Introduction
Breast cancer (BrCa) is the most prevalent
malignancy among women. Approximately 252,710 new cases of invasive
BrCa and 40,610 cancer-related deaths are expected to occur among
American women in 2017 (1). BrCa is
a heterogametic disease that can be categorized into luminal A,
luminal B, human epithelial growth factor receptor-2 (HER-2)
overexpressing and basal/triple negative (TN) molecular subtypes on
the basis of gene expression profiling (2–5). Given
the high morbidity and poor prognosis of patients with BrCa,
additional research is required to understand the potential
molecular mechanism that mediates BrCa tumorigenesis. To date, a
variety of biological factors have been identified to be involved
in the development of this disease. Therefore, it is imperative to
identify potential therapeutic targets such as progesterone
receptor (PR) and estrogen receptor (ER), human epidermal growth
factor receptor 2 (HER2) among the various biological factors
involved in BrCa development (6–8).
The Arp2/3 protein complex consists of 7 subunits:
The actin-related proteins ARP2 (ACTR2) and ARP3 (ACTR3), ARPC1B
(ARC41), ARPC2 (ARC34), ARPC3 (ARC21), ARPC4 (ARC20), and ARPC5
(ARC16) (9,10). Among these subunits, ARPC2 (subunit
2, 34 kDa) has a fundamental role in the nucleation of actin
filaments (AFs) and participates in the polarization of the Golgi
apparatus (11). Al Ghouleh et
al demonstrated that ARPC2 participates in smooth muscle cell
migration (12), Zhang et al
also reported that ARPC2 may be involved in human gastric cancer
cell proliferation or migration (13). Thus ARPC2 is critical to cell
migration and invasion (14).
Activation of epithelial-mesenchymal transition (EMT) has been
implicated in the metastasis of human tumors in previous studies
(15–17).
EMT is a specific physiological or
pathophysiological phenomenon that is characterized by the loss of
the epithelial phenotype and gain of mesenchymal characteristics by
epithelial cells. These processes are evidenced by the decrease in
E-cadherin expression and increase in vimentin/N-cadherin
expression (18). The activation of
EMT has been implicated in the metastasis of human tumors. In
cancer, the frequent occurrence of EMT is associated with poor
prognosis and distant metasis. TGF-β is a pathway involved in EMT
regulation (19,20). The downstream factors of the TGF-β
pathway include N-cadherin, vimentin, ZEB1, MMP-9 and MMP-3, and
these factors have important roles in EMT regulation (21,22).
However, whether the TGF-β/EMT pathway is involved in
ARPC2-mediated carcinogenesis has never been reported.
The present study was aimed to determine whether
ARPC2 is a BrCa oncogene and to further understand the mechanisms
that underlie its in vitro and in vivo effect. By
searching the Oncomine database and performing immunohistochemistry
(IHC), we found that ARPC2 mRNA and protein expression levels were
upregulated in BrCa. The results of the cell cycle, wound-healing,
migration and invasion assays revealed that ARPC2 significantly
promoted the proliferation, migration and invasion of BrCa cells.
Clinical data, revealed that ARPC2 was associated with lymph node
invasion and tumor stage. High ARPC2 expression levels were
associated with a low overall survival (OS) rate. Lastly, we
performed western blot analysis to further explore the potential
mechanism of ARPC2 in BrCa. We found that ARPC2 increased the
expression of EMT-inducing transcription factors (EMT-TFs), such as
N-cadherin, vimentin, ZEB1, MMP-9 and MMP-3. Notably, ARPC2 may
contribute to the malignant progression of BrCa.
Materials and methods
Patients and tissue collection
We obtained 172 paraffin-embedded surgical BrCa
tissue specimens from the Department of Pathology at the First
Affiliated Hospital of Anhui Medical University (Hefei, Anhui,
China). We recruited 68 female patients with benign breast diseases
from the same hospital. The 68 female patients were used as the
control group. No patients had received preoperative adjuvant
therapy. Two pathologists confirmed the diagnoses of breast tumors
in all the samples used. All cases were followed-up for 60 months.
Overall survival (OS) was calculated on the basis of data. Patient
samples were categorized into two groups in accordance with median
expression (high vs. low expression) (Table I) and assessed using a Kaplan-Meier
survival plot. The protocol of the present study was approved by
the Institutional Review Board of Anhui Medical University.
Detailed information of patients including ER/PR expression, lymph
node metastasis and age are listed in Table II. The agreement on the use of
these tissue samples was approved by the Biomedical Ethics
Committee of Anhui Medical University and the confidentiality of
patient information was maintained. We obtained informed consent
from all patients before enrolling participants in the present
study.
 | Table I.Expression of ARPC2 in breast cancer
and normal tissues. |
Table I.
Expression of ARPC2 in breast cancer
and normal tissues.
|
|
| ARPC2
expression |
|---|
|
|
|
|
|---|
| Group | n | Negative/low
positive, n (%) | High positive, n
(%) |
|---|
| Cancer | 172 | 76 (44.0) | 96
(56.0)a |
| Normal | 68 | 48 (72.0) | 20
(28.0)a |
| Table II.Association of ARPC2 expression with
clinicopathological parameters from breast cancer patients. |
Table II.
Association of ARPC2 expression with
clinicopathological parameters from breast cancer patients.
|
|
| ARPC2
expression |
|
|---|
|
|
|
|
|
|---|
| Parameter | n | High | Low or none | P-value |
|---|
| Age (years) |
|
≤35 | 6 | 4 | 2 | 0.075 |
|
36–55 | 111 | 57 | 54 |
|
|
>60 | 55 | 28 | 27 |
|
| Tumor size
(cm) |
| ≤2 | 41 | 16 | 25 | <0.05 |
|
3–5 | 110 | 51 | 59 |
|
|
>6 | 21 | 15 | 6 |
|
| Lymph node
involvement |
|
pN0 | 79 | 35 | 44 | <0.05 |
|
pN1 | 45 | 24 | 21 |
|
|
pN2 | 48 | 35 | 13 |
|
| Grade |
| I | 79 | 38 | 41 | <0.05 |
| II | 45 | 25 | 20 |
|
|
III | 48 | 32 | 16 |
|
| Stage |
| I | 24 | 9 | 15 | 0.65 |
| II | 132 | 66 | 66 |
|
|
III–IV | 16 | 7 | 9 |
|
| ER |
| + | 106 | 37 | 69 | 0.864 |
| − | 66 | 24 | 42 |
|
| PR |
| + | 96 | 46 | 50 | 0.65 |
| − | 76 | 37 | 40 |
|
| HER-2 |
| + | 47 | 25 | 22 | 0.288 |
| − | 125 | 70 | 55 |
|
Oncomine, The cancer genome atlas data
and cBioPortal analysis
The Oncomine microarray database (http://www.oncomine.org) was used for analysis. We
screened for relevant entries on ARPC2 expression in BrCa tissues
and normal breast tissues. With ‘Cancer vs. normal’ analysis as the
primary filter. Data sets were used in the present study in
accordance with Ma et al (23), Curtis et al (24), Perou et al (25) and Zhao et al (26). We screened for differentially
expressed genes (Pearson's correlation >0.4) in several
important node locations of ARPC2 in BrCa (Pearson's correlation
>0.4) by using BioPortal for Cancer Genomics (http://www.cbioportal.org). All searches were
performed according to the cBioPortal's online instructions.
