Introduction
Hepatocellular carcinoma (HCC) is one of the most
lethal malignancies worldwide due to its aggressive properties and
limited therapeutic options. Thus, one important aim of the present
study was to identify biomarkers for predicting therapeutic
response and prognosis, which may provide a deeper understanding of
the tumor biology (1,2). To date, numerous molecular events,
including those at the epigenetic, pre-transcriptional,
transcriptional, translational and post-translational levels, have
been identified and validated as important prognostic biomarkers
(3), such as the Ki-67 protein and
TP53 gene mutations. However, the functional significance of
cancer-specific alternative splicing (AS) events is largely
unexplored. AS of pre-mRNA enables a gene to produce multiple
protein isoforms, usually with distinct functions. This mechanism
generates transcriptomic and proteomic diversity in different
tissues and cell types (4,5), however, in some disease states, for
example in cancerous conditions, the damaged or diseased cells take
this advantage to produce aberrant protein isoforms that contribute
to tumorigenesis (6).
In the last decade, growing data has indicated that
the dysregulation of AS is a crucial event in carcinogenesis and
tumor progression (3,7–9). Genes
with aberrant AS events are involved in almost every aspect of
cancer, including proliferation, differentiation, cell cycle
control, metabolism, apoptosis, motility, invasion and angiogenesis
(10). Although several studies
have revealed multiple tumor-associated AS variants in a wide
variety of cancers (10–14), there is still much research to be
conducted at the splicing level in HCC. In HCC, various aberrant AS
events were found to promote the generation of oncogenic isoforms,
whereas tumor suppressors were self-inactivated by aberrant AS
(15). As a result, the
identification and validation of isoforms as novel biomarkers at
the splicing level in HCC may be of great significance for
revealing novel drug targets for therapeutic intervention.
In the present study, we analyzed the published
RNA-Seq datasets of three paired HCC and adjacent non-tumor tissues
and summarized the aberrant gene expression and splicing patterns
in HCC samples. We focused on the putative novel biomarkers of
these three aberrant AS events in HCC, and their association with
clinical features and prognosis was further investigated.
Materials and methods
RNA-Seq data download and
processing
The RNA-Seq data of three paired HCC patients were
downloaded from NCBI GEO database with the accession number of
GSE33294 (16). Read alignment was
performed using TopHat2 (17) with
the known reference genes from the GENCODE v22 annotation
(http://www.gencodegenes.org/human/release_22.html).
Gene expression was analyzed using Cufflinks v2.2.1 (http://cole-trapnell-lab.github.io/cufflinks/) and
significantly differential genes were identified by the Cuffdiff
module at the FDR of 5% (18).
Alternative splicing analysis was carried out using the MATS
software v3.0.8 (http://rnaseq-mats.sourceforge.net/) with the
parameters of ‘-t single-len 58 -a 8 -c 0.0001 -analysis U’. Only
the significantly differential events of the SE type were used for
subsequent analysis.
Clinical samples and ethical
statement
Forty-five paired tumor and normal liver tissues
from HCC patients, with an average age of 45 years old, including
28 males and 17 females, were collected from April 2010 to June
2014 at the China-Japan Union Hospital, Jilin University. All
patients consented to the use of their clinical specimens for
medical and biological study. The present study was approved by the
Clinical Research Ethics Committee of China-Japan Union Hospital,
Jilin University (2015-wjw002).
RT-qPCR validation and inclusion level
(IL) calculation
Fresh samples were frozen in liquid nitrogen and
total RNA was extracted using a TRIzol Isolation kit. cDNA was
synthesized using the PrimeScript™ RT Master Mix (Takara Bio, Inc.,
Otsu, Japan). RT-PCR was performed on the Illumina ECO system
(Illumina, Inc., San Diego, CA, USA). ILs of skipped exons in every
clinical sample were estimated by the ΔΔCq approach (19). Each splicing event used two paired
primers, one representing the inclusion isoform expression and the
other representing the total expression. The inclusion ratio was
calculated using the following equation: IL=2-(Inclusion
ΔCT−TotalΔCT). Thus, the
‘InclusionΔCT-TotalΔCT’
was used to denote the IL of each skipped exon.
Association analysis between AS and
clinical features
Clinical features for the association analysis
included age, sex, viral status, cell differentiation, tumor size,
vascular invasion, alpha-fetoprotein (AFP) level, metastasis and
recurrence, all of which were converted to the quantitative data.
The correlation between these quantitative clinical features and
splicing changes was calculated using the maximal information
coefficient (MIC) in the MINE toolkit (20) with the parameter of ‘-allPairs’. The
confidence for various MIC values was evaluated by the P-value
table at the sample size of 45.
Statistical analysis and
visualization
Heatmaps of expression and splicing were plotted by
the ‘pheatmap’ package in R language with the parameter of
‘scale’=‘row’. Three DS genes were visualized using the sashimi
plotting function in IGV software (21). Student's t-test was used in the
differential analysis of RT-PCR values using GraphPad Prism version
5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Fisher's exact
test was applied to the association test of contingency table
analysis. The network of AS and clinical features was visualized
using Cytoscape software v3.3.0 (http://cytoscape.org). Enrichment analysis was
performed using the ‘ClusterProfiler’ package (22) in R language.
