NMR) investigation, molecular dynamics (MD) and
docking simulations (22). We here
provide in vitro biological data on the effects of some of
these molecules on acute leukemia cell survival.
Materials and methods
Peptides and small molecules
Peptides were synthesized by solid-phase synthesis
using standard Fmoc chemistry in a Syro multiple peptide
synthesizer (MultiSynTech GmbH, Witten, Germany). The final product
was cleaved from the solid support, deprotected by treatment with
trifluoroacetic acid containing tri-isopropylsilane and water
(95/2.5/2.5), and precipitated with diethyl ether. Crude peptides
were purified by reverse-phase chromatography. The final peptide
purity and identity were confirmed by reverse-phase chromatography
on a Phenomenex Jupiter C18 analytical column and by mass
spectrometry with a Bruker Daltonics ultraflex matrix assisted
laser desorption/ionization-time of flight tandem system (Bruker
Corporation, Billerica, MA, USA). Peptide 4-175-185 MS: Calculated
for C79H129N21O28S was 1,853.05; detected 1,854.19 m/z;
high-performance liquid chromatography (HPLC) room temperature (RT)
[80% eluent A (DDW)-5% eluent A (DDW)], 18.37 min. Peptide 4-29-35
MS: Calculated for C63H102N18O21S was 1,479.65; detected 1,480.96
m/z; HPLC RT (80%A-5%A) 14.92 min. Peptide 4-1-17 MS: Calculated
for C108H170N24O40S3 was 2,540.83; detected 250.96; HPLC RT
(80%A-1%A) 21.64 min. Peptide 7-1-17 MS: Calculated for
C117H174N26O37S2 was 2,600.91; detected 2,598.29 m/z. HPLC RT (from
80%A-20%A) 23.33 min. Positive ion mode was used in all cases. All
surface plasmon resonance experiments were performed on a BIA T100
system (GE Healthcare, Chicago, IL, USA). The binding of CXCL12 was
performed on a streptavidin (SA)-sensor chip previously coated with
biotinylated peptides. The immobilization of the biotinylated
peptide(s) was achieved by injecting peptide(s) diluted at 50 µg/ml
in HBS-EP+ (10 mM HEPES, 150 mM NaCl, 3.4 m EDTA, 0.05%
polysorbate 20; pH 7.4) for 60 sec over an SA-coated flow cell at
the flow rate of 10 µl/min. The binding of CXCL12 with immobilized
peptides was investigated using injections for 180 sec at chemokine
concentrations between 1 and 100 nM in HBS-EP+, at a
flow rate of 30 µl/min. The regeneration of the matrix was achieved
by flushing a short pulse of 1 M NaCl-10 mM NaOH. For small
molecule binding, CXCL12 was immobilized on a dextran matrix of a
CM4 sensor chip flow cell via a standard amino coupling procedure.
A flow cell was used as a reference surface following a blank
immobilization. Small molecules were injected over a CXCL12-coated
chip for 60 sec at a flow rate of 30 µl/min and diluted at 100 µM
in 5% dimethyl sulfoxide-HBS-EP+, which was also used as
the running buffer. Molecular docking simulation was performed
using the AutoDock Vina package v 1.1.2 (23) using a ZINC-derived library
(http://zinc.docking.org) of small molecules
probed against a CXCL12 pocket region previously identified by NMR
experiments and MD simulations (22), and consisting of residues V23, K24,
H25, K27, A40, R41 and K43. Standard Vina parameters were used for
the simulation runs. Selected molecules were purchased from
ChemBridge Corporation (San Diego, CA, USA).
Cell culture
Leukemia cell lines [B-cell precursor-ALL (BCP-ALL)
697 cells and AML FLG 29.1 cells] and normal Epstein-Barr virus
(EBV)-infected B lymphocytes were cultured in RPMI-1640 medium
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) supplemented with 2
mM L-glutamine (EuroClone SpA, Pero, Italy) and 10% fetal calf
serum (FCS; EuroClone). Human BM-derived MSCs immortalized by
enforcing the expression of telomerase reverse transcriptase in
primary MSCs were established in the laboratory of Dr D. Campana
(Department of Pediatrics, National University of Singapore,
Singapore) and maintained as described previously (7).
MSCs were seeded in 96-well flat-bottomed plates
coated with fibronectin (1 µg/well) and grown until confluence
prior to undertaking the co-culture experiments.
Primary samples
BM samples from children with newly diagnosed AML
were analyzed at the Hematology-Oncology Laboratory of the
Department of Pediatrics, University of Padua (Padua, Italy).
Diagnoses were made according to standard cytomorphology,
cytochemistry, an immunophenotypic criteria as described previously
(12). Patients studied were
enrolled in the AIEOP-BFM ALL 2009 therapy protocol, approved by
the local ethical committee (Comitato Etico per la Sperimentazione
dell'Azienda Ospedaliera di Padova; no. 0002862-18/01/2012). The
parents or legal guardians of the patients provided written
informed consent, following the tenets of the Declaration of
Helsinki.
Trypan blue assay
Cell viability was assessed by trypan blue exclusion
assay. In brief, 20 µl 0.4% trypan blue solution was added to 20-µl
cell suspensions in culture medium. The suspension was gently mixed
and transferred to a hemocytometer. Viable and dead cells were
identified and counted under a light microscope (×10 magnification)
Blue cells failing to exclude the dye were considered nonviable,
and transparent cells were considered viable. The percentage of
viable cells was calculated on the basis of the total number of
cells (viable and non-viable). The median lethal dose
(LD50) value was calculated by fitting the data points
(following 24 h of incubation) with a sigmoidal curve using
OriginPro 2015 (OriginLab, Northampton, MA, USA).
Pharmacology experiments
Leukemic cells were serum- starved for 16 h in RPMI
medium and seeded in 96-well flat-bottomed plates (Corning-Costar;
Corning Incorporated, Corning, NY, USA) at a cell density of
2×105 cells/well in RPMI containing 10% FCS.
Small molecules and peptides were used at the
LD50 values indicated in the figures following three
different schedules: i) Single treatment (added at timepoint 0);
ii) double treatment (added at timepoints 0 and 12 h); and iii)
triple treatment (added at timepoints 0, 12 and 24 h).
Following 24, 48 and 72 h of incubation, viable
cells (determined by the trypan blue exclusion test) were counted
in triplicate using a hemocytometer. Each experimental point
represents the mean of four samples from three independent
experiments.
Co-culture experiments
Cell suspensions (at a cell density of
2×105 cells/well) were placed in a 96-well flat-bottomed
plate, with or without bone marrow-derived MSCs and treated with
4-1-17 or 8673 at the LD50 dose alone, or in combination
with cytarabine (45 nM) or doxorubicin (0.1 µg/ml). Cultures were
maintained for 48 h at 37°C, 5% CO2 and 90% humidity.
Following incubation, cells were separated from MSCs by pipetting
with ice-cold PBS, and were processed for apoptosis or caspase
activity analyses.
Apoptosis analysis
The Annexin V/propidium iodide (PI) (Annexin V-FLUOS
staining kit; Roche Diagnostics, Basel, Switzerland) test was
applied to measure apoptosis. Leukemia cells were washed twice with
PBS, resuspended in 100 µl assay buffer, and incubated with
fluorescein isothiocyanate-conjugated Annexin V and PI. The mixture
was incubated at room temperature for 15 min prior to flow
cytometric analysis. Data were analyzed using BD FACSDiva Software
6.1.3 (BD Biosciences, Franklin Lakes, NJ, USA) as described in
(7).
Caspase activity assay
The generic caspase activity assay kit
(Fluorometric-Green; cat. no. ab112130; Abcam, Cambridge, UK) was
used to detect the activity of caspases 1, 3, 4, 5, 6, 7, 8 and 9.
Leukemic cells were treated for 48 h (as discussed above) and
subsequently incubated at 37°C, 5% CO2 for 2 h with 2 µl
500X TF2-VAD-FMK. Following PBS washing, cells were resuspended in
0.5 ml assay buffer and analyzed with BD FACSDiva Software 6.1.3
(BD Biosciences).
Hypoxia experiments
Exponentially growing cells were treated as
described above and incubated at 37°C in 0.1% O2
(water-saturated atmosphere containing 94.9% N2 and 5%
CO2) in a DG250 Anaerobic Workstation (Don Whitley
Scientific, Ltd., Bingley, UK) for 48 h.
