Introduction
The lungs are the leading site of cancer in males.
Notably, lung cancer contributes to 17% of new cancer cases and 23%
of cancer fatalities (1). Due to
increases in cigarette smoking and environmental pollution, novel
cases and lung cancer-associated mortalities have increased in
China (2). Surgery and chemotherapy
are two major treatment strategies for patients with lung cancer,
and are performed according to their pathological type (3). However, advancements in therapy have
led to improvements in overall survival, and more lung
cancer-associated fatalities are ascribed to distant metastases
rather than the primary tumor (4).
The treatment of metastatic lung cancer has represented a challenge
for clinicians and researchers. Unfortunately, the molecular
mechanisms underlying lung cancer metastases remain poorly
understood. This knowledge gap prevents the development of a
potential marker and therapeutic target for lung cancer prediction
and treatment, respectively.
Chemokines are classified into four highly conserved
groups according to the mutual arrangements of cysteine residues
and disulfide bridges: CXC, CC, C and CX3C (5). As the name suggests, the CX3C subgroup
contains conserved cysteine residues at positions 8, 12, 34 and 50
in humans and other species (6).
C-X3-C motif chemokine ligand 1 (CX3CL1), the only member of the
CX3C subgroup, is a transmembrane protein, containing a mucin-like
stalk with a chemokine domain on the top and a short intracellular
C terminus on the bottom (7,8).
Since the stalk in this setting appears to serve as
a domain extender, the chemokine domain assumes the CX3CL1-C-X3-C
motif chemokine receptor 1 (CX3CR1) interaction. There are two
forms of CX3CL1, the membrane-attached form and the shed form.
Multiple cleavage sites exist in the CX3CL1 stalk structure, which
results in multiple distinct shed forms of this chemokine (9). Furthermore, the various shed forms of
CX3CL1 may be associated with the specific cell types within the
different tissues (10).
As indicated by various studies, the CX3CL1-CX3CR1
interaction is involved in various clinical diseases, including
cancer (11,12). However, reports on the clinical role
of CX3CL1 in tumors are contradictory because CX3CL1 exerts
pro-tumor and antitumor effects. The CX3CL1-CX3CR1 interaction has
demonstrated pro-tumor effects in multiple types of cancer,
including breast cancer (12,13),
B-cell lymphoma, colon (14),
ovarian (15), prostate (16), pancreatic (17) and renal cell cancer (18). This discrepancy may be ascribed to
the dual functions of CX3CL1 as a chemoattractant for leukocytes
and an adhesion molecule for tumor cells. However, there are
relatively few studies on the effects of CX3CL1 on lung cancer, let
alone the molecular mechanism involved.
In the present study, it was investigated whether
CX3CR1 was expressed by the lung cancer cell lines. Experiments
were performed to investigate the role of CX3CL1 in the
proliferation and movement of lung cancer cells. Furthermore, the
molecular mechanism was also studied. The findings provide a basis
for further study in this field.
Materials and methods
Cell lines and cell culture
Six human lung cancer cell lines (H1650, H292, H460,
A549, HCC827, SK-MES-1) and one human bronchial epithelial cell
line (BEAS-2B) were purchased from the Type Culture Collection of
the Chinese Academy of Sciences (Shanghai, China). Cell lines were
cultivated in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum (FBS) at 37°C in a
humidified atmosphere (5% CO2/95% air).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) was used to extract total
cellular RNA from the cells. Following this, the total RNA was
converted to cDNA with the RT reagent Kit with gDNA Eraser (Takara
Bio, Inc., Otsu, Japan) according to the manufacturer's
instructions. Subsequently, RT-qPCR was performed using the ABI
7500 Real-Time PCR System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). All primers were purchased from Sangon Biotech
Co. Ltd. (Shanghai, China) and the sequences were as follows:
β-actin, sense 5′-CAACCGCGAGAAGATGACCC-3′ and antisense
5′-GAGGCGTACAGGGATAGCAC-3′; CX3CR1, sense
5′-AGTGTCACCGACATTTACCTCC-3′ and antisense
5′-AAGGCGGTAGTGAATTTGCAC-3. qPCR was performed using SYBR-Green Mix
(Takara Bio, Inc.). The following PCR conditions were used: Initial
denaturation, 1 cycle of 95°C for 30 sec, followed by 40 cycles of
denaturation at 95°C for 5 sec, and annealing and extension at 60°C
for 34 sec. Relative quantification of the aforementioned genes was
determined using the 2−∆∆Cq method with β-actin as an
endogenous control (19).
Immunofluorescence microscopy
Cells were fixed with 4% paraformaldehyde for 10 min
at room temperature. Following this, cells were rinsed three times
with PBS, overlaid with 5% protease-free bovine serum albumin
(Sangon Biotech Co. Ltd.) for 1 h at room temperature, rinsed with
PBS and incubated with primary antibody against CX3CR1 (1:1,000,
cat. no. ab8021, Abcam, Cambridge, MA, USA) at 4°C overnight. Cells
were incubated with fluorescein-conjugated goat anti-rabbit IgG
antibody (1:1,000, cat. no. ab150077, Abcam, Cambridge, MA, USA)
for 1 h at room temperature following washing with PBS.
Subsequently, cells were washed three times with PBS and mounted in
Hoechst solution (cat. no. ab228551; Abcam, Cambridge, MA, USA) for
0.5 h at room temperature. Slides were viewed with a confocal laser
scanning microscope (Leica Microsystems GmbH, Wetzlar,
Germany).
Western blotting
Cells were harvested at 80% confluence using lysis
buffer with phosphatase and protease inhibitor cocktails (Cell
Lysis Buffer; Cell Signaling Technology, Inc., Danvers, MA, USA).