Results
Comparison between gene expression and
splicing profiles of HCC
We reanalyzed the RNA-Seq datasets from three
previously published paired HCC tumors and adjacent non-tumor
tissues (16). After reads
alignment and expression quantification, a total of 13,327
protein-coding genes were identified with FPKM≥1. Of these, we
detected 1,246 upregulated genes and 1,197 downregulated genes in
the HCC group at a false discovery rate (FDR) of 5% using the
Cufflinks software (18). The
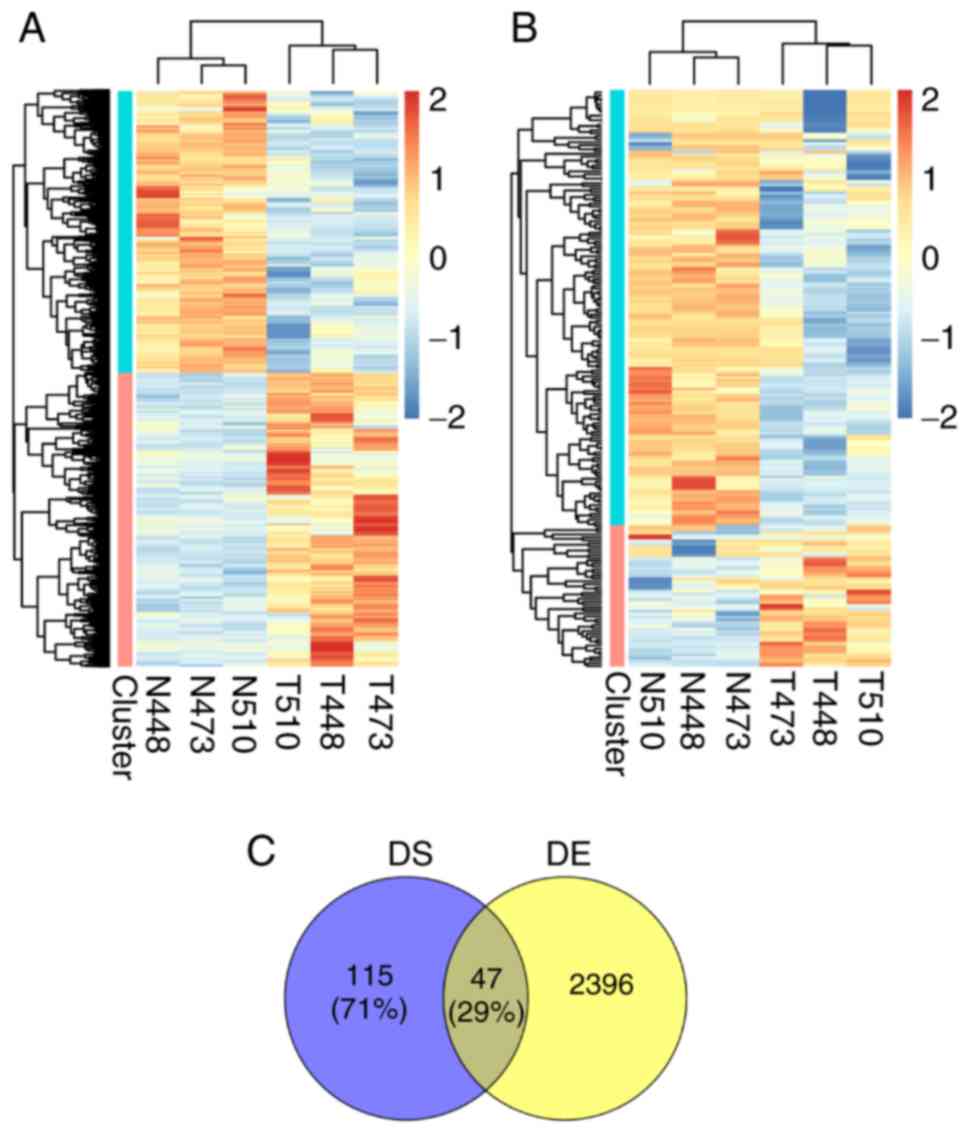
expression pattern for these differentially expressed (DE) genes in
three paired samples is presented in Fig. 1A, and it was revealed that the
non-tumor and tumor samples were clearly separated and the size of
upregulated and downregulated gene clusters were nearly equal.
Additionally, there were similar expression patterns between three
samples in the same group of tumor or non-tumor samples.
Besides gene expression changes, AS, as an important
post-transcriptional modification, also caused differences between
tumor and non-tumor tissues. The splicing differences were analyzed
using MATS (23,24) for five common splicing types,
including skipped exon (SE), retained intron (RI), mutually
exclusive exons (MXEs), alternative 5′splice site (A5SS), and
alternative 3′splice site (A3SS). The most frequent splicing type
was the SE type, which had 197 splicing events with statistically
significant differences. The percent-spliced-in (PSI) value,
representing the fraction of the exon-inclusion variant, was used
to estimate the splicing level. As in the expression patterns of DE
genes, the hierarchical clustering for PSIs of differential skipped
exons still illustrated the separation between the tumor and
non-tumor samples (Fig. 1B).
However, PSI-decreased exons were obviously more than PSI-increased
exons, suggesting that HCC broke the splicing balance of exon
inclusion and exclusion on the whole. The differentially spliced
(DS) genes were fewer in number than the DE genes, and only 29% of
the former overlapped with the latter (Fig. 1C). Moreover, the splicing
specificity of samples also existed in the same group. These
splicing-changed genes in HCC revealed a new layer of regulation in
liver carcinogenesis.
