Introduction
Glioma occurs in the glial cells of the spine or
brain, and is classified according to cell type, location and grade
(1). The incidence rate of
glioblastoma multiforme is the highest (3.20 per 100,000
population) of all malignant central nervous system tumors in the
United States (2). Gliomas in
different locations exhibit distinct symptoms, and the tumor
metastasizes via the cerebrospinal fluid, rather than the
bloodstream (3). The majority of
gliomas are incurable; in particular, the prognosis for older
patients with advanced glioma is poorer (4). The O6-methylguanine-DNA
methyltransferase (MGMT) gene, which is located on
chromosome 10q26 and codes for a DNA repair enzyme, can eliminate
the efficacy of alkylating chemotherapy (5). Promoter methylation of MGMT has
been detected in 51–66% of cases of glioblastoma; this methylation
inactivates the MGMT gene and promotes the chemosensitivity
and survival of patients with glioblastoma (6,7).
Therefore, exploring the key genes associated with MGMT
promoter methylation in glioma is important.
In recent years, the mechanisms underlying glioma
prognosis have been investigated. For example, enhancer of zeste 2
polycomb repressive complex 2 subunit (EZH2) overexpression
is associated with tumor grade and predicts a short survival;
therefore, EZH2 is a promising prognostic factor and
therapeutic target in patients with glioblastoma (8). Chitinase 3-like 1 (CHI3L1)
functions in mediating local invasiveness and malignant
transformation; therefore, CHI3L1 may be considered a
potential target for the treatment of glioma (9,10).
Furthermore, large tumor suppressor kinase 1 is a critical tumor
suppressor, the inhibition of which may promote the progression of
glioma (11). Conversely, the
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4
(PFKFB4) gene serves an important role in maintaining cancer
stem-like cells in the brain, and high PFKFB4 expression is
correlated with unfavorable survival of patients with glioblastoma
(12). Nevertheless, the mechanisms
underlying the effects of MGMT promoter methylation on the
prognosis of patients with glioma have not been thoroughly
studied.
Expression profile analysis is widely used for
selecting meaningful and important genes from large amounts of data
via the evaluation of gene expression levels (13,14).
In the present study, RNA-sequencing (RNA-seq) data and methylation
data of glioma were downloaded, and were then analyzed through a
series of bioinformatics analyses. The present study aimed to
reveal the key genes involved in MGMT promoter methylation
in patients with glioma, in order to provide potential targets for
promoting MGMT promoter methylation and improving the
chemosensitivity of patients with glioma.
Materials and methods
Data source and preprocessing
For glioma, RNA-seq data (platform: IlluminaHiSeq),
methylation data (platform: HumanMethylation27) and clinical
follow-up data were obtained from The Cancer Genome Atlas (TCGA;
http://cancergenome.nih.gov/) database
(updated March 1, 2017). From a total of 295 glioma samples, 77
samples (61 samples from patients that had succumbed, 15 samples
from patients that had survived, and 1 sample from a patient for
whom there was no survival information) with RNA-seq data and
methylation data were selected, and their clinical follow-up data
were downloaded. RNA-seq data and methylation data were normalized
using the quantile normalization method (15).
Identification of feature genes
between high and low methylation samples
The methylation levels of CpG islands in the
promoter region of MGMT in the 77 samples were analyzed and
ranked. The median methylation level of the CpG sites in CpG
islands was determined, and samples with methylation levels
>0.35 were defined as high methylation samples, whereas those
with methylation levels <0.35 were considered low methylation
samples. Methylation levels in the high and low methylation groups
were compared using independent samples t-test; P<0.05 was
considered to indicate a statistically significant difference. High
and low methylation levels indicated patients with high and low
MGMT promoter methylation; these patients have differences
in prognosis (16). To identify the
feature genes that could differentiate high and low methylation
samples, the expression levels of each gene (i) in high (H) and low
(L) methylation samples were calculated to obtain the difference
index (Fi) using R software (https://www.r-project.org/). The formula used was as
follows:
Fi=(MeaniH-MeaniL)/(SDiH+SDiL)
To identify the optimal gene combination that could
distinguish between high and low methylation samples, the permTS
function in the R package perm (https://cran.r-project.org/web/packages/perm/)
(17) was used to calculate the
significant P-value of each gene in the two groups of samples. The
step size for setting the threshold k for Fi was 0.01.
The gene combination with |Fi| > k and P<0.01 was
selected, and its corresponding expression profile was obtained.
Afterwards, the following classifier models were used for
performing sample classification:
bi=(MeaniH+MeaniL)/2
Vi=Fi(ei-bi)
PSj=∑i=0NVi/∑i=0N|Vi|
Bi refers to the mean value
between high and low methylation samples; ei
represents the expression level of gene i; Vi is the product
of Fi and the difference between ei and
Bi; N indicates the number of genes under
the present threshold; PSj represents the
classification score of sample j. Finally, the samples were divided
into two groups according to their positive or negative scores.
samples with positive scores were divided into a new high
methylation group, whereas samples with negative scores were
divided into a new low methylation group. Meanwhile, the
consistency ratio of the new groups and the previous groups (high
and low methylation groups) was calculated. The gene combination
that corresponded to the highest consistency ratio was selected as
the feature gene set.
Expression characteristic analysis and
differential expression analysis of the feature genes
To determine the stability of the feature gene set
for classifying samples, five-fold cross validation (18) was performed 10 times. Under each
cross validation, the consistency ratio of the new high and low
methylation groups, and the previous high and low methylation
groups was calculated. Based on the expression levels of the
feature genes in each sample, unsupervised hierarchical clustering
was performed to observe the expression characteristics of the
feature genes using heatmap function (https://stat.ethz.ch/R-manual/R-devel/library/stats/html/heatmap.html)
in R package. In addition, the prognostic differences of the
samples in different clusters were analyzed using the R package
Kaplan Meier (19). Furthermore,
Wilcoxon rank sum test (20) was
performed in R software to observe the differential expression of
the feature genes in high and low methylation samples.
Multivariate survival analysis
Multivariate survival analysis was performed for the
feature genes to examine the effects of the feature genes on
prognosis. Using the R package survivalROC (https://cran.r-project.org/web/packages/survivalroc/index.html)
(21), a receiver operating
characteristic (ROC) curve was generated.
Validation of the feature genes using
independent datasets
To confirm that the feature genes were repeatable,
the validation datasets GSE7696 (22) (platform: Affymetrix Human Genome
U133 Plus 2.0 Array; comprised 84 samples, including 78 glioma
samples with clinical follow-up data) and GSE42669 (23) (platform: Affymetrix Human Gene 1.0
ST Array; comprised 58 samples, including 55 glioma samples with
clinical follow-up data) were downloaded from the Gene Expression
Omnibus (https://www.ncbi.nlm.nih.gov/geo/) database. Using the
R package survival (http://cran.r-project.org/package=survival) (24), analysis of the two datasets was
conducted with Cox regression analysis.
