Introduction
Lung cancer is the most frequent malignancy, causing
1.6 million deaths/year worldwide (1). Approximately 85% of lung cancers
consist of non-small cell lung carcinoma (NSCLC). NSCLC is
generally categorized into three major histological subtypes: Lung
adenocarcinoma, lung squamous cell carcinoma and large cell
carcinoma, and lung adenocarcinoma is the most prevalent form of
lung cancer (2). Although treatment
strategies for NSCLC have markedly increased in recent years, the
estimated 5-year overall survival (OS) remains only 16% (3). Thus, understanding the molecular
mechanisms which regulate the progression of lung adenocarcinoma
would lead to improved treatment of patients.
MicroRNAs (miRNAs) are a class of small non-coding
RNAs that negatively regulate genes at the post-transcriptional
level by directly binding to the 3′-untranslated region (UTR) of
target mRNAs; resulting in the degradation of mRNA molecules or the
inhibition of their translation (4,5).
Moreover, miRNAs are involved in the regulation of multiple
biological processes involving cancer, such as tumorigenesis and
development, cell proliferation, metastasis, invasion and apoptosis
(6,7). In recent years, increasing evidence
has revealed the relationships between miRNAs and disease
progression of NSCLC. For example, miR-30a/c reduction maintained
self-renewal and promoted tumorigenesis in NSCLC-initiating cells
by targeting oncogene TM4SF1 (8),
while Song et al reported that miR-409 inhibited human
non-small cell lung cancer progression by directly targeting SPIN1
(9). Although the function of
numerous miRNAs has been reported in lung cancer, the molecular
regulatory mechanisms of miRNAs and their effects on lung cancer
progression are still not well understood.
Recently, miR-888 was revealed to be upregulated in
several types of cancer, such as endometrial and colorectal cancer,
and the upregulated levels of miR-888 were correlated with the poor
outcome in patients with endometrial and colorectal cancer
(10–12). In addition, several studies revealed
that miR-888 functioned as an oncogene and modulated cancer cell
proliferation, invasion and migration (13–16).
However, the biological role of miR-888 in lung adenocarcinoma is
not fully elucidated.
In the present study, the expression, roles, and
mechanisms of miR-888 in the progression of lung adenocarcinoma in
A549 cells and human lung adenocarcinoma tissues were investigated,
and the results revealed that miR-888 may be a potential new
therapeutic target in lung adenocarcinoma.
Materials and methods
Tissue samples for patients with lung
adenocarcinoma
A total of 38 pairs of primary lung adenocarcinoma
and adjacent non-tumor tissues were obtained from the Affiliated
Zhongshan Hospital of Xiamen University (between January 2014 and
June 2016). The clinical characteristics of 38 lung adenocarcinoma
patients were as follows: Mean age (range): 62.9 years (45–81
years). Sex: Male, 26 cases; female, 12 cases. Clinical staging:
Stage I–II, 16 cases; and stage III–IV, 22 cases. All patients
provided informed consent, and all specimens were confirmed by a
pathologist. The study was approved by the Research Ethics
Committee of Xiamen University (Xiamen, China).
Immunohistochemistry (IHC)
For IHC, 4-µm thick slides were deparaffinized in
xylene and rehydrated in a descending graded series of alcohol
dilutions. Antigen retrieval was performed in 10 mM citrate buffer
(pH 6.0) in a microwave oven at maximum power (800 W) for 3 min,
followed by 15 min at medium power, and cooling to room
temperature. After phosphate-buffered saline (PBS) washes, the
slides were blocked with 3% H2O2 for 10 min
and blocked with goat serum for 30 min at room temperature. The
sections were incubated with a primary mouse anti-human antibody
against cell division cycle 7 (CDC7) (dilution 1:100; cat. no.
sc-56275; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight
at 4°C. The slides were washed thrice with PBS and then incubated
with an HRP-conjugated secondary antibody (dilution 1:250; cat. no.
ZDR-5307; ZSGB-BIO, Beijing, China) for 30 min at room temperature.
For all slides, the immune reaction was assessed using
diaminobenzidine, and the sections were then counterstained with
hematoxylin. For the semiquantitative analysis of CDC7
immunoreactivity, the immunohistochemical score (IHS) was used.