Western blotting
Protein extraction and western blotting were
performed largely as described in (7). Leukemic cells following treatment were
washed with cold PBS and immediately extracted with 1% NP-40 lysis
buffer (1% NP-40, 150 mM NaCl, 50 mM Tris-HCl, pH 8, 5 mM EDTA and
10 mM Na4P2O7) supplemented with a
tablet of a complete mix of protease inhibitors (Roche Complete
Mini; Roche Diagnostics). Cell lysates were centrifuged at 13,000 ×
g for 10 min (4°C), and the supernatants were collected and assayed
for protein concentration with the Bradford protein assay method
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Proteins were
eluted by boiling the samples in Laemmli buffer, analyzed by
SDS-PAGE (7.5%) under reducing conditions, and transferred to a
nitrocellulose membrane (Hybond P; Amersham; GE Healthcare). The
membrane was incubated for 4 h at room temperature with 0.1%
Tween-20 in PBS (T-PBS) containing 5% bovine serum albumin,
Sigma-Aldrich; Merck KGaA) and incubated overnight at 4°C with the
appropriate primary antibodies at the concentrations listed below.
Membranes were washed three times with T-PBS and incubated with the
appropriate secondary antibodies for 45 min at room temperature.
Following three washes with T-PBS, the immunoreactivity was
determined by an enhanced chemiluminescence reaction (SuperSignal;
Pierce; Thermo Fisher Scientific, Inc., Waltham, MA, USA). For the
stripping of the membranes, the ReBlot WB recycling kit (Chemicon;
EMD Millipore, Billerica, MA, USA) was used, according to
manufacturer's protocol.
The following primary antibodies were used:
Anti-phospho-p44/42 MAPK (Thr202/Tyr204) (Cell Signaling
Technology, Inc., Danvers, MA, USA; cat. no. 9101; dilution, 1:500)
and anti-pAkt1/2/3 (Thr308)R (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA; cat. no. sc-271966; dilution, 1:500).
Anti-α-tubulin mouse monoclonal (Sigma-Aldrich; Merck KGaA; cat.
no. T9026; dilution, 1:500), anti-tAkt1/2/3 (H-136) (Santa Cruz
Biotechnology, Inc.; cat. no. sc8312; dilution, 1:500),
anti-extracellular signal-regulated kinase (ERK)1 (C-16) (Santa
Cruz Biotechnology, Inc.; cat. no. sc-292838; dilution, 1:500)
antibodies were used as loading controls. Secondary antibodies for
western blotting included anti-rabbit peroxidase-conjugate
(Sigma-Aldrich; Merck KGaA; cat. no. A0545; dilution, 1:10,000) and
anti-mouse peroxidase conjugate (Sigma-Aldrich; Merck KGaA; cat.
no. A4416; dilution, 1:5,000).
Western blotting images were acquired with an Epson
3200 scanner, and the relative bands analyzed with Scion Image
software version 4.0 (Scion Corporation, Frederick, MD, USA). The
intensity of the bands was normalized to the intensity of the bands
that corresponded to the total protein. The control cell ratio was
set as 1.
Statistical analysis
Graphs and statistical analyses were prepared using
Prism 4.00 (GraphPad Software, Inc., La Jolla, CA, USA). Values in
all panels are the mean ± standard deviation of three independent
experiments. The normality of the data distribution was checked
with the Kolmogorov-Smirnov test. In the case of normal
distributions, each dataset was first checked for variance
homogeneity, using the Brown-Forsythe test for multiple
comparisons. For multiple comparisons, one-way analysis of variance
followed by Bonferroni's post hoc test was performed to derive the
P-values. P<0.05 was considered to indicate a statistically
significant difference.
Results
Selection of peptides from CXCR4
Amino acid (aa) residues involved in, and putatively
modulating, the CXCR4/CXCL12 interaction were identified from CXCR4
fragments located in regions in which the interaction with CXCL12
had been inferred either by mutagenesis (24), structural modeling of the ligand
receptor/complex (25), NMR
evaluation of the binding of CXCR4 N-terminal peptides spanning aa
1–27 (26), or the NMR-derived
structure of CXCL12 in complex with a CXCR4 N-terminal fragment
spanning aa 1–38 (27). Comparison
of the two NMR studies allowed for the accurate selection of a
shorter (thus more suitable as a molecular tool) peptide, strongly
interacting with CXCL12 and spanning aa 1–17 of CXCR4 (termed
peptide 4-1-17). Such a fragment exhibits a large interaction
surface in the complex CXCL12/CXCR41-38 (27) (Fig.
1; left) and marked binding (in terms of NMR chemical shift
perturbation) when used as a fragment against CXCL12 (26). The left panel of Fig. 1 illustrates the mode of interaction
of peptide 1–17 with the chemokine as derived from the complex
CXCL12/CXCR41-38 (Protein Data Bank ID 2K04; http://www.rcsb.org); a strong interaction between the
peptide and the crevice surface formed by the N-terminal and the
central β-sheet of CXCL12 is apparent.
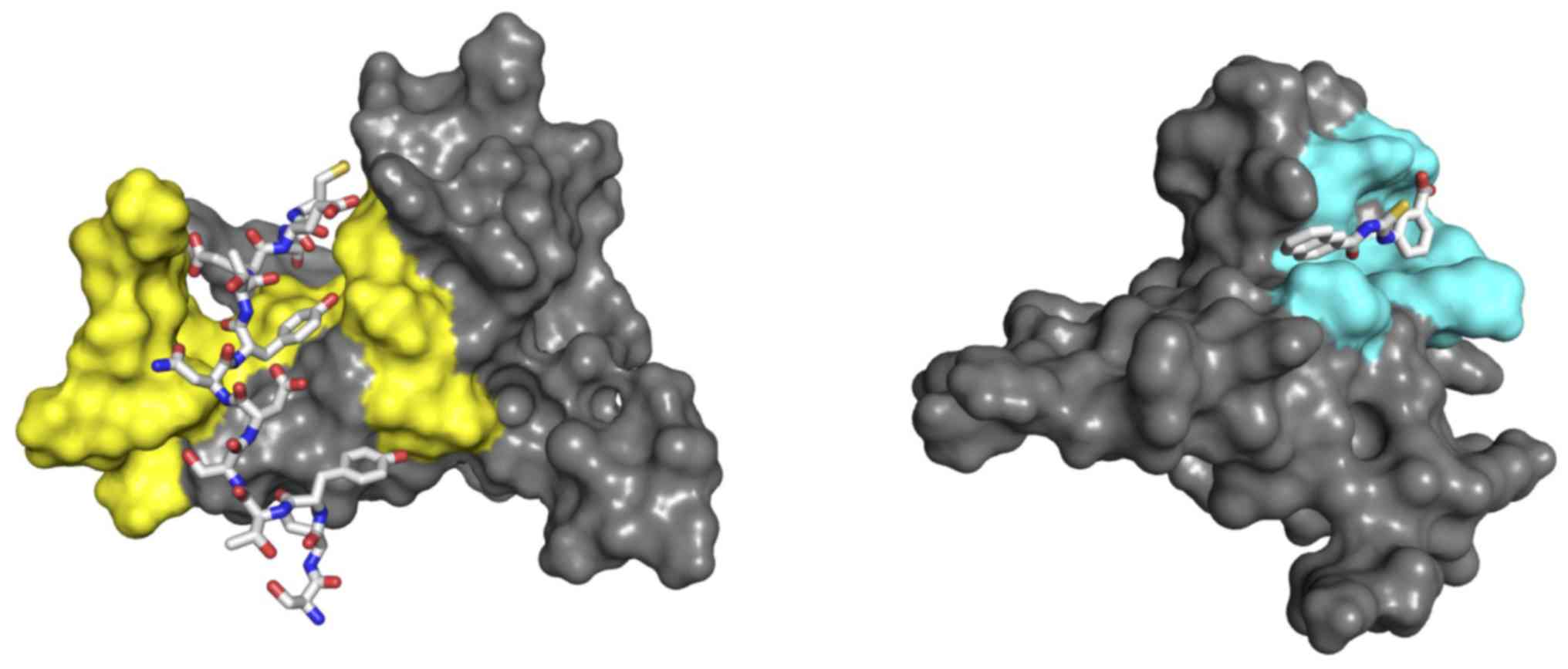 | Figure 1.Molecular models of CXCL12 bound to
4-1-17 and 8673. Molecular models of CXCL12 (represented as grey
surfaces) bound to 4-1-17 (sticks, left) and 8673 (sticks, right),
as derived from the nuclear magnetic resonance structure and
molecular docking simulations, respectively. The interacting
surfaces were formed by the N-terminal segment and the central
beta-sheet (yellow, left), and an open-state transient pocket
involving residues V23, K24, H25, K27, A40, R41 and K43 (cyan,
right). CXCL12, stromal cell-derived factor 12. |
The CXCR418-27 sequence, on the contrary,
exhibited no binding to CXCL12 when used as a fragment. Due to the
lack of binding data for the remaining sequence
CXCR428-38 and the large conformational variability in
the CXCL12/CXCR41-38 structure, the peptide spanning aa
29–35 (termed 4-29-35) of CXCR4 was selected for the evaluation of
binding capability of this region. Although no other experimental
structural data are available for CXCR4, mutagenesis and modeling
studies (24,25) indicated the second extracellular
loop ECD2 as being involved in ligand binding and signaling, due to
its interaction with the CXCL12 loop linking β-strands I and II.