Results
Identification of feature genes
between high and low methylation samples
The methylation levels of CpG islands in the
promoter region of MGMT were markedly varied in the
different samples, and the 77 samples were ranked according to
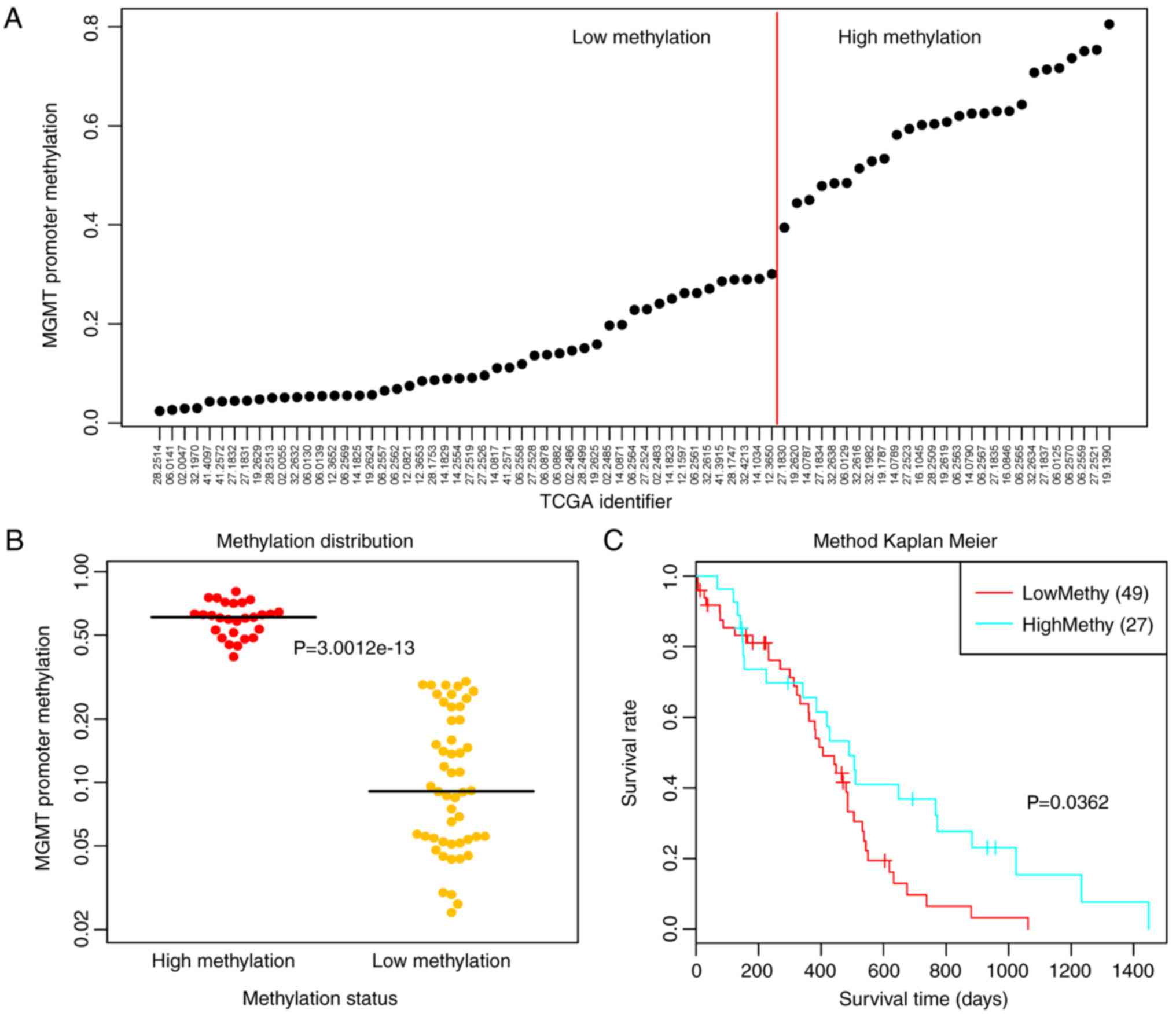
methylation levels (Fig. 1A).
Subsequently, the samples were divided into high and low
methylation groups using 0.35 as the cut-off point for methylation
levels. The results demonstrated that the two groups of samples had
significant MGMT methylation differences (P<0.0001;
Fig. 1B). Meanwhile, univariate
survival analysis revealed that the low methylation group had a
significantly poorer prognosis (P=0.0362; Fig. 1C).
There were 24,991 genes expressed in the 77 samples.
A total of 19,659 genes were expressed in >50% of samples and
were selected for the following analysis. Briefly, the difference
index and P-value of the permutation test were calculated for each
gene. The fold change between high and low methylation groups and
P-value for MGMT were 1.23 and 9.26×10−11,
respectively. Combined with different classifier models, sample
classification was performed using gene combinations with different
cut-off values. Subsequently, the consistency ratios of the new
