Introduction
Non-small cell lung cancer (NSCLC) is the most
common form of lung cancer and is notable for being resistant to
chemotherapy. Epidermal growth factor receptor (EGFR) is a
molecular oncotherapeutic target for many types of cancer,
particularly NSCLC (1). In
particular, EGFR tyrosine kinase inhibitors (EGFR-TKIs) have shown
excellent efficacy for NSCLCs with EGFR-activating mutations, such
as exon 19 deletions (Del19) or L858R substitutions (2,3). These
mutations alter EGFR, leading to preferential binding between ATP
and the kinase domain, causing spontaneous activation (1). Furthermore, EGFR-TKI combinations,
such as those involving VEGF antibody or chemotherapy, have
recently improved treatment efficacy compared to that with EGFR-TKI
alone (4,5). However, EGFR-linked cancers ultimately
become refractory to EGFR-TKI treatment. Previous research has
elucidated several mechanisms that underlie resistance, revealing
that approximately 50% of EGFR-TKI-resistant NSCLCs with
EGFR-activating mutations possess a T790M secondary mutation
(6). It is therefore critical that
further investigation be undertaken to identify new strategies to
overcome EGFR-TKI resistance, particularly those involving
T790M.
Afatinib is a second-generation EGFR-TKI that
irreversibly binds to the ATP-binding pocket of EGFR tyrosine
kinase more strongly than first-generation reversible EGFR-TKIs,
leading to efficient inhibition (7). In phase II clinical trials, afatinib
was shown to improve progression-free survival in patients with
advanced NSCLC compared to first-generation EGFR-TKI gefitinib
(8). Additionally,
second-generation EGFR-TKI dacomitinib was more effective than
first-generation EGFR-TKIs for patients with advanced NSCLC with
EGFR-activating mutations (9).
Therefore, second-generation EGFR TKIs, particularly afatinib, are
now recommended for NSCLCs with EGFR-activating mutations.
Pre-clinical studies suggest that second-generation EGFR-TKIs are
also effective at treating cancer cells with T790M. This
substitution has been shown to associate with the development of
resistance to first-generation EGFR-TKIs (6). However, clinical observations found
little efficacy against NSCLC with the T790M mutation (10). Specifically, afatinib had an 8%
response rate for patients who progressed from prior treatment with
first-generation EGFR-TKIs. This suggests that afatinib is
insufficient to overcome T790M-induced resistance.
In contrast, third-generation EGFR-TKI osimertinib
irreversibly binds EGFR with T790M, in addition to EGFR with L858R
and Del19 (11). Importantly,
osimertinib does not bind wild-type EGFR as strongly as these
mutants so is highly specific for NSCLC with EGFR-activating
mutations and is consequently much less toxic. Finally, osimertinib
is more effective against NSCLCs with T790M compared to other
cytotoxic chemotherapies (12).
Although first- and second-generation EGFR-TKIs only marginally
reduced tumor sizes (if at all), osimertinib demonstrated a 61%
response rate for NSCLCs with T790M (12). Furthermore, osimertinib improved
progression-free survival in patients with EGFR-TKI-naïve NSCLC
harboring EGFR-activating mutations compared to other TKIs
(13). Osimertinib is therefore a
standard therapy for NSCLC with EGFR-activating mutations,
particularly those with T790M.
Despite the unique properties of osimertinib,
including its ability to inhibit the tyrosine kinase activity of
EGFR with T790M, cells still eventually acquire resistance. To
investigate how this occurs in more detail, and develop new
treatment regimens for NSCLC with EGFR-activating mutations, we
investigated the underlying mechanisms of afatinib and osimertinib
resistance using Ba/F3 cells with Del19. Using these data, we
explored potential new therapeutic strategies for overcoming
resistance to EGFR-TKI therapy that will contribute to improving
patient outcomes.
Materials and methods
Cell culture and reagents
Murine pro-B Ba/F3 cells were purchased from the
Riken BioResource Center (Tsukuba, Japan) and cultured in RPMI-1640
medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) supplemented
with 10% fetal bovine serum (FBS) and
penicillin-streptomycin-amphotericin B (Wako Pure Chemical
Industries, Ltd., Osaka, Japan) at 37°C in a humidified atmosphere
with 5% CO2. Cells were analyzed using a previously
validated short-tandem repeat method (14). Ba/F3 cells with Del19 were stably
transfected with a full-length cDNA fragment encoding the human
EGFR Del19 mutant (del E746_A750), as previously described
(15). Afatinib and the osimertinib
were purchased from Selleck Chemicals (Houston, TX, USA). Stock
solutions of 10 mM afatinib and osimertinib were prepared in
dimethyl sulfoxide (DMSO) and stored at −20°C.
Establishment of afatinib and
osimertinib-resistant clones through N-ethyl-N-nitrosourea (ENU)
mutagenesis
Ba/F3 cells with EGFR Del19 were exposed to
100 mg/ml ENU (Sigma-Aldrich; Merck KGaA) for 24 h and then washed
and cultured in RPMI-1640 (Sigma-Aldrich; Merck KGaA) with 10% FBS
for 24 h. Similar to previous studies (14,15),
5×104 cells were plated on 96-well plates. The
concentrations of afatinib and osimertinib used were 100 nM when
used singularly, or 50 nM when used in combination, to mimic plasma
concentrations achieved in-clinic (15). Media were changed weekly and cell
growth was observed visually until confluence achieved.
EGFR mutation analyses
Total RNA from resistant cells was isolated using an
RNeasy Mini kit (Qiagen, Hilden, Germany). cDNA was transcribed
from total RNA using a High Capacity RNA-to-cDNA kit (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
tyrosine kinase domains of EGFR (exons 18 to 21) were
amplified with previously reported primers (15). Sanger sequencing was performed using
a Genetic Analyzer 3,130 or 3,500 ×l (Applied Biosystems; Thermo
Fisher Scientific, Inc.).