Briefly, IHS=SI (staining intensity) × PP (percentage of positive
cells). The staining intensity (SI) was categorized into 4 groups
(0–3), where 0 was negative, 1 was weak, 2 was moderate, and 3 was
strong. The PP was estimated and classified on a five-point
positive range score as follows: 0, ≤5% staining; 1, 6–25%
staining; 2, 26–50% staining; 3, 51–74% staining; and 4, ≥75%
staining. Cases were categorized into two groups: IHS=0, negative
and IHS ≥1, positive (17).
Cell culture
The human non-small cell lung cancer (NSCLC) cell
line A549 was obtained from the American Type Culture Collection
(ATCC; Manassas, VA, USA). Cells were maintained in Dulbecco's
modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS;
HyClone, Thermo Scientific, Inc., Waltham, MA, USA) and grown at
37°C in 5% CO2.
Plasmid generation, small interfering
RNA (siRNAs), and transfection
The pcDNA3.1-CDC7 construct was kindly provided by
Dr Peter Cherepanov (Imperial College London, London, UK). For the
construction of luciferase reporter plasmids, the full length CDC7
3′-UTR, truncated versions of this UTR, or constructs containing
3′-UTR point mutations were amplified by PCR and inserted into the
pMiR-reporter plasmid. Primers were as follows: CDC7
full-length 3′-UTR forward,
5′-GGACTAGTCCTAATGGATCTTCATTTAATGTTTAC-3′ and reverse,
5′-CCCAAGCTTGGGTAAAAAATATAAAAGGATAACTTTATTG-3′; Fragment A forward,
5′-GGACTAGTCCTAATGGATCTTCATTTAATGTTTAC-3′ and reverse,
5′-CCCAAGCTTAACAGAAACTTTGTGGTCAG-3′; Mut A forward,
5′-CTAACAACATGATCTTCTTTCCTTTAAACCTACCTAAGTA-3′ and reverse,
5′-TACTTAGGTAGGTTTAAAGGAAAGAAGATCATGTTGTTAG-3′; Fragment B forward,
5′-GGACTAGTAAGTTTCTGGATGTTTTA-3′ and reverse,
5′-CCCAAGCTTGGGTAAAAAATATAAAAGGATAACTTTATTG-3′; Mut B forward,
5′-CCAAATGCTTTTCTTTTTTCCTTTGTATATTTTTTCACAC-3′ and reverse,
5′-GTGTGAAAAAATATACAAAGGAAAAAAGAAAAGCATTTGG-3′. The CDC7,
E-cadherin and TIMP2 siRNA target sequences, miR-888 mimics and
inhibitors were synthesized by Shanghai GenePharma Co., Ltd.,
(Shanghai, China), and the sequences were as follows: CDC7,
5′-AAGCAGUCAAAGACUGUGGAU-3′; E-cadherin, 5′-CAGACAAAGACCAGGACUA-3′;
TIMP2-1, 5′-GGAAAGAAGGAAUAUCUCA-3′; TIMP2-2,
5′-GGAAGUGGACUCUGGAAAC-3′, both TIMP2-1 and TIMP2-2 targeted TIMP2,
and in order to enhance the efficacy of knockdown, TIMP2-1 and
TIMP2-2 were mixed together. For siRNA, miR-888 mimic and inhibitor
transfection, 50 nM of synthesized sequences were delivered to the
cells using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
RNA isolation and RT-qPCR
Total RNA was isolated from tissues and cell lines
using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.). For mRNA
detection, cDNA was prepared using RevertAid™ First Strand cDNA
Synthesis kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Gene expression was quantified by
using the Power SYBR-Green PCR Master Mix (Applied Biosystems;
Thermo Fisher Scientific, Inc.). Relative gene expression was
assessed by standard curves and quantified using the
2−ΔΔCq method (18). The
primers were as follows: CDC7 forward,
5′-AGTGCCTAACAGTGGCTGG-3′ and reverse,
5′-CACGGTGAACAATACCAAACTGA-3′; GAPDH forward,
5′-TGTCAGTGGTGGACCTGACCT-3′ and reverse,
5′-AGGGGAGATTCAGTGTGGTG-3′; miR-888 forward,
5′-ACACTCCAGCTGGGTACTCAAAAAGCTGTC-3′ and reverse,
5′-TGGTGTCGTGGAGTCG-3′; U6 forward, 5′-CCTGCTTCGGCAGCACA-3′ and
reverse, 5′-TGGAACGCTTCACGAA-3′. A stem-loop RT primer for miR-888,
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTGACTGAC-3′ and a specific
RT primer for U6, 5′-AAAATATGGAACGCTTCACGAATTTGC-3′ were used.