groups (divided according to the positive and negative
PSj classification scores) and the previous
groups (divided according to high and low methylation) were
calculated. The results revealed that the consistency ratios under
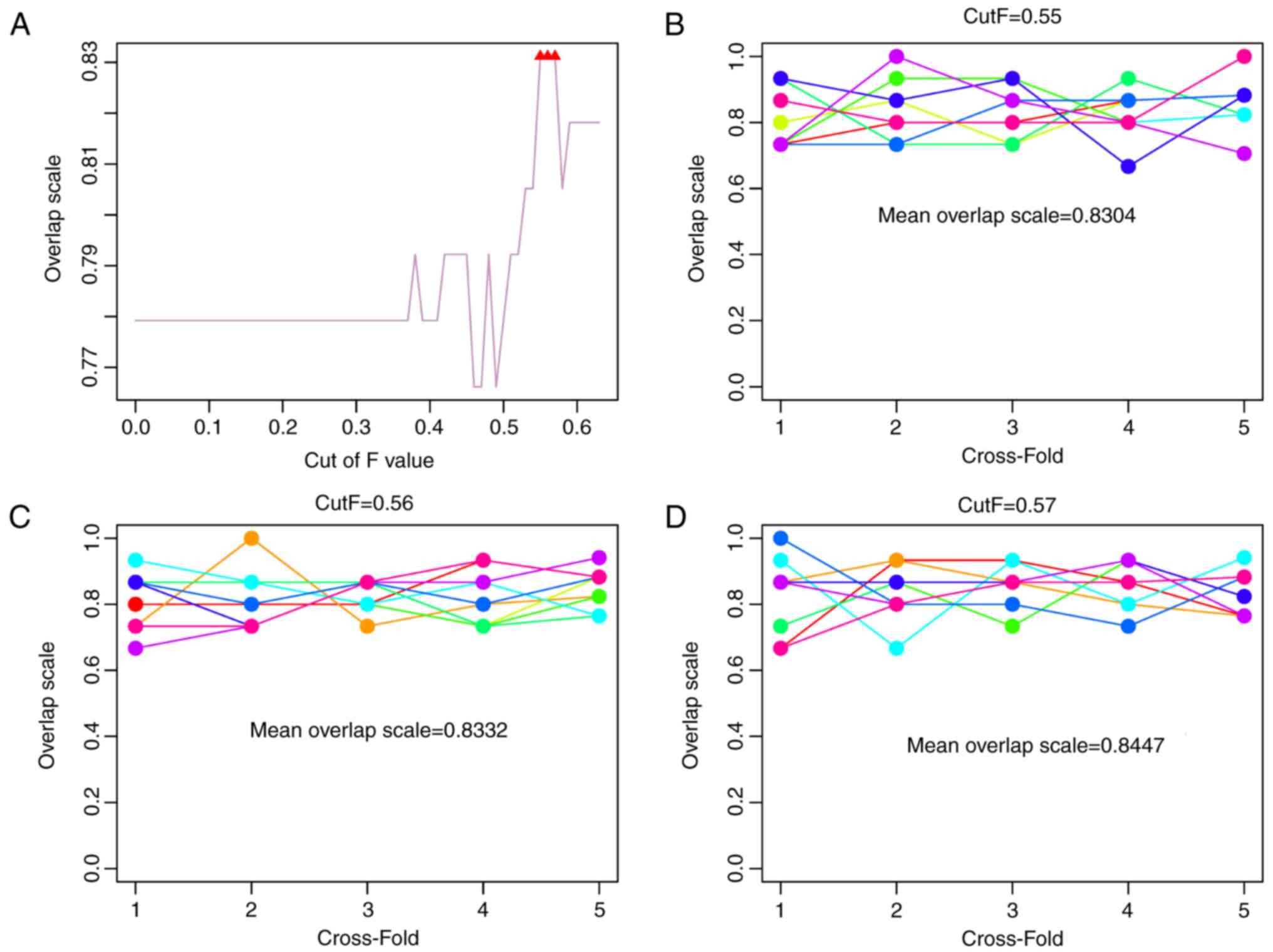
different cut-off values were different. In particular, the
consistency ratios were higher when the cut-off values were 0.55,
0.56 and 0.57 (Fig. 2A).
Furthermore, five-fold cross validation was
performed 10 times to detect the consistency ratios under the three
cut-off values. The results indicated that the consistency ratios
under the cut-off values of 0.55 (Fig.
2B), 0.56 (Fig. 2C) and 0.57
(Fig. 2D) were high and were all
>0.83.
Expression characteristic analysis and
differential expression analysis of the feature genes
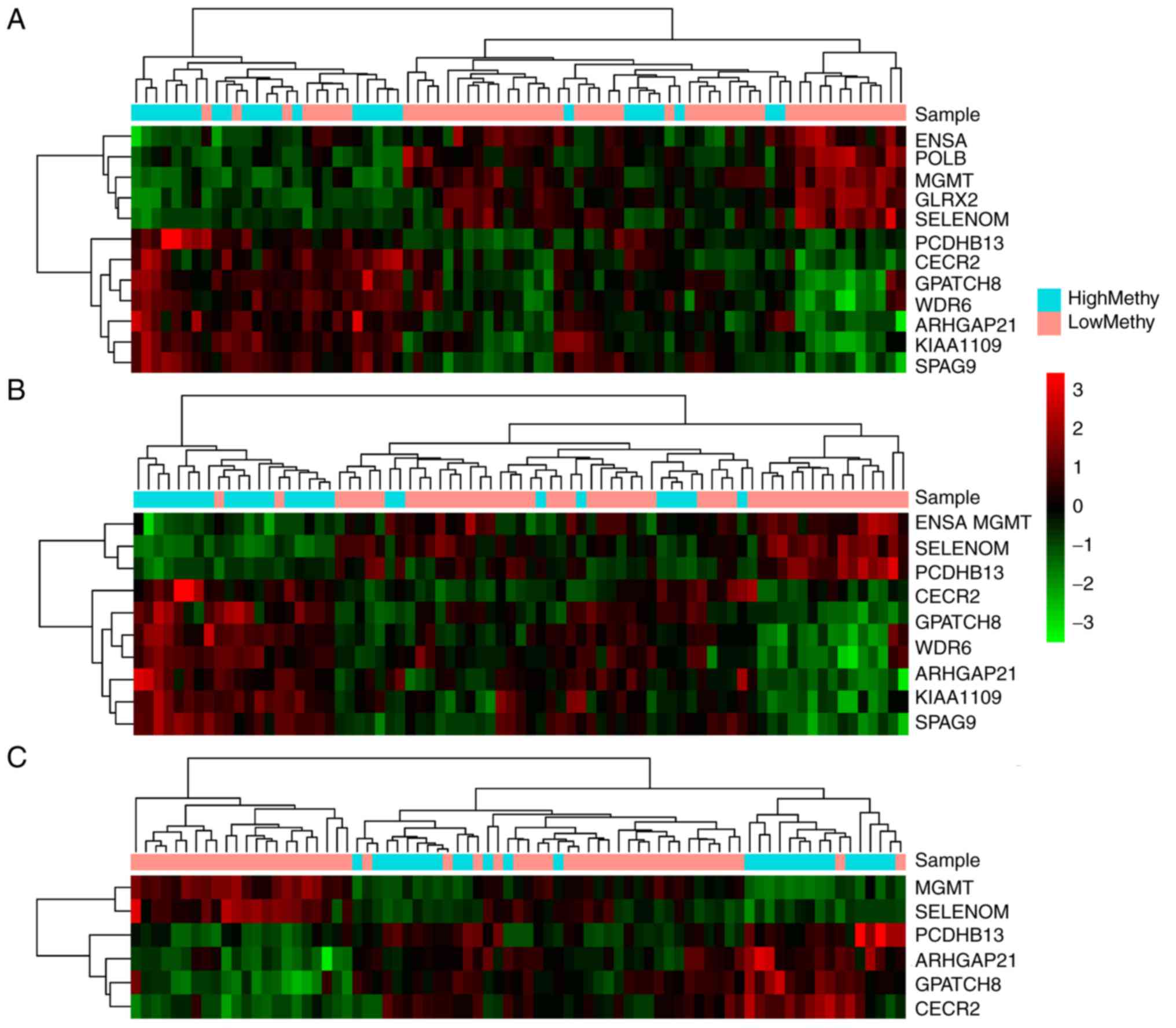
Based on the three cut-off values, three feature
gene sets were selected. Subsequently, unsupervised hierarchical
clustering analysis was performed for the feature gene sets, which
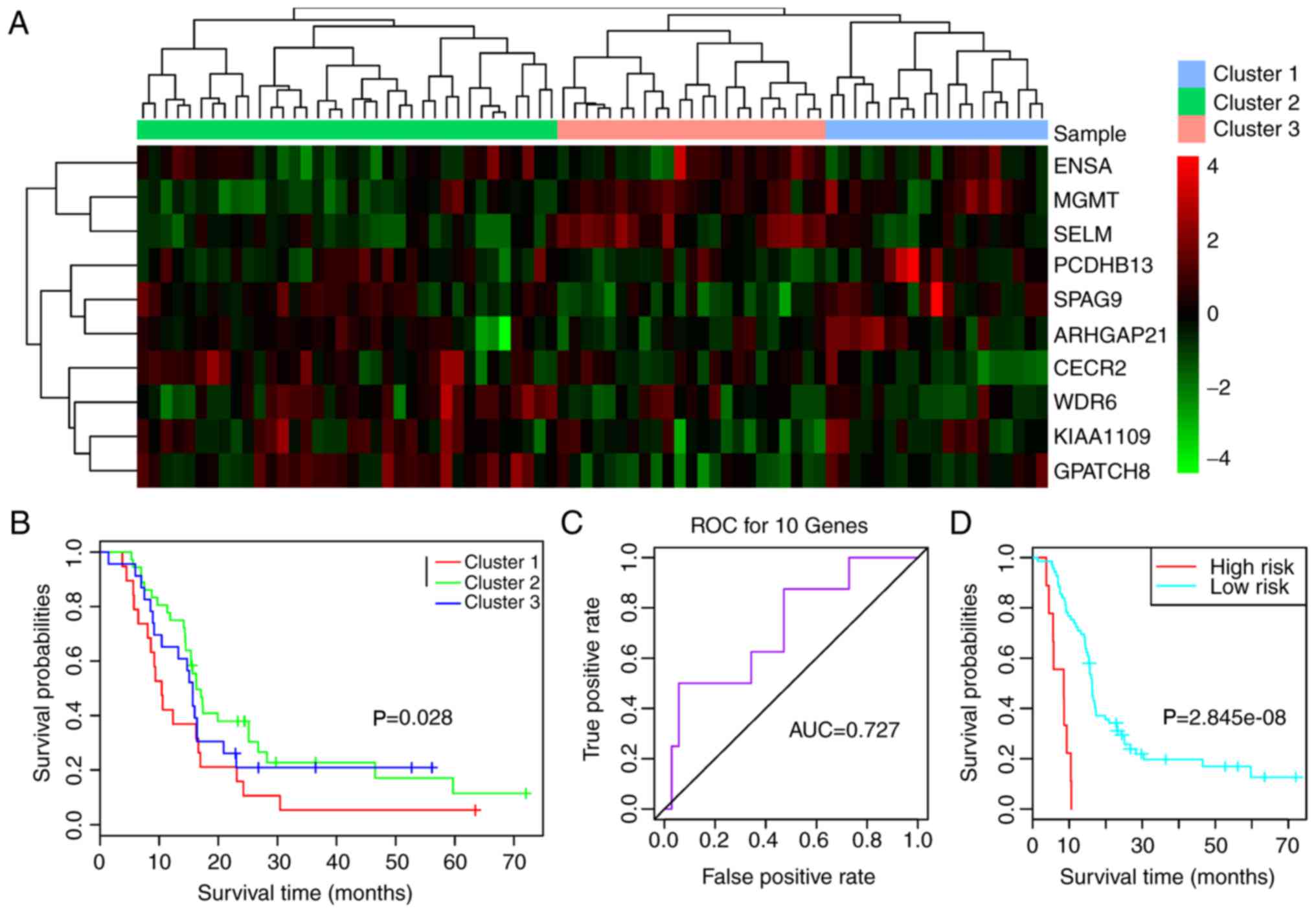
corresponded to the cut-off values of 0.55 (Fig. 3A), 0.56 (Fig. 3B) and 0.57 (Fig. 3C). As shown in Fig. 3A, 11 genes corresponding to cut-off
values of 0.55 had high distinction degrees for the high and low
methylation groups. Meanwhile, there were 10 genes corresponding to
cut-off values of 0.56 (Fig. 3B),
which were able to clearly distinguish between the high and low
methylation groups. However, the six genes corresponding to cut-off
values of 0.57 (Fig. 3C) could not
efficiently differentiate between the two groups.
According to the clustering results, the samples
were divided into two clusters: High methylation and low
methylation. Survival analysis for the clustering results revealed
that only the 10 genes corresponding to cut-off values of 0.56
exhibited significant correlation with prognosis (Fig. 4B, P=0.0074). Therefore, the 10 genes
were selected as feature genes [Rho GTPase-activating protein 21
(ARHGAP21), CECR2, histone acetyl-lysine reader
(CECR2), endosulfine α (ENSA), G patch
domain-containing 8 (GPATCH8), KIAA1109, MGMT,
protocadherin β 13 (PCDHB13), selenoprotein M (SELM)
sperm-associated antigen 9 (SPAG9) and WD repeat domain 6
(WDR6)]. Subsequently, the differential expression of the 10
feature genes in high and low methylation samples was analyzed. As
a result, all of the 10 feature genes exhibited significant
differential expression between the high and low methylation
samples (Fig. 5A-J).