Thus, the CXR4175-185 fragment spanning the ECD2
sequence was selected for evaluation (termed 4-175-185). CXCL12
binding to another seven-transmembrane span receptor, CXCR7
(28), suggested the selection of
an additional peptide from the latter one. Although the structure
of CXCR7 remains to be resolved, comparative molecular modelling
predicted that interacting regions of CXCR4 and CXCR7 with CXCL12
are similarly located in the N-terminal region (29). Hence, to exploit the
CXCR4/CXCR7/CXCL12 chemokine axis, the sequence segment spanning
the N-terminal residues 1 to 17 of the CXCR7 was selected to obtain
a peptide (termed 7-1-17) homologous to 4-1-17.
Selection of small molecules
Small molecules modulating the CXCR4/CXCL12
interaction were selected from a virtual library [Clean Leads
subset from ZINC database; (30)]
using experimental data as constraints for a molecular docking
simulation. In particular, all molecules were docked to the
transient binding pocket opening on Val23/Ala40 (22). Transient pockets, indeed, refer to
their rapid appearance and disappearance on flat protein surfaces
as a consequence of fluctuations in conformational dynamics [hence
the definition of dynamic drug design (DDD)]. The presence of such
a pocket was inferred by an NMR paramagnetic perturbation study,
while the open conformation was determined by trajectory analysis
of molecular dynamics simulation of CXCL12 (22). The trajectory frame representing the
pocket open conformation was used for the docking run, and the
simulation box was narrowed down to the involved residues V23, K24,
H25, K27, A40, R41 and K43. A total of one out of the three chosen
small molecules, henceforth termed 8673, had already been reported
to interact with CXCL12 in previous investigations (31), and its binding geometry is reported
in Fig. 1 (right panel).
Selected peptides and small molecules were tested
for chemokine binding by surface plasmonic resonance (see Tables I and II).
 | Table I.Peptides. |
Table I.
Peptides.
|
|
|
|
|
|
| LD50,
µM |
|---|
|
|
|
|
|
|
|
|
|---|
| Code | CXCR4 sequence
span | CXCR7 sequence
span | Sequence | MM | Kd, 298
K | FLG 29.1 | 697 |
|---|
| 4-1-17 | 1–17 | – |
MEGISIYTSDNYTEEMG | 1,940 |
7.51×10−7 | 17.0±1.2 | 14±1.1 |
| 4-175-185 | 175–185 | – | ANVSEADDRYI | 1,253 | No binding | >100 | >100 |
| 4-29-35 | 29–35 | – | FREENAN | 879 | No binding | Nd | Nd |
| 7-1-17 | – | 1–17 |
MDLHLFDYSEPGNFSDI | 2,000 |
4.57×10−8 | 66.7±2.3 | 28±1.4 |
| Table II.Small molecules. |
Effect of peptides and small molecules
targeting CXCL12 on leukemia cell vitality and proliferation
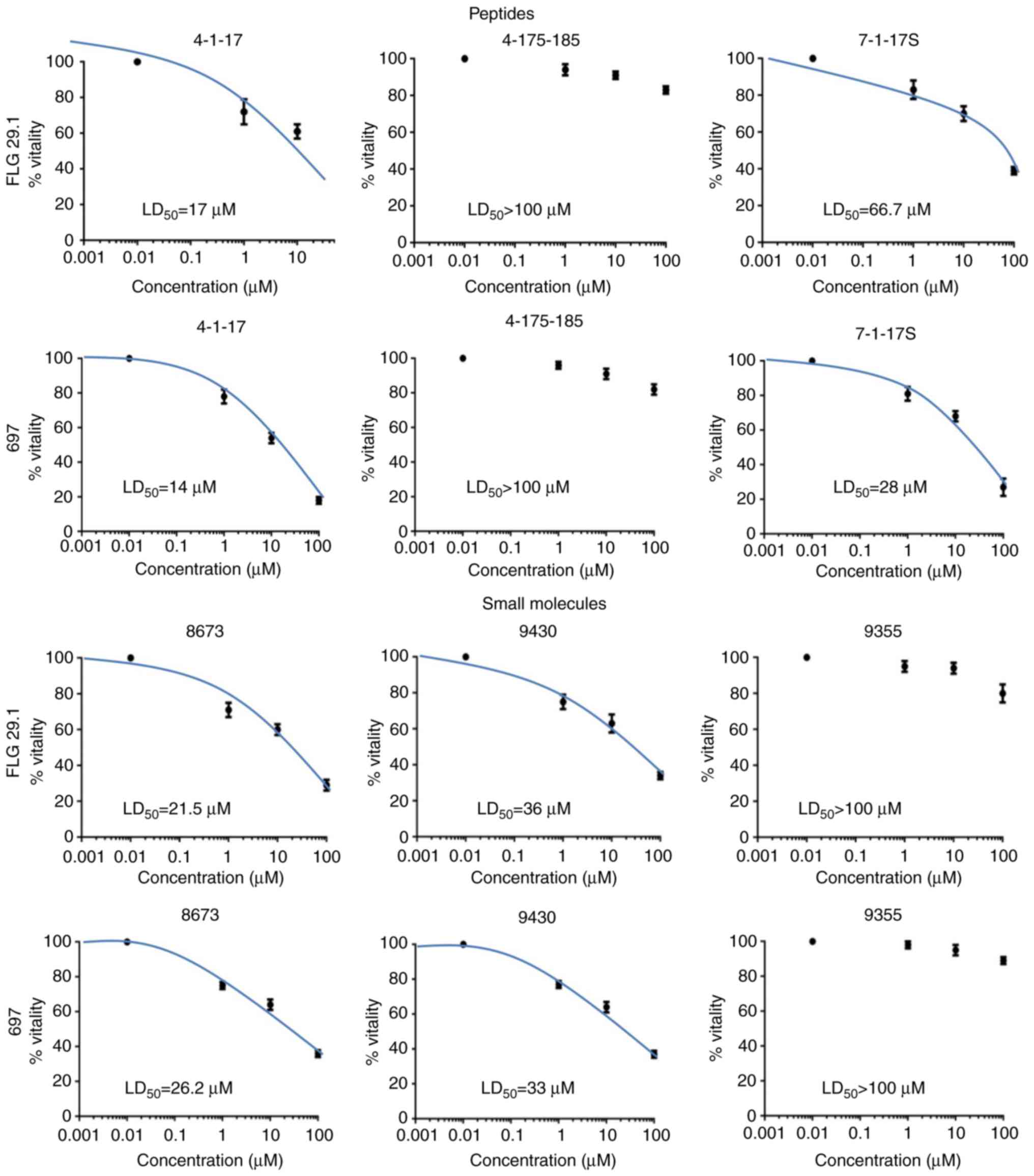
The panel of molecules reported above was tested in
a viability assay. For each molecule, the LD50 in AML
(FLG 29.1) and ALL cell lines (697) were determined.
Dose-dependence curves and LD50 values measured
following 24 h of treatment are presented in Fig. 2 and Table I, respectively. Peptide 4-1-17 and
the small molecule 8673 had a strong anti-proliferative effect on
the two leukemic cell lines at micromolar concentrations. Similar
effects were observed for small molecule 9430, although to a lesser
extent. On the other hand, 4-175-185 and 9355, which did not bind
CXCL12 in the SPR assay (see Table
II), did not exert any effects on cell viability at
concentrations up to 100 µM.