Cell culture
Human BrCa cell lines (MCF-10A, MCF-7, T47D, BT474
and MDA-MB-231) were obtained from the American Type Culture
Collection (ATCC; Manassas, VA, USA) and cultured under
ATCC-recommended conditions. Cells were routinely incubated at 37°C
and 5% CO2 under a humidified atmosphere.
ARPC2 overexpression and
downregulation
The full length of Homo sapiens ARPC2 was
synthesized and subcloned into an entry vector by OriGene
Technologies Technologies, Inc., (Beijing, China). siRNA-ARPC2
(siARPC2) was purchased from Shanghai GenePharma Co., Ltd.
(Shanghai, China). MCF-7 and MDA-MB-231 cells were transfected
using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) in accordance to the manufacturer's
instructions. Transfection efficiency was confirmed by performing
reverse transcription-quantitative real-time polymerase chain
reaction (qRT-PCR) and western blotting.
Western blotting and RT-PCR
We used RT-PCR to evaluate the mRNA levels of ARPC2.
Total RNA was isolated from cultured cells using TRIzol reagent.
RNA was reverse transcribed into cDNA using the PrimeScript RT
reagent kit (Takara Biotechnology Co., Ltd., Dalian, China).
qRT-PCR was performed using the SYBR Prime Script kit (Takara Bio,
Inc., Otsu, Japan). ARPC: Forward primer,
5′-TCCGACTCTACCAGCTGATGC-3′ and reverse primer,
5′-AAGCTGGACTCATCCCACAGC-3′. PCR was conducted at 95°C for 15 min,
followed by 40 cycles of 94°C for 15 sec, 55°C for 30 sec, and 64°C
for 30 sec. Experiment was repeated three times and the expression
of ARPC2 was quantified using the 2−ΔΔCq method
(27). Cells were lysed via a lysis
buffer which consisted of RIPA (Beyotime Institute of
Biotechnology, Haimen, China). The protein concentration was
determined using BCA kit. Equal amounts of total protein (10 µg)
were separated by 10% SDS polyacrylamide gels and transferred onto
polyvinylidene fluoride (PVDF) membranes, followed by blocking with
5% non-fat milk for 1 h. The membranes were incubated with primary
antibodies overnight at 4°C. Protein expression was quantified by
densitometry (Quantity One software; Bio-Rad Laboratories,
Hercules, CA, USA). The antibodies used for western blotting were:
N-cadherin (cat. no. 4061; Cell Signaling Technology, Inc.,
Danvers, MA, USA), E-cadherin (cat. no. 5296; Cell Signaling
Technology, Inc.), vimentin (cat. no. 5296; Cell Signaling
Technology, Inc.), ZEB1 (cat. no. 3396; Cell Signaling Technology,
Inc.) and MMP-9 (cat no. 3852; Cell Signaling Technology, Inc.),
MMP-3 (cat. no. ab52915; Abcam, Cambridge, UK), β-actin (cat. no.
ab5694; Abcam), ARPC2 (cat. no. ab11798; Abcam) and GAPDH (cat. no.
g9545; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). The bands
were displayed by ECL illumination and analyzed by the Tanon 5200
image acquisition system (Tanon Science and Technology Co., Ltd.,
Shanghai, China).
Colony formation assays
For the colony formation assay, transfected cells
were grown in a fresh 6-well plate with RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum. For the cell colony formation assay, the transfected cells
were seeded in 6-well plates (1×103/well) and cultured
for 14 days at 37°C. After 14 days, all visible colonies per well
were manually counted. Assays were performed in triplicate, and the
results were calculated as the mean ± standard deviation (SD).
Transwell assay
The siRNA-control (siControl) and siRNA-ARPC2
(siARPC2) were transfected into MCF-7 and MDA-MB-231 cells for 48
h. For the migration assay, 5×104 cells were seeded with
serum-free RPMI-1640 medium in the upper chamber of Transwell
inserts (8-µm; Corning Incorporated, Corning, NY, USA). For the
invasion assay, inserts were coated with Matrigel (Sigma-Aldrich;
Merck KGaA). For the migration and invasion assays, the lower
chamber of the Transwell unit was filled with culture medium
supplemented with 10% serum. After 36–48 h, the cell filters were
fixed using methanol and stained with 0.1% crystal violet. The
number of cells was manually counted. Each experiment was assessed
in triplicate.
Wound healing assay
Cells (5×106) were seeded in 6-well
plates. Once the cells achieved 100% confluence, the culture plate
was scratched along its central axis of with 200-µl sterile pipette
tips. An inverted microscope was used to observe wound healing at
three time-points (0, 24 and 48 h) after wounding. The wound
healing rate was calculated and wounds were observed and images
were captured. The assays were performed three times.
In vivo tumor formation
Five-week-old, female, and BALB/c nude mice (7 in
each group) were used for the xenograft tumor formation assay. All
animal testing was performed at the Model Animal Research Center of
The University of Science and Technology of China (Hefei, China).
All of the experiments were approved by the Institute Research
Ethics Committee of Anhui Medical University (Hefei, China). The
suspended cells (5×106) were injected into each mouse on
the right side of the posterior flank. Seven days after injections,
tumor nodules were checked every 5 days and tumor volumes were
calculated by width × length × (width + length)/2. All tumors were
removed from the nude mice after 40 days.
IHC staining
For IHC staining, 4-µm sections of the TMA blocks
were incubated overnight with a pre-diluted goat antihuman ARPC2
monoclonal antibody (Fuzhou Maixin Biotech Co., Ltd., Fuzhou,
China). Immunoreactivity was evaluated semi-quantitatively, and the
result was calculated on the basis of the percentage of positive
cells. Immunostaining was considered as low positive when the
percentage of the stained tumor cells was <30%, and as high
positive when the percentage of stained tumor cells exceeded
30%.
Cell cycle and apoptosis analyses
Cells (0.5×106−1×106) were
washed twice with cold phosphate-buffered saline. Cells were
stained with propidium iodide (PI; Cycletest™ Plus DNA reagent kit;
BD Biosciences, Franklin Lakes, NJ, USA) in accordance with the
manufacturer's protocol. Analyses and measurements were performed
using FACSVerse flow cytometer (BD Biosciences). The percentage of
cells in the S, G0/G1 and G2/M phases were counted and compared.
Similarly, cell apoptosis was analyzed by flow cytometry using the
Annexin V-FITC/PI Apoptosis Detection kit (BD Biosciences) 48 h
after transfection.