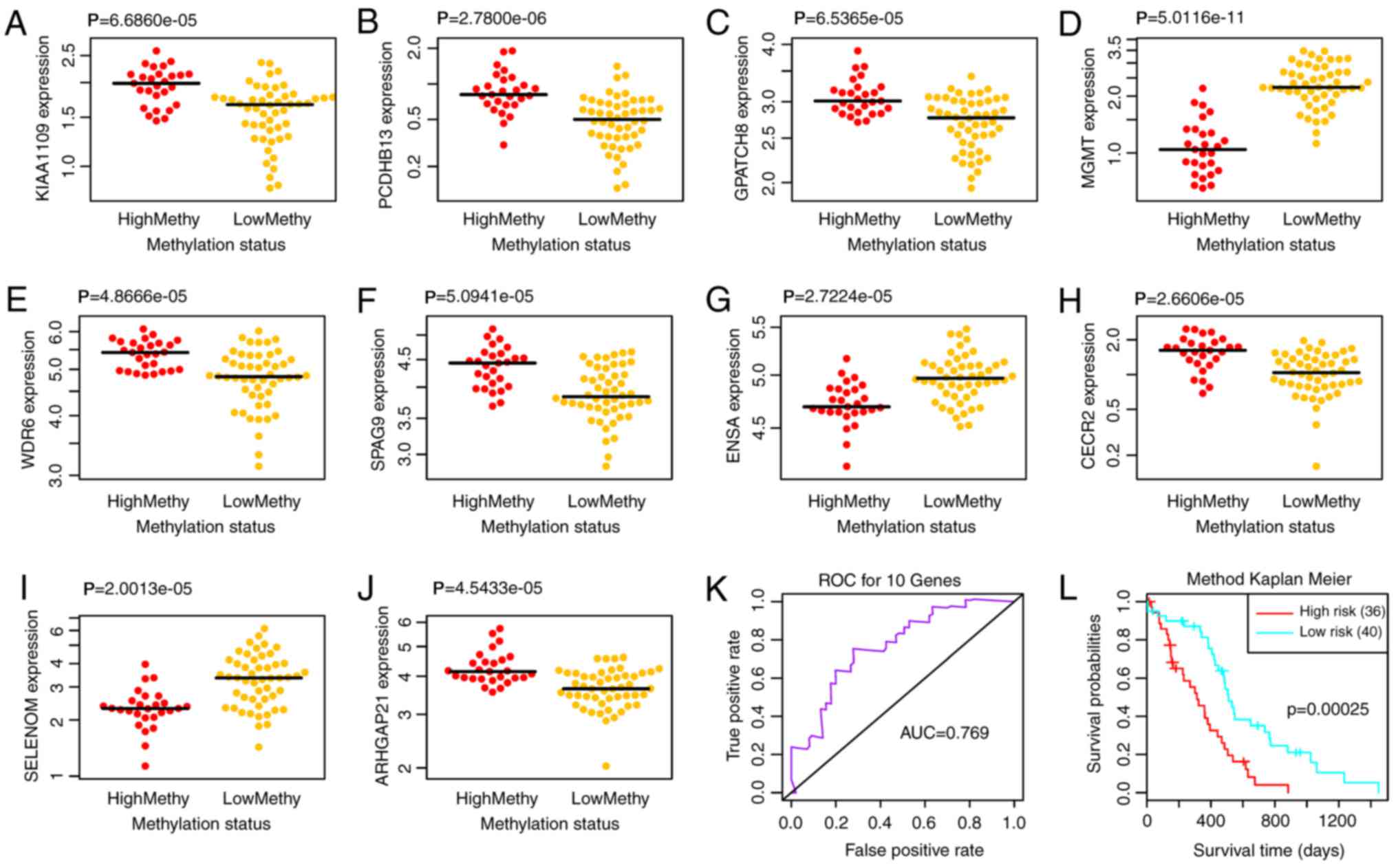 | Figure 5.Differential expression of (A)
KIAA1109, (B) PCDHB13, (C) GPATCH8, (D)
MGMT, (E) WDR6, (F) SPAG9, (G) ENSA,
(H) CECR2, (I) SELM and (J) ARHGAP21 in high
and low methylation samples. (K) ROC curve and (L) prognostic
differences of the two risk groups. AUC, area under the curve; ROC,
receiver operating characteristic. |
Multivariate survival analysis
To examine the effects of the 10 feature genes on
prognosis, a multivariate survival analysis was conducted. Based on
the feature genes, the samples were classified into high and low
risk groups. The area under the ROC curve (AUC) was 0.769 (Fig. 5K) and the two risk groups had a
significant difference in survival probability (Fig. 5L; P=0.0003), thus indicating that
the 10 feature genes could effectively perform classification and
prognostic prediction for the samples.
Validation of the feature genes using
independent datasets
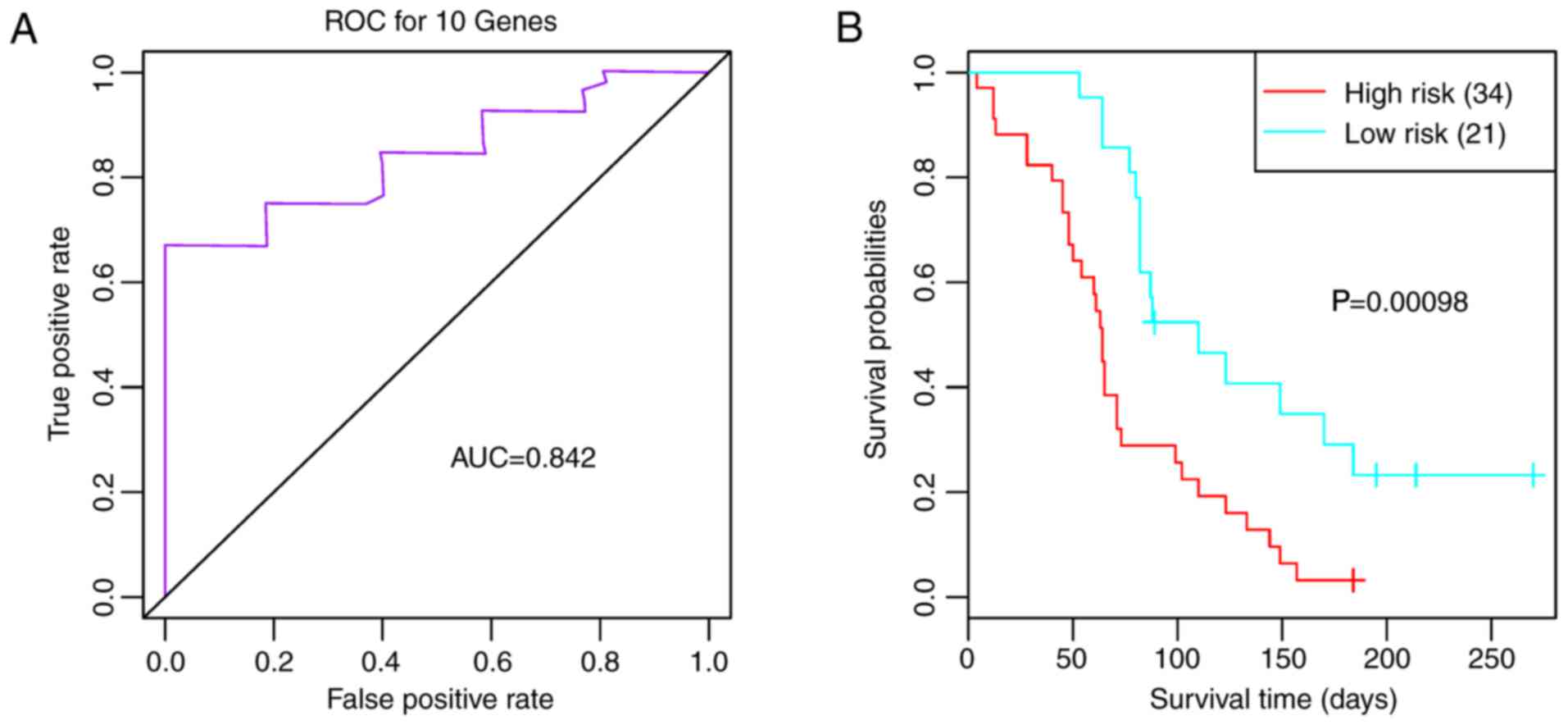
The validation datasets GSE7696 and GSE42669 were
downloaded to confirm that the feature genes were repeatable.