Cell growth inhibition assays
Afatinib and osimertinib sensitivity was evaluated
using CellTiter-Glo Luminescent Cell Viability Assays (Promega
Corporation, Madison, WI, USA). Briefly, cells were resuspended in
medium containing 10% FBS in 96-well plates at 2×103
cells/well. After overnight incubation at 37°C, afatinib or
osimertinib were added at concentrations ranging from 0.0033 to 1
µM. Cells were left for 72 h and viability was quantified based on
luminescence after the addition of CellTiter-Glo reagent (Promega
Corporation). Experimental sampling was performed for 6-wells for
each treatment.
Antibodies and western blotting
Cells were seeded at a density of 1×106
cells/plate in 60-mm plates and allowed to grow overnight in medium
containing 0.5% FBS before the addition of afatinib or osimertinib.
Cells were incubated for 3 h, harvested, and then lysed in buffer
containing 25 mM Tris (pH 8.3), 192 mM glycine, 0.1% sodium dodecyl
sulfate and 1 mM phenylmethylsulfonyl fluoride (PMSF). Cell lysates
were centrifuged at 15,000 × g for 10 min at 4°C, and the
supernatant was collected for subsequent procedures. Western
blotting was performed following a standard protocol; a total of 30
µg sample was resolved by 7.5% sodium dodecyl sulfate
polyacrylamide gel electrophoresis and transferred to
nitrocellulose membranes, which were probed with antibodies against
phospho-EGFR (cat. no. 2234; Cell Signaling Technology, Inc.,
Danvers, MA, USA), EGFR (cat. no. 4267; Cell Signaling Technology,
Inc.), phospho-ERK1/2 (cat. no. 16982; Santa Cruz Biotechnology,
Santa Cruz, CA, USA), ERK1/2 (cat. no. 9102; Cell Signaling
Technology, Inc.) and β-actin (cat. no. 4970; Cell Signaling
Technology, Inc.) diluted at 1:1,000 at 4°C overnight. Secondarily
those membranes were probed with anti-rabbit antibody (cat. no.
NA934V; GE Healthcare Life Sciences, Little Chalfont, UK) diluted
at 1:2,500. Immune complexes were detected with ECL detection
reagents (GE Healthcare). Protein bands were quantified using
ImageJ version 1.52 software (NIH; National Institutes of Health,
Bethesda, MD, USA) and normalized against β-actin.
Statistical analyses
Statistical analyses were performed using SPSS 22.0
(IBM Corp., Armonk, NY, USA). For statistical hypothesis testing,
Fisher's exact test was used. One-way analysis of variance (ANOVA)
with Dunnett's post hoc test was applied for analysis of western