Statistical analysis
Data were analyzed with SPSS 19.0 software (SPSS
Inc., Chicago, IL, USA). The association between the ARPC2
expression level and the clinopathological characteristics of
patients was analyzed by the Chi-square test. Student's t-test was
used for comparing the differences between groups. Cumulative
recurrence and survival rates were analyzed by the Kaplan-Meier
method. P<0.05 was considered to indicate a statistically
significant difference.
Results
ARPC2 expression levels in different
BrCa cell lines
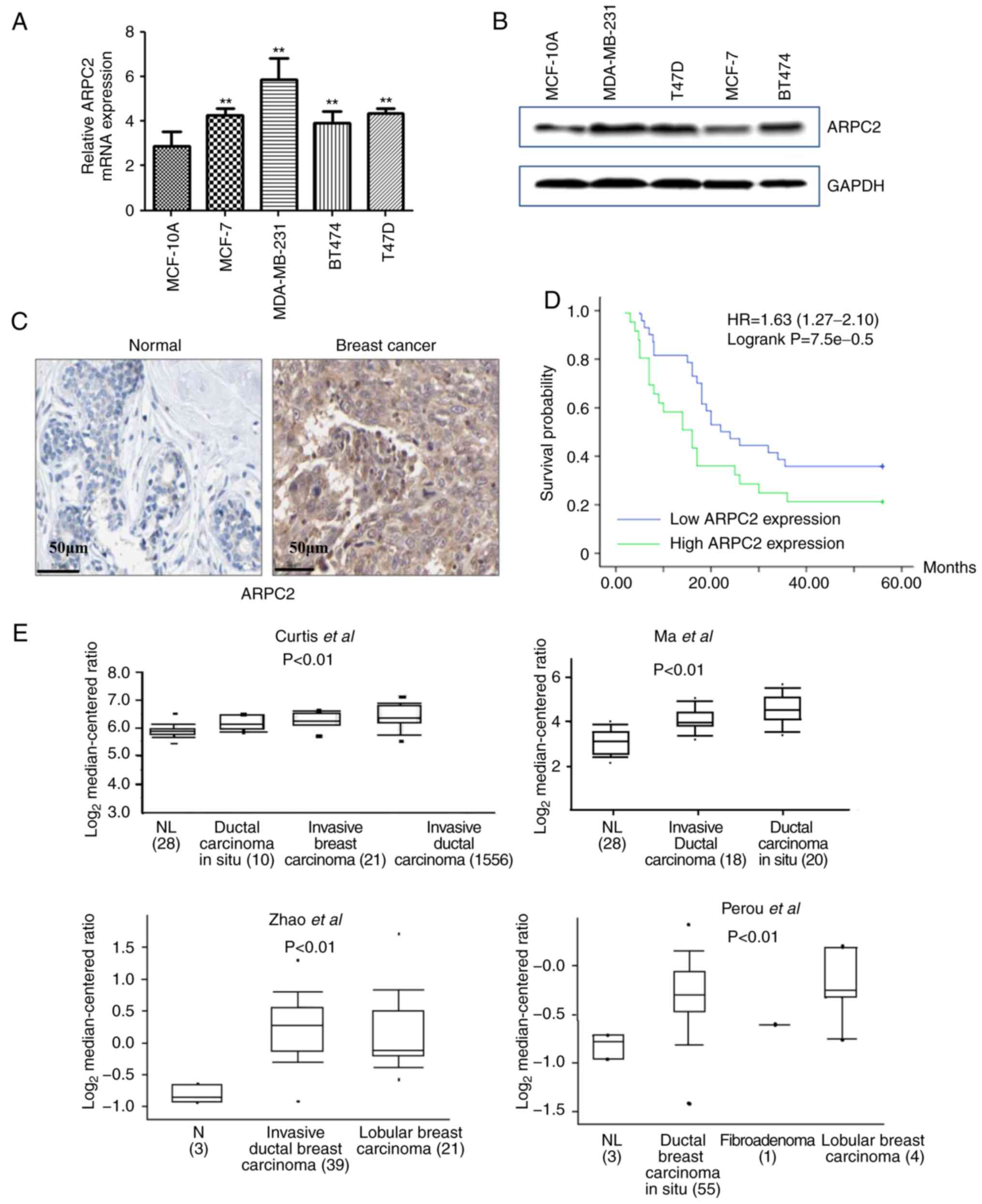
We quantified the expression of ARPC2 in different
types of human BrCa cell lines (MCF-7, T47D, BT474 and MDA-MB-231)
and human breast epithelial cell line (MCF-10A) by RT-PCR and
western blotting. As revealed in Fig.
1A and B, we found that ARPC2 was highly expressed in BrCa cell
lines. However, changes in ARPC2 expression in four types of BrCa
cell lines did not exhibit a consistent pattern. Since MCF-7 and
MDA-MB-231 cell lines had a good tumorigenic effect, we selected
MCF-7 and MB-MDA-231 cell lines as experimental models.
ARPC2 mRNA and protein expression in
BrCa and normal tissues
We performed IHC to assess ARPC2 protein expression
in different types of BrCa (n=172) and paired normal breast (n=68)
tissues. As revealed in Fig. 1C,
the stain for ARPC2 protein expression was predominantly located in
the cytosol. Our results demonstrated that the expression level of
ARPC2 protein in normal tissues was significantly lower than in the
cancer tissues (Table I). The
Kaplan-Meier curve and log-rank test analyses revealed that ARPC2
expression was associated with the OS of patients with BrCa. We
evaluated the prognostic value of ARPC2 using the protein
expression data and survival information of 172 patients with BrCa.
We divided patient samples into two groups in accordance with the
median expression (high vs. low expression). As revealed in
Fig. 1D, high ARPC2 levels were
associated with a low OS rate. Moreover, to illustrate the
potential relationship between ARPC2 and BrCa progression, we used
the Oncomine database (http://www.oncomine.org) for the analysis of ARPC2
mRNA expression. We found that ARPC2 mRNA expression was
significantly upregulated in BrCa relative to that in normal breast
tissues (Fig. 1E).
Relationship of ARPC2 with the
clinicopathological characteristics and poor prognosis of patients
with BrCa
We analyzed the relationship between ARPC2 protein
expression and the clinicopathological parameters of patients with
BrCa. As presented in Table II,
ARPC2 protein expression was significantly and positively
associated with lymph node metastasis, tumor size and grade of BrCa
(P<0.05). The protein expression of ARPC2, however, was not
significantly associated with other clinical parameters, including
age, HER-2 expression, and estrogen and progesterone receptor
status of patients with BrCa.