Function and pathway enrichment of DE
and DS genes
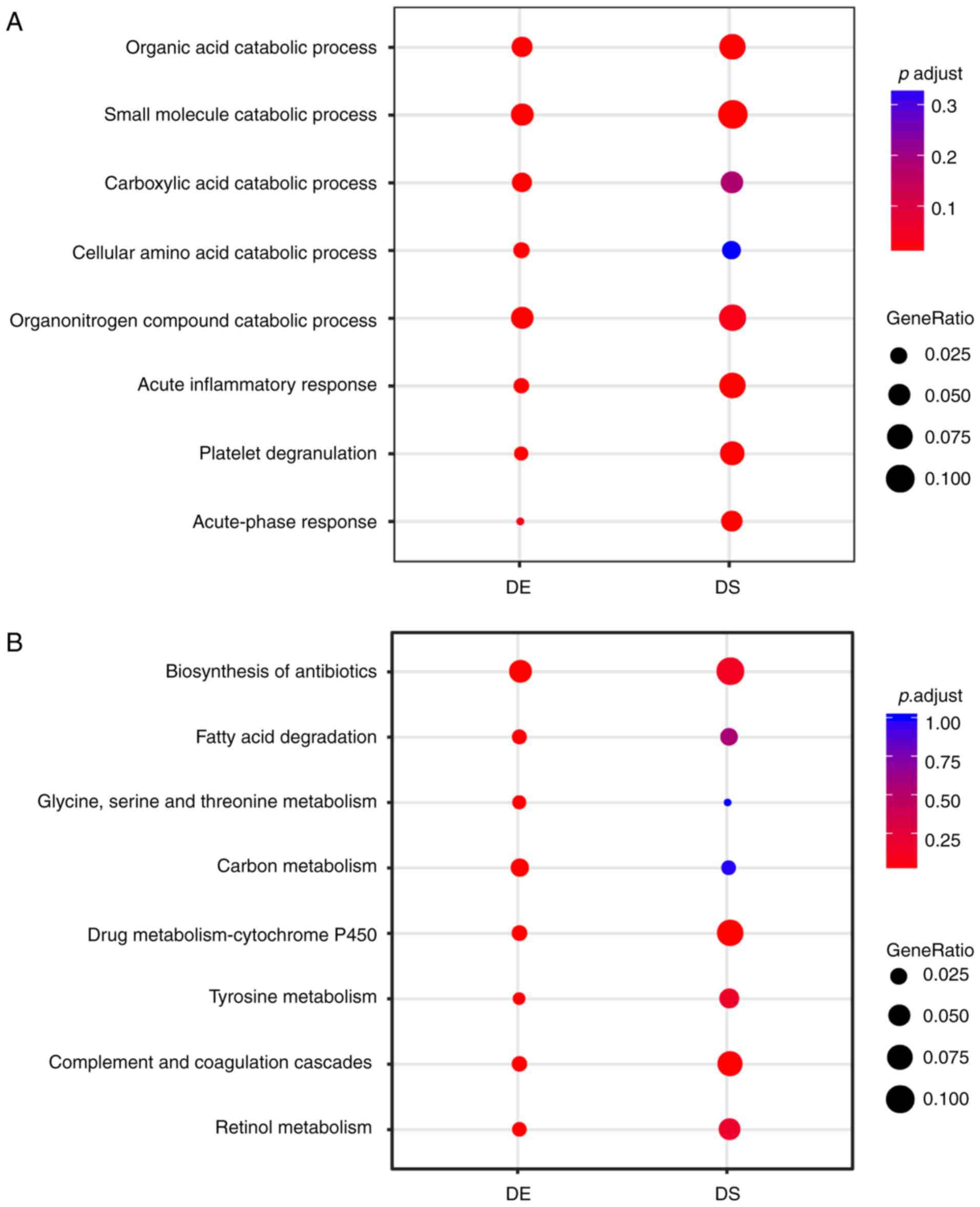
Next, an enrichment analysis was performed on DE and
DS genes between HCC tumor and non-tumor samples to observe whether
the splicing-induced changes were clustered in the main functions
and processes of the liver and liver cancer. Top
differentially-enriched terms of biological processes (P<0.05)
between DE and DS genes are presented in Fig. 2A. It was revealed that the DE
gene-specific-enriched terms included carboxylic acid catabolic
process and cellular amino acid catabolic process. DS genes were
primarily enriched in Gene Ontology (http://geneontology.org) terms involving acute-phase
response, platelet degranulation and acute inflammatory response,
which were identical to the core roles of the liver in
immunological effects. Similarly, enrichment analysis of the KEGG
pathway (https://www.genome.jp/kegg/pathway.html) revealed the
enriched pathways of liver characteristics, such as cytochrome P450
metabolism, and complement and coagulation cascades in DS genes
(Fig. 2B). These results indicated
that the DS genes reflected the important functions and processes
of the liver even though they were far fewer than the DE genes.