The total protein was detected using BCA methods. Cell lysates
containing equivalent amounts of proteins (30 µg/lane) were
subjected to SDS-PAGE using 10% polyacrylamide gels. Following
this, the proteins were transferred onto polyvinylidene difluoride
membranes (EMD Millipore, Billerica, MA, USA). The membrane was
blocked with 5% skim milk in Tris-buffered saline with Tween-20 for
1 h at room temperature. Monoclonal antibodies recognizing CX3CR1
(cat. no. WH0001524M1; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) and Tubulin (cat. no. AT819; Beyotime Institute of
Biotechnology, Haimen, China) were used to blot the membrane at 4°C
overnight (all 1:1,000). Horseradish peroxidase-conjugated
anti-rabbit or anti-mouse IgG (cat. no. A0208/A0216; Beyotime
Institute of Biotechnology) were used as the secondary antibodies
to detect the primary antibody for 2 h at room temperature (1:500).
The bands were detected using chemiluminescence reagents (Thermo
Fisher Scientific, Inc.). ImageJ 1.51m (National Institutes of
Health, Bethesda, MD, USA) was used to analyze the bands.
Notably, preliminary experiments with 20–200 nM
CX3CL1 treatment (PeproTech, Inc., Rocky Hill, NJ, USA)
demonstrated that 100 nM CX3CL1 produced the strongest effect.
Therefore, for CX3CL1-induced signaling, cells were stimulated with
100 nmol/l of CX3CL1 for 15, 30 and 60 min. For signaling pathway
inhibition, cells were pre-treated with 100 nmol/l of the Src
inhibitor, saracatinib (Selleck Chemicals, Houston, TX, USA), for 1
h prior to stimulation with the same amount of CX3CL1 for 15, 30
and 60 min. Monoclonal antibodies for the unphosphorylated and
phosphorylated forms of Src (phosphorylation of tyrosine 416) and
focal adhesion kinase (FAK) (phosphorylation of tyrosine 576/577)
were purchased from Cell Signaling Technology, Inc., to evaluate
the activation status of the Src/FAK signaling pathway. Anti-GAPDH
(Beyotime Institute of Biotechnology) was used to detect GAPDH,
which served as the internal reference.
Cell counting Kit-8 (CCK-8)
Cells were seeded onto a 96-well plate at
2×103 cells/well. Following 24 h of incubation, the
cells were stimulated with 50 nmol/l of CX3CL1 in the experimental
group. In the control group, the same amount of distilled water was
added to the wells. After 1, 2 and 3 days, CCK-8 (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) was added to the corresponding