ARPC2 silencing inhibits the migration
and invasion capabilities of BrCa cell lines
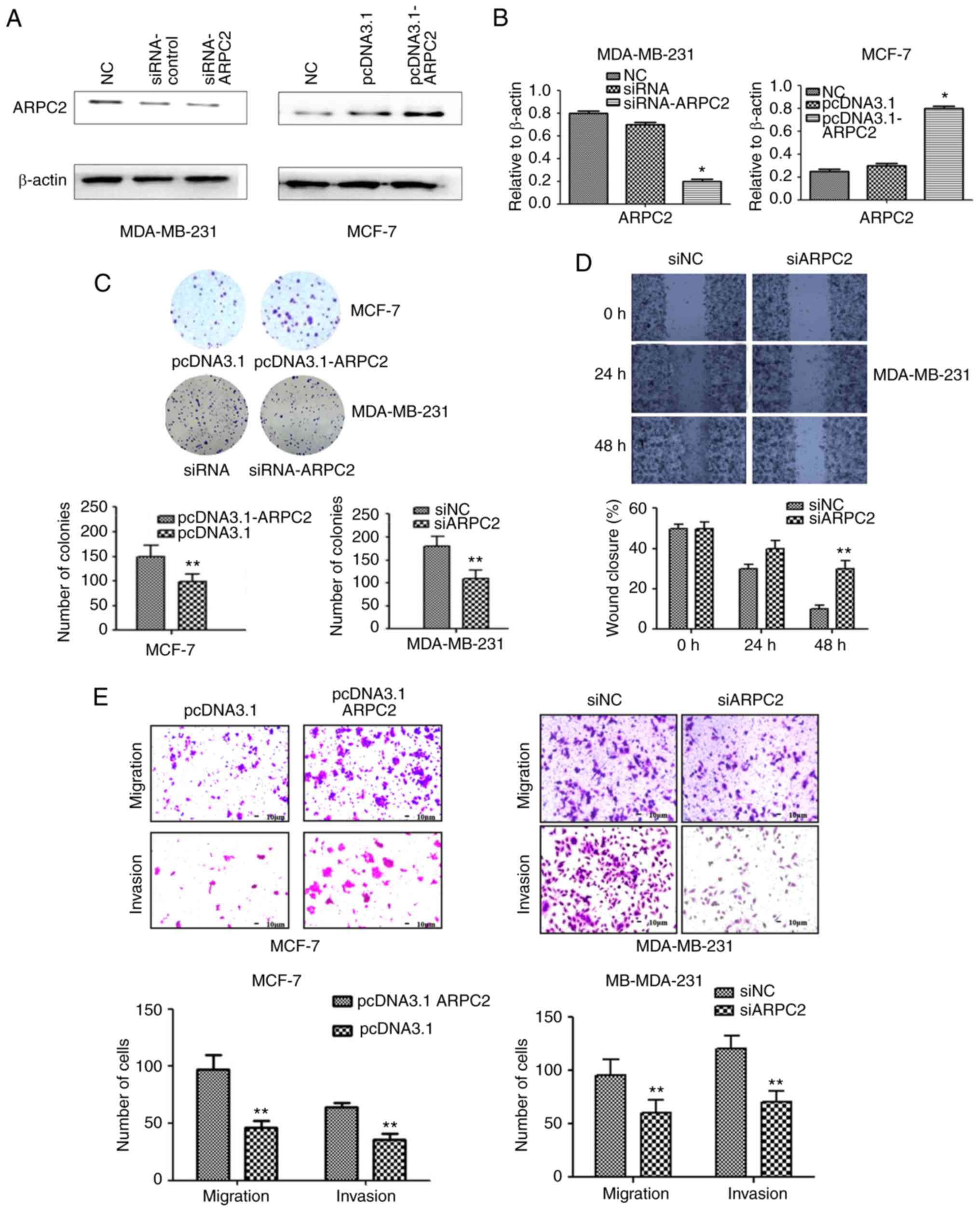
To delineate the role of ARPC2 in BrCa
tumorigenesis, we knocked out ARPC2 in MDA-MB-231 cells and
overexpressed ARPC2 expression in MCF-7 cell lines. We confirmed
ARPC2 expression by performing western blotting and qRT-PCR
(Fig. 2A and B). To further confirm
the role of ARPC2 in BrCa migration and invasion, we used a colony
formation and scratch wound healing assays (Fig. 2C and D). The colony forming ability
of cells was decreased and the percentage of wound closure was
increased by ARPC2 silencing in MDA-MB-231 cells. Similarly, the
migration and the invasion of MDA-MB-231 was also inhibited by
ARPC2 silencing as determined by Transwell assays. Thus, the
inducive effect of ARPC2 in BrCa cell migration and invasion was
revealed.
ARPC2 overexpression inhibits the
growth of BrCa cells
We assessed the effects of pcDNA3.1-ARPC2
transfection on cellular proliferation and growth through colony
formation assays. Our results demonstrated that pcDNA3.1-ARPC2
displayed a relatively rapid proliferation rate compared to control
cells when MCF-7 cells were transfected with pcDNA3.1-ARPC2. The
colony forming ability (Fig. 2C) as
well as the migration and invasion abilities (Fig. 2E) were enhanced as determined by
colony formation and Transwell assays.
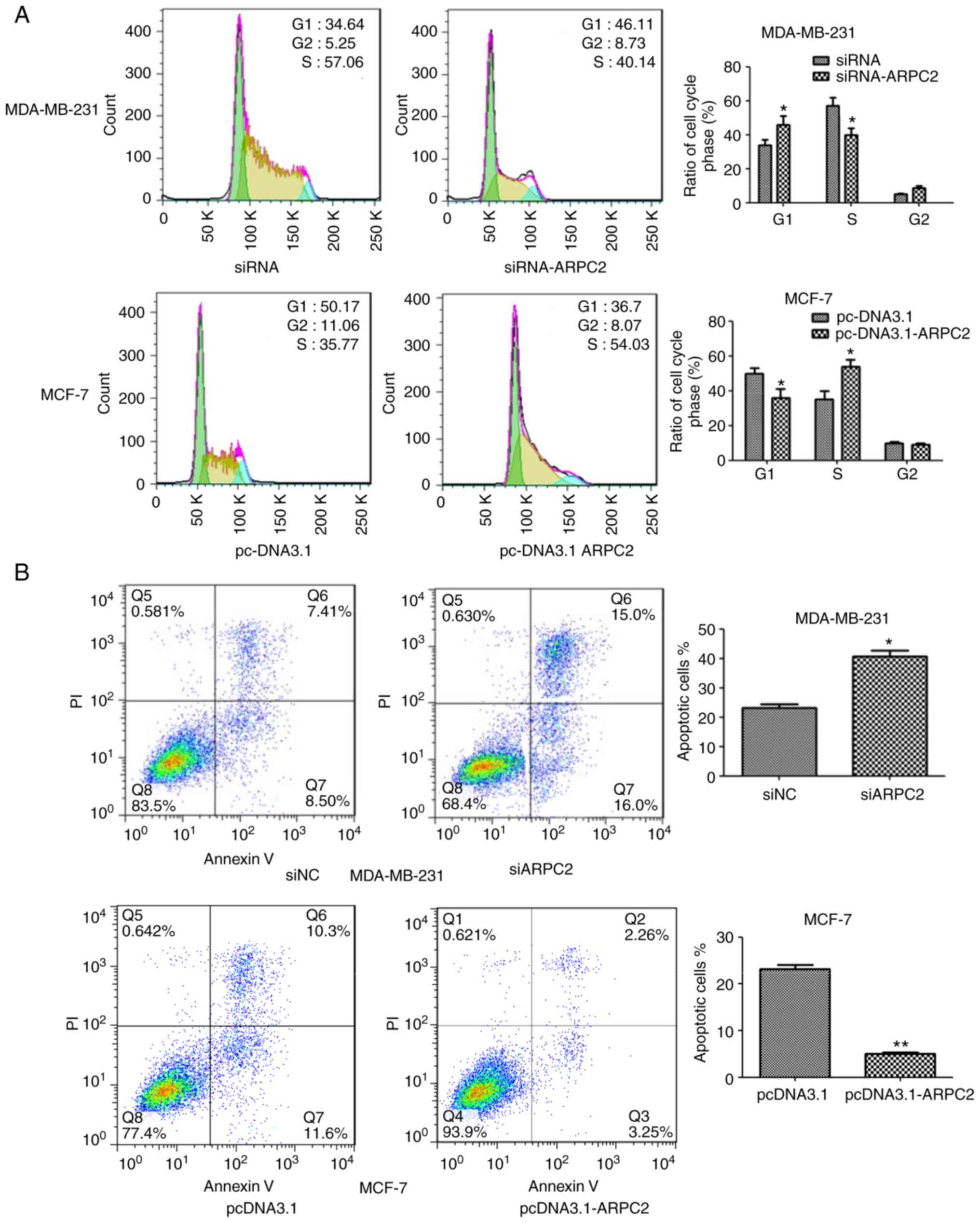
ARPC2 silencing inhibits BrCa cell
proliferation and induces apoptosis
We downregulated ARPC2 expression in the MDA-MB-231
cell line through transfection with an ARPC2-specific siRNA or a
non-specific siRNA control. As revealed in Fig. 3A, ARPC2 silencing altered the cell