Clinical sample validation for AS
isoforms as novel candidate biomarkers
As aforementioned, the splicing changes were
indispensable for investigating the differences between HCC tumors
and non-tumors as well as exploring HCC biomarkers. Among the 197
DS events, we chose three AS events from both the PSI values and
the functional importance of the related genes to validate their
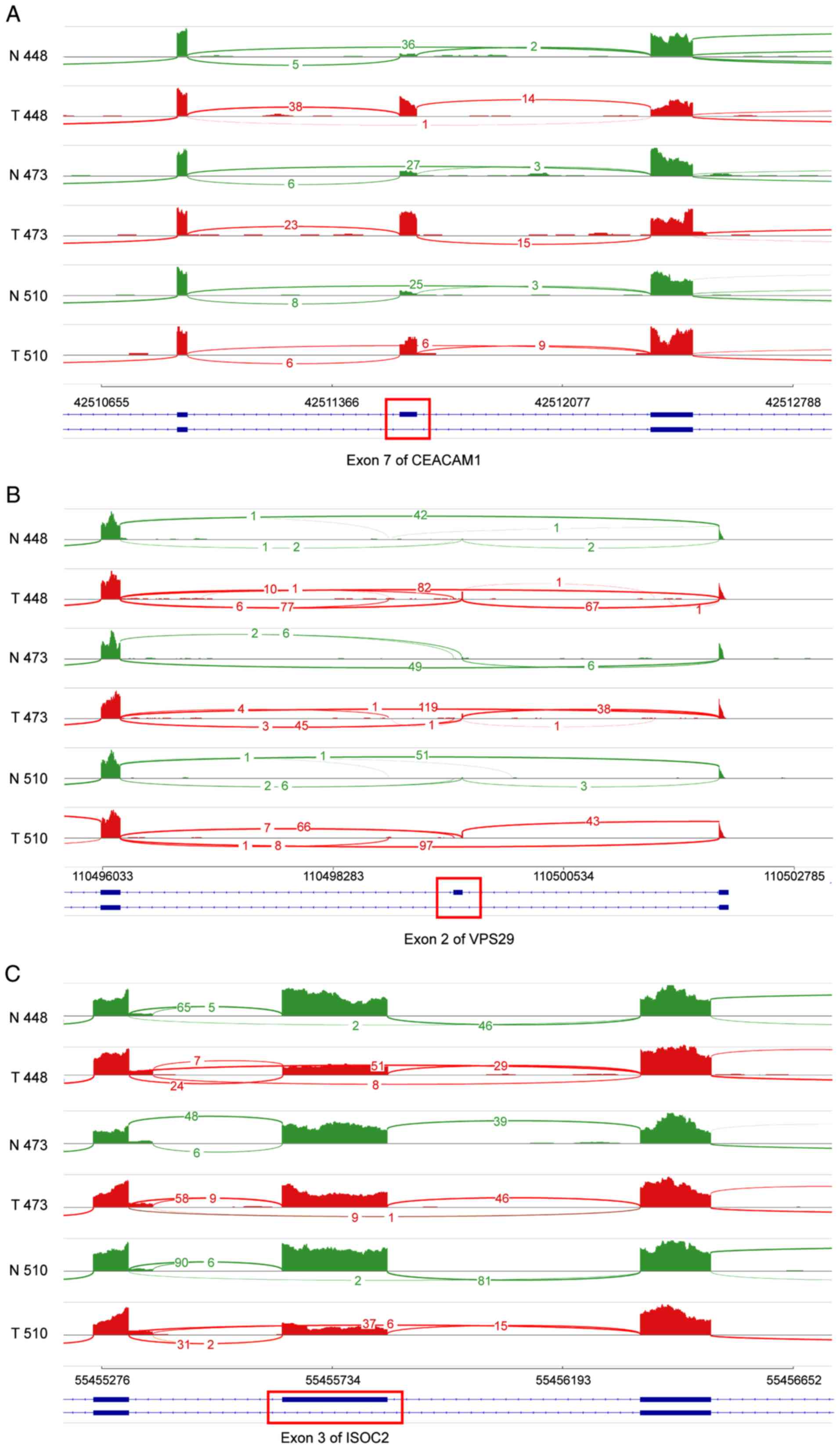
potential as novel splicing biomarkers in HCC. Reads alignments and
coverages in three paired samples illustrated the splicing
junctions of these three DS events (Fig. 3). We found that exon 7 of
carcinoembryonic antigen-related cell adhesion molecule 1
(CEACAM1) and exon 2 of VPS29 revealed an increase in
exon inclusion in HCC samples. CEACAM1 encodes
carcinoembryonic antigen-related cell adhesion molecule 1, and the
isoform with exon 7 inclusion is the longest transcript of
CEACAM1 and plays a role as a co-inhibitory receptor in the
immune response (25). VPS29
is a vacuolar protein sorting-associated protein and the exon
2-included isoform adds four amino acids to the protein initiation
position. Moreover, exon 3 of ISOC2 revealed an increase in
exon exclusion in HCC samples, the exclusion of which can alter the
core isochorismatase domain of ISOC2.
Although the splicing differences were identified by
mRNA sequencing from only three paired samples, a large number of