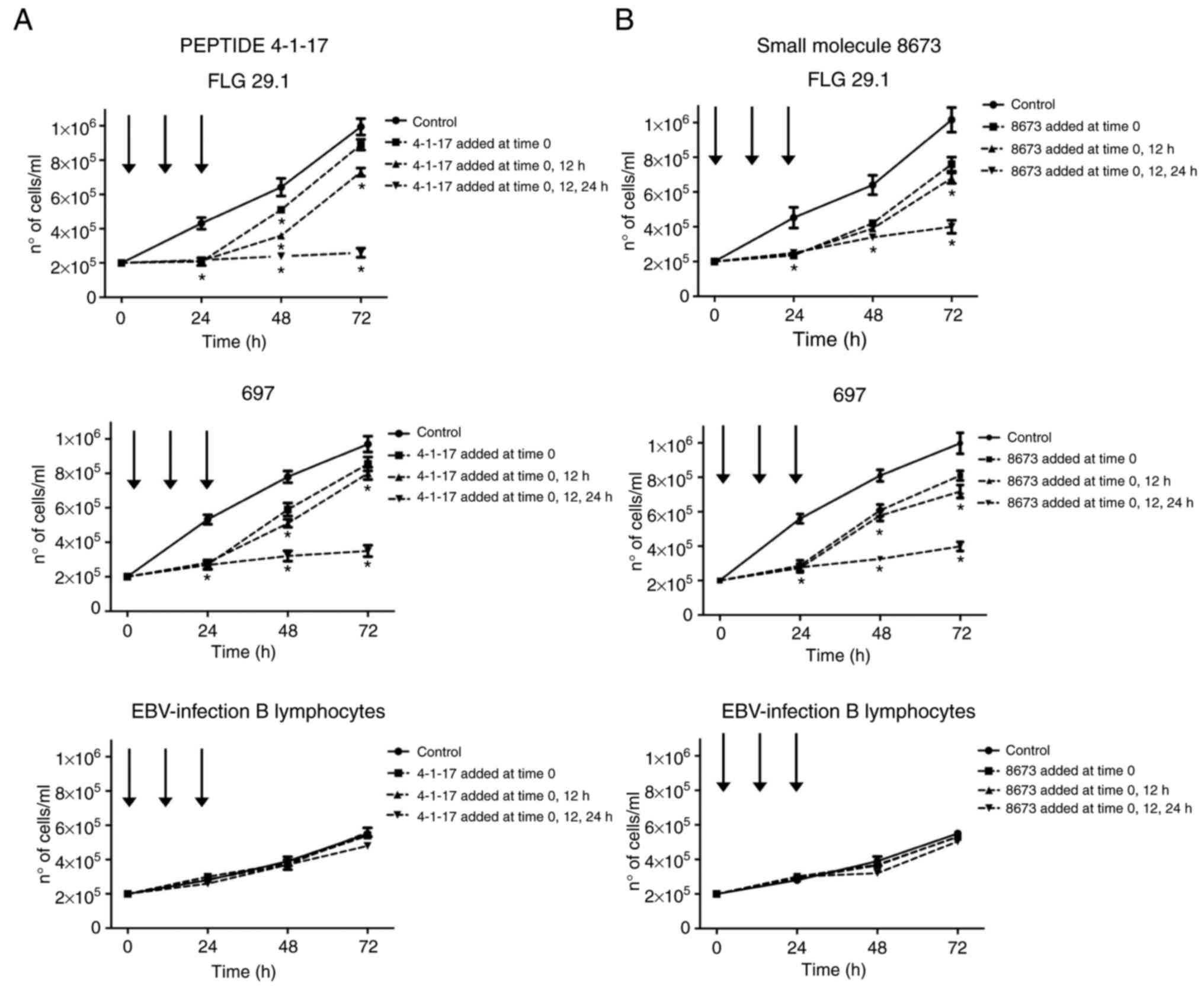
Fig. 3 illustrates
the effects of 4-1-17 (Fig. 3A) and
8673 (Fig. 3B), tested at their
LD50 value, on the proliferation of FLG 29.1 and 697
leukemic cells, in addition to that of normal EBV-infected B
lymphocytes. The two molecules almost completely inhibited cell
proliferation in either leukemic cell lines in the first 24 h of
incubation. Subsequently, cells recommenced proliferation, although
at a lower rate. However, when the two compounds were re-added to
the cells following 12 and 24 h of incubation, leukemia cell
proliferation was almost abolished. On the contrary, 4-1-17 and
8673 did not affect the cell viability of normal EBV-infected B
lymphocytes (Fig. 3A and B lower
panels).
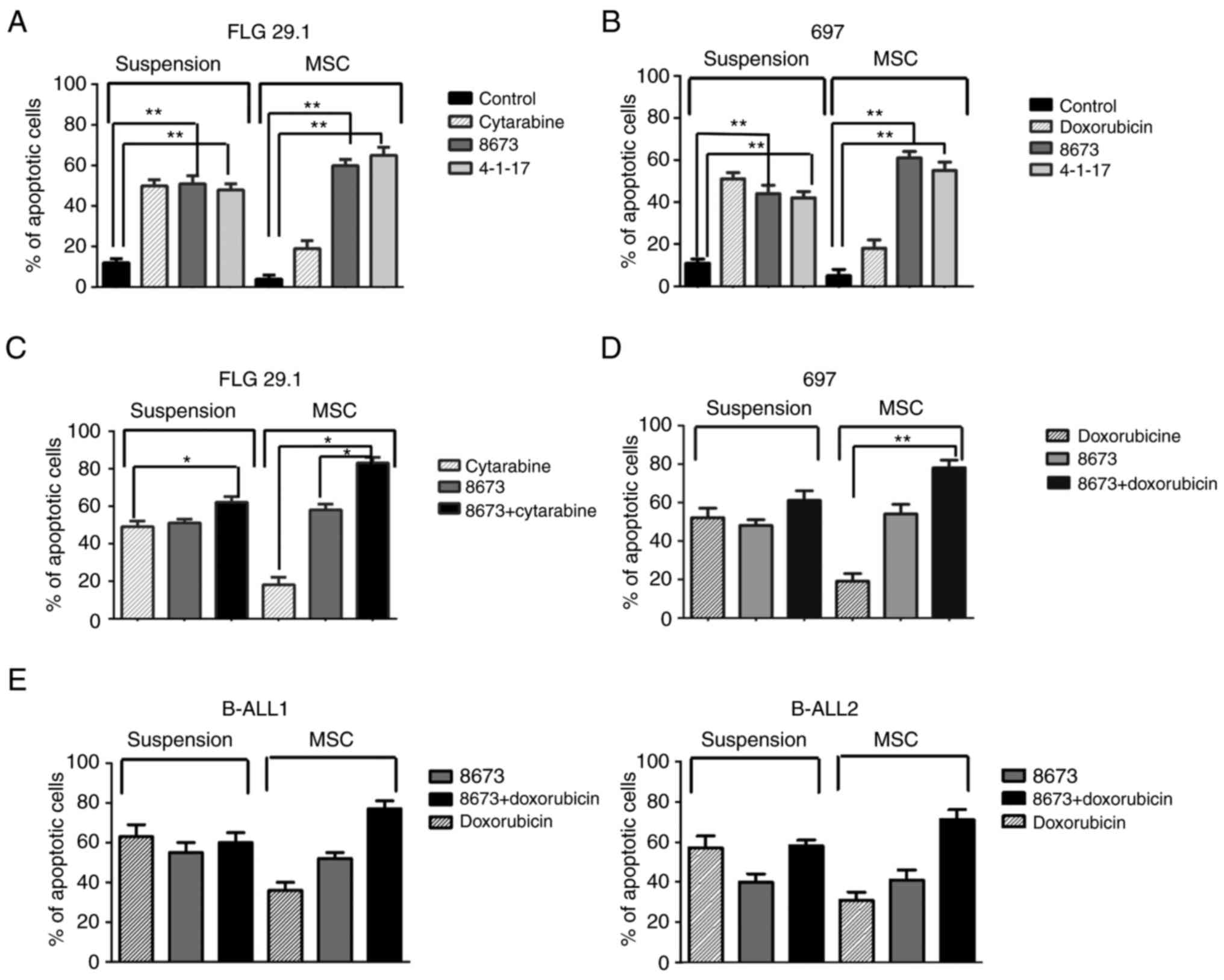
Effect of peptides and small molecules
targeting CXCL12 on leukemia cell apoptosis
The effects of 4-1-17 and 8673 on cellular apoptosis
were also tested (at their LD50 value) on the two
leukemia cell lines (FLG 29.1 and 697) and on primary BCP-ALL
samples, in suspension or co-culture with MSCs (as in 7) and in the
absence or presence of classical chemotherapeutic drugs (cytarabine
in AML, or doxorubicin in ALL). As expected (7,32,33),
MSCs significantly protected leukemic cells from either
spontaneous, or cytarabine-(in AML) or doxorubicin-(in ALL) induced
apoptosis (Fig. 4). On the
contrary, the pro-apoptotic effects exerted by either 4-1-17 or
8673 were not reduced by MSCs in AML and ALL cells (Fig. 4).