cycle of the MDA-MB-231 cell line. We found that a higher number of
siARPC2 MDA-MB-231 cells were in the G1 phase compared with the
control cells. In addition, the transfection of siARPC2 increased
apoptosis of MDA-MB-231 cells, as determined by performing Annexin
V-FITC/PI staining (Fig. 3B).
TGF-β signaling is activated in
ARPC2-induced EMT
ARPC2 overexpression significantly increased the
level of phosphorylated Smad2 (p-Smad2) protein, a downstream
effector of the TGF-β pathway (Fig.
4A). Moreover, ARPC2 overexpression efficiently increased
phosphorylated Smad2 levels. To further validate that TGF-β
signaling is responsible for ARPC2-induced EMT, we used TGF-β
receptor kinase inhibitor SB43 to block TGF-β signaling. The
results revealed that suppression of TGF-β signaling reduced Smad2
phosphorylation levels and downregulated vimentin expression
(Fig. 4B). To investigate whether
ARPC2 is required for TGF-β-induced EMT, we transfected MDA-MB-231
cells with siARPC2 or non-target control siRNA and examined their
responses to TGF-β treatments after 48 h. It was determined that
TGF-β stimulation induced EMT transfer in MDA-MB-231-siCtrl cells,
but not in MB-MDA-231-siARPC2 cells. TGF-β treatment inhibited
E-cadherin expression and increased vimentin expression. However,
under the same conditions E-cadherin and vimentin expression levels
were not altered in MB-MDA-231-siARPC2 cells (Fig. 4C). These experiments suggested that
ARPC2 is involved in TGF-β-induced EMT.
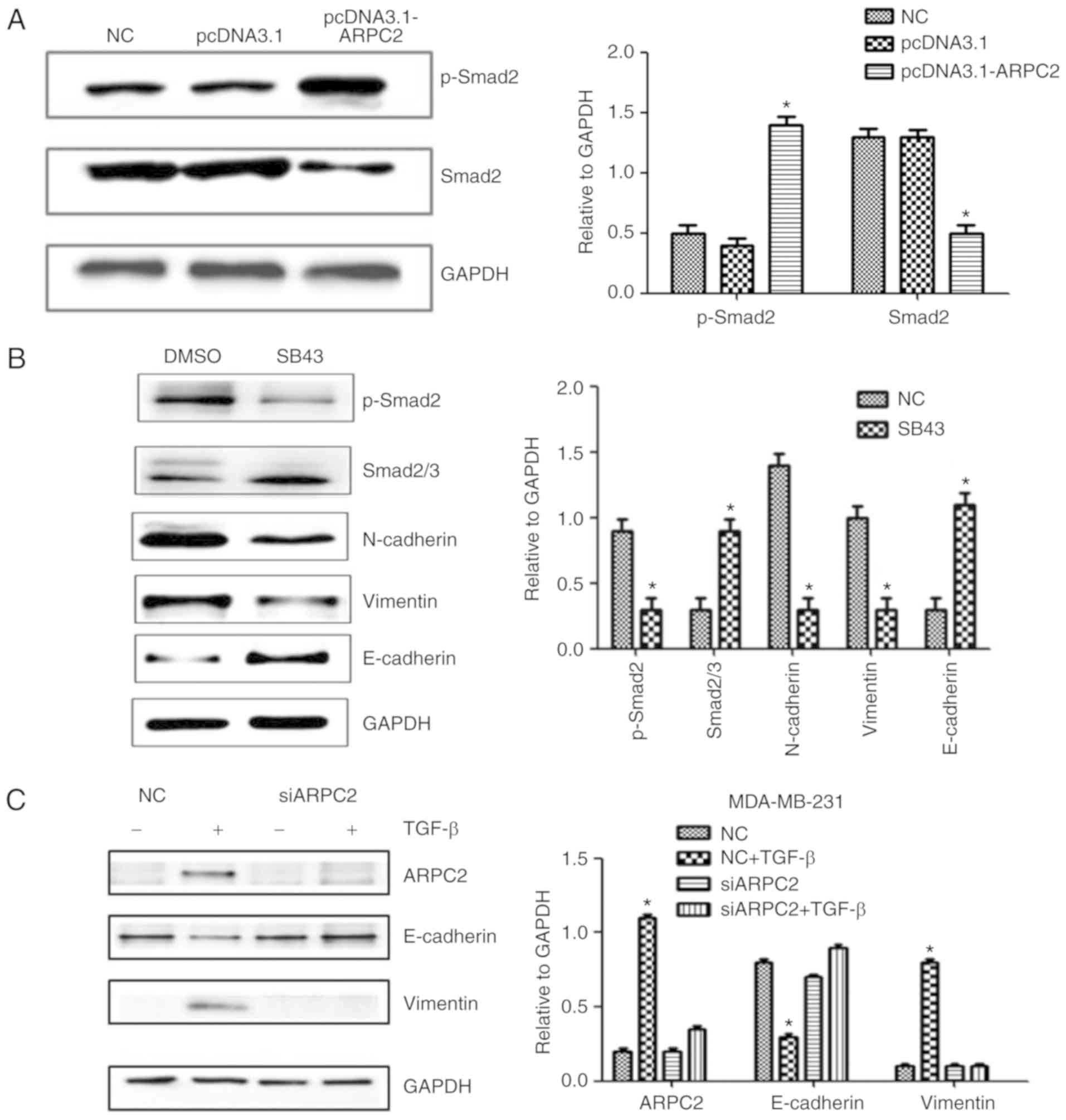 | Figure 4.Western blotting detects EMT-related
protein expression levels after ARPC2 knockdown or overexpression.