wells, and the plate was incubated at 37°C for 2 h. A multi-well
spectrophotometer was used to measure the absorbance at 450 nm.
In vitro migration and invasion
assay
The migration and invasion assays were performed
using 24-well Transwell chambers with 8-µm pores (Corning
Incorporated, Corning, NY, USA); however, the chambers were coated
with Matrigel for the invasion assay.
Briefly, after extensive washing, 3×104
(migration assay) or 6×104 (invasion assay) cells were
suspended in DMEM without FBS and plated on each upper chamber. The
lower chambers were filled with DMEM containing 0.1% bovine serum
albumin, which was supplemented with or without 100 nmol/l of
CX3CL1. For inhibition of the assays, the upper chambers were
treated with 100 nmol/l of saracatinib for 1 h prior to the assays.
Following 24 h of incubation at 37°C, the cells in the upper
chamber of the membrane were removed with a cotton swab, and the
cells on the underside were fixed with paraformaldehyde and stained
with 0.1% crystal violet. The cells were counted in three randomly
selected fields under light microscope (magnification, ×40).
Statistical analysis
Data were expressed as the mean ± standard
deviation. Significant differences were identified using the
one-way analysis of variance with subsequent use of post hoc LSD
tests to differentiate between two groups when needed. P<0.05
was considered to indicate a statistically significant
difference.
Results
Expression of CX3CR1 in the cell
lines
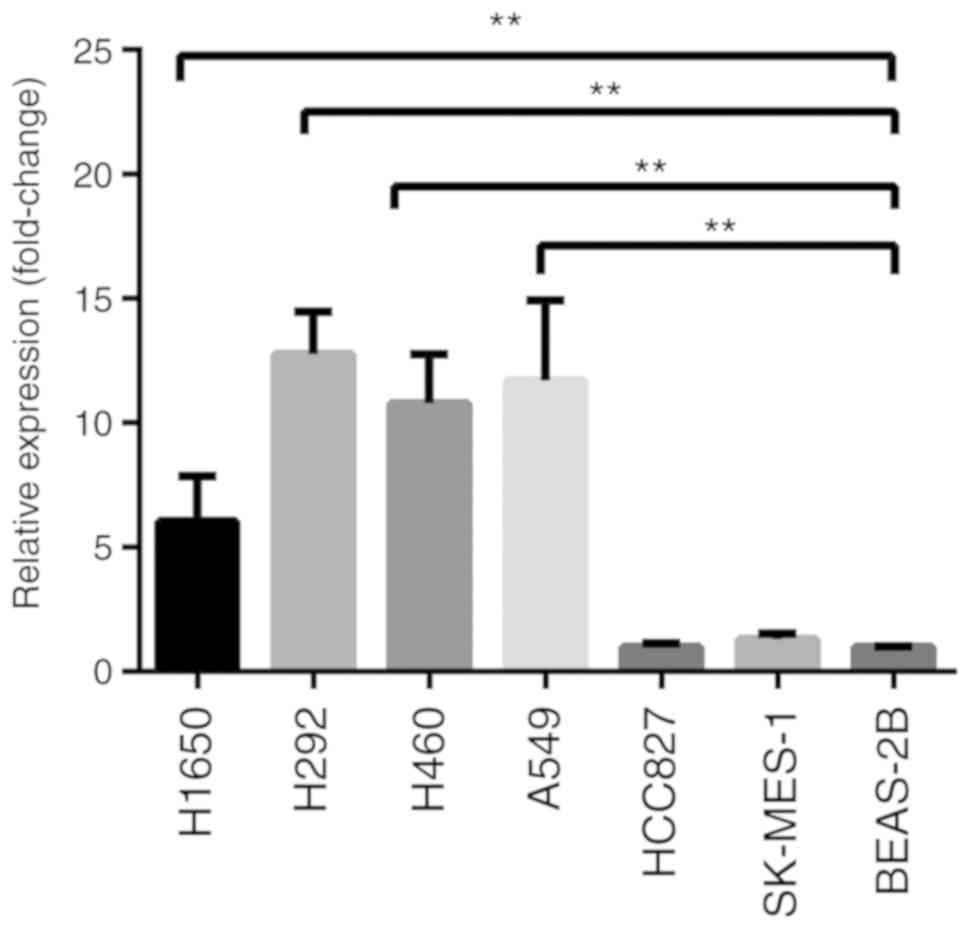
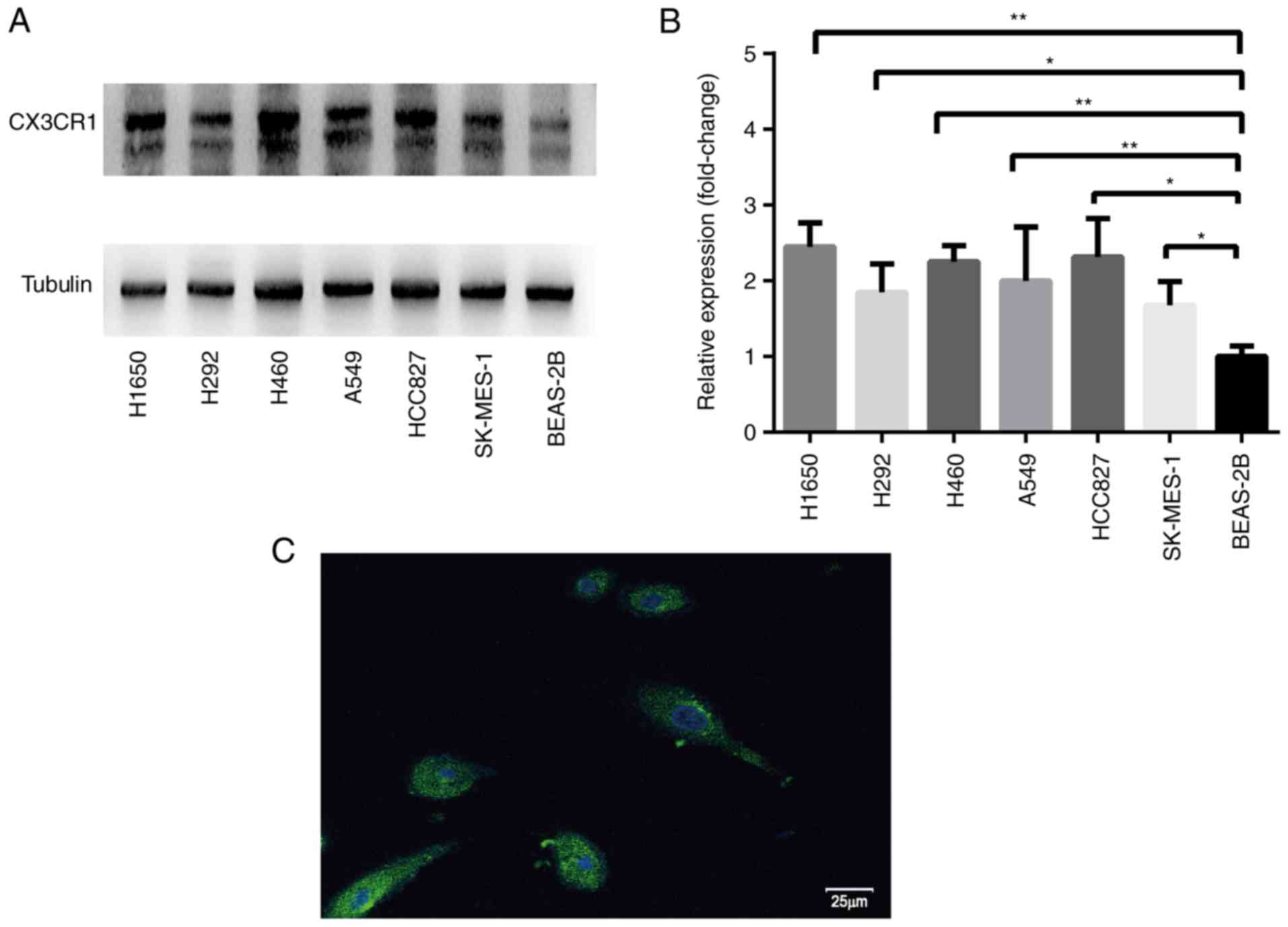
RT-qPCR and western blotting were used to analyze
CX3CR1 mRNA and protein expression levels, respectively. RT-qPCR
results revealed that the CX3CR1 mRNA levels were significantly
increased in the H1650, H292, H460 and A549 cells compared with
BEAS-2B cells (Fig. 1). The protein
expression levels of CX3CR1 were significantly increased in all six
lung cancer cell lines when compared with BEAS-2B (Fig. 2). Notably, the two CX3CR1 bands
exhibited in Fig. 2A can be
attributed to various factors. Firstly, CX3CR1 may have isomers.
Secondly, there was slight degradation of CX3CR1 during detection.
The upper band with the anticipated molecular mass was used to
calculate the expression. Notably, there was a discrepancy between
the expression levels of CX3CR1 mRNA and protein. Based on the data
from both evaluations, H460 was selected for further study.