GAPDH or U6 were used for normalization.
Western blotting
Whole-cell extracts were prepared in RIPA lysis
buffer (Beyotime Institute of Biotechnology, Beijing, China). The
protein concentrations were determined using the BCA Protein Assay
reagent kit (Thermo Fisher Scientific, Inc.). Thirty micrograms of
the sample were separated by 10% SDS-PAGE and transferred onto
nitrocellulose membranes. Then, the membranes were incubated in
blocking solution consisting of 5% w/v non-fat milk in TBST at room
temperature for 1 h, immunoblotted with primary antibodies at 4°C
overnight, and subsequently incubated with a secondary antibody
(dilution 1:1,000; cat. nos. ZDR-5306 and ZDR-5307; ZSGB-BIO) for 1
h at room temperature. The protein band was visualized using
SuperSignal West Pico Chemiluminescent Substrate (Pierce
Biotechnology, Inc.; Thermo Fisher Scientific, Inc.) and the
chemiluminescent image system (ChemiDoc Touch; Bio-Rad
Laboratories, Hercules, CA, USA) and the densitometry of western
blotting was quantified using the ImageJ 1.52a software program
(NIH; National Institutes of Health, Bethesda, MD, USA). The
antibodies used for western blotting were as follows: CDC7
(dilution 1:1,000; cat. no. sc-56275; Santa Cruz Biotechnology),
TIMP2 (dilution 1:1,000; cat. no. CST-5738; Cell Signaling
Technology, Danvers, MA, USA), E-cadherin (dilution 1:1,000; cat.
no. CST-14472; Cell Signaling Technology) and α-tubulin (dilution
1:2,000; cat. no. PM054; MBL, Medical & Biological
Laboratories, Nagoya, Japan).
Luciferase reporter assay
For the luciferase reporter assay, A549 cells were
co-transfected with luciferase reporter plasmids and miR-888 mimics
in 24-well plates. Cells were harvested 48 h after transfection.
The luciferase reporter assay was performed using the
Dual-Luciferase reporter assay system (Promega Corporation,
Madison, WI, USA) according to the manufacturer's instructions.
Wound-healing assay
Cell migration was assayed using a wound healing
assay. A549 cells were transfected with the indicated molecules,
such as miR-888 mimics, miR-888 inhibitors and CDC7 siRNA.
Forty-eight hours after transfection, A549 cells were seeded into
6-well plates. A scratch was generated in the cell monolayer using
a 10-µl pipette tip after A549 cells had grown to a confluence of
80–90%. Cells were gently washed three times with PBS to remove the
cellular debris and incubated in serum-free DMEM. Wound areas were
captured at the indicated time-points (0 and 24 h) using light
microscopy, and the migration distance was quantified using the
ImageJ 1.52a software program (NIH; National Institutes of
Health).
Invasion assay
Cell invasion was assayed using 24-well Transwell
chambers coated with 250 µg/ml Matrigel. A549 cells were
transfected with the indicated molecules, such as miR-888 mimics,
miR-888 inhibitors and CDC7 siRNA. Forty-eight hours after
transfection, a total of 1×104 cells in 200 µl
serum-free medium were added into the upper chamber, and 500 µl
DMEM with 10% FBS was added to the lower well. After 24 h of
incubation, the non-invaded cells on the upper chamber membrane
were removed, and the invaded cells on the lower surface of the
chamber membrane were fixed with 4% paraformaldehyde and stained
with 0.1% crystal violet. Cells were counted in five random fields
using an inverted microscope. Each assay was repeated at least
three times.