blotting results. All statistical tests were two-sided. P-values
<0.05 were considered to indicate statistically significant
results. Data are graphically displayed using GraphPad Prism 5.0
for Windows (GraphPad Software, Inc., La Jolla, CA, USA).
Results
An afatinib and osimertinib
combination prevents the appearance of drug resistant clones in
Ba/F3 cells with Del19
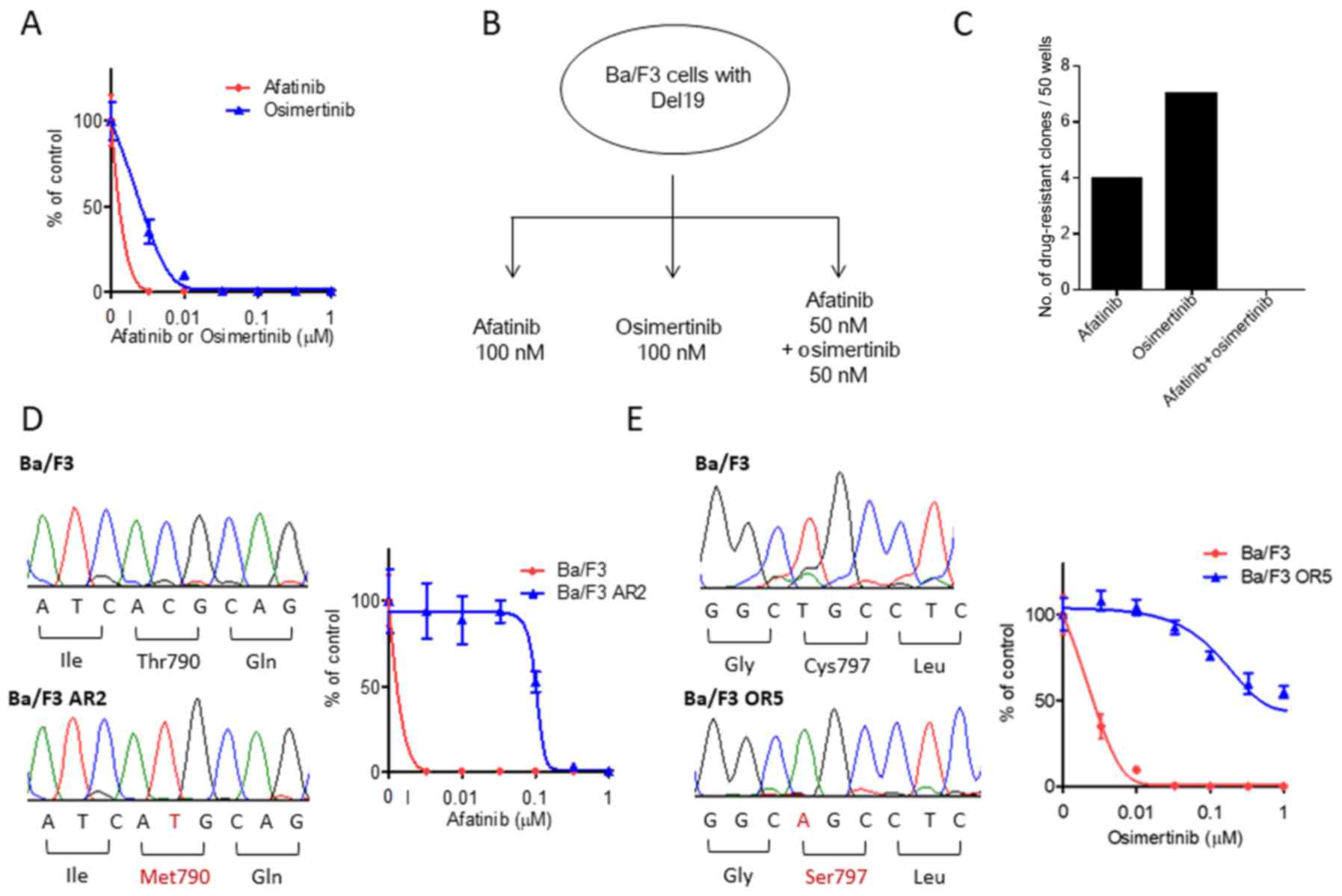
We first evaluated the efficacy of afatinib and
osimertinib for treating Ba/F3 cells with Del19, revealing that
both strongly inhibited cell proliferation. The afatinib and
osimertinib IC50 values were both <10 nM (Fig. 1A), markedly lower than values that
would be clinically available (15). We next examined whether Ba/F3 cells
with Del19 acquired resistance to afatinib or osimertinib. Ba/F3
cells with Del19 were exposed to 100 mg/ml of ENU mutagen for 24 h
and then treated with 100 nM afatinib, 100 nM osimertinib, or 50 nM
afatinib and 50 nM osimertinib in combination for 14 days (Fig. 1B). For each group, 50 wells
containing 5×104 cells/well were examined for resistant
clones. For cells treated with afatinib alone, we observed cell
confluency (suggesting drug resistance) in 4 of the 50 wells
(Fig. 1C). Osimertinib alone led to
confluency in 7 of the 50 wells (afatinib vs. osimertinib, P=0.52;
Fig. 1C). Notably, treatment with
afatinib and osimertinib did not lead to any resistant clones among
the 50 wells (Fig. 1C). We repeated
these observations across 500 wells but were unable to find any
resistant clones for the afatinib and osimertinib combination.
As we expected that secondary mutations in EGFR lead
to EGFR-TKI resistance, we sequenced the kinase domains of
EGFR exons 18 and 21. All four of the afatinib-resistant
clones, including Ba/F3 AR2, had the secondary EGFR mutation T790M
(Fig. 1D). In vitro growth
inhibition assays showed that the IC50 values for these
resistant cells were >30-fold higher than Ba/F3 cells with only
Del19 (Fig. 1D). We also examined
the seven osimertinib-resistant clones, including Ba/F3 OR5,
finding that they all had the secondary EGFR mutation C797S
(Fig. 1E). In vitro growth
inhibition assays showed that the IC50 values for these
cells were >100-fold higher than Ba/F3 cells with only Del19
(Fig. 1E). These results suggest
that secondary EGFR mutations, such as T790M or C797S, can cause
resistance to afatinib or osimertinib, whereas both drugs used in