samples were employed to strictly validate the possibilities of
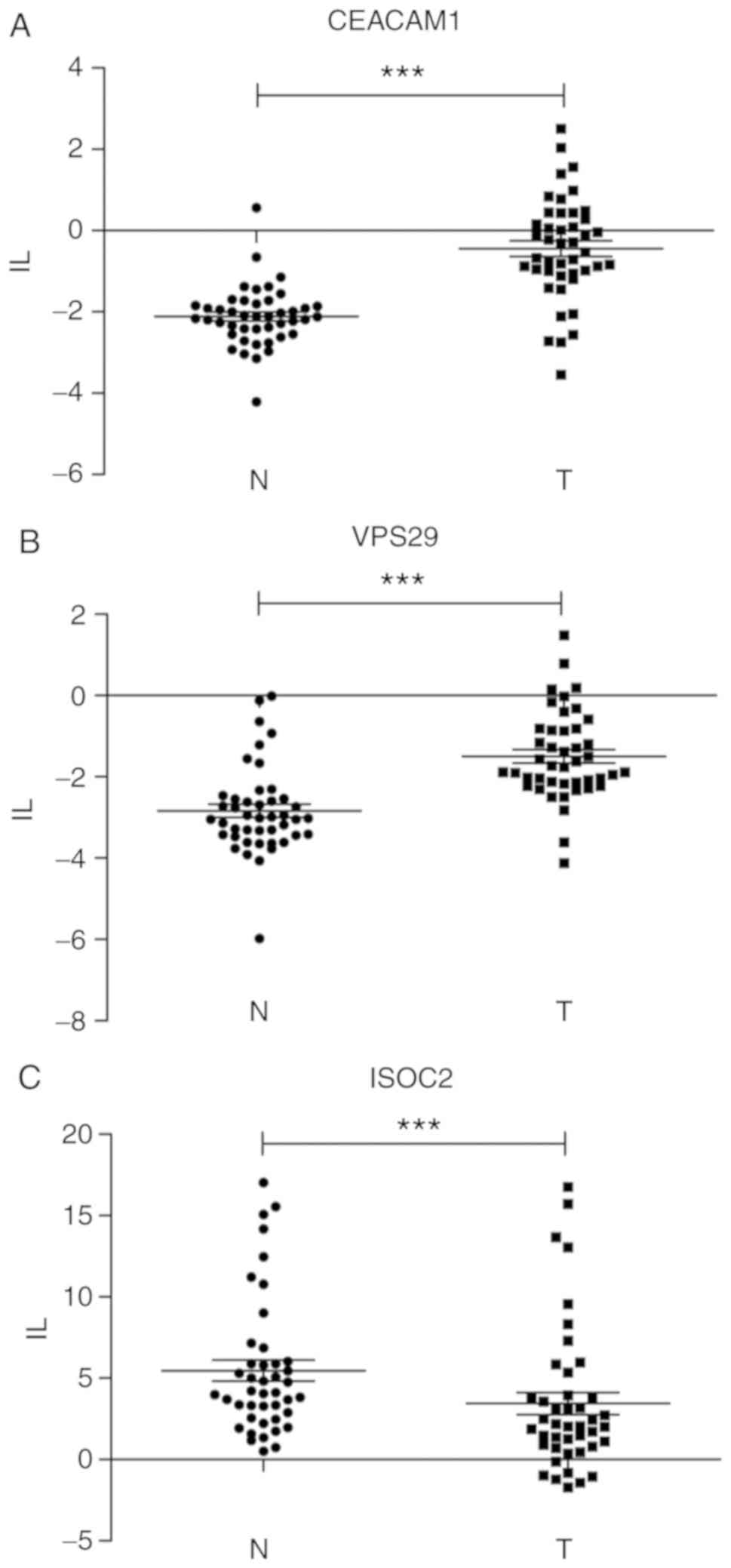
splicing isoforms as biomarkers of HCC. The exon inclusion level
(IL) differences were assessed by RT-qPCR in a total of 45 paired
HCC tumor and adjacent non-tumor samples (Fig. 4). The ΔΔCq values of DS exons were
significantly different between tumor and non-tumor tissues
(P<0.01) and the splicing variation tendency was the same as the
evidence from the RNA sequencing analysis. These splicing switches
were expected to be closely associated with HCC pathology.
Association of DS with clinical
features of HCC
Clinicopathological analysis of the three DS genes
in 45 HCC patients was performed to evaluate which features were
significantly associated with splicing switching. Contingency table
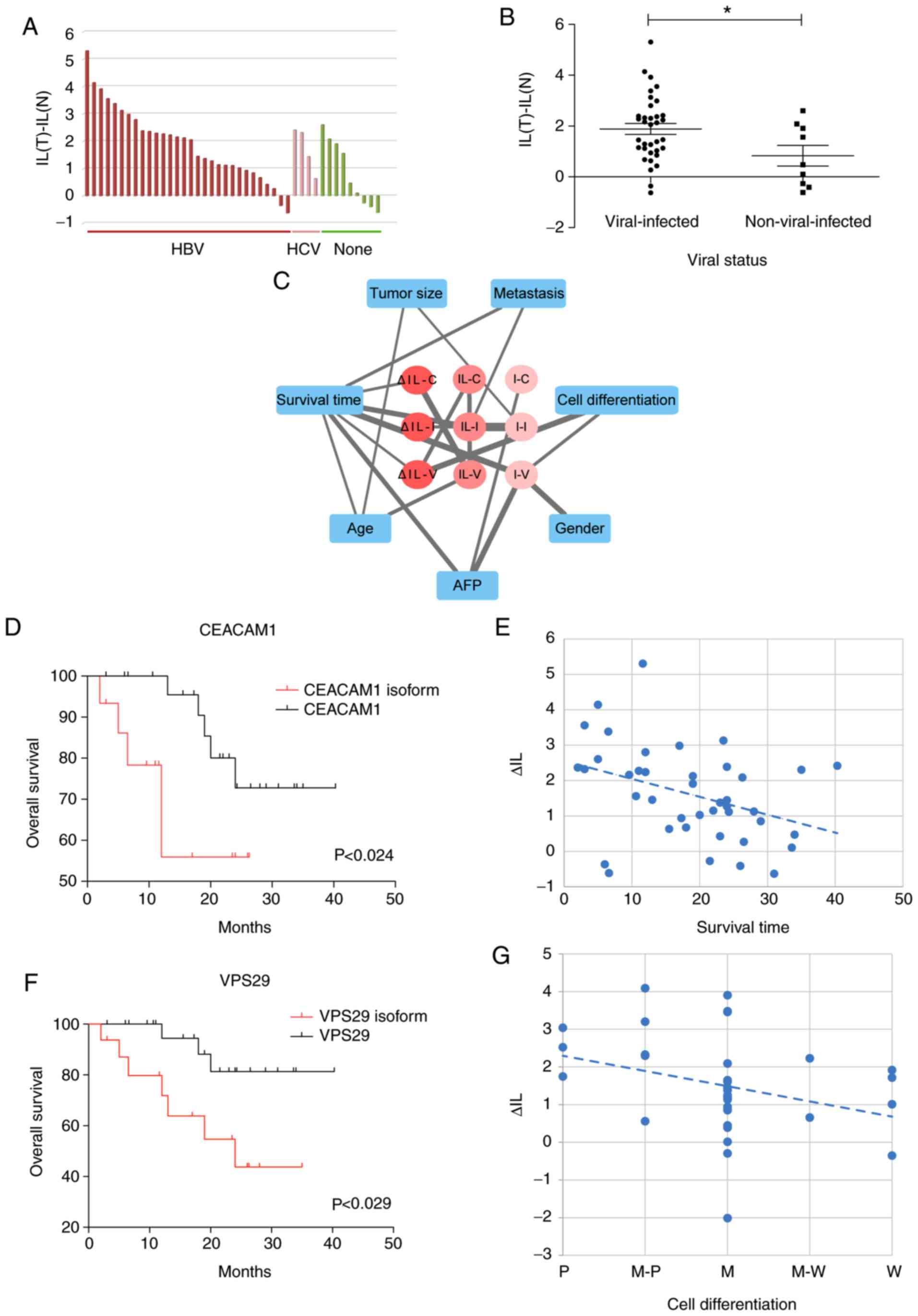
analysis and Fisher's exact test revealed that the exon 7 IL of
CEACAM1 was significantly associated with the viral status
of clinical samples (Table I and
Fig. 5A). HBV-/HCV-infected
patients were more likely to express the isoform with exon 7
inclusion. The difference test also represented the significant
difference of ΔIL, that is IL(tumor)-IL(non-tumor), between the
HBV-/HCV- and non-infected patients (Fig. 5B).
 | Table I.Contingency table analysis of
CEACAM1, VPS29 and ISOC2 splicing changes. |
Table I.