Furthermore, the cell membrane was not permeated by 0.1% Triton
X-100. Therefore, the signal represented the cell surface CX3CR1
expression (Fig. 2C).
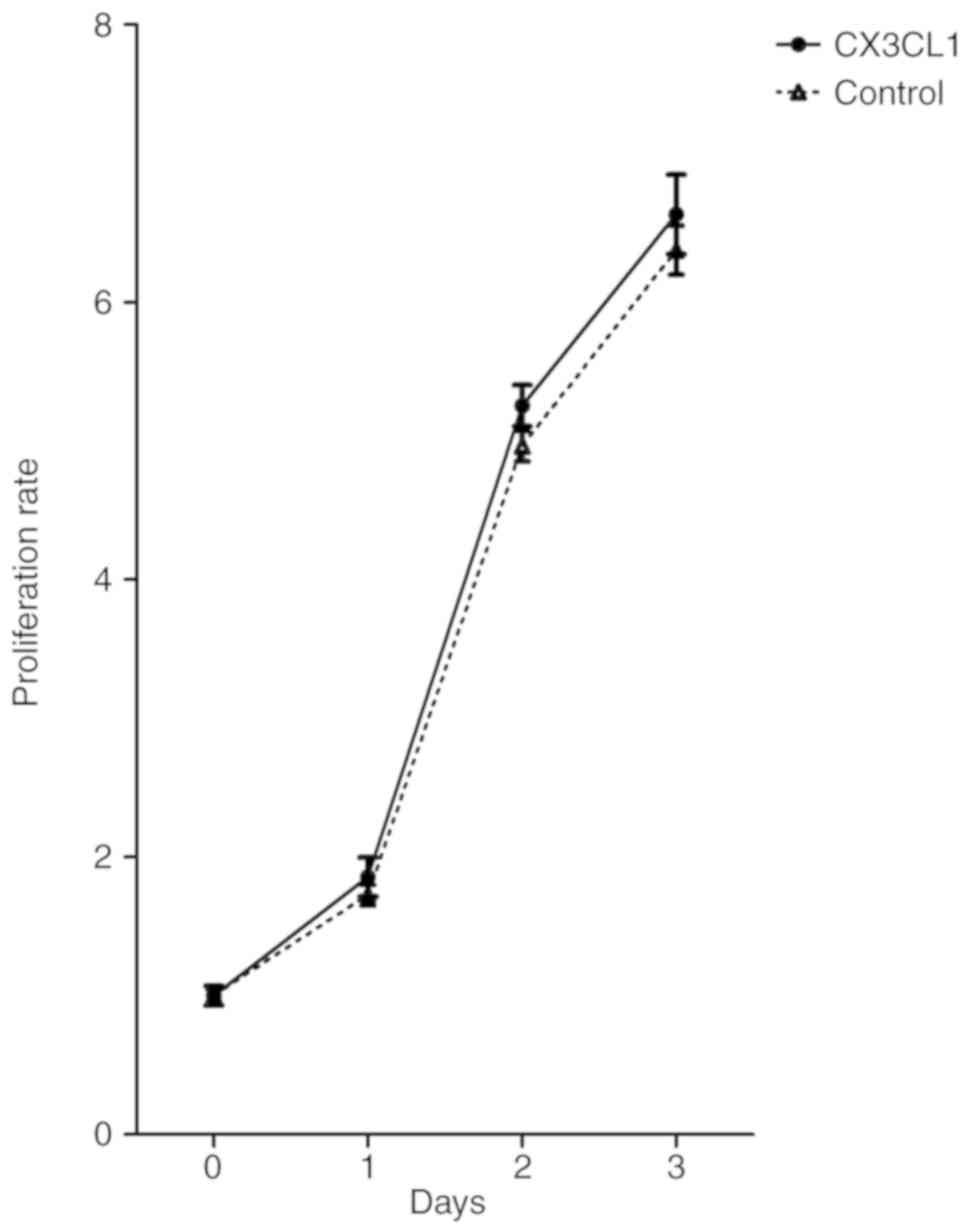
Impact of CX3CL1 on tumor cell
proliferation
To investigate whether CX3CL1 has a direct impact on
tumor cell proliferation, the CCK-8 assay was used to evaluate the
growth of H460 with and without CX3CL1. As indicated in Fig. 3, CX3CL1 did not significantly affect
the proliferative capacity of H460.