Cell proliferation assay
A549 cells were seeded into 96-well plates and
treated with the indicated molecules, such as transfected with
miR-888 mimics, miR-888 inhibitors, or cotransfected with miR-888
mimics and pcDNA3.1-CDC7. The effect of miR-888 on cell
proliferation was measured by CellTiter 96®
AQueous Non-Radioactive Cell Proliferation Assay (MTS)
(Promega Corporation) according to the manufacturer's
instructions.
Colony formation assay
For the colony formation assay, A549 cells after
transfection were seeded in 6-well plates (5×102
cells/well). After incubation for 10 days, the colonies were washed
three times with PBS and fixed with 4% paraformaldehyde for 15 min,
then stained with 0.1% crystal violet for 15 min. Visible cell
colonies (ranging in number from 65 to 362) were imaged and
counted.
Statistical analysis
All statistical analyses were performed using the
software GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA,
USA). Data are presented as the mean ± SD. The paired Student's
t-test was used to compare the difference in paired lung
adenocarcinoma tissue samples and adjacent non-tumor tissues
obtained from the same patient, and other statistical analyses were
conducted using unpaired Student's t-test for two comparisons.
One-way analysis of variance (ANOVA) followed with Bonferroni post
hoc test was used for three and more comparisons. All statistical
tests were two-tailed. A P-value of <0.05 was considered to
indicate a statistically significant difference.
Results
miR-888 is upregulated in the tumors
of patients with lung adenocarcinoma
To explore the biological function of miR-888 in
lung adenocarcinoma, the expression levels of miR-888 in 38 pairs
of lung adenocarcinoma tissues was detected. RT-qPCR revealed that
the expression levels of miR-888 in lung adenocarcinoma tissues
were significantly higher than in adjacent non-tumor tissues
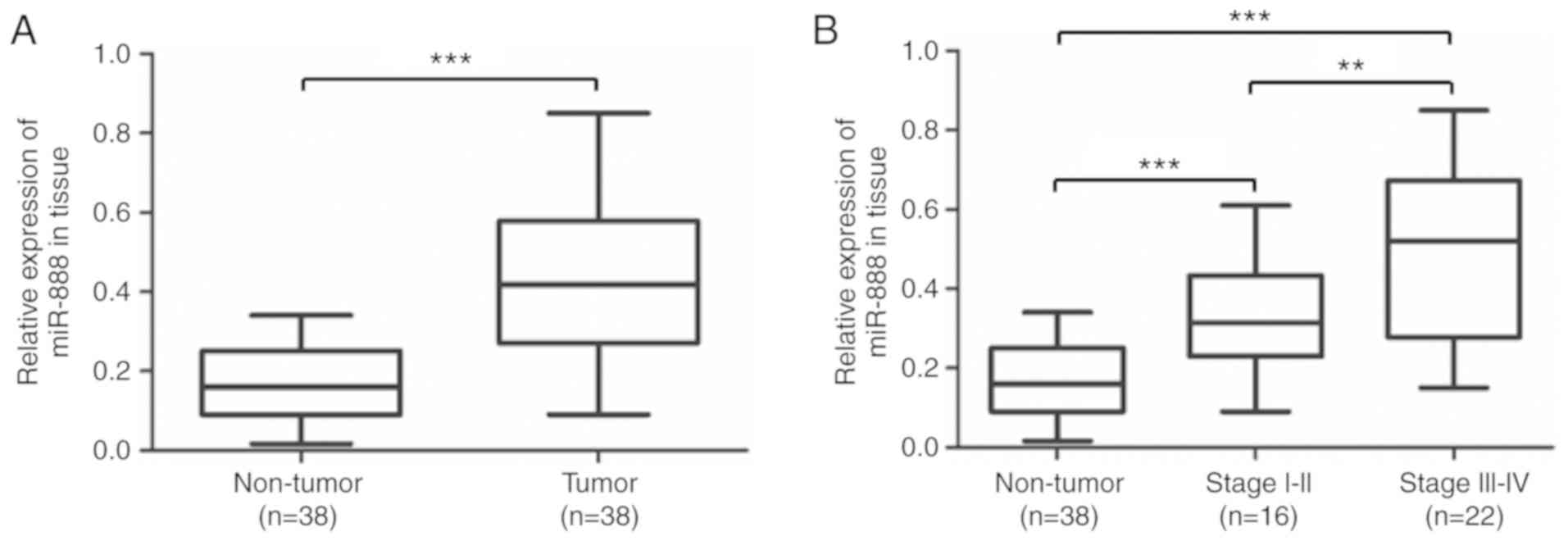
(median=0.427 and 0.163 respectively, Fig. 1A; P<0.001). In addition, an
increase in the level of miR-888 was significantly associated with
the clinical stage of patients. The results revealed that patients
in advanced stages (stage III and IV, n=22) had a much higher
miR-888 expression (median=0.163, 0.330 and 0.498, respectively)
than patients in early stages (stage I and II, n=16; P<0.001,
P=0.0063, P<0.001, respectively; Fig. 1B). These results indicated that
miR-888 may play a crucial role in lung adenocarcinoma
progression.