combination prevented resistant clones.
Afatinib- or osimertinib-resistant
clones maintain sensitivity to other EGFR-TKIs
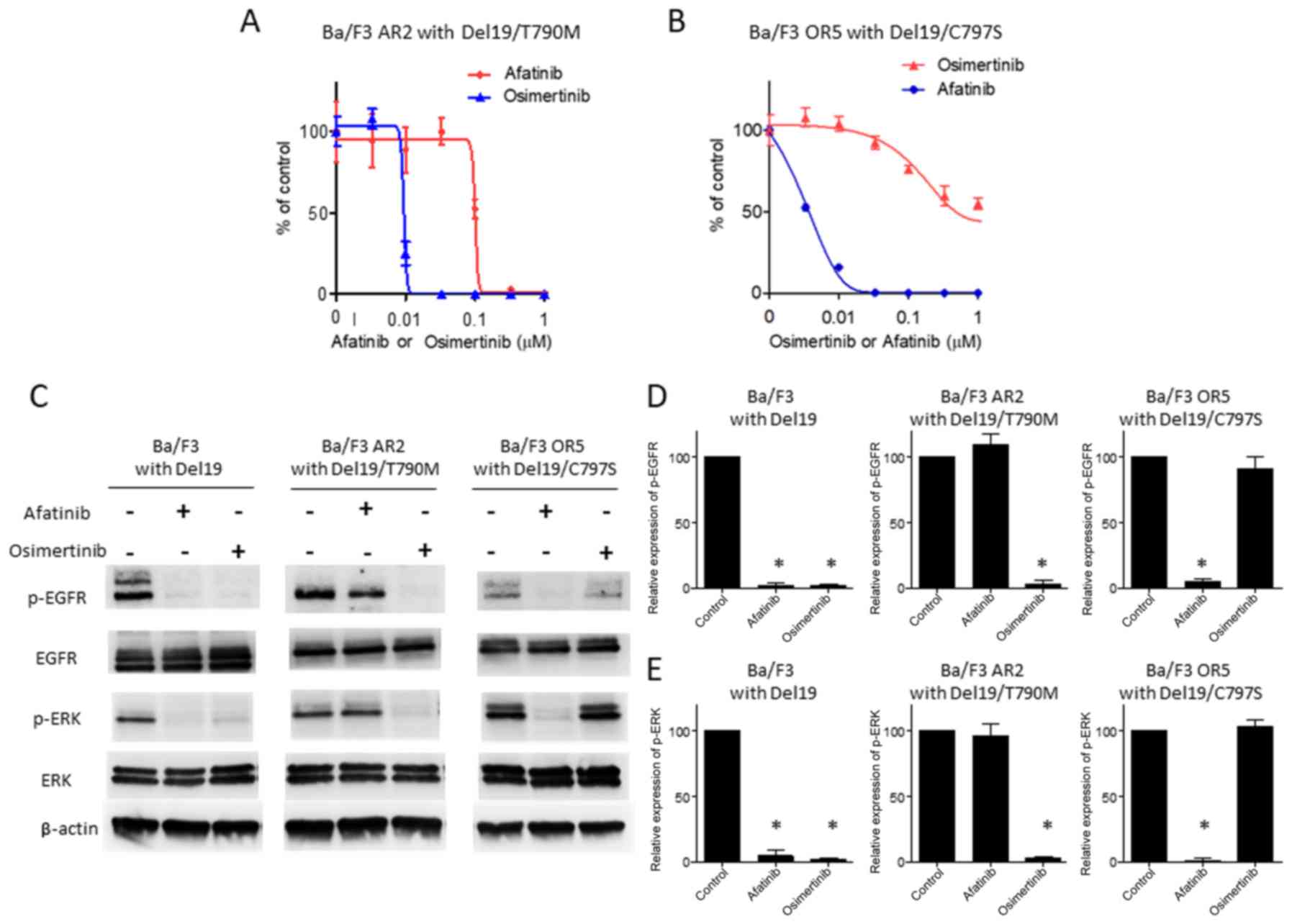
Based on our previous experiment, we hypothesized
that afatinib prevents the appearance of cells with C797S in a
Ba/F3 cell population with Del19, whereas osimertinib prevents the
appearance of cells with T790M. To test this hypothesis, we
examined the efficacy of osimertinib against T790M
afatinib-resistant cells. In contrast to afatinib treatment,
osimertinib dose-dependently inhibited cell-proliferation in
Del19/T790M cells, almost completely eradicating viable cells at 50
nM (Fig. 2A). Conversely, we
examined the efficacy of afatinib in osimertinib-resistant cells
with C797S. In contrast to osimertinib treatment, afatinib
moderately inhibited proliferation, almost completely eradicating
viable cells at 50 nM (Fig. 2B).
Then, we examined signaling molecules in cells treated with
afatinib or osimertinib. Parental Ba/F3 cells exhibited significant
decreases in the phosphorylation of EGFR upon afatinib and
osimertinib treatment (Fig. 2C and
D). Additionally, these cells exhibited significant decreases
in the phosphorylation of ERK, downstream of EGFR, induced by
either drug (Fig. 2C and E). In
contrast to parental Ba/F3 cells, Ba/F3 cells with Del19/T790M did
not exhibit downregulation of EGFR or ERK phosphorylation upon
afatinib treatment (Fig. 2C-E).
Furthermore, Ba/F3 cells with Del19/C797S did not exhibit decreases
in the phosphorylation of EGFR or ERK upon osimertinib treatment
(Fig. 2C-E). Combined, these
results suggest that osimertinib and afatinib may block EGFR-ERK
signaling in Ba/F3 cells with Del19/T790M and Del19/C797S,
respectively, eradicating those cells. Therefore, the combination
of osimertinib and afatinib prevents the appearance of resistant
clones.
Treatment with an afatinib and
osimertinib combination does not eradicate resistant clones after
the generation of C797S mutations in Ba/F3 cells with Del19
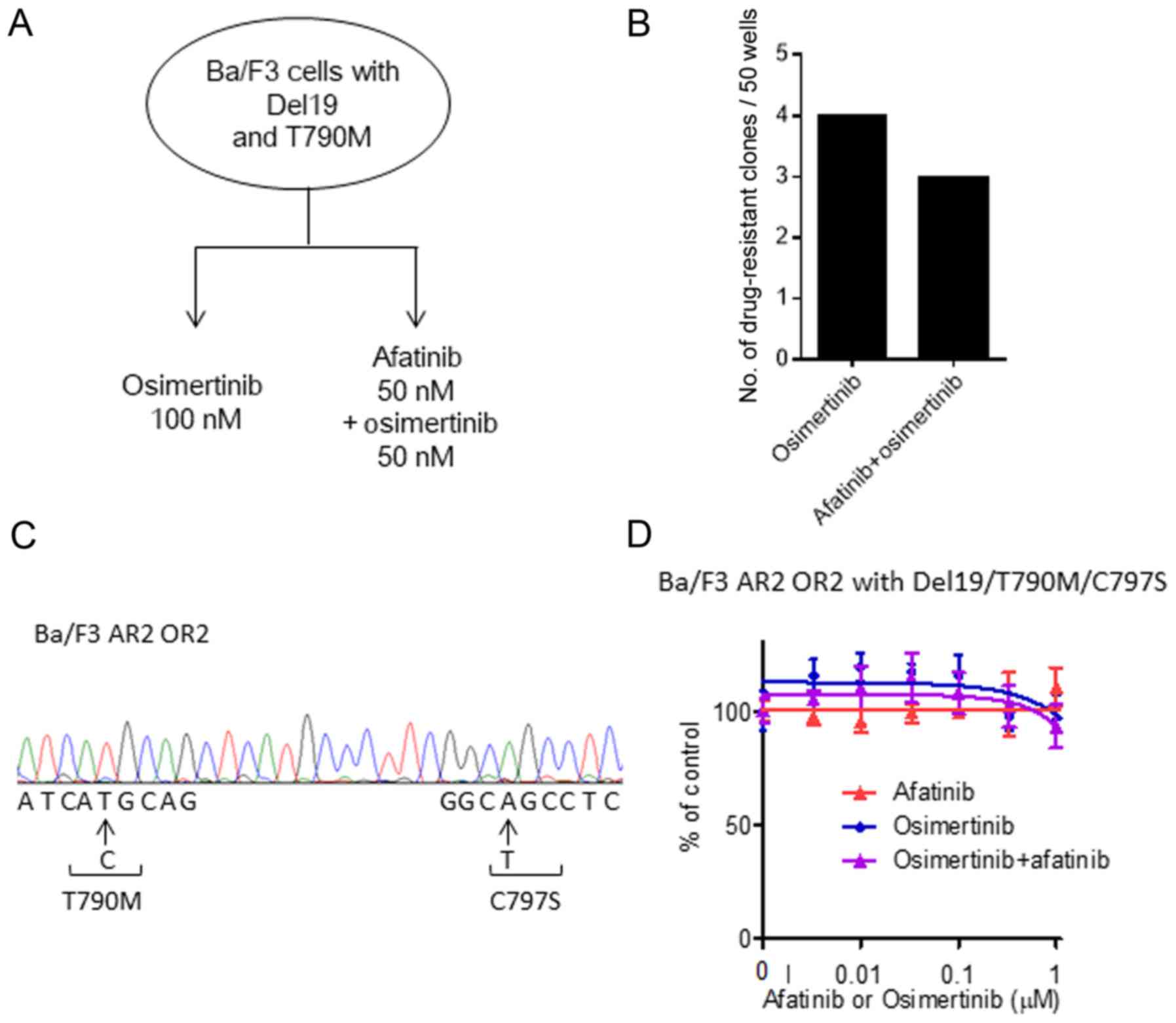
Then, we speculated that afatinib-resistant clones
with T790M (generated after afatinib monotherapy) may be eradicated
by osimertinib or an osimertinib and afatinib combination. We
therefore treated afatinib-resistant cells that possessed T790M
with either 100 nM osimertinib or 50 nM osimertinib with 50 nM
afatinib for 14 days (Fig. 3A).