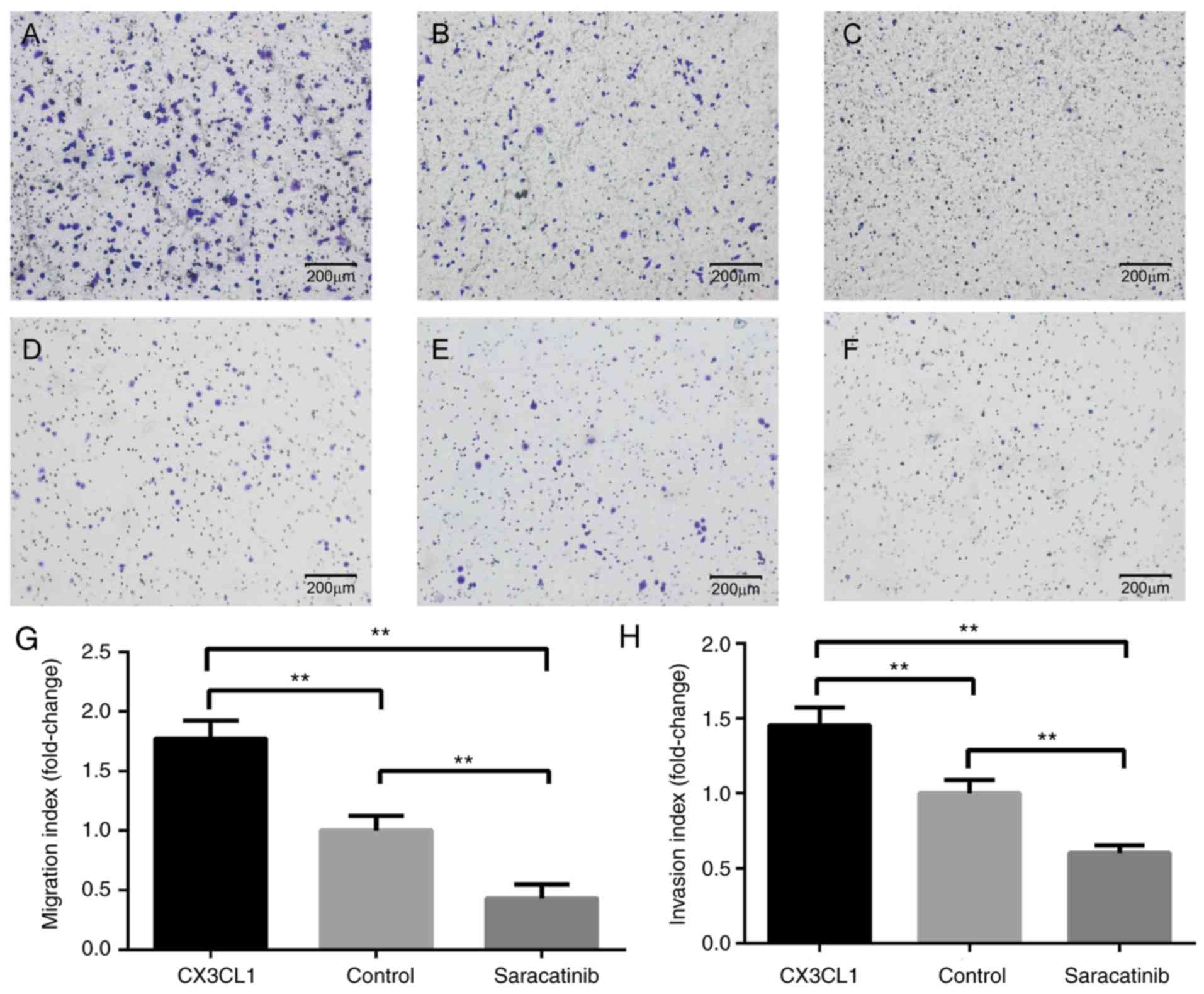
Impact on tumor cell movement
As indicated in Fig.
4, Transwell invasion and migration assays were performed.
Compared with the cells without stimulation of CX3CL1, the
migration and invasion abilities of H460 were significantly
increased following 24 h of exposure to CX3CL1 (Fig. 4A-C and G). However, saracatinib
significantly inhibited the enhanced CX3CL1-induced migration and
invasion abilities of H460 to lower than basal level (Fig. 4D-F and H). Notably, CX3CL1 did not
elicit a significant response in H292 and A549 cells (data not
shown).
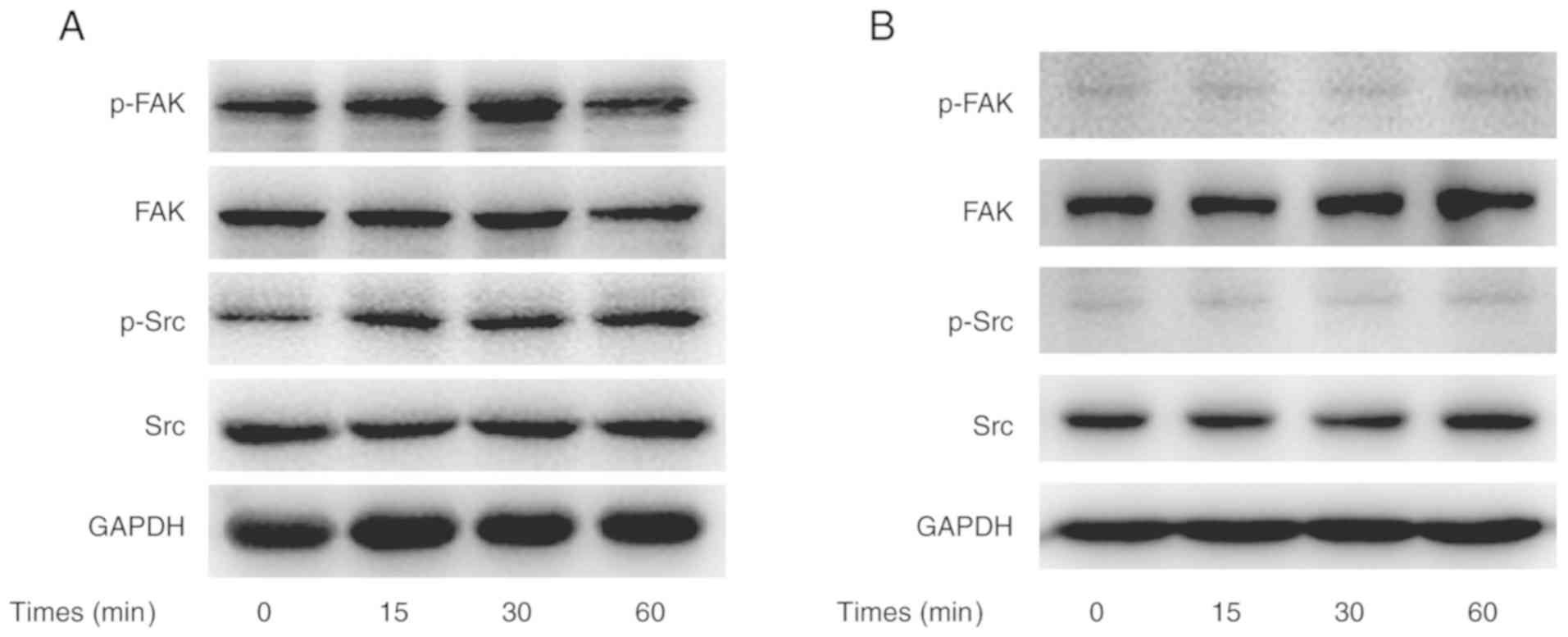
CX3CL1-induced activation of the
Src/FAK pathway
To obtain evidence that CX3CL1 promoted H460 cell
migration and invasion via the Src/FAK signaling pathway, the
phosphorylation of Src and FAK was analyzed in CX3CL1-stimulated
cells. In this experiment, CX3CL1-induced Src phosphorylation
peaked 15 min post-stimulation. In addition, CX3CL1-induced FAK
phosphorylation peaked 30 min post-stimulation (Fig. 5A). However, saracatinib hindered
CX3CL1-induced Src and FAK phosphorylation (Fig. 5B).