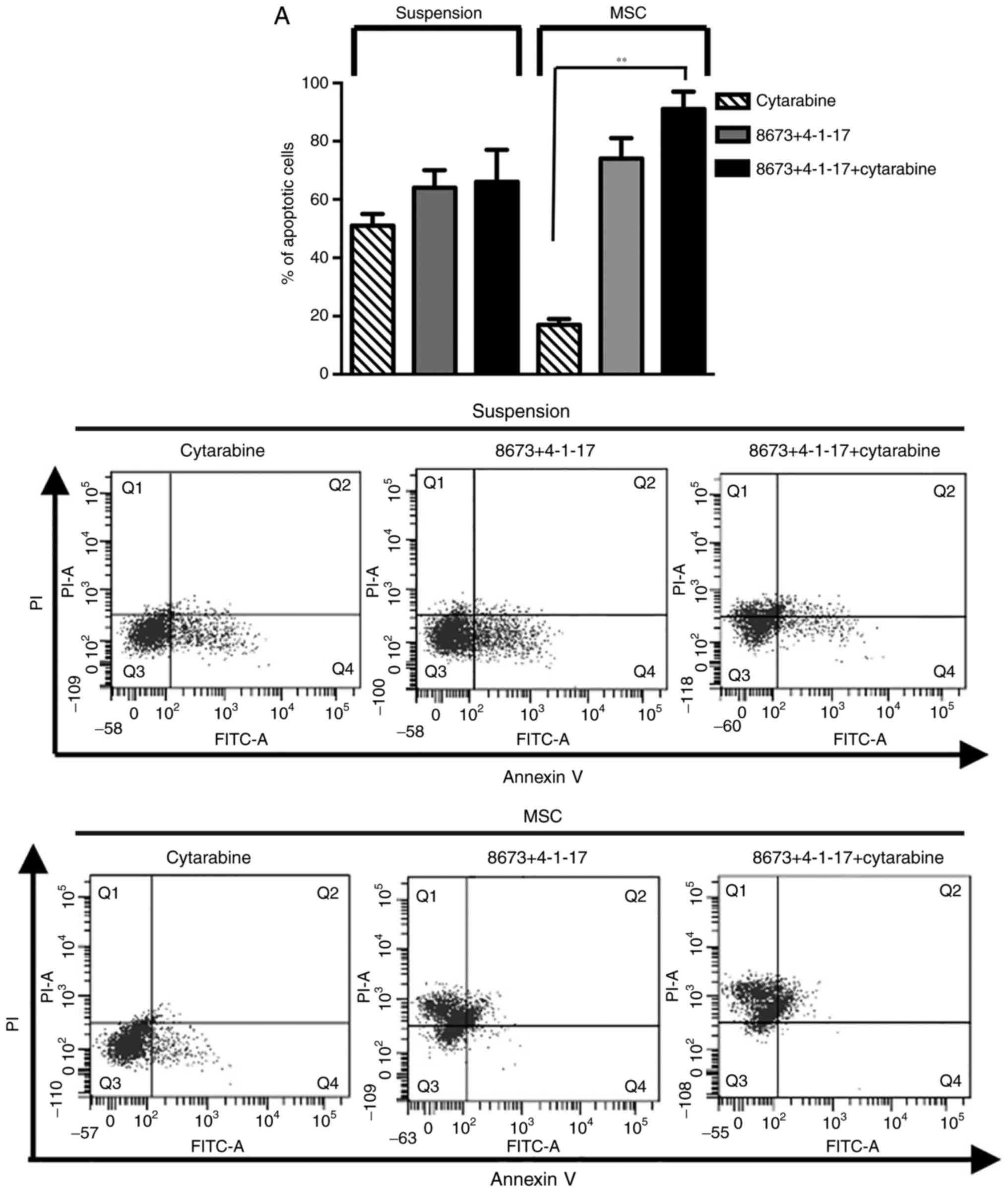
Since 4-1-17 and 8673 bind CXCL12 at different
sites, the effects of the combination of the two molecules were
tested in FLG 29.1 cells cultured either in suspension or onto
MSCs. The combined treatment had a strong pro-apoptotic effect,
evidenced by the increase in the percentage of apoptotic cells and
by caspase activation (Fig. 5A and
B). This effect was more evident in cells cultured on MSCs, and
even more when cells were treated with cytarabine. Overall, these
data indicated that peptide 4-1-17 and small molecule 8673 overcame
MSC-induced resistance to cytarabine (Fig. 5).
CXCL12/CXCR4 interactions have been demonstrated to
trigger Akt and ERK signaling (33); the present study therefore tested
the effects of 4-1-17 and 8673, alone or in combination, on the
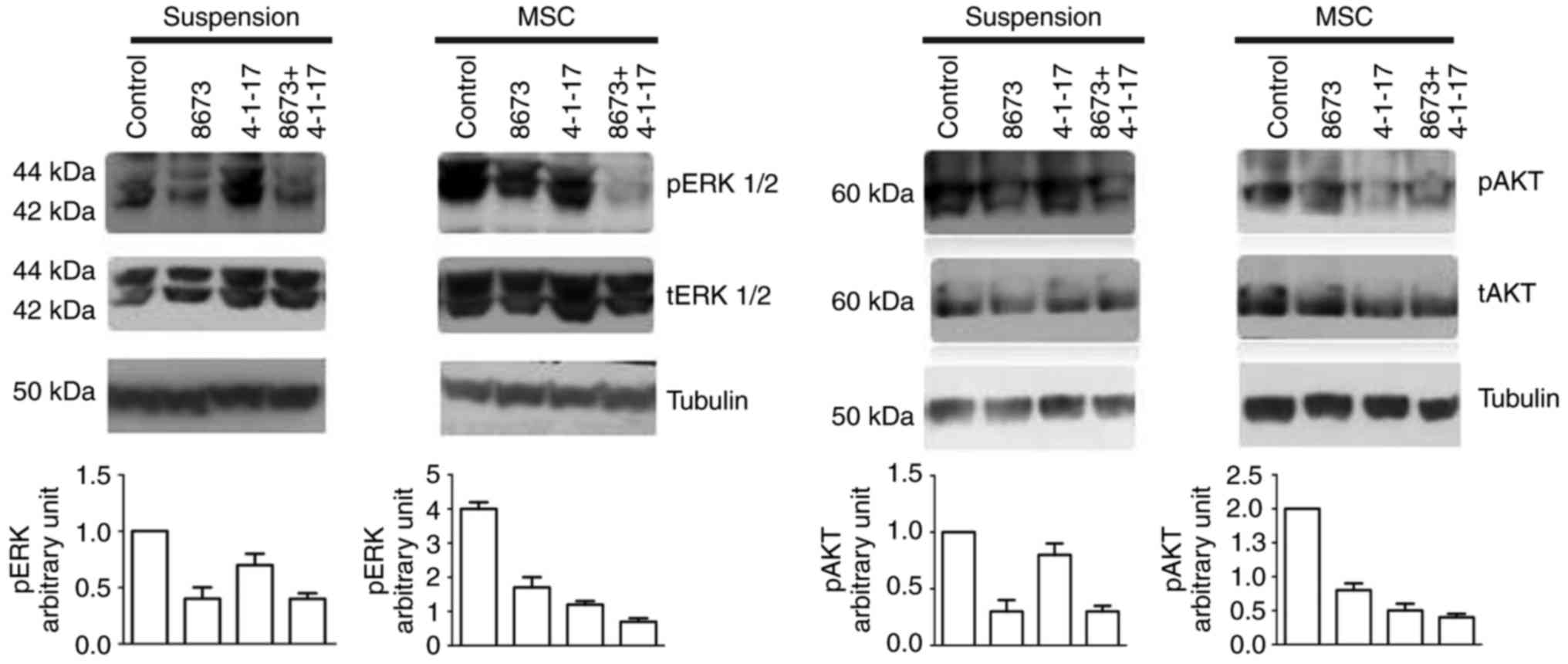
MSC-induced activation of Akt and ERK in FLG 29.1 cells. Fig. 6 demonstrates that 4-1-17 and 8673
downregulated ERK phosphorylation induced by MSCs (left panels),
and that the combined treatment abrogated MSC-induced ERK
phosphorylation. Similarly, the phosphorylation of Akt was
downregulated following treatment with either 4-1-17 or 8673, an
effect particularly evident when MSCs were added to the leukemia
cells (right panels).