However, contrary to expectations, treatment with 100 nM
osimertinib led to 4 of the 50 wells having resistant cells,
including Ba/F3 AR2 OR2 (Fig. 3B).
Furthermore, treatment with 50 nM osimertinib and 50 nM afatinib
led to 3 of the 50 wells having resistant cells (P>0.05;
Fig. 3B). To investigate the
underlying mechanisms, we sequenced the kinase domain of
EGFR. All the resistant clones that emerged after treatment
with osimertinib or the osimertinib with afatinib combination had a
Del19/T790M/C797S triple mutation in the EGFR kinase domain
(Fig. 3C). In vitro growth
inhibition assays revealed that the resistant Ba/F3 AR2 OR2 cells
maintained strong growth in 100 nM osimertinib, 100 nM afatinib,
and the 50 nM osimertinib and 50 nM afatinib combination (Fig. 3D). These observations suggested that
afatinib-resistant cells with T790M were more sensitive to
sequential osimertinib treatment or an osimertinib with afatinib
combination. However, a small population of cells with
Del19/T790M/C797S survived and proliferated despite drug
treatment.
Treatment with an afatinib and
osimertinib combination does not eradicate resistant clones after
the generation of C797S mutations in Ba/F3 cells with Del19
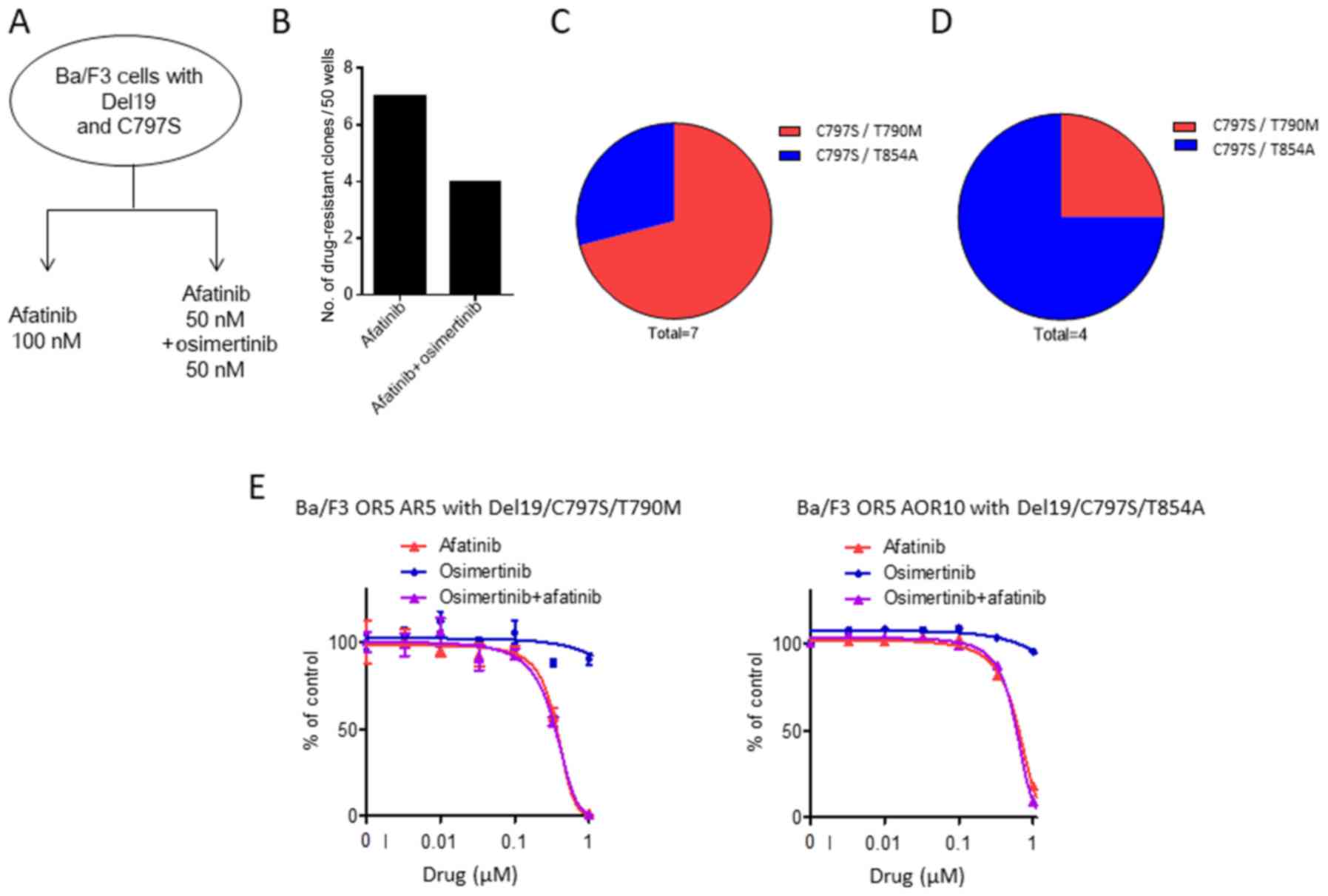
Finally, we examined whether osimertinib-resistant
clones with C797S could be eradicated by treatment with afatinib or
an afatinib and osimertinib combination. To assess this, we treated
C797S osimertinib-resistant cells with either 100 nM afatinib or a
50 nM afatinib with 50 nM osimertinib combination for 14 days
(Fig. 4A). This revealed that
treatment with 100 nM afatinib led to 7 of 50 wells with resistant
cells (Fig. 4B). In addition, the
50 nM afatinib with 50 nM osimertinib combination generated a
similar number, with 4 resistant wells from the total 50 (Fig. 4B). We again sequenced EGFR
exons 18 and 21, showing that two distinct triple mutations had
emerged in cells resistant to sequential afatinib single therapy,
Del19/C797S/T790M and Del19/C797S/T854A (Fig. 4C). In cells resistant to the
afatinib and osimertinib combination, we observed two triple
mutations, including Del19/C797S/T790M and Del19/C797S/T854A
(Fig. 4D). In vitro growth
inhibition assays indicated that cells with the triple mutation
(Ba/F3 OR5 AR5 with Del19/C797S/T790M and Ba/F3 OR5 AOR10 with
Del19/C797S/T854A) proliferated in 100 nM osimertinib, 100 nM
afatinib, and a combination of 50 nM osimertinib and 50 nM afatinib
(Fig. 4E). These data suggest that
these mutations lead to EGFR-TKI resistance in cancer cells, in
addition to the mutations identified in previous reports (16,17).
In summary, our observations suggest that osimertinib-resistant
cells with C797S are sensitive to sequential afatinib treatment and
afatinib with osimertinib combination therapy. However, a small
population acquire additional mutations (typically T790M or T854A)
that allows them to proliferate despite drug treatment.
Discussion
The present study found that epidermal growth factor
receptor (EGFR) dual-blockade using afatinib and osimertinib
eradicated Ba/F3 cancer cells with Del19, indicating that this
combination prevents the generation of resistance to these two
drugs. In contrast to combination treatment, osimertinib alone
primarily generated resistant clones with the C797S mutation, and
afatinib alone generated resistant clones with T790M. In fact,
tissue samples from tumors resistant to afatinib are reported to
frequently carry secondary T790M EGFR mutations at a rate of 50%
(18). In addition, 20–30% of
patients treated with osimertinib acquire resistance with an