Contingency table analysis of
CEACAM1, VPS29 and ISOC2 splicing changes.
|
| CEACAM1 | ISOC2 | VPS29 |
|---|
|
|
|
|
|
|---|
| Clinical
features | Number | + | − | P-value | + | − | P-value | + | − | P-value |
|---|
| Age (years) |
|
|
|
|
|
|
|
|
|
|
|
>50 | 25 | 23 | 2 | 0.6378 | 6 | 18 | 0.2696 | 22 | 2 | 1 |
|
≤50 | 20 | 16 | 3 |
| 2 | 17 |
| 18 | 2 |
|
| Sex |
|
|
|
|
|
|
|
|
|
|
|
Male | 28 | 23 | 5 | 0.1414 | 4 | 24 | 0.4188 | 24 | 4 | 0.2797 |
|
Female | 17 | 16 | 0 |
| 4 | 11 |
| 16 | 0 |
|
| Viral status |
|
|
|
|
|
|
|
|
|
|
|
Yes | 36 | 34 | 2 | 0.0471 | 7 | 28 | 1 | 32 | 4 | 0.5687 |
| No | 9 | 6 | 3 |
| 1 | 8 |
| 9 | 0 |
|
| Cell
differentiation |
|
|
|
|
|
|
|
|
|
|
|
Poorly | 3 | 2 | 1 | 0.3188 | 0 | 3 | 0.8444 | 3 | 0 | 0.6662 |
|
Poorly-moderately | 5 | 4 | 0 |
| 1 | 3 |
| 5 | 0 |
|
|
Moderately | 23 | 21 | 2 |
| 4 | 18 |
| 20 | 2 |
|
|
Moderately-well | 2 | 1 | 1 |
| 0 | 2 |
| 2 | 0 |
|
|
Well | 4 | 3 | 1 |
| 1 | 3 |
| 3 | 1 |
|
| Tumor size
(cm) |
|
|
|
|
|
|
|
|
|
|
|
>5 | 17 | 15 | 1 | 0.638 | 3 | 13 | 1 | 15 | 2 | 0.6337 |
| ≤5 | 28 | 24 | 4 |
| 5 | 22 |
| 25 | 2 |
|
| Vascular
invasion |
|
|
|
|
|
|
|
|
|
|
|
Yes | 9 | 8 | 1 | 1 | 2 | 6 | 0.6354 | 9 | 0 | 0.5661 |
| No | 36 | 31 | 4 |
| 6 | 28 |
| 31 | 4 |
|
| AFP |
|
|
|
|
|
|
|
|
|
|
| + | 13 | 13 | 0 | 0.5382 | 3 | 9 | 0.6639 | 11 | 1 | 1 |
| − | 25 | 21 | 3 |
| 4 | 20 |
| 23 | 2 |
|
| Metastasis |
|
|
|
|
|
|
|
|
|
|
|
Yes | 6 | 6 | 0 | 1 | 0 | 6 | 0.3058 | 6 | 0 | 1 |
| No | 29 | 25 | 4 |
| 7 | 21 |
| 25 | 3 |
|
| Recurrence |
|
|
|
|
|
|
|
|
|
|
|
Yes | 8 | 7 | 1 | 1 | 0 | 8 | 0.1497 | 8 | 0 | 0.5548 |
| No | 25 | 22 | 3 |
| 7 | 17 |
| 21 | 3 |
|
| Survival |
|
|
|
|
|
|
|
|
|
|
|
Yes | 31 | 26 | 5 | 0.3102 | 7 | 23 | 0.1612 | 26 | 4 | 0.5558 |
| No | 10 | 10 | 0 |
| 0 | 10 |
| 10 | 0 |
|
Furthermore, the maximal information coefficient
(MIC) was used to assess the dependence between clinical features
and the three DS genes. We focused on three classes of splicing
changes in HCC tumors, including inclusion isoform expression, IL
and ΔIL. The former two only considered the degree of AS in HCC
tumor samples, while ΔIL reflected the AS changes between HCC
tumors and non-tumors. Then we identified their significant
association with clinical features at P<0.05 (Fig. 5C). Notably, ΔIL of CEACAM1
exon 7 was significantly associated with survival time
(MIC=0.38915, P<0.024; Fig. 5D).
The period of overall survival time was significantly shorter in
patients with increased inclusion levels of exon 7 in HCC samples
(P<0.014; Fig. 5E). Moreover,
ΔIL of VPS29 exon 2 was closely associated with overall
survival time (MIC=0.38142, P<0.029; Fig. 5F) and cell differentiation stages
(MIC=0.47849, P<0.002; Fig.
5G).
Discussion
AS plays a critical role in both physiological and
pathological processes. The dysregulation of AS is highly
associated with diseases such as cancer and neurodegenerative
diseases (26). Recently,
significant progress has been made in deciphering the role of
aberrant AS events in carcinogenesis and drug resistance, and
several novel molecules have been identified as possible
therapeutic targets in clinical HCC research. Splicing-based
prognostic biomarkers as well as therapeutic options hold great
potential towards improvements in cancer therapy (27).