Effect of miR-888 on the proliferation
of lung adenocarcinoma cells
To investigate the biological function of miR-888 in
the progression of lung adenocarcinoma, miR-888 was overexpressed
or silenced in A549 cells transfected with miR-888 mimics and
inhibitor. MTS assays were performed to assess the effect of
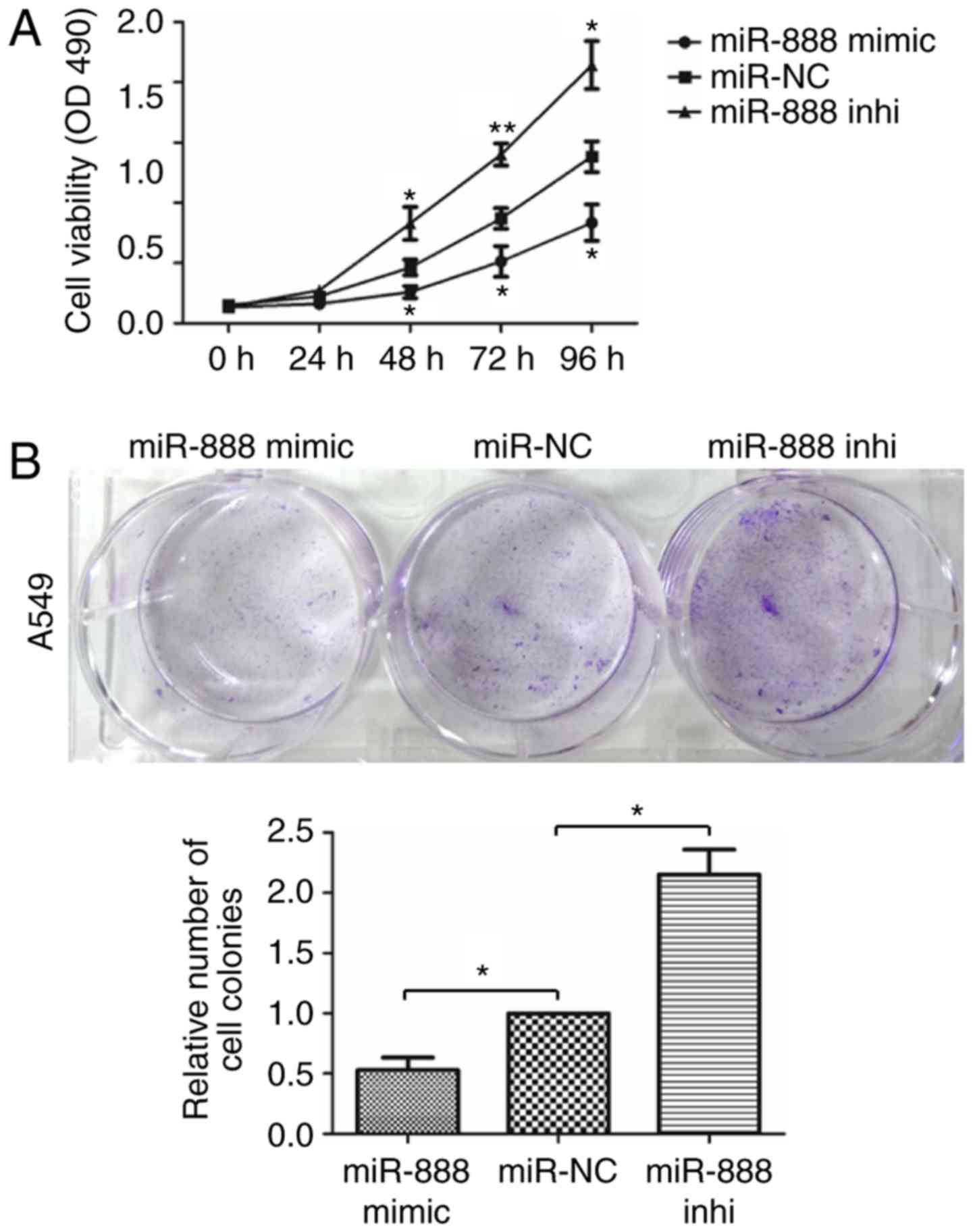
miR-888 on lung adenocarcinoma cell proliferation. The results
revealed that overexpression of miR-888 significantly inhibited the
proliferation of A549 cells (Fig.