(A) MCF-7 cells were stably transfected with NC or pcDNA3.1-ARPC2,
and the levels of p-Smad2 and Smad2/3 were assessed by western
blotting. *P<0.05. (B) MCF-7 cells were treated with 10 ng/l
SB43 or DMSO for 48 h, and the protein expression levels of EMT
markers, p-Smad2 and total Smad2/3 protein were assessed by western
blotting. *P<0.05. (C) Western blotting was performed to examine
ARPC2, E-cadherin, and vimentin expression after 48 h of treatment
with TGF-β in siCtrl and siARPC2. *P<0.05. (D) Effect of ARPC2
downregulation, analysis of the expression of ARPC2, E-cadherin,
vimentin, MMP-9, MMP-3 and ZEB-1 by western blotting. *P<0.05.
(E) Effect of ARPC2 overexpression. Analysis of the level of ARPC2,
E-cadherin, vimentin, MMP-9, MMP-3 and ZEB-1 by western blotting.
*P<0.05. (F) ARPC2 mRNA expression levels in MDA-MB-231 cells
treated with different concentrations of TGF-β at 48 h and (10
ng/ml) TGF-β at different time-points. EMT, epithelial-mesenchymal
transition; ARPC2, actin-related protein 2/3 complex. *P<0.05,
**P<0.01. |
ARPC2 regulates EMT-related protein
expression
Cellular adhesive ability is associated with EMT. To
determine whether ARPC2 expression triggers EMT in BrCa cells, we
examined MMP-3, MMP-9 (cell migration marker), E-cadherin
(epithelial cell marker), N-cadherin, vimentin (mesenchymal marker)
and ZEB-1 protein expression after transfection with siARPC2 and
pcDNA3.1-ARPC2 (Fig. 4D and E).
Compared to the control cell line, the expression levels of,
vimentin, MMP-3, MMP-9 and ZEB-1 were upregulated by transfection
with pcDNA3.1-ARPC2 and the level of E-cadherin was downregulated.
These results demonstrated that ARPC2 stimulated the expression of
EMT-related proteins and EMT-TFs. The transfection of
pcDNA3.1-ARPC2 resulted in a markedly high invasiveness of BrCa.
Knockdown of ARPC2 by siRNA resulted in the inhibition of invasion
and metastasis of BrCa. To examine whether the TGF-β could
stimulate the expression of ARPC2 in MDA-MB-231 cells, cells were
incubated with the presence 10 ng/ml TGF-β for 12, 24 and 48 h.
Subsequently, we used 2.5, 5, and 10 ng/ml TGF-β to culture cells
for 48 h. qPCR analysis revealed that ARPC2 mRNA expression was
time- and dose-dependent on TGF-β stimulation (Fig. 4F).
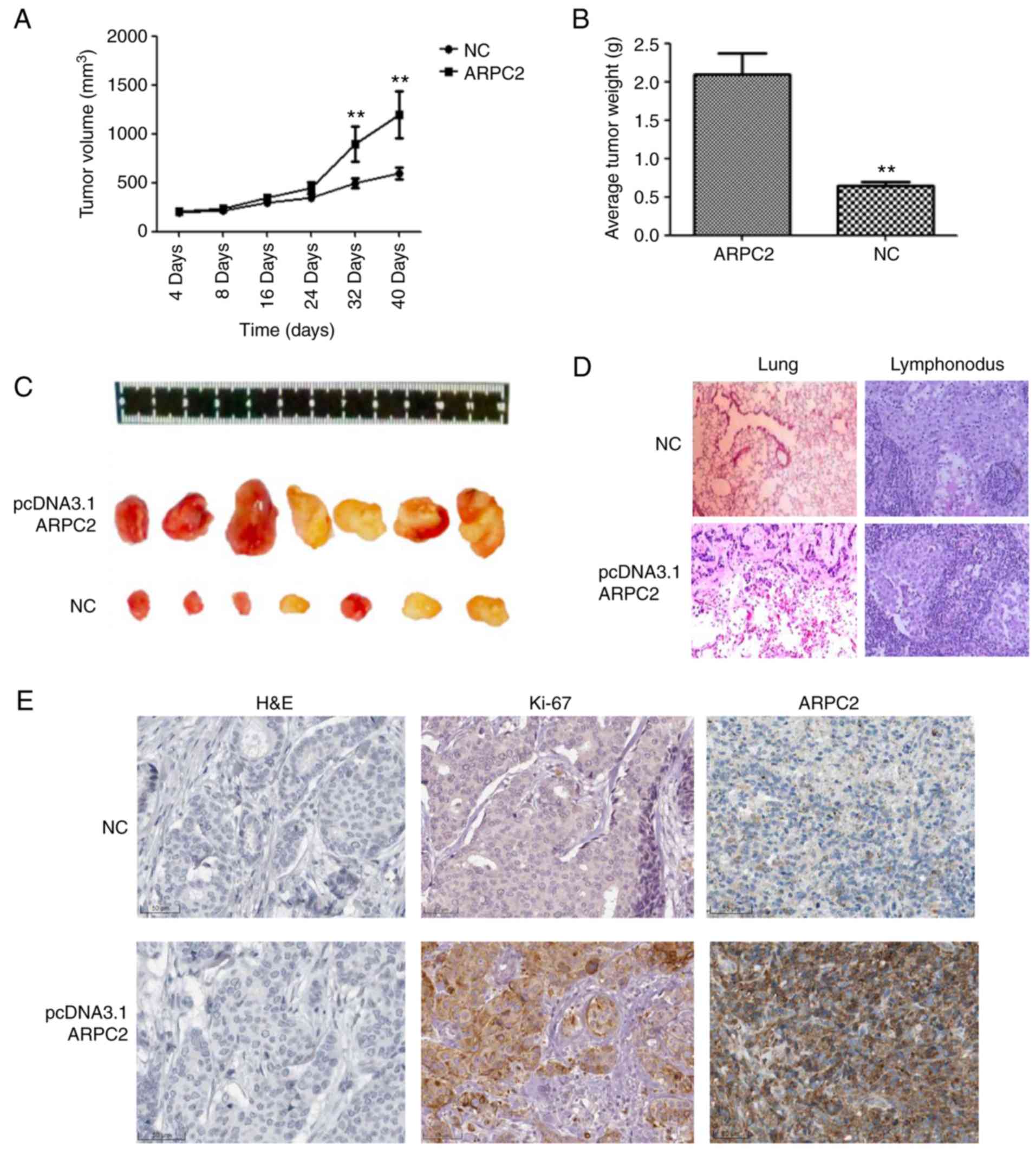
ARPC2 overexpression promotes mammary
tumor growth in a xenograft model
We demonstrated the biological role of ARPC2 in
MDA-MB-231 cells through an in vivo experiment. We
transfected the ARPC2 vector into MDA-MB-231 cells which were then
subcutaneously injected into BALB/c nude mice. MDA-MB-231 and
ARPC2-MDA-MB-231 groups were examined 40 days after implantation
(Fig. 5). The ARPC2-MDA-MB-231
group of mice formed significantly larger tumor sizes than the
MDA-MB-231 control group (Fig. 5C).