accompanying C797S mutation (19).
Considering these results, EGFR dual-blockade using afatinib and
osimertinib may be a clinically relevant solution for preventing
secondary EGFR mutation-dependent resistance.
Previously, several combination therapies have been
reported to overcome the resistance to epidermal growth factor
receptor tyrosine kinase inhibitors (EGFR-TKIs), which depends on
EGFR secondary mutations or other mechanisms such as bypass
signaling (Table I) (20–28).
Another EGFR-TKI, brigatinib, or allosteric EGFR inhibitor EAI045
could overcome both T790M- and C797S-positive triple-mutant
resistant clones, particularly when combined with the anti-EGFR
antibody cetuximab (21,22). Alternatively, we and others have
suggested that a combination of two different types of EGFR-TKIs
may prevent the appearance of secondary EGFR mutation-dependent
resistant clones (23–25). Ercan et al and Uchibori et
al also reported that a gefitinib and osimertinib combination
prevented the appearance of resistant Ba/F3 clones with the EGFR
mutation L858R (23,25). Third-generation EGFR-TKIs generate
covalent bonds with cysteine 797 in EGFR; therefore, this could
prevent the increased ATP affinity mediated by the T790M mutation
(11). Alternatively,
first-generation or second-generation EGFR-TKIs may form hydrogen
bonds with methionine 793 in EGFR and competitively inhibit ATP
binding in the ATP binding pocket, regardless of the presence of
the C797S mutation (15). The
second-generation EGFR-TKI afatinib and third-generation EGFR-TKI
osimertinib used in the present study may competitively bind to the
ATP binding pockets of the mutant EGFR in the combination
treatment, enabling these drugs to complement each other in terms
of their disadvantages mediated by secondary EGFR mutation.
 | Table I.EGFR tyrosine kinase inhibitor
combination therapy. |
Table I.
EGFR tyrosine kinase inhibitor
combination therapy.
| Combination
therapy | Resistance
mechanism | Resistance
mutation | Cell line
model | (Refs.) |
|---|
| Afatinib +
cetuximab | EGFR secondary
mutation | EGFR T790M | H1975 | (20) |
| Brigatinib +
cetuximab |
| EGFR
T790M/C797S | PC9, MGH121,
Ba/F3 | (21) |
| EAI045 +
cetuximab |
| EGFR
T790M/C797S | H1975, Ba/F3 | (22) |
| Gefitinib +
osimertinib |
| EGFR
T790M/C797S | Ba/F3 | (23) |
| Gefitinib +
WZ4002 |
| EGFR
T790M/C797S | Ba/F3 | (24) |
| Gefitinib +
osimertinib |
| EGFR
T790M/C797S | Ba/F3 | (25) |
| Afatinib +
osimertinib |
| EGFR
T790M/C797S | Ba/F3 | Present study |
| EGFR-TKI +
sonidegib | Others | Hedgehog signal
activation | HCC827 | (26) |
| EGFR-TKI + MET
inhibitor |
| MET
amplification | HCC827 | (27) |
| EGFR-TKI + MEK
inhibitor |
| MAPK1
amplification | PC9 | (28) |
In contrast to combination therapy with osimertinib
and afatinib, sequential therapy with osimertinib after resistance
to afatinib did not completely eliminate Ba/F3 cells, nor reduce
development of resistant clones. This also occurred after
sequential therapy with afatinib following the emergence of
osimertinib resistance. Clinical observation found that afatinib
followed by osimertinib achieved long-term overall survival in the
case of secondary T790M-positive non-small cell lung cancers
(NSCLC) (29,30). However, another report indicated
that 20–30% of patients treated secondarily with osimertinib
acquired resistance with an accompanying C797S mutation (19). Therefore, to totally eradicate
mutant EGFR-dependent cells, the present study suggests initial
treatment with an osimertinib and afatinib combination would have
greater efficacy than sequential usage, and this should be tested
in future clinical trials.