2A; miR-888 mimics vs miR-NC: P=0.0117, P=0.0158, P=0.0286;
miR-888 inhibitor vs miR-NC: P=0.0187, P=0.0048, P=0.0165,
respectively), while knockdown of miR-888 in A549 cells revealed
the opposite effect. In addition, colony formation assays also
revealed that upregulation of miR-888 inhibited the colony
formation capacity of A549 cells and knockdown of miR-888 promoted
the colony formation capacity of A549 cells (Fig. 2B; P=0.0460, P=0.0305, respectively).
Collectively, these results indicated that miR-888 suppressed the
proliferation of lung adenocarcinoma cells.
Effect of miR-888 on invasion and
migration of lung adenocarcinoma cells
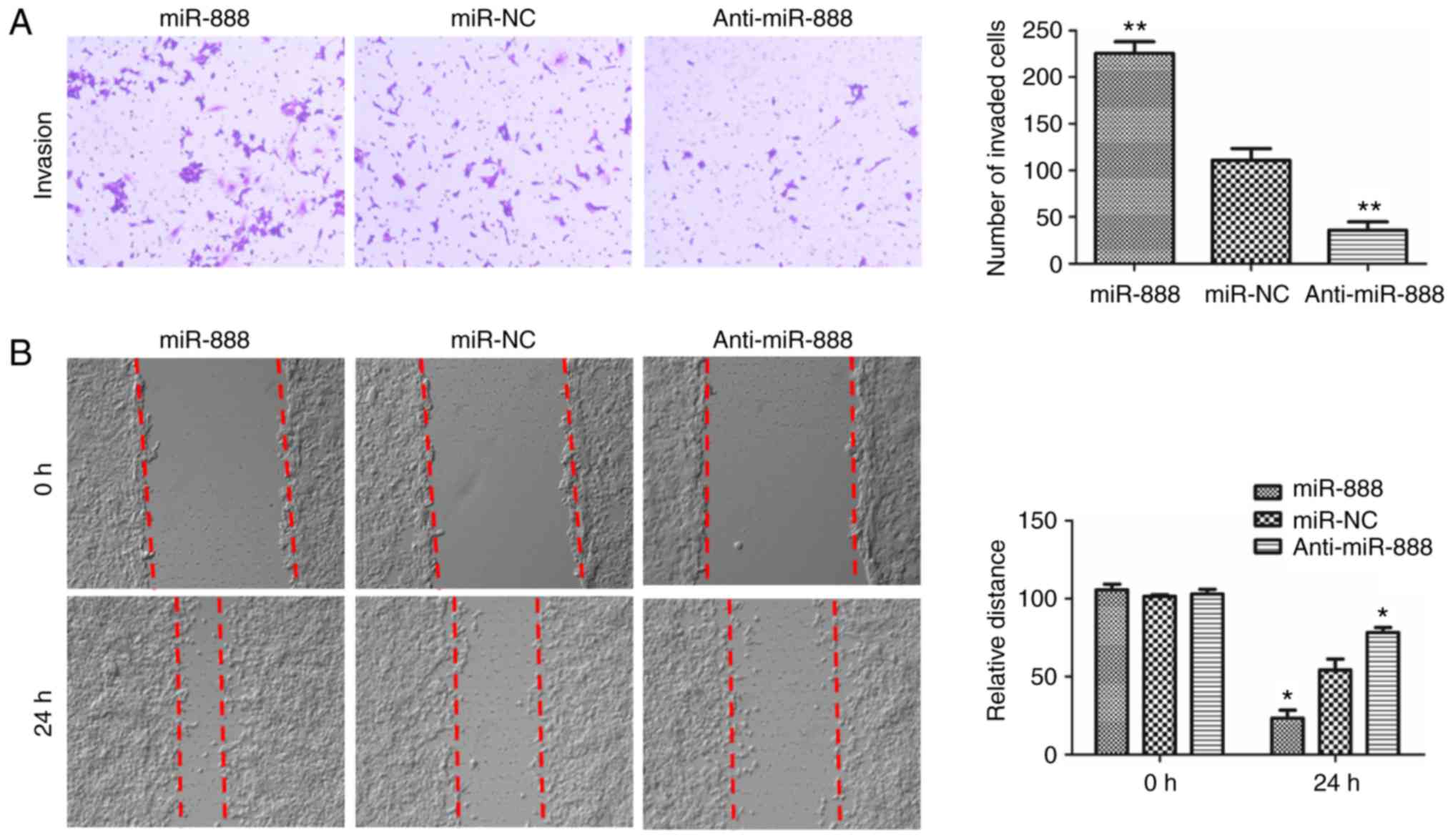
To further assess the effect of miR-888 on the
invasion and migration potential of A549 cells, Transwell and
wound-healing assays were performed. As revealed in Fig. 3A, the Transwell assay revealed that
overexpression of miR-888 in A549 cells significantly increased the
invasive abilities, while downregulation of miR-888 markedly
decreased the invasive abilities (Fig.
3A; P=0.0028, P=0.0081, respectively). In addition,
wound-healing assays revealed that overexpression of miR-888 in
A549 cells significantly increased the wound-healing in the miR-888
mimic-transfected A549 cells after 24 h, while downregulation of
miR-888 in A549 cells revealed the opposite effect (Fig. 3B, P=0.0439, P=0.0252, respectively).
Collectively, these results demonstrated that overexpression of
miR-888 promoted the invasion and migration of lung adenocarcinoma
A549 cells.
miR-888 downregulates CDC7 expression
by targeting the CDC7 3′-UTR
To further explore the mechanisms underlying
miR-888-mediated inhibition of proliferation in lung adenocarcinoma
A549 cells, four bioinformatics tools (miRanda, MirTarget2, PITA
and RNA hybrid) in the miRecords databases (http://c1.accurascience.com/miRecords/) were used to
predict its potential target genes and focused on CDC7 as a
potential target gene for miR-888. It has been reported that CDC7
was involved in the proliferation of cancer cells and can act as an
oncogene in cancer progression (19–21).