Discussion
Although the CX3CL1-CX3CR1 interaction is well known
in the metastatic process of variety of cancer types, the present
study provided insight into the role of CX3CL1 as an enhancer of
metastasis in lung cancer. The present findings provided evidence
that CX3CL1 promotes the chemotaxis ability of lung cancer cells by
binding to CX3CR1 and activating the Src/FAK signaling pathway.
The precursor of CX3CL1 is synthesized as an
intracellular 50–75 kDa protein that is rapidly processed and
transported to the cell surface (20). A soluble 85-kDa fragment of CX3CL1,
containing a 76-amino acid chemokine domain, can be cleaved from
the cell surface under normal growth conditions. However, this
process may be accelerated by stimuli, such as cancer (21). Therefore, under physiological and
pathological conditions, CX3CL1 is allowed to mediate the
chemotaxis and firm capture of CX3CR1-expressing cells via the
soluble and membrane-attached form of CX3CL1, respectively
(22). CX3CR1, a
seven-transmembrane G-protein-coupled receptor, mediates the
activation of the downstream signaling pathway (c-Raf,
mitogen-activated protein kinase kinase, extracellular
signal-regulated kinase and nuclear factor-κB) through its ligand,
CX3CL1 (23).
The present study investigated CX3CR1 expression at
mRNA and protein levels. Although all six lung cancer cell lines
demonstrated overexpression of CX3CR1 at protein levels, some of
the lung cancer cells lines were not upregulated at mRNA levels.
The discrepancy between the mRNA and protein expression levels may
be due to posttranscriptional and posttranslational control
mechanisms. Thus, the amount and frequency of protein synthesis may
not simply coincide with the amount of mRNA (24). Since H460 exhibited higher CX3CR1
expression at mRNA and protein levels, this cell line was selected
for further experiments in the present study.
To study the potential effects of CX3CL1 on lung
cancer cell proliferation, the proliferation rate of H460 with and
without CX3CL1 was assessed using CCK-8 in vitro. No
significant differences were observed between the two groups.
However, Tardaguila et al (25) reported that CX3CL1 contributes to
tumorigenesis in breast cancer. Through proteolytic shedding of an
ErbB ligand, CX3CL1 triggered cell proliferation by transactivating
the ErbB receptors. Thus, the increased tumor multiplicity was a
consequence of CX3CL1 acting as a positive modifier of breast
cancer in concert with ErbB receptors rather than CX3CL1-induced
metastatic dissemination of the primary tumor (25). This inconformity may be associated
with the different molecular mechanisms of tumor heterogeneity.
Based upon the observations from the in vitro
migration and invasion assays in the present study, CX3CL1
significantly promoted the chemotaxis ability of lung cancer cells.
Notably, the results were negative for H292 and A549 cells, which
can be attributed to tumor heterogeneity. Furthermore, the present
study revealed that the molecular mechanisms, following CX3CL1
activation in lung cancer cells, involved the Src/FAK signaling
pathway. Src, a proto-oncogene, is a non-receptor protein-tyrosine
kinase that is a critical regulator of signal transduction induced
by a variety of cell-surface receptors. Src serves a key role in
cell growth, division, migration and survival signaling pathways.