Combined with the 10 feature genes, survival analysis was performed
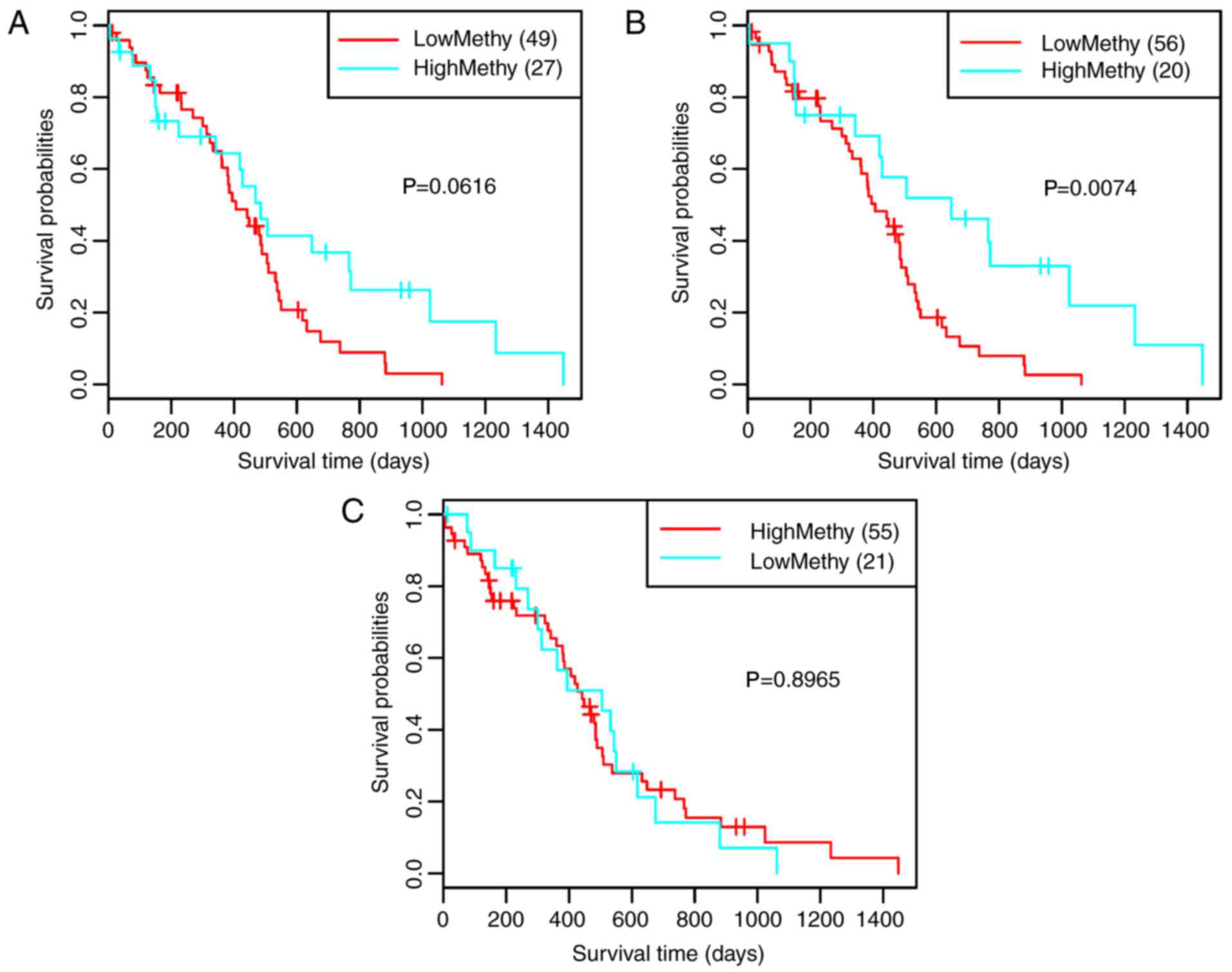
for GSE7696. As shown in Fig. 6A,
the 10 feature genes divided the samples into three clusters
(clusters 1, 2 and 3) (Fig. 6A).
Survival analysis for the three clusters suggested that prognosis
was significantly different between clusters 1 and 2 (P=0.0280;
Fig. 6B). The AUC was 0.727
(Fig. 6C) and the two risk groups
had a significant difference (Fig.
6D; P<0.0001), suggesting that the 10 feature genes had good
classification and prognostic effects for the samples. For the
validation dataset GSE42669, the AUC was 0.842 (Fig. 7A), and the high and low risk groups
had a significant difference (Fig.
7B; P=0.0010). These findings indicated that the 10 feature
genes were key genes that may affect the prognosis of glioma.
Discussion
In the present study, 77 samples were divided into
high and low methylation groups. Subsequently, 11, 10 and six genes
were revealed to have high distinction degrees for the high and low
methylation groups. However, the results of a survival analysis
revealed that only the gene set containing 10 genes had a
significant result; therefore, the 10 genes were selected as
feature genes (including ARHGAP21, CECR2, PCDHB13, MGMT, SELM,
SPAG9 and WDR6). Multivariate survival analysis
indicated that the 10 feature genes were effective in performing
sample classification and prognostic prediction. Furthermore, the
10 feature genes were confirmed by the validation datasets GSE7696
and GSE42669.
ARHGAP21 is considered a potential tumor
suppressor gene that may act in mediating the migration of various
glial tumor types (25). Chromatin
remodeling complexes are important for development, and the
transcription factor CECR2 is associated with neurulation
and is a composition of the chromatin remodeling complex
CECR2-containing remodeling factor (26). Protocadherins (PCDHs) belong to the
cadherin superfamily, and are transmembrane proteins that affect
brain development and are associated with some neuronal diseases
(27). PCDH dysregulation has been
detected in numerous malignant tumors, and their downregulation or
absence is correlated with tumor progression (28,29).
These results suggested that ARHGAP21, CECR2 and
PCDHB13 may be implicated in the development and progression
of glioma.
Glioma stem-like cells (GSCs) are associated with
the recurrence and chemoresistance of glioma, and MGMT
overexpression can promote the resistance of GSCs to temozolomide
(30,31). Selenoproteins (SELs) are highly
expressed in astrocytes prior to brain injury, and their impaired
biosynthesis can lead to neurological dysfunction (32). SELs can resist oxidative stress
through neutralizing reactive oxygen species (ROS), and some SELs
are highly expressed in the brain and are necessary for brain
development (33). As a
selenium-rich plasma protein, selenoprotein P serves an
antioxidative role in selenium-deficient astrocytes (34,35).