As revealed in Fig 5, a high ARPC2
level increased tumor growth and weight (Fig. 5A and B), and generated lung or
lymphonodus metastases in nude mice (Fig. 5D). We subsequently explored the
effects of ARPC2 on tumor growth in vivo by IHC, the results
of which revealed that tumour growth and tumor weight were
significantly increased in the ARPC2-overexpression group compared
with the negative control group and that the Ki-67 expression
levels in the ARPC2-overexpression group were higher than those in
the negative control group.
Discussion
Recurrent and metastatic BrCa gravely threatens the
health of women worldwide. Thus, intensive studies on the potential
candidate molecules involved in the metastasis of BrCa are urgently
required for the development of BrCa treatment. Previous studies on
microarray profiling have revealed that ARPC2 is aberrantly
expressed during BrCa progression. In the present study, it was
revealed that ARPC2 expression was increased in BrCa cell lines
(MCF-7, BT474, MDA-MB-231 and T47D). Furthermore, ARPC2 expression
was uniquely linked with the invasion, apoptosis and proliferation
of mammary carcinoma cells. This positive association was further
supported by evidence obtained through in vitro studies and
a nude mouse model. The analysis of clinical specimens, IHC
results, and clinicopathological and prognostic information
indicated that ARPC2 was significantly associated with tumor size,
lymph node metastasis, and tumor grade. Moreover, Kaplan-Meier
analyses revealed that ARPC2 expression was linked with the poor OS
of patients with BrCa. Therefore, we proposed that ARPC2 has
regulatory functions in BrCa development and that it can be treated
as an independent prognostic marker for BrCa.
The evolutionarily conserved Arp2/3 complex consists
of five subunits (ARPC1-5) (28,29),
and two actin-related proteins ARP2 and ARP3. ARPC2 can promote
actin assembly in lamellipodia and participate in lamellipodial
protrusions, which promote cell shape change and locomotion. ARPC2,
along with some co-expressed genes, plays a crucial role in
invasion and metastasis (30–32).
To explore the regulatory network of ARPC2, we found that ARPC2 may
be involved in EMT, a key early event in breast tumor invasion and
metastasis. A growing body of evidence has revealed that EMT, a
specific physiological or pathophysiological phenomenon, is
characterized by the conversion of epithelial cells into
mesenchymal-like cells through the loss of polarity and disruption
of cell-cell connections (33,34).
Although the significance of EMT-induced carcinogenesis has been
defined, its molecular mechanism remains unclear.
EMT involves the dissolution of epithelial tight
junctions, the modulation of adherent junctions, the remodeling of
the cytoskeleton, and the loss of apical-basal polarity. Matrix
metalloproteinases (MMPs) are EMT-related proteins (35) and ZEB1 is crucial for the
transcriptional regulation of EMT (14). N-cadherin and vimentin are present
in the microfilaments (actins) and microtubules (tubulins)
(36,37). Arp2/3 subunits exhibit a
tissue-specific expression pattern characterized by the increased
expression of mesenchymal proteins. To directly test this
hypothesis, we quantified the expression levels of ARPC2,
E-cadherin, vimentin, ZEB1, MMP-9 and MMP-3 proteins. We found that
in vitro ARPC2 increased breast tumor cell migration and
invasion, as well as vimentin, ZEB1, MMP-9 and MMP-3 proteins. The
results demonstrated that ARPC2 is an oncogene or has oncogenic
effects on BrCa cell growth and metastasis by stimulating EMT.
Recently, a study revealed that a TGF-β/Smad-dependent gene
regulated fibronectin and smooth muscle actin (38) in human lung mesenchymal cells. In
the present study, we found that TGF-β signaling was activated in
ARPC2-induced EMT. In addition, TGF-β signaling induced ARPC2
expression in breast epithelial cells, and the knockdown of ARPC2
blocked TGF-β-induced EMT. We proposed that a feedback loop exists
between ARPC2 and TGF-β expression. This feedback loop could
regulate EMT and BrCa progression.
To the best of our knowledge, we are the first to
report that ARPC2 plays a critical role in BrCa in vitro and
in vivo. ARPC2 overexpression promoted BrCa cell
proliferation, invasion, and metastasis through EMT. Our results
indicated that ARPC2 is a potential biomarker or therapeutic target
for BrCa.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific
Research Foundation of the Institute for Translational Medicine of
Anhui Province, SRFITMAP (no. 2017zhyx36).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
QW conceived and designed the study and the
experiments and acquired the funding. ZC, WW and YH performed the
experiments. JW selected the patients and collected the clinical
samples; YS, YH and JW analyzed the data. ZW, XD and YS collected
data. ZC and WW wrote the manuscript. ZW and XD reviewed and edited
the manuscript. All authors have read and approved the final
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work re appropriately investigated and resolved.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Anhui Medical University (Hefei, China) and consent
was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
There authors declare that they have no competing
interests.
References
|
1
|
DeSantis CE, Ma J, Goding Sauer A, Newman
LA and Jemal A: Breast cancer statistics, 2017, racial disparity in
mortality by state. CA Cancer J Clin. 67:439–448. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ngamcherdtrakul W, Castro DJ, Gu S, Morry
J, Reda M, Gray JW and Yantasee W: Current development of targeted
oligonucleotide-based cancer therapies: Perspective on
HER2-positive breast cancer treatment. Cancer Treat Rev. 45:19–29.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maruti SS, Willett WC, Feskanich D, Rosner
B and Colditz GA: A prospective study of age-specific physical
activity and premenopausal breast cancer. J Natl Cancer Inst.
100:728–737. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hiatt RA, Klabunde C, Breen N, Swan J and
Ballard-Barbash R: Cancer screening practices from National Health
Interview Surveys: Past, present, and future. J Natl Cancer Inst.