Furthermore, Src activity is regulated by tyrosine phosphorylation
of two chief sites, pTyr416 and pTyr530. Phosphorylation of Tyr416,
located in the activation loop of the kinase domain, by an adjacent
Src molecule results in upregulated enzyme activity. However,
phosphorylation of Tyr530, located in the regulatory tail, by
C-terminal Src kinase or Csk homologous kinase renders the enzyme
less active (26,27). FAK is another subgroup of the
non-receptor protein tyrosine kinases, and it is regulated by
phosphorylation and dephosphorylation. FAK has been identified to
participate, through various pathways, in a diverse spectrum of
receptor-induced biological activities, particularly the activation
of cell spreading and migration through integrin-mediated signal
transduction (28). Specifically,
integrin clustering results in the phosphorylation of Tyr397, which
prepares the binding site for Src family kinases. Following this,
the recruitment of Src family kinases leads to the phosphorylation
of Tyr576/577 in the catalytic domain (29). Therefore, when deregulated, the
Src/FAK complex indicates strong oncogenic activity. Based on the
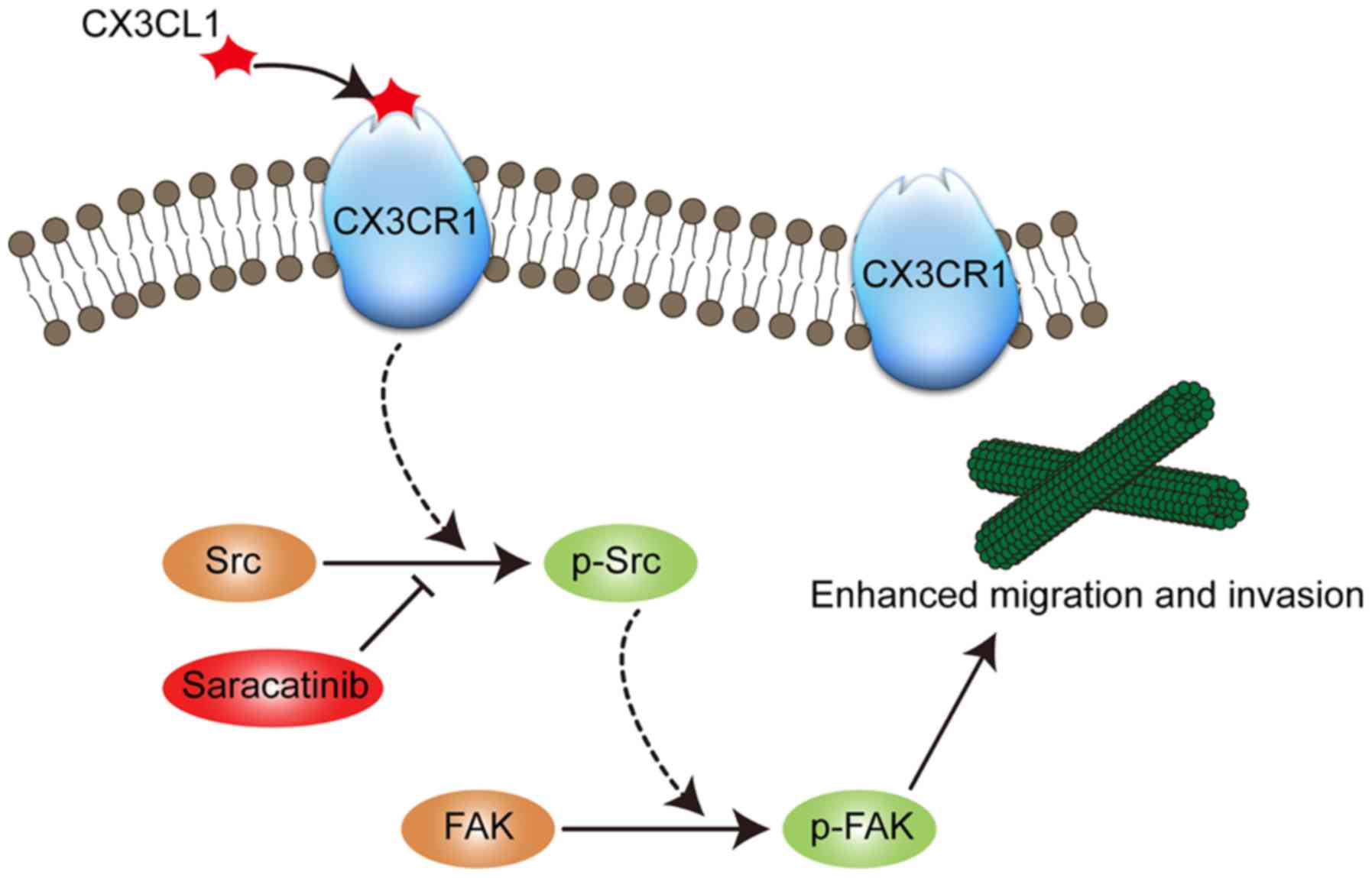
present study, a model was proposed in which CX3CL1-CX3CR1
interaction first phosphorylates Src at Tyr416. Subsequently, the
phosphorylated Src binds with FAK and phosphorylates FAK at
Tyr576/577 (Fig. 6). Saracatinib, a
Src inhibitor, could abrogate the effect of CX3CL1 on chemotaxis.
Saracatinib has also been reported to inhibit cell movement in
prostate cancer (30) and bladder
cancer (31), which is consistent
with the present study. Therefore, saracatinib may translate as a
clinical therapy that inhibits cancer metastasis; however, further
studies are required.
In conclusion, CX3CL1 enhanced the migration and
invasion of lung cancer cells. It was proposed that the Src/FAK
signaling pathway is involved in CX3CL1-CX3CR1 interaction, which
causes enhanced migration and invasion. The present results also
highlight the potent anti-migratory and anti-invasive effects of
saracatinib in vitro.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81572629).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
JD conceived the study and revised the manuscript.
WL and YL performed the experiments and wrote the manuscript. QC
and LJ collected and analyzed the data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Zeng H and Zhang S:
Epidemiology of lung cancer in China. Thorac Cancer. 6:209–215.
2007. View Article : Google Scholar
|
|
3
|
Arriagada R, Bergman B, Dunant A, Le
Chevalier T, Pignon JP and Vansteenkiste J; International Adjuvant
Lung Cancer Trial Collaborative Group, : Cisplatin-based adjuvant
chemotherapy in patients with completely resected non-small-cell
lung cancer. N Engl J Med. 350:351–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang T, Nelson RA, Bogardus A and Grannis
FW Jr: Five-year lung cancer survival: Which advanced stage
nonsmall cell lung cancer patients attain long-term survival?
Cancer. 116:1518–1525. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luster AD: Chemokines-chemotactic
cytokines that mediate inflammation. N Engl J Med. 338:436–445.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Harrison JK, Fong AM, Swain PA, Chen S, Yu
YR, Salafranca MN, Greenleaf WB, Imai T and Patel DD: Mutational
analysis of the fractalkine chemokine domain. Basic amino acid
residues differentially contribute to CX3CR1 binding, signaling,
and cell adhesion. J Biol Chem. 276:21632–21641. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ludwig A and Weber C: Transmembrane
chemokines: Versatile ‘special agents’ in vascular inflammation.