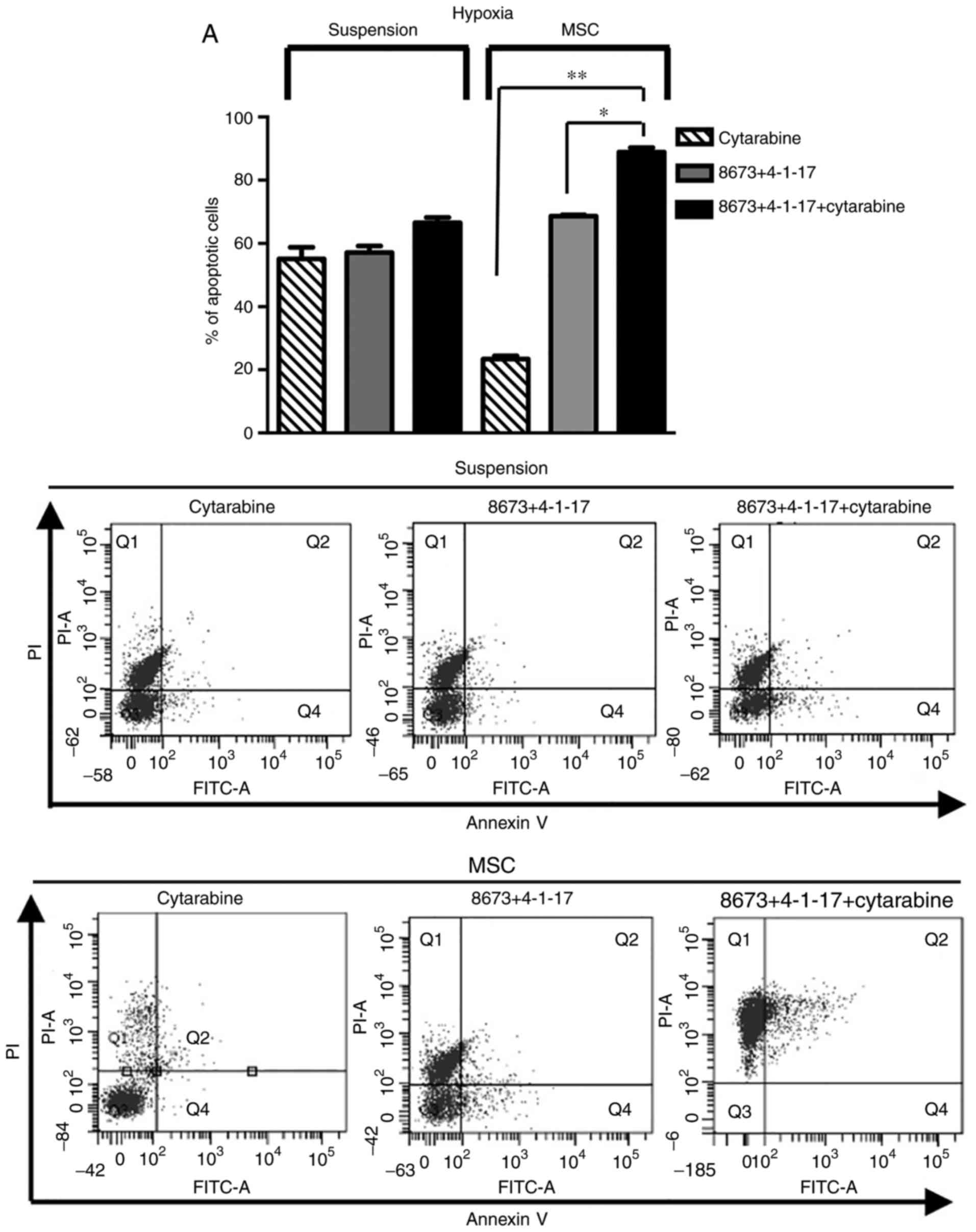
Since hypoxia has a relevant impact on leukemic cell
survival (34), the effect of the
combined treatment was assessed in FLG 29.1 cells cultured either
in suspension or onto MSCs in hypoxic conditions. Similar to what
was observed in normoxia, the addition of 4-1-17 and 8673 induced a
pro-apoptotic effect and overcame MSC-mediated chemoresistance
(Fig. 7).
Discussion
The present study provided evidence that two
different molecular tools targeting the CXCL12/CXCR4 complex,
peptide 4-1-17 and the small molecule 8673, inhibited the
proliferation of AML and ALL cell lines by inducing apoptosis. In
contrast to what occurs with chemotherapeutic drugs, this effect
was not reduced by the presence of MSCs. Furthermore, the two tools
significantly increased the sensitivity of leukemic cells to
cytarabine in the presence of MSCs.
The principal cause of treatment failure in acute
leukemia, particularly in the pediatric setting, which is generally
associated with better outcomes, is chemotherapy resistance
(3,35). Leukemic cells have been reported to
take refuge within the BM niche (36), which thus leads to survival of
residual leukemic cells following chemotherapy, resulting in
disease relapse. In other words, leukemic cells that adhere to MSCs
through CXCR4 are protected from the effects of cytotoxic
chemotherapy and represent a reservoir for minimal residual disease
and relapse (37). CXCL12 and its
cognate receptor, CXCR4, are two key mediators in the cross-talk
between leukemia cells and their microenvironment.
A novel mechanism of chemoresistance in ALL was
previously elucidated, centered on a plasma membrane macromolecular
complex consisting of the hERG1 potassium channel, the CXCR4
chemokine receptor and the β1 integrin subunit (7). The role of hERG1 was critical, as
hERG1 inhibitors abrogated the protective effect of MSCs and
enhanced the cytotoxicity of drugs commonly used to treat leukemia.
However, the targeting of hERG1 channels with classical
hERG1-specific blockers is difficult to propose in the clinical
setting, due to the potential cardiotoxicity displayed by classical
hERG1 blockers (13,14,38).
For this reason, aside from using non-cardiotoxic hERG1 blockers
(15,38), an alternative therapeutic strategy
was proposed to specifically target the hERG1/β1/CXCR4 complex, and
in turn overcome chemoresistance in leukemia. Such a strategy
consisted of inhibition of the interaction between CXCL12 and its
receptor CXCR4 with either peptides or small molecules developed
following a previously described procedure (22), and targeting the chemokine
CXCL12.
Previous studies provided evidence that targeting
CXCR4 with different agents (AMD3100, AMD3465, BKT140, RCP168 and
TN140) increased the sensitivity of leukemia cells to chemotherapy
(33,39,40).
AMD3100 was approved by the FDA in 2008 for the mobilization of
hematopoietic stem cells as an injectable agent for short-term
treatments (in patients with non-Hodgkin's lymphoma and multiple
myeloma) and is currently in phase I clinical trials for AML and
chronic myeloid leukemia. The safety and feasibility of combining
AMD3100 with chemotherapy in patients with myeloma and lymphoma has
already been demonstrated. On the contrary, concerns remain
regarding the administration of AMD3100 to patients with acute
leukemia. Concerns are primarily associated with BM aplasia,
delayed hematopoietic recovery following chemotherapy and CXCR4
upregulation (41). In addition,
such treatment may result in a preferential mobilization of
leukemic blasts over normal cells. Another compound, the orally
active AMD11070, which binds overlapping non-identical residues in
the binding pocket of the receptor, is currently in phase I
clinical trials for cancer (42,43).
Furthermore, a number of novel tools to block CXCR4/CXCL12
interactions are under development, being either peptides or small
molecules. Among the peptide class, T140 and its stable derivative
BKT140 have recently been reported to target AML anchorage in the
BM, in addition to the differentiation and survival of leukemic
cells (44–46). The same molecule is undergoing
further development via substitution of uncharged and
negatively-charged side chains with positively-charged ones. Still
in the ‘biological’ domain, the fully human anti-CXCR4 antibody
BMS-936564 has exhibited exerting effects similar to those of the
AMD3100 small molecule drug, in addition to pro-apoptotic activity
(47). Regarding small molecules,
the isothiourea derivative IT1t has been proven to be an antagonist
for CXCR4, and the structure of IT1t/CXR4 complex has been resolved
by X-ray crystallography (25),
opening the way to rational design of molecules with improved
efficacy (48).