94:1837–1846. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Slatkin M: Linkage
disequilibrium-understanding the evolutionary past and mapping the
medical future. Nat Rev Genet. 9:477–485. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim IA, No M, Lee JM, Shin JH, Oh JS, Choi
EJ, Kim IH, Atadja P and Bernhard EJ: epigenetic modulation of
radiation response in human cancer cells with activated EGFR or
HER-2 signaling: Potential role of histone deacetylase 6. Radiother
Oncol. 92:125–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lakhani SR, Van De Vijver MJ, Jacquemier
J, Anderson TJ, Osin PP, McGuffog L and Easton DF: The pathology of
familial breast cancer: Predictive value of immunohistochemical
markers estrogen receptor, progesterone receptor, HER-2, and p53 in
patients with mutations in BRCA1 and BRCA2. J Clin
Oncol. 20:2310–2318. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kyndi M, Sørensen FB, Knudsen H, Overgaard
M, Nielsen HM and Overgaard J; Danish Breast Cancer Cooperative
Group, : Estrogen receptor, progesterone receptor, HER-2, and
response to postmastectomy radiotherapy in high-risk breast cancer:
The Danish Breast Cancer Cooperative Group. J Clin Oncol.
26:1419–1426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jeng RL, Goley ED, D'Alessio JA, Chaga OY,
Svitkina TM, Borisy GG, Heinzen RA and Welch MD: A
Rickettsia WASP-like protein activates the Arp2/3 complex
and mediates actin-based motility. Cell Microbiol. 6:761–769. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goley ED and Welch MD: The ARP2/3 complex:
An actin nucleator comes of age. Nat Rev Mol Cell Biol. 7:713–726.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Frank DJ, Hopmann R, Lenartowska M and
Miller KG: Capping protein and the Arp2/3 complex regulate
nonbundle actin filament assembly to indirectly control actin
bundle positioning during Drosophila melanogaster bristle
development. Mol Biol Cell. 17:3930–3939. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Al Ghouleh I, Rodríguez A, Pagano PJ and
Csányi G: Proteomic analysis identifies an NADPH oxidase 1
(Nox1)-mediated role for actin-related protein 2/3 complex subunit
2 (ARPC2) in promoting smooth muscle cell migration. Int J Mol Sci.
14:20220–20235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang J, Liu Y, Yu CJ, Dai F, Xiong J, Li
HJ, Wu ZS, Ding R and Wang H: Role of ARPC2 in human gastric
cancer. Mediators Inflamm. 2017:54328182017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moreno-Bueno G, Portillo F and Cano A:
Transcriptional regulation of cell polarity in EMT and cancer.
Oncogene. 27:6958–6969. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wright JA, Richer JK and Goodall GJ:
microRNAs and EMT in mammary cells and breast cancer. J Mammary
Gland Biol Neoplasia. 15:213–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Creighton CJ, Chang JC and Rosen JM:
Epithelial-mesenchymal transition (EMT) in tumor-initiating cells
and its clinical implications in breast cancer. J Mammary Gland
Biol Neoplasia. 15:253–260. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hardy KM, Booth BW, Hendrix MJ, Salomon DS
and Strizzi L: ErbB/EGF signaling and EMT in mammary development
and breast cancer. J Mammary Gland Biol Neoplasia. 15:191–199.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Doble BW and Woodgett JR: Role of glycogen
synthase kinase-3 in cell fate and epithelial-mesenchymal
transitions. Cells Tissues Organs. 185:73–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kudo-Saito C, Shirako H, Takeuchi T and
Kawakami Y: Cancer metastasis is accelerated through
immunosuppression during snail-induced EMT of cancer cells. Cancer
Cell. 15:195–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gavert N and Ben-Ze'ev A:
Epithelial-mesenchymal transition and the invasive potential of
tumors. Trends Mol Med. 14:199–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ma XJ, Dahiya S, Richardson E, Erlander M
and Sgroi DC: Gene expression profiling of the tumor
microenvironment during breast cancer progression. Breast Cancer
Res. 11:R72009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Curtis C, Shah SP, Chin SF, Turashvili G,
Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et
al: The genomic and transcriptomic architecture of 2,000 breast
tumours reveals novel subgroups. Nature. 486:346–352. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Perou CM, Sørlie T, Eisen MB, van de Rijn
M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA,
et al: Molecular portraits of human breast tumours. Nature.
406:747–752. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao H, Langerod A, Ji Y, Nowels KW,
Nesland JM, Tibshirani R, Bukholm IK, Kåresen R, Botstein D,
Børresen-Dale AL, et al: Different gene expression patterns in
invasive lobular and ductal carcinomas of the breast. Mol Biol
Cell. 15:2523–2536. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Abella JV, Galloni C, Pernier J, Barry DJ,
Kjær S, Carlier MF and Way M: Isoform diversity in the Arp2/3
complex determines actin filament dynamics. Nat Cell Biol.
18:76–86. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Robinson R, Turbedsky K, Kaiser D,
Marchand J, Higgs H, Choe S and Pollard T: Crystal structure of
Arp2/3 complex. Science. 294:1679–1684. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Daugherty KM and Goode BL: Functional
surfaces on the p35/ARPC2 subunit of Arp2/3 complex required for
cell growth, actin nucleation, and endocytosis. J Biol Chem.
283:16950–16959. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Saedler R, Mathur N, Srinivas BP,
Kernebeck B, Hülskamp M and Mathur J: Actin control over
microtubules suggested by DISTORTED2 encoding the
Arabidopsis ARPC2 subunit homolog. Plant Cell Physiol.
45:813–822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Franke A, Balschun T, Karlsen TH,
Sventoraityte J, Nikolaus S, Mayr G, Domingues FS, Albrecht M,
Nothnagel M, Ellinghaus D, et al: Sequence variants in IL10,
ARPC2 and multiple other loci contribute to ulcerative colitis
susceptibility. Nat Genet. 40:1319–1323. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shapiro IM, Cheng AW, Flytzanis NC,
Balsamo M, Condeelis JS, Oktay MH, Burge CB and Gertler FB: An
EMT-driven alternative splicing program occurs in human breast
cancer and modulates cellular phenotype. PLoS Genet.
7:e10022182011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cannito S, Novo E, di Bonzo LV, Busletta
C, Colombatto S and Parola M: Epithelial-mesenchymal transition:
From molecular mechanisms, redox regulation to implications in
human health and disease. Antioxid Redox Signal. 12:1383–1430.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kessenbrock K, Plaks V and Werb Z: Matrix
metalloproteinases: Regulators of the tumor microenvironment. Cell.
141:52–67. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mendez MG, Kojima S and Goldman RD:
Vimentin induces changes in cell shape, motility, and adhesion
during the epithelial to mesenchymal transition. FASEB J.
24:1838–1851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Prasain N and Stevens T: The actin
cytoskeleton in endothelial cell phenotypes. Microvasc Res.
77:53–63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hur W, Rhim H, Jung CK, Kim JD, Bae SH,
Jang JW, Yang JM, Oh ST, Kim DG, Wang HJ, et al: SOX4
overexpression regulates the p53-mediated apoptosis in
hepatocellular carcinoma: Clinical implication and functional
analysis in vitro. Carcinogenesis. 31:1298–1307. 2010. View Article : Google Scholar : PubMed/NCBI
|