The role of SELM in the pathogenesis of glioma has not been
reported; however, the Sec-to-Cys mutant SELM is capable of
binding transition metal ions and modulating
Zn2+-mediated Aβ aggregation, ROS production and
neurotoxicity (36). Therefore,
MGMT and SELM may serve roles in the pathogenesis of
glioma.
SPAG9 is overexpressed in astrocytomas and
can act as a critical oncoprotein by mediating cell proliferation
and invasion (37). SPAG9
contributes to the invasion of astrocytoma cells through improving
the expression of podocalyxin-like via a c-Jun N-terminal
kinase-dependent mechanism (38,39).
WDR1 overexpression is an independent predictor of
unfavorable prognosis for the patients with primary glioblastoma,
indicating that WDR1 is a promising prognostic marker and a
candidate therapeutic target for the disease (40). WDR6 is implicated in the cell
growth inhibitory pathway of threonine kinase 11 via regulation of
p27 (Kip1) (41). These findings
indicated that SPAG9 and WDR6 may be associated with
the prognosis of patients with glioma. In the present study, the
expression levels of SPAG9 and WDR6 were reduced in
the low methylation group, thus indicating that the mechanism
regulating the expression of these genes is not limited to
MGMT promoter methylation; therefore, the complexity of
these gene regulatory mechanisms should be considered.
At present, the role of epigenetic mechanisms in
carcinogenesis is well documented; CpG island hypomethylation
promotes the transcriptional activation of oncogenes and induces
chromosomal instability, whereas hypermethylation silences tumor
suppressor genes (42,43). Etcheverry et al (44) used array technology for quantitative
expression and methylation profiling in a well-characterized cohort
of patients with glioblastoma. This previous study identified
frequent tumor-specific methylation alterations in glioblastoma,
some of which directly affected gene expression. In the present
study, the 10 feature genes were selected based on MGMT
promoter methylation, thus suggesting that associations may exist
between MGMT promoter methylation and these genes; however,
to the best of our knowledge, there is currently no report
regarding these relationships. Given the role of DNA methylation in
gene expression, it was hypothesized that MGMT promoter
methylation may influence the survival of patients with glioma by
regulating the expression levels of these genes; however, this
requires further investigation.
There are numerous limitations to the present study.
The present results were acquired using bioinformatics analyses,
not experimental research. In addition, data heterogeneities and
platform differences among the datasets may affect the accuracy of
the results. In the future, comprehensive experiments should be
designed and performed to validate these findings.
In conclusion, 10 feature genes (ARHGAP21, CECR2,
ENSA, GPATCH8, KIAA1109, MGMT, PCDHB13, SELM, SPAG9 and
WDR6) that corresponded to a cut-off value of 0.56 were
selected as the optimal gene combination in the present study. The
10-gene combination may be mediated by MGMT promoter
methylation and could be associated with the prognosis of patients
with glioma.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ performed data analyses and wrote the manuscript.
JZ conceived and designed the study. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mamelak AN and Jacoby DB: Targeted
delivery of antitumoral therapy to glioma and other malignancies
with synthetic chlorotoxin (TM-601). Expert Opin Drug Deliv.
4:175–186. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ostrom QT, Gittleman H, Fulop J, Liu M,
Blanda R, Kromer C, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: Primary brain and central nervous system
tumors diagnosed in the United States in 2008–2012. Neuro Oncol. 17
Suppl 4:iv1–iv62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wen PY and Reardon DA: Neuro-oncology in
2015: Progress in glioma diagnosis, classification and treatment.
Nat Rev Neurol. 12:69–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McGuire S: World cancer report 2014.
Geneva, Switzerland: World health organization, international
agency for research on cancer, WHO Press, 2015. Adv Nutr.
7:418–419. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weller M, Stupp R, Reifenberger G, Brandes
AA, van den Bent MJ, Wick W and Hegi ME: MGMT promoter
methylation in malignant gliomas: Ready for personalized medicine?
Nat Rev Neurol. 6:39–51. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wick W, Weller M, van den Bent M, Sanson
M, Weiler M, von Deimling A, Plass C, Hegi M, Platten M and
Reifenberger G: MGMT testing - the challenges for
biomarker-based glioma treatment. Nat Rev Neurol. 10:372–385. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
O'Hagan HM, Mohammad HP and Baylin SB:
Double strand breaks can initiate gene silencing and
SIRT1-dependent onset of DNA methylation in an exogenous promoter
CpG island. PLoS Genet. 4:e10001552008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang J, Chen L, Han L, Shi Z, Zhang J, Pu
P and Kang C: EZH2 is a negative prognostic factor and exhibits
pro-oncogenic activity in glioblastoma. Cancer Lett. 356:929–936.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ku BM, Lee YK, Ryu J, Jeong JY, Choi J,
Eun KM, Shin HY, Kim DG, Hwang EM, Yoo JC, et al: CHI3L1 (YKL-40)
is expressed in human gliomas and regulates the invasion, growth
and survival of glioma cells. Int J Cancer. 128:1316–1326. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Steponaitis G, Skiriutė D, Kazlauskas A,
Golubickaitė I, Stakaitis R, Tamašauskas A and Vaitkienė P: High
CHI3L1 expression is associated with glioma patient
survival. Diagn Pathol. 11:422016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ji T, Liu D, Shao W, Yang W, Wu H and Bian
X: Decreased expression of LATS1 is correlated with the progression
and prognosis of glioma. J Exp Clin Cancer Res. 31:672012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Goidts V, Bageritz J, Puccio L, Nakata S,
Zapatka M, Barbus S, Toedt G, Campos B, Korshunov A, Momma S, et
al: RNAi screening in glioma stem-like cells identifies PFKFB4 as a
key molecule important for cancer cell survival. Oncogene.
31:3235–3243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li PCH: Overview of microarray technology.
Microarray Technology: Methods and Applications. Li PCH, Sedighi A
and Wang L: 1368. 1st. Humana Press, Inc.; Totowa, NJ: pp. 3–4.
2016, View Article : Google Scholar
|
|
14
|
Naidu CN and Suneetha Y: Review article:
Current knowledge on microarray technology-an overview. Trop J
Pharm Res. 11:1532012. View Article : Google Scholar
|
|
15
|
Teschendorff AE, Marabita F, Lechner M,
Bartlett T, Tegner J, Gomez-Cabrero D and Beck S: A beta-mixture
quantile normalization method for correcting probe design bias in
Illumina Infinium 450 k DNA methylation data. Bioinformatics.