In the present study, we examined the relationship
between AS events and human HCC and described several aberrant AS
events in clinical HCC samples, such as increased inclusion of exon
2 of VPS29 and the exclusion of exon 3 of ISOC2,
which could aid in HCC diagnosis and therapy. One important
aberrant AS event existed in CEACAM1, exon 7 which revealed
increased inclusion levels in HCC samples. CEACAM1 is a member of
the immunoglobulin superfamily and a type-I transmembrane
glycoprotein of the carcinoembryonic antigen family (28). In HCC, the loss of CEACAM1 indicates
high metastatic potential and is associated with poor survival even
after hepatectomy (29). However,
research on the genome scale indicated that the different CEACAM1
isoforms had divergent roles during tumorigenesis. CEACAM1-L
(CEACAM1 long cytoplasmic domain isoform) is a risk factor for HCC
recurrence and its overexpression is involved in tumor cell
invasion and metastasis (30).
More specifically, in this study, we demonstrated
that exon 7 of CEACAM1 was the critical hub of the
functional differences between the CEACAM1 isoforms. Patients with
increased inclusion levels of CEACAM1 exon 7 in HCC samples
had significantly shorter survival times. Exon 7 is located on the
3′terminal of the CEACAM1 transcript, although it does not
encode the immunoglobulin-like domain, the inclusion or exclusion
of which may influence the subcellular location of the protein,
which could remodel the tumorigenicity of CEACAM1 isoforms
(30–32).
Previous association analyses between candidate
biomarkers and clinical features mainly utilized contingency tables
and Fisher's exact test (15,33,34).
Some qualitative features, such as sex, viral status and cell
differentiation could not well observe the potential relevant
trends with expression or splicing changes. In the present study,
the MIC was first applied to the clinicopathological analysis,
including both qualitative and quantitative features. This MIC
method efficiently characterized the relationships for all clinical
features and is expected to extend the applications in the
biomarker research field.
In summary, the present study undertook a strategy
of ‘small sample sequencing and large sample validation’, which
means that a small number of samples for deep sequencing provided
the differential spliced biomarker candidates, and a large number
of samples for validation experiments confirmed the significantly
differential splicing. The development of high-throughput
technology has led to the generation of huge amounts of data on HCC
and other cancer types. We anticipate that our method could be
widely applied to thoroughly research these big datasets and
discover more valuable biomarkers for HCC. The novel splicing
biomarkers in this study could also provide a new understanding of
HCC research.
Acknowledgements
Not applicable.
Funding
The present study was partially supported by the
National Key R&D Program of China (nos. 2016YFC0902400 and
2017YFC0906603), the National Natural Science Foundation of China
(nos. 81770581, 81570526 and 81123001), the Innovation project
(16CXZ027), the Program of International S&T Cooperation (nos.
2014DFB30020 and 2014DFB30010), the Beijing Science and Technology
Project (Z161100002616036), the Open Project Program of the State
Key Laboratory of Proteomics (Academy of Military Medical Sciences,
SKLP-O201509). All funding was received during this study, and
there was no additional external funding received for this
study.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
PW and DZ designed the study, analyzed the results
and created a draft of the manuscript; YW and WL collected the
experimental samples and performed the qPCR experiment; AS, HW and
YF prepared the experimental materials and performed the
statistical analysis; XC and YJ conceived the study, performed the
data interpretation, revised and approved the manuscript. All
authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Clinical Research Ethics Committee of China-Japan Union Hospital,
Jilin University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
A3SS
|
alternative 3′splice site
|
|
A5SS
|
alternative 5′splice site
|
|
AS
|
alternative splicing
|
|
DE
|
differentially expressed
|
|
DS
|
differentially spliced
|
|
FDR
|
false discovery rate
|
|
FPKM
|
fragments per kilobase of transcript
per million mapped reads
|
|
HBV
|
hepatitis B virus
|
|
HCC
|
hepatocellular carcinoma
|
|
HCV
|
hepatitis C virus
|
|
IL
|
inclusion level
|
|
MIC
|
maximal information coefficient
|
|
MXE
|
mutually exclusive exons
|
|
PSI
|
percent-spliced-in
|
|
RI
|
retained intron
|
|
RNA-Seq
|
RNA sequencing
|
|
RT-PCR
|
reverse transcription quantitative
polymerase chain reaction
|
|
SE
|
skipped exon
|
References
|
1
|
Lou J, Zhang L, Lv S, Zhang C and Jiang S:
Biomarkers for hepatocellular carcinoma. Biomark Cancer. 9:1–9.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Burkhart RA, Ronnekleiv-Kelly SM and
Pawlik TM: Personalized therapy in hepatocellular carcinoma:
Molecular markers of prognosis and therapeutic response. Surg
Oncol. 26:138–145. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Luz FA, Brigido PC, Moraes AS and Silva
MJ: Aberrant splicing in cancer: Mediators of malignant progression
through an imperfect splice program shift. Oncology. 92:3–13. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ding C, Li Y, Guo F, Jiang Y, Ying W, Li
D, Yang D, Xia X, Liu W, Zhao Y, et al: A cell-type-resolved liver
proteome. Mol Cell Proteomics. 15:3190–3202. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Merkin J, Russell C, Chen P and Burge CB:
Evolutionary dynamics of gene and isoform regulation in Mammalian
tissues. Science. 338:1593–1599. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang J and Manley JL: Misregulation of
pre-mRNA alternative splicing in cancer. Cancer Discov.