All of the tools reported above target the CXCR4
receptor, since chemokines are generally viewed as ‘undruggable’
proteins. The two molecular tools here proposed target the
CXCL12/CXCR4 complex, although they bind to the ligand, CXCL12,
rather than the receptor. Notably, targeting a specific ligand may
facilitate regulation rather than the elimination of receptor
activity. The two compounds, 4-1-17 and 8673 are very different in
nature, the former being a 17-mer peptide derived from the
N-terminal domain of CXCR4, and the latter, a small drug-like
molecule selected by virtual screening. The two compounds target
CXCL12, although they use distinct binding sites. Standard
screening for potential drugs is usually performed by examining the
static features of target protein surfaces. On the other hand, in
the present study, CXCL12 structural dynamics were used as a
rational framework for drug selection. Accordingly, disruptors of
the CXCL12/CXCR4 interaction were predicted and tested for their
activity. Thus, one peptide, which was shorter than those
previously suggested, and one small molecule, which interacted with
a transient hot spot, were highlighted by the present DDD approach.
The fact that 4-1-17 and 8673 bind CXCL12 at different sites
suggests that the conjugation of the two molecules may exhibit
stronger biological activity. Taken together, the present findings
demonstrated the efficacy of double targeting of CXCL12 in leukemic
cells co-cultured with MSCs, either in normoxic or hypoxic
conditions. In this respect, the structural features of peptide
4-1-17 and 8673 binding with CXCL12 may be used to further optimize
more of the active fragments to assemble novel drugs.
Overall, the present results provided data for a
novel approach to the treatment of chemoresistant acute leukemia,
based on: i) The simultaneous blockade of one of the members of the
CXCR4/ hERG1/β1 complex on the plasma membrane of
leukemic cells, i.e. the CXCR4/CXCL12 axis; and ii) a novel
approach to drug selection, based on structural dynamics analyses.
Further conjugation of the two molecules targeting CXCL12 reported
in the present study (4-1-17 and 8673) may be proposed to induce
stronger antileukemic activity.
Acknowledgements
The authors would like to thank Professor Persio
dello Sbarba (Department of Clinical and Experimental Biomedical
Sciences, University of Florence, Florence, Italy) for allowing the
use of the DG250 Anaerobic Workstation.
Funding
The present study was supported by grants from the
Associazione Noi per Voi Onlus, Associazione Italiana per la
Ricerca sul Cancro (grant nos. 15627 and 1662) to AA, Istituto
Toscano Tumori 2009 to NN and ex 60% University of Firenze 2014 to
SP.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author on reasonable request.
Authors' contributions
BL performed the peptide synthesis. AB conceived the
peptide and small molecules approach, performed modelling and
docking work and contributed to writing the manuscript. OS
performed the bioinformatics analysis. LB and NN contributed to the
analysis of the results. SP performed the cell viability assay,
pharmacology experiments, western blotting and hypoxia experiments.
GP performed the caspase activity assay. AA, NN and SP designed the
study. AA supervised the study and wrote the paper. SP and AB
revised the paper. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Patients studied were enrolled in the AIEOP-BFM ALL
2009 therapy protocol, approved by the local ethical committee
(Comitato Etico per la Sperimentazione dell'Azienda Ospedaliera di
Padova; no. 0002862-18/01/2012). The parents or legal guardians of
the patients provided written informed consent, following the
tenets of the Declaration of Helsinki.
Patient consent for publication
The parents or legal guardians of the patients
provided written informed consent, following the tenets of the
Declaration of Helsinki.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pui CH, Schrappe M, Ribeiro RC and
Niemeyer CM: Childhood and adolescent lymphoid and myeloid
leukemia. Hematology Am Soc Hematol Educ Program. 118–145. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
August KJ, Narendran A and Neville KA:
Pediatric relapsed or refractory leukemia: New pharmacotherapeutic
developments and future directions. Drugs. 73:439–461. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Konopleva MY and Jordan CT: Leukemia stem
cells and microenvironment: Biology and therapeutic targeting. J
Clin Oncol. 29:591–599. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Konopleva M, Tabe Y, Zeng Z and Andreeff
M: Therapeutic targeting of microenvironmental interactions in
leukemia: Mechanisms and approaches. Drug Resist Updat. 12:103–113.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tavor S and Petit I: Can inhibition of the
SDF-1/CXCR4 axis eradicate acute leukemia? Semin Cancer Biol.
20:178–185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Peled A and Tavor S: Role of CXCR4 in the
pathogenesis of acute myeloid leukemia. Theranostics. 3:34–39.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pillozzi S, Masselli M, De Lorenzo E,
Accordi B, Cilia E, Crociani O, Amedei A, Veltroni M, D'Amico M,
Basso G, et al: Chemotherapy resistance in acute lymphoblastic
leukemia requires hERG1 channels and is overcome by hERG1 blockers.
Blood. 117:902–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li H, Du YM, Guo L, Jie S, Zhang S, Du W,
Chen X, Liu W, Fan L, Zhu J, et al: The role of hERG1 K+
channels and a functional link between hERG1 K+ channels
and SDF-1 in acute leukemic cell migration. Exp Cell Res.
315:2256–2264. 2008. View Article : Google Scholar
|
|
9
|
Pillozzi S, Brizzi MF, Balzi M, Crociani
O, Cherubini A, Guasti L, Bartolozzi B, Becchetti A, Wanke E,
Bernabei PA, et al: HERG potassium channels are constitutively
expressed in primary human acute myeloid leukemias and regulate
cell proliferation of normal and leukemic hemopoietic progenitors.
Leukemia. 16:1791–1798. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pillozzi S, Brizzi MF, Bernabei PA,
Bartolozzi B, Caporale R, Basile V, Pegoraro L, Becchetti A and
Arcangeli A: VEGFR-1 (FLT-1), β1 integrin, and hERG
K+ channel for a macromolecular signaling complex in
acute myeloid leukemia: Role in cell migration and clinical
outcome. Blood. 110:1238–1250. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Erdem M, Tekiner TA, Fejzullahu A, Akan G,
Anak S, Saribeyoglu ET, Ozbek U and Atalar F: herg1b expression as
a potential specific marker in pediatric acute myeloid leukemia
patients with HERG 897K/K genotype. Pediatr Hematol Oncol.
32:182–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pillozzi S, Accordi B, Rebora P, Serafin
V, Valsecchi MG, Basso G and Arcangeli A: Differential expression
of hERG1A and hERG1B genes in pediatric acute lymphoblastic
leukemia identifies different prognostic subgroups. Leukemia.
28:1352–1355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Redfern WS, Carlsson L, Davis AS, Lynch
WG, MacKenzie I, Palethorpe S, Siegl PK, Strang I, Sullivan AT,
Wallis R, et al: Relationships between preclinical cardiac
electrophysiology, clinical QT interval prolongation and torsade de
pointes for a broad range of drugs: evidence for a provisional
safety margin in drug development. Cardiovasc Res. 58:32–45. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vandenberg JI, Perry MD, Perrin MJ, Mann
SA, Ke Y and Hill AP: hERG K+ channels: Structure,
function, and clinical significance. Physiol Rev. 92:1393–1478.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gasparoli L, D'Amico M, Masselli M,
Pillozzi S, Caves R, Khuwaileh R, Tiedke W, Mugridge K, Pratesi A,
Mitcheson JS, et al: New pyrimido-indole compound CD-160130
preferentially inhibits the KV11.1B isoform and produces
antileukemic effects without cardiotoxicity. Mol Pharmacol.
87:183–196. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Spoo AC, Lübbert M, Wierda WG and Burger
JA: CXCR4 is a prognostic marker in acute myelogenous leukemia.