29:189–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brandes AA, Franceschi E, Paccapelo A,
Tallini G, De Biase D, Ghimenton C, Danieli D, Zunarelli E, Lanza
G, Silini EM, et al: Role of MGMT methylation status at time
of diagnosis and recurrence for patients with glioblastoma:
Clinical implications. Oncologist. 22:432–437. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pesarin F and Salmaso L: The permutation
testing approach: A review. Statistica. 70:481–509. 2013.
|
|
18
|
Wiens TS, Dale BC, Boyce MS and Kershaw
GP: Three way k-fold cross-validation of resource selection
functions. Ecological Modelling. 212:244–255. 2008. View Article : Google Scholar
|
|
19
|
Lacny S, Wilson T, Clement F, Roberts DJ,
Faris PD, Ghali WA and Marshall DA: Kaplan-meier survival analysis
overestimates the risk of revision arthroplasty: A meta-analysis.
Clin Orthop Relat Res. 473:3443–3445. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Perolat J, Couso I, Loquin K and Strauss
O: Generalizing the Wilcoxon rank-sum test for interval data. Int J
Approximate Reasoning. 56:108–121. 2015. View Article : Google Scholar
|
|
21
|
Heagerty PJ, Lumley T and Pepe MS:
Time-dependent ROC curves for censored survival data and a
diagnostic marker. Biometrics. 56:337–344. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lambiv WL, Vassallo I, Delorenzi M, Shay
T, Diserens AC, Misra A, Feuerstein B, Murat A, Migliavacca E,
Hamou MF, et al: The Wnt inhibitory factor 1 (WIF1) is targeted in
glioblastoma and has a tumor suppressing function potentially by
induction of senescence. Neuro Oncol. 13:736–747. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Murat A, Migliavacca E, Gorlia T, Lambiv
WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven
MC, et al: Stem cell-related ‘self-renewal’ signature and high
epidermal growth factor receptor expression associated with
resistance to concomitant chemoradiotherapy in glioblastoma. J Clin
Oncol. 26:3015–3024. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mizukami A, Matsue Y, Naruse Y, Kowase S,
Kurosaki K, Suzuki M, Matsumura A, Nogami A, Aonuma K and Hashimoto
Y: Kaplan-Meier survival analysis and Cox regression analyses
regarding right ventricular septal pacing: Data from Japanese
pacemaker cohort. Data Brief. 8:1303–1307. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bigarella CL, Borges L, Costa FF and Saad
ST: ARHGAP21 modulates FAK activity and impairs glioblastoma cell
migration. Biochim Biophys Acta. 1793:806–816. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Banting GS, Barak O, Ames TM, Burnham AC,
Kardel MD, Cooch NS, Davidson CE, Godbout R, McDermid HE and
Shiekhattar R: CECR2, a protein involved in neurulation, forms a
novel chromatin remodeling complex with SNF2L. Hum Mol Genet.
14:513–524. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hirabayashi T and Yagi T: Protocadherins
in neurological diseases. Adv Neurobiol. 8:293–314. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shan M, Su Y, Kang W, Gao R, Li X and
Zhang G: Aberrant expression and functions of protocadherins in
human malignant tumors. Tumor Biol. 37:12969–12981. 2016.
View Article : Google Scholar
|
|
29
|
Sui X, Wang D, Geng S, Zhou G, He C and Hu
X: Methylated promoters of genes encoding protocadherins as a new
cancer biomarker family. Mol Biol Rep. 39:1105–1111. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Villalva C, Cortes U, Wager M, Tourani JM,
Rivet P, Marquant C, Martin S, Turhan AG and Karayan-Tapon L:
O6-Methylguanine-methyltransferase (MGMT) promoter methylation
status in glioma stem-like cells is correlated to temozolomide
sensitivity under differentiation-promoting conditions. Int J Mol
Sci. 13:6983–6994. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fiano V, Trevisan M, Trevisan E, Senetta
R, Castiglione A, Sacerdote C, Gillio-Tos A, De Marco L, Grasso C,
Magistrello M, et al: MGMT promoter methylation in plasma of glioma
patients receiving temozolomide. J Neurooncol. 117:347–357. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Steinbrenner H and Sies H: Selenium
homeostasis and antioxidant selenoproteins in brain: Implications
for disorders in the central nervous system. Arch Biochem Biophys.
536:152–157. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pitts MW, Byrns CN, Ogawa-Wong AN, Kremer
P and Berry MJ: Selenoproteins in nervous system development and
function. Biol Trace Elem Res. 161:231–245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Steinbrenner H, Alili L, Bilgic E, Sies H
and Brenneisen P: Involvement of selenoprotein P in protection of
human astrocytes from oxidative damage. Free Radic Biol Med.
40:1513–1523. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang X, Hill KE, Maguire MJ and Burk RF:
Synthesis and secretion of selenoprotein P by cultured rat
astrocytes. Biochim Biophys Acta. 1474:390–396. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Du X, Li H, Wang Z, Qiu S, Liu Q and Ni J:
Selenoprotein P and selenoprotein M block Zn2+ -mediated
Aβ42 aggregation and toxicity. Metallomics. 5:861–870.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yi F, Ni W, Liu W, Pan X, Han X, Yang L,
Kong X, Ma R and Chang R: SPAG9 is overexpressed in human
astrocytoma and promotes cell proliferation and invasion. Tumour
Biol. 34:2849–2855. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jiang J, Liu Y, Fang W and Liu F:
Sperm-associated antigen 9 promotes astrocytoma cell invasion
through the upregulation of podocalyxin. Mol Med Rep. 10:417–422.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ando K, Uemura K, Kuzuya A, Maesako M,
Asada-Utsugi M, Kubota M, Aoyagi N, Yoshioka K, Okawa K, Inoue H,
et al: N-cadherin regulates p38 MAPK signaling via association with
JNK-associated leucine zipper protein: Implications for
neurodegeneration in Alzheimer disease. J Biol Chem. 286:7619–7628.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu H, Chen Y, Tan C, Xu T, Yan Y, Qin R,
Huang Q, Lu C, Liang C, Lu Y, et al: High expression of WDR1 in
primary glioblastoma is associated with poor prognosis. Am J Transl
Res. 8:1253–1264. 2016.PubMed/NCBI
|
|
41
|
Xie X, Wang Z and Chen Y: Association of
LKB1 with a WD-repeat protein WDR6 is implicated in cell growth
arrest and p27Kip1 induction. Mol Cell Biochem.
301:115–122. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome-biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Karpf AR and Matsui S: Genetic disruption
of cytosine DNA methyltransferase enzymes induces chromosomal
instability in human cancer cells. Cancer Res. 65:8635–8639. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Etcheverry A, Aubry M, de Tayrac M,
Vauleon E, Boniface R, Guenot F, Saikali S, Hamlat A, Riffaud L,
Menei P, et al: DNA methylation in glioblastoma: Impact on gene
expression and clinical outcome. BMC Genomics. 11:7012010.
View Article : Google Scholar : PubMed/NCBI
|