3:1228–1237. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Villegas-Ruiz V, Hendlmeier F,
Buentello-Volante B, Rodriguez-Loaiza JL, Miranda-Duarte A and
Zenteno JC: Genome-wide mRNA analysis reveals a TUBD1 isoform
profile as a potential biomarker for diabetic retinopathy
development. Exp Eye Res. 155:99–106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Y, Sun N, Lu Z, Sun S, Huang J, Chen Z
and He J: Prognostic alternative mRNA splicing signature in
non-small cell lung cancer. Cancer Lett. 393:40–51. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Singh B and Eyras E: The role of
alternative splicing in cancer. Transcription. 8:91–98. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
He C, Zhou F, Zuo Z, Cheng H and Zhou R: A
global view of cancer-specific transcript variants by subtractive
transcriptome-wide analysis. PLoS One. 4:e47322009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Laversin SA, Phatak VM, Powe DG, Li G,
Miles AK, Hughes DC, Ball GR, Ellis IO, Gritzapis AD, Missitzis I,
et al: Identification of novel breast cancer-associated transcripts
by UniGene database mining and gene expression analysis in normal
and malignant cells. Genes Chromosomes Cancer. 52:316–329. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang X, Sheng P, Guo X, Wang J, Hou L, Hu
G, Luo C, Dong Y and Lu Y: Identification and expression of a novel
MDM4 splice variant in human glioma. Brain Res 1537. 260–266.
2013.
|
|
13
|
Arafat H, Lazar M, Salem K, Chipitsyna G,
Gong Q, Pan TC, Zhang RZ, Yeo CJ and Chu ML: Tumor-specific
expression and alternative splicing of the COL6A3 gene in
pancreatic cancer. Surgery. 150:306–315. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim YJ and Kim HS: Alternative splicing
and its impact as a cancer diagnostic marker. Genomics Inform.
10:74–80. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lin KT, Shann YJ, Chau GY, Hsu CN and
Huang CY: Identification of latent biomarkers in hepatocellular
carcinoma by ultra-deep whole-transcriptome sequencing. Oncogene.
33:4786–4794. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chan TH, Lin CH, Qi L, Fei J, Li Y, Yong
KJ, Liu M, Song Y, Chow RK, Ng VH, et al: A disrupted RNA editing
balance mediated by ADARs (Adenosine DeAminases that act on RNA) in
human hepatocellular carcinoma. Gut. 63:832–843. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Trapnell C, Pachter L and Salzberg SL:
TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics.
25:1105–1111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Trapnell C, Roberts A, Goff L, Pertea G,
Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL and Pachter L:
Differential gene and transcript expression analysis of RNA-seq
experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reshef DN, Reshef YA, Finucane HK,
Grossman SR, McVean G, Turnbaugh PJ, Lander ES, Mitzenmacher M and
Sabeti PC: Detecting novel associations in large data sets.
Science. 334:1518–1524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thorvaldsdottir H, Robinson JT and Mesirov
JP: Integrative genomics viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu G, Wang LG, Han Y and He QY:
ClusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen S, Park JW, Huang J, Dittmar KA, Lu
ZX, Zhou Q, Carstens RP and Xing Y: MATS: A Bayesian framework for
flexible detection of differential alternative splicing from
RNA-Seq data. Nucleic Acids Res. 40:e612012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shen S, Park JW, Lu ZX, Lin L, Henry MD,
Wu YN, Zhou Q and Xing Y: rMATS: Robust and flexible detection of
differential alternative splicing from replicate RNA-Seq data. Proc
Natl Acad Sci USA. 111:E5593–E5601. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen Z, Chen L, Qiao SW, Nagaishi T and
Blumberg RS: Carcinoembryonic antigen-related cell adhesion
molecule 1 inhibits proximal TCR signaling by targeting ZAP-70. J
Immunol. 180:6085–6093. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Le KQ, Prabhakar BS, Hong WJ and Li LC:
Alternative splicing as a biomarker and potential target for drug
discovery. Acta Pharmacol Sin. 36:1212–1218. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wojtuszkiewicz A, Assaraf YG, Maas MJ,
Kaspers GJ, Jansen G and Cloos J: Pre-mRNA splicing in cancer: The
relevance in oncogenesis, treatment and drug resistance. Expert
Opin Drug Metab Toxicol. 11:673–689. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Williams AF and Barclay AN: The
immunoglobulin superfamily-domains for cell surface recognition.
Annu Rev Immunol. 6:381–405. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cruz PV, Wakai T, Shirai Y, Yokoyama N and
Hatakeyama K: Loss of carcinoembryonic antigen-related cell
adhesion molecule 1 expression is an adverse prognostic factor in
hepatocellular carcinoma. Cancer. 104:354–360. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kiriyama S, Yokoyama S, Ueno M, Hayami S,
Ieda J, Yamamoto N, Yamaguchi S, Mitani Y, Nakamura Y, Tani M, et
al: CEACAM1 long cytoplasmic domain isoform is associated with
invasion and recurrence of hepatocellular carcinoma. Ann Surg
Oncol. 21 Suppl 4:S505–S514. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lawson EL, Mills DR, Brilliant KE and
Hixson DC: The transmembrane domain of CEACAM1-4S is a determinant
of anchorage independent growth and tumorigenicity. PLoS One.
7:e296062012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu J, Yang Y, Ma C, Zhang G, Wang K and
Hu S: CEACAM1 cytoplastic expression is closely related to tumor
angiogenesis and poorer relapse-free survival after curative
resection of hepatocellular carcinoma. World J Surg. 35:2259–2265.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kobel M, Kalloger SE, Boyd N, McKinney S,
Mehl E, Palmer C, Leung S, Bowen NJ, Ionescu DN, Rajput A, et al:
Ovarian carcinoma subtypes are different diseases: Implications for
biomarker studies. PLoS Med. 5:e2322008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Viasus D, Del Rio-Pertuz G, Simonetti AF,
Garcia-Vidal C, Acosta-Reyes J, Garavito A and Carratalà J:
Biomarkers for predicting short-term mortality in
community-acquired pneumonia: A systematic review and
meta-analysis. J Infect. 72:273–282. 2016. View Article : Google Scholar : PubMed/NCBI
|