The present study also revealed that cells with the
Del19/T790M/C797S triple mutation were resistant to even
osimertinib and afatinib in combination. Niederst et al
previously reported that a combination of first- and
third-generation EGFR-TKIs can prevent proliferation of NSCLC cells
with a double T790M/C797S mutation when T790M and C797S are located
on different alleles (i.e., a trans position) (24). However, in the case of T790M and
C797S located on the same allele (i.e., a cis position), a
first- and third-generation EGFR-TKI combination had limited
efficacy (24). Piotrowska et
al reported that T790M and C797S were on the same allele in
44/46 of evaluable patients (98%), indicating that the resistant
cis position is more common (31). Based on these observations, we
hypothesize that both the afatinib- and osimertinib-resistant cells
in the present study had T790M and C797S mutations in a cis
location. This would mean that an afatinib and osimertinib
combination could not eradicate resistant cells following the
development of afatinib-resistance and the T790M mutation.
Ba/F3 cells are a limited model for investigating
EGFR secondary mutation-dependent resistance to EGFR-TKIs. In
actual clinical settings, other resistance mechanisms, such as
bypass signaling, epithelial-mesenchymal transition, or
EGFR-downstream activation, may occur. Therefore, the EGFR-dual
blockade strategy may be less useful in these situations, and other
combination strategies may be required.
Acknowledgements
We thank Haruka Yamaguchi, Yume Shinkai, Michiko
Kitano and Mami Kitano at the Department of Medical Oncology,
Kindai University Faculty of Medicine, for their technical
support.
Funding
The present study was financially supported by
Boehringer Ingelheim Co. Ltd.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KY, YK and HH were involved in the conception and
design of the study, the performance of the experiments, the data
analysis and interpretation and the manuscript writing. YC was
involved in the data analysis and their interpretation. TM and KN
were involved in the conception and the design of the study and the
interpretation of the date. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
KY, HH, TM and KN received research funding from
Boehringer Ingelheim Co., Ltd. The other authors declare that they
have no competing interests.
References
|
1
|
Mitsudomi T, Kosaka T and Yatabe Y:
Biological and clinical implications of EGFR mutations in lung
cancer. Int J Clin Oncol. 11:190–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al: Gefitinib or chemotherapy for non-small-cell lung cancer
with mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mitsudomi T, Morita S, Yatabe Y, Negoro S,
Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, et
al: Gefitinib versus cisplatin plus docetaxel in patients with
non-small-cell lung cancer harbouring mutations of the epidermal
growth factor receptor (WJTOG3405): An open label, randomised phase
3 trial. Lancet Oncol. 11:121–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakamura A, Inoue A, Morita S, Hosomi Y,
Kato T, Fukuhara T, Gemma A, Takahashi K, Fujita Y, Harada T, et
al: Phase III study comparing gefitinib monotherapy (G) to
combination therapy with gefitinib, carboplatin, and pemetrexed
(GCP) for untreated patients (pts) with advanced non-small cell
lung cancer (NSCLC) with EGFR mutations (NEJ009). J Clin Oncol. 36
Suppl 15:S90052018. View Article : Google Scholar
|
|
5
|
Furuya N, Fukuhara T, Saito H, Watanabe K,
Sugawara S, Iwasawa S, Tsunezuka Y, Yamaguchi O, Okada M, Yoshimori
K, et al: Phase III study comparing bevacizumab plus erlotinib to
erlotinib in patients with untreated NSCLC harboring activating
EGFR mutations: NEJ026. J Clin Oncol. 36 Suppl 15:S90062018.
View Article : Google Scholar
|
|
6
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
75:75ra262011.
|
|
7
|
Li D, Ambrogio L, Shimamura T, Kubo S,
Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A,
Himmelsbach F, et al: BIBW2992, an irreversible EGFR/HER2 inhibitor
highly effective in preclinical lung cancer models. Oncogene.
27:4702–4711. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park K, Tan EH, O'Byrne K, Zhang L, Boyer
M, Mok T, Hirsh V, Yang JC, Lee KH, Lu S, et al: Afatinib versus
gefitinib as first-line treatment of patients with EGFR
mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase
2B, open-label, randomised controlled trial. Lancet Oncol.
17:577–589. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa
K, Niho S, Tsuji F, Linke R, Rosell R, Corral J, et al: Dacomitinib
versus gefitinib as first-line treatment for patients with
EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A
randomised, open-label, phase 3 trial. Lancet Oncol. 18:1454–1466.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Katakami N, Atagi S, Goto K, Hida T, Horai
T, Inoue A, Ichinose Y, Koboyashi K, Takeda K, Kiura K, et al:
LUX-Lung 4: A phase II trial of afatinib in patients with advanced
non-small-cell lung cancer who progressed during prior treatment
with erlotinib, gefitinib, or both. J Clin Oncol. 31:3335–3341.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cross DA, Ashton SE, Ghiorghiu S, Eberlein
C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ,
et al: AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated
resistance to EGFR inhibitors in lung cancer. Cancer Discov.