Blood. 109:786–791. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fecteau JF, Messmer D, Zhang S, Cui B,
Chen L and Kipps TJ: Impact of oxygen concentration on growth of
mesenchymal stromal cells from the marrow of patients with chronic
lymphocytic leukemia. Blood. 121:971–974. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sison EA, McIntyre E, Magoon D and Brown
P: Dynamic chemotherapy-induced upregulation of CXCR4 expression: A
mechanism of therapeutic resistance in pediatric AML. Mol Cancer
Res. 11:1004–1016. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baba M, Takashima K, Miyake H, Kanzaki N,
Teshima K, Wang X, Shiraishi M and Iizawa Y: TAK-652 inhibits
CCR5-mediated human immunodeficiency virus type 1 infection in
vitro and has favorable pharmacokinetics in humans. Antimicrob
Agents Chemother. 49:4584–4591. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nichols WG, Steel HM, Bonny T, Adkison K,
Curtis L, Millard J, Kabeya K and Clumeck N: Hepatotoxicity
observed in clinical trials of aplaviroc (GW873140). Antimicrob
Agents Chemother. 52:858–865. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hachet-Haas M, Balabanian K, Rohmer F,
Pons F, Franchet C, Lecat S, Chow KY, Dagher R, Gizzi P, Didier B,
et al: Small neutralizing molecules to inhibit actions of the
chemokine CXCL12. J Biol Chem. 283:23189–23199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bernini A, Henrici De Angelis L, Morandi
E, Spiga O, Santucci A, Assfalg M, Molinari H, Pillozzi S,
Arcangeli A and Niccolai N: Searching for protein binding sites
from Molecular Dynamics simulations and paramagnetic fragment-based
NMR studies. Biochim Biophys Acta. 1844:561–566. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Trott O and Olson AJ: AutoDock Vina:
Improving the speed and accuracy of docking with a new scoring
function, efficient optimization, and multithreading. J Comput
Chem. 31:455–461. 2010.PubMed/NCBI
|
|
24
|
Zhou N, Luo Z, Luo J, Liu D, Hall JW,
Pomerantz RJ and Huang Z: Structural and functional
characterization of human CXCR4 as a chemokine receptor and HIV-1
co-receptor by mutagenesis and molecular modeling studies. J Biol
Chem. 276:42826–42833. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu B, Chien EY, Mol CD, Fenalti G, Liu W,
Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, et al: Structures
of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide
antagonists. Science. 330:1066–1071. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gozansky EK, Louis JM, Caffrey M and Clore
GM: Mapping the binding of the N-terminal extracellular tail of the
CXCR4 receptor to stromal cell-derived factor-1alpha. J Mol Biol.
345:651–658. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Veldkamp CT, Seibert C, Peterson FC, De la
Cruz NB, Haugner JC III, Basnet H, Sakmar TP and Volkman BF:
Structural basis of CXCR4 sulfotyrosine recognition by the
chemokine SDF-1/CXCL12. Sci Signal. 1:ra42008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun X, Cheng G, Hao M, Zheng J, Zhou X,
Zhang J, Taichman RS, Pienta KJ and Wang J: CXCL12/CXCR4/CXCR7
chemokine axis and cancer progression. Cancer Metastasis Rev.
29:709–722. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Costantini S, Raucci R, De Vero T,
Castello G and Colonna G: Common structural interactions between
the receptors CXCR3, CXCR4 and CXCR7 complexed with their natural
ligands, CXCL11 and CXCL12, by a modeling approach. Cytokine.
64:316–321. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Irwin JJ, Sterling T, Mysinger M, Bolstad
ES and Coleman RG: ZINC: A free tool to discover chemistry for
biology. J Chem Inf Model. 52:1757–1768. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Veldkamp CT, Ziarek JJ, Peterson FC, Chen
Y and Volkman BF: Targeting SDF-1/CXCL12 with a ligand that
prevents activation of CXCR4 through structure-based drug design. J
Am Chem Soc. 132:7242–7243. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Konopleva M, Konoplev S, Hu W, Zaritskey
AY, Afanasiev BV and Andreeff M: Stromal cells prevent apoptosis of
AML cells by up-regulation of anti-apoptotic proteins. Leukemia.
16:1713–1724. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zeng Z, Samudio IJ, Munsell M, An J, Huang
Z, Estey E, Andreeff M and Konopleva M: Inhibition of CXCR4 with
the novel RCP168 peptide overcomes stroma-mediated chemoresistance
in chronic and acute leukemias. Mol Cancer Ther. 5:3113–3121. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rashidi A and DiPersio JF: Targeting the
leukemia-stroma interaction in acute myeloid leukemia: Rationale
and latest evidence. Ther Adv Hematol. 7:40–51. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Estey E and Döhner H: Acute myeloid
leukemia. Lancet. 368:1894–1907. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Colmone A, Amorim M, Pontier AL, Wang S,
Jablonski E and Sipkins DA: Leukemic cells create bone marrow
niches that disrupt the behavior of normal hematopoietic progenitor
cells. Science. 322:1861–1865. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lagneaux L, Delforge A, Bron D, De Bruyn C
and Stryckmans P: Chronic lymphocytic leukemia B cells but not
normal B cells are rescued from apoptosis by contact with normal
bone marrow stromal cells. Blood. 91:2387–2396. 1998.PubMed/NCBI
|
|
38
|
Pillozzi S, Masselli M, Gasparoli L,
D'Amico M, Polletta L, Veltroni M, Favre C, Basso G, Becchetti A
and Arcangeli A: Macrolide antibiotics exert antileukemic effects
by modulating the autophagic flux through inhibition of hERG1
potassium channels. Blood Cancer J. 6:e4232016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Beider K, Begin M, Abraham M, Wald H,
Weiss ID, Wald O, Pikarsky E, Zeira E, Eizenberg O, Galun E, et al:
CXCR4 antagonist 4F-benzoyl-TN14003 inhibits leukemia and multiple
myeloma tumor growth. Exp Hematol. 39:282–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang Y, Patel S, Abdelouahab H, Wittner
M, Willekens C, Shen S, Betems A, Joulin V, Opolon P, Bawa O, et
al: CXCR4 inhibitors selectively eliminate CXCR4-expressing human
acute myeloid leukemia cells in NOG mouse model. Cell Death Dis.
3:e3962012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Uy GL, Rettig MP, Stone RM, Konopleva MY,
Andreeff M, McFarland K, Shannon W, Fletcher TR, Reineck T, Eades
W, et al: A phase 1/2 study of chemosensitization with plerixafor
plus G-CSF in relapsed or refractory acute myeloid leukemia. Blood
Cancer J. 7:e5422017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Parameswaran R, Yu M, Lim M, Groffen J and
Heisterkamp N: Combination of drug therapy in acute lymphoblastic
leukemia with a CXCR4 antagonist. Leukemia. 25:1314–1323. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
O'Boyle G, Swidenbank I, Marshall H,
Barker CE, Armstrong J, White SA, Fricker SP, Plummer R, Wright M
and Lovat PE: Inhibition of CXCR4-CXCL12 chemotaxis in melanoma by
AMD11070. Br J Cancer. 108:1634–1640. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Feng Z, Dubyak GR, Jia X, Lubkowski JT and
Weinberg A: Human β-defensin-3 structure motifs that are important
in CXCR4 antagonism. FEBS J. 280:3365–3375. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Beider K, Darash-Yahana M, Blaier O,
Koren-Michowitz M, Abraham M, Wald H, Wald O, Galun E, Eizenberg O,
Peled A, et al: Combination of imatinib with CXCR4 antagonist
BKT140 overcomes the protective effect of stroma and targets CML in
vitro and in vivo. Mol Cancer Ther. 13:1155–1169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Abraham M, Klein S, Bulvik B, Wald H,
Weiss ID, Olam D, Weiss L, Beider K, Eizenberg O, Wald O, et al:
The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by
downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered
miR-15a/16-1 expression. Leukemia. 31:2336–2346. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kashyap MK, Kumar D, Jones H,
Amaya-Chanaga CI, Choi MY, Melo-Cardenas J, Ale-Ali A, Kuhne MR,
Sabbatini P, Cohen LJ, et al: Ulocuplumab (BMS-936564 / MDX1338): A
fully human anti-CXCR4 antibody induces cell death in chronic
lymphocytic leukemia mediated through a reactive oxygen
species-dependent pathway. Oncotarget. 7:2809–2822. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Das D, Maeda K, Hayashi Y, Gavande N,
Desai DV, Chang SB, Ghosh AK and Mitsuya H: Insights into the
mechanism of inhibition of CXCR4: identification of
Piperidinylethanamine analogs as anti-HIV-1 inhibitors. Antimicrob
Agents Chemother. 59:1895–1904. 2015. View Article : Google Scholar : PubMed/NCBI
|