Thromb Haemost. 97:694–703. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bazan JF, Bacon KB, Hardiman G, Wang W,
Soo K, Rossi D, Greaves DR, Zlotnik A and Schall TJ: A new class of
membrane- bound chemokine with a CX3C motif. Nature. 385:640–644.
1997. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haskell CA, Cleary MD and Charo IF: Unique
role of the chemokine domain of fractalkine in cell capture.
Kinetics of receptor dissociation correlate with cell adhesion. J
Biol Chem. 275:34183–34189. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fonovic UP, Jevnikar Z and Kos J:
Cathepsin S generates soluble CX3CL1 (fractalkine) in vascular
smooth muscle cells. Biol Chem. 394:1349–1352. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marchesi F, Locatelli M, Solinas G, Erreni
M, Allavena P and Mantovani A: Role of CX3CR1/CX3CL1 axis in
primary and secondary involvement of the nervous system by cancer.
J Neuroimmunol. 224:39–44. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsang JY, Ni YB, Chan SK, Shao MM, Kwok
YK, Chan KW, Tan PH and Tse GM: CX3CL1 expression is associated
with poor outcome in breast cancer patients. Breast Cancer Res
Treat. 140:495–504. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Andre F, Cabioglu N, Assi H, Sabourin JC,
Delaloge S, Sahin A, Broglio K, Spano JP, Combadiere C, Bucana C,
et al: Expression of chemokine receptors predicts the site of
metastatic relapse in patients with axillary node positive primary
breast cancer. Ann Oncol. 17:945–951. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng J, Yang M, Shao J, Miao Y, Han J and
Du J: Chemokine receptor CX3CR1 contributes to macrophage survival
in tumor metastasis. Mol Cancer. 12:1412013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim M, Rooper L, Xie J, Kajdacsy-Balla AA
and Barbolina MV: Fractalkine receptor CX3CR1 is
expressed in epithelial ovarian carcinoma cells and required for
motility and adhesion to peritoneal mesothelial cells. Mol Cancer
Res. 10:11–24. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shulby SA, Dolloff NG, Stearns ME, Meucci
O and Fatatis A: CX3CR1-fractalkine expression regulates cellular
mechanisms involved in adhesion, migration, and survival of human
prostate cancer cells. Cancer Res. 64:4693–4698. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Celesti G, Di Caro G, Bianchi P, Grizzi F,
Marchesi F, Basso G, Rahal D, Delconte G, Catalano M, Cappello P,
et al: Early expression of the fractalkine receptor CX3CR1 in
pancreatic carcinogenesis. Br J Cancer. 109:2424–2433. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao X, Qi L, Chen X, Du J, Zhang Z and Liu
S: Expression of CX3CR1 associates with cellular migration,
metastasis, and prognosis in human clear cell renal cell carcinoma.
Urol Oncol. 32:162–170. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Garton KJ, Gough PJ, Blobel CP, Murphy G,
Greaves DR, Dempsey PJ and Raines EW: Tumor necrosis
factor-alpha-converting enzyme (ADAM17) mediates the cleavage and
shedding of fractalkine (CX3CL1). J Biol Chem. 276:37993–38001.
2001.PubMed/NCBI
|
|
21
|
Shiraishi K, Fukuda S, Mori T, Matsuda K,
Yamaguchi T, Tanikawa C, Ogawa M, Nakamura Y and Arakawa H:
Identification of fractalkine, a CX3C-type chemokine, as a direct
target of p53. Cancer Res. 60:3722–3726. 2000.PubMed/NCBI
|
|
22
|
Imai T, Hieshima K, Haskell C, Baba M,
Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ,
et al: Identification and molecular characterization of fractalkine
receptor CX3CR1, which mediates both leukocyte migration
and adhesion. Cell. 91:521–530. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hou SM, Hou CH and Liu JF: CX3CL1 promotes
MMP-3 production via the CX3CR1, c-Raf, MEK, ERK, and NF-κB
signaling pathway in osteoarthritis synovial fibroblasts. Arthritis
Res Ther. 19:2822017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ideker T, Thorsson V, Ranish JA, Christmas
R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R and Hood
L: Integrated genomic and proteomic analyses of a systematically
perturbed metabolic network. Science. 292:929–934. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tardaguila M, Mira E, Garcia-Cabezas MA,
Feijoo AM, Quintela-Fandino M, Azcoitia I, Lira SA and Mañes S:
CX3CL1 promotes breast cancer via transactivation of the EGF
pathway. Cancer Res. 73:4461–4473. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thomas SM and Brugge JS: Cellular
functions regulated by Src family kinases. Annu Rev Cell Dev Biol.
13:513–609. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roskoski R Jr: Src protein-tyrosine kinase
structure, mechanism, and small molecule inhibitors. Pharmacol Res.
94:9–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Parsons JT, Martin KH, Slack JK, Taylor JM
and Weed SA: Focal adhesion kinase: A regulator of focal adhesion
dynamics and cell movement. Oncogene. 19:5606–5613. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang X, Chattopadhyay A, Ji QS, Owen JD,
Ruest PJ, Carpenter G and Hanks SK: Focal adhesion kinase promotes
phospholipase C-gamma1 activity. Proc Natl Acad Sci USA.
96:9021–9026. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chang YM, Bai L, Liu S, Yang JC, Kung HJ
and Evans CP: Src family kinase oncogenic potential and pathways in
prostate cancer as revealed by AZD0530. Oncogene. 27:6365–6375.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Green TP, Fennell M, Whittaker R, Curwen
J, Jacobs V, Allen J, Logie A, Hargreaves J, Hickinson DM,
Wilkinson RW, et al: Preclinical anticancer activity of the potent,
oral Src inhibitor AZD0530. Mol Oncol. 3:248–261. 2009. View Article : Google Scholar : PubMed/NCBI
|