4:1046–1061. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Janne PA, Yang JC, Kim DW, Planchard D,
Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, et al: AZD9291
in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J
Med. 372:1689–1699. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim
HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WS, et
al: Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung
cancer. N Engl J Med. 376:629–640. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kobayashi Y, Togashi Y, Yatabe Y, Mizuuchi
H, Jangchul P, Kondo C, Shimoji M, Sato K, Suda K, Tomizawa K, et
al: EGFR exon 18 mutations in lung cancer: Molecular predictors of
augmented sensitivity to afatinib or neratinib as compared with
first- or third-generation TKIs. Clin Cancer Res. 21:5305–5313.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kobayashi Y, Azuma K, Nagai H, Kim YH,
Togashi Y, Sesumi Y, Chiba M, Shimoji M, Sato K, Tomizawa K, et al:
Characterization of EGFR T790M, L792F, and C797S mutations as
mechanisms of acquired resistance to afatinib in lung cancer. Mol
Cancer Ther. 16:357–364. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bean J, Riely GJ, Balak M, Marks JL,
Ladanyi M, Miller VA and Pao W: Acquired resistance to epidermal
growth factor receptor kinase inhibitors associated with a novel
T854A mutation in a patient with EGFR-mutant lung
adenocarcinoma. Clin Cancer Res. 14:7519–7525. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Avizienyte E, Ward RA and Garner AP:
Comparison of the EGFR resistance mutation profiles generated by
EGFR-targeted tyrosine kinase inhibitors and the impact of drug
combinations. Biochem J. 415:197–206. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tanaka K, Nosaki K, Otsubo K, Azuma K,
Sakata S, Ouchi H, Morinaga R, Wataya H, Fujii A, Nakagaki N, et
al: Acquisition of the T790M resistance mutation during afatinib
treatment in EGFR tyrosine kinase inhibitor-naïve patients with
non-small cell lung cancer harboring EGFR mutations.
Oncotarget. 8:68123–68130. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thress KS, Paweletz CP, Felip E, Cho BC,
Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et
al: Acquired EGFR C797S mutation mediates resistance to AZD9291 in
non-small cell lung cancer harboring EGFR T790M. Nat Med.
21:560–562. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Regales L, Gong Y, Shen R, de Stanchina E,
Vivanco I, Goel A, Koutcher JA, Spassova M, Ouerfelli O,
Mellinghoff IK, et al: Dual targeting of EGFR can overcome a major
drug resistance mutation in mouse models of EGFR mutant lung
cancer. J Clin Invest. 119:3000–3010. 2009.PubMed/NCBI
|
|
21
|
Uchibori K, Inase N, Araki M, Kamada M,
Sato S, Okuno Y, Fujita N and Katayama R: Brigatinib combined with
anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated
non-small-cell lung cancer. Nat Commun. 8:147682017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jia Y, Yun CH, Park E, Ercan D, Manuia M,
Juarez J, Xu C, Rhee K, Chen T, Zhang H, et al: Overcoming
EGFR(T790M) and EGFR(C797S) resistance with mutant-selective
allosteric inhibitors. Nature. 534:129–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ercan D, Choi HG, Yun CH, Capelletti M,
Xie T, Eck MJ, Gray NS and Jänne PA: EGFR mutations and resistance
to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res.
21:3913–3923. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Niederst MJ, Hu H, Mulvey HE, Lockerman
EL, Garcia AR, Piotrowska Z, Sequist LV and Engelman JA: The
allelic context of the C797S mutation acquired upon treatment with
third-generation EGFR inhibitors impacts sensitivity to subsequent
treatment strategies. Clin Cancer Res. 21:3924–3933. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Uchibori K, Inase N, Nishio M, Fujita N
and Katayama R: Identification of mutation accumulation as
resistance mechanism emerging in first-line osimertinib treatment.
J Thorac Oncol. 13:915–925. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Della Corte CM, Malapelle U, Vigliar E,
Pepe F, Troncone G, Ciaramella V, Troiani T, Martinelli E, Belli V,
Ciardiello F, et al: Efficacy of continuous EGFR-inhibition and
role of Hedgehog in EGFR acquired resistance in human lung cancer
cells with activating mutation of EGFR. Oncotarget. 8:23020–23032.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in
lung cancer by activating ERBB3 signaling. Science. 316:1039–1043.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tricker EM, Xu C, Uddin S, Capelletti M,
Ercan D, Ogino A, Pratilas CA, Rosen N, Gray NS, Wong KK, et al:
Combined EGFR/MEK inhibition prevents the emergence of resistance
in EGFR mutant lung cancer. Cancer Discov. 5:960–971. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Paz-Ares L, Tan EH, O'Byrne K, Zhang L,
Hirsh V, Boyer M, Yang JC, Mok T, Lee KH, Lu S, et al: Afatinib
versus gefitinib in patients with EGFR mutation-positive
advanced non-small-cell lung cancer: Overall survival data from the
phase IIb LUX-Lung 7 trial. Ann Oncol. 28:270–277. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park K, Tan E, O'Byrne K, Zhang L, Boyer
M, Mok T, Hirsh V, Yang JC, Schuler M, Yamamoto N, et al: P3.01–039
Sequential afatinib-osimertinib therapy in EGFR mutation-positive
(EGFRm+) NSCLC: Analysis of time on treatment and OS. J Thorac
Oncol. 12 Suppl 2:S2215–S2216. 2017. View Article : Google Scholar
|
|
31
|
Piotrowska Z, Nagy RJ, Fairclough S,
Lanman R, Marcoux N, Gettinger S, Owonikoko T, Ramalingam S and
Sequist L: OA 09.01 Characterizing the genomic landscape of EGFR
C797S in lung cancer using ctDNA next-generation sequencing. J
Thorac Oncol. 12 Suppl 2:S17672017. View Article : Google Scholar
|