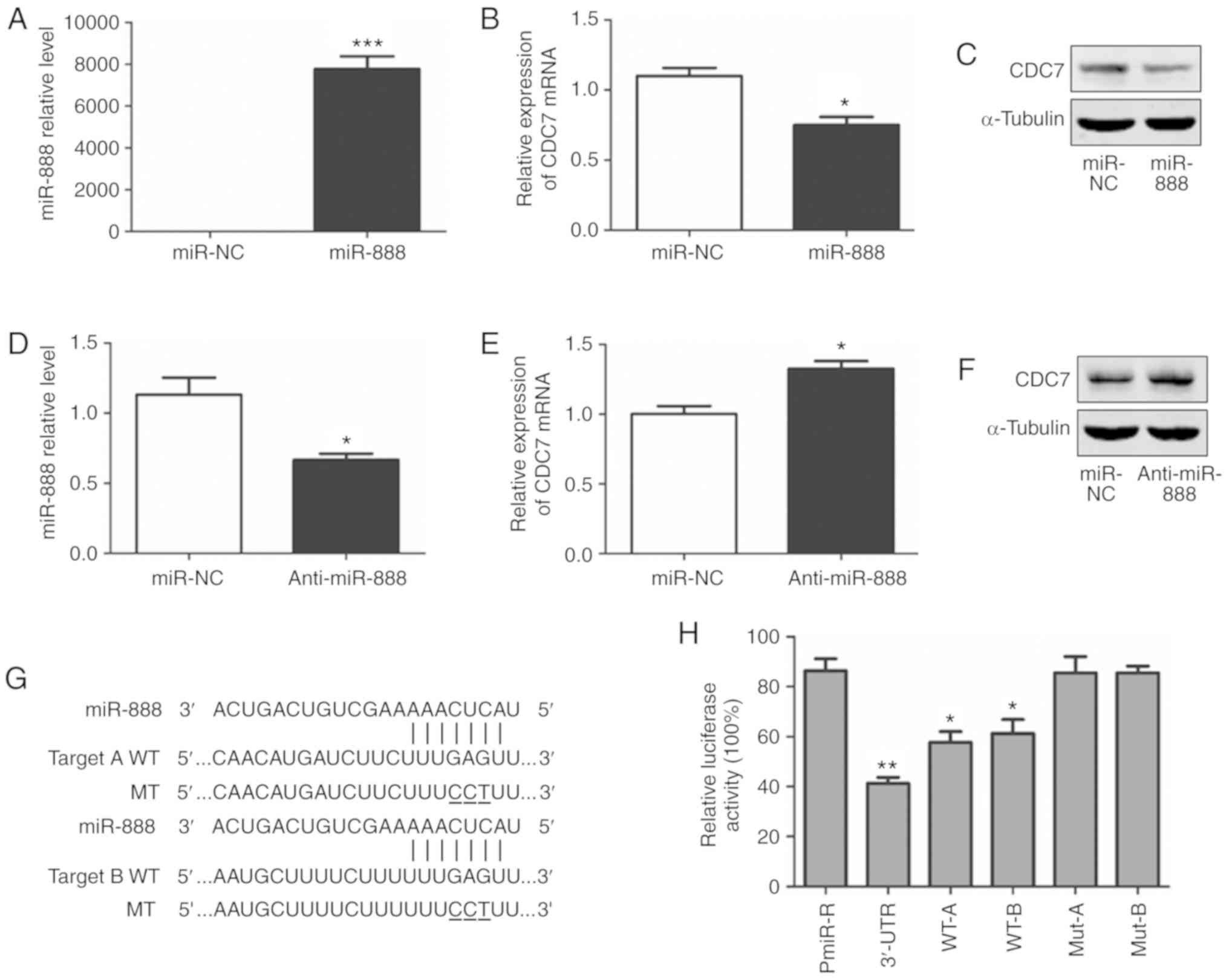
To validate whether miR-888 could target CDC7, A549 cells were
transfected with miR-888 mimics and miR-888 inhibitors. RT-qPCR was
employed to ascertain the transfection efficiency (Fig. 4A and D; P=0.0002, P=0.0219,
respectively) and CDC7 expression. RT-qPCR and western blotting
revealed that both the CDC7 mRNA and protein levels were
significantly reduced following miR-888 overexpression (Fig. 4B and C; P=0.0218) whereas they were
increased following miR-888 inhibition (Fig. 4E and F; P=0.0219).
To further assess whether CDC7 is a target of
miR-888, luciferase reporter assays were performed. Wild-type (WT)
CDC7 containing a full-length 3′-UTR, the 3′-UTR fragments ‘A’, ‘B’
(containing one putative miR-888 target site, respectively) and
their mutants (Fig. 4G) were
inserted downstream of the pMiR-Reporter vector to generate a
series of reporter constructs. Each of these constructs was
co-transfected with scrambled siRNA or miR-888 mimics. It was
observed that miR-888 mimics significantly decreased luciferase
activity in cells transfected with the WT miR-888 binding site, but
not in cells transfected with a construct containing the mutant
CDC7 3′-UTR (Fig. 4H; P=0.0012,
P=0.0119, P=0.0279, respectively). These results indicated that
miR-888 downregulates CDC7 expression by directly binding to the
CDC7 3′-UTR.
CDC7 is required for miR-888-mediated
inhibition of proliferation in lung adenocarcinoma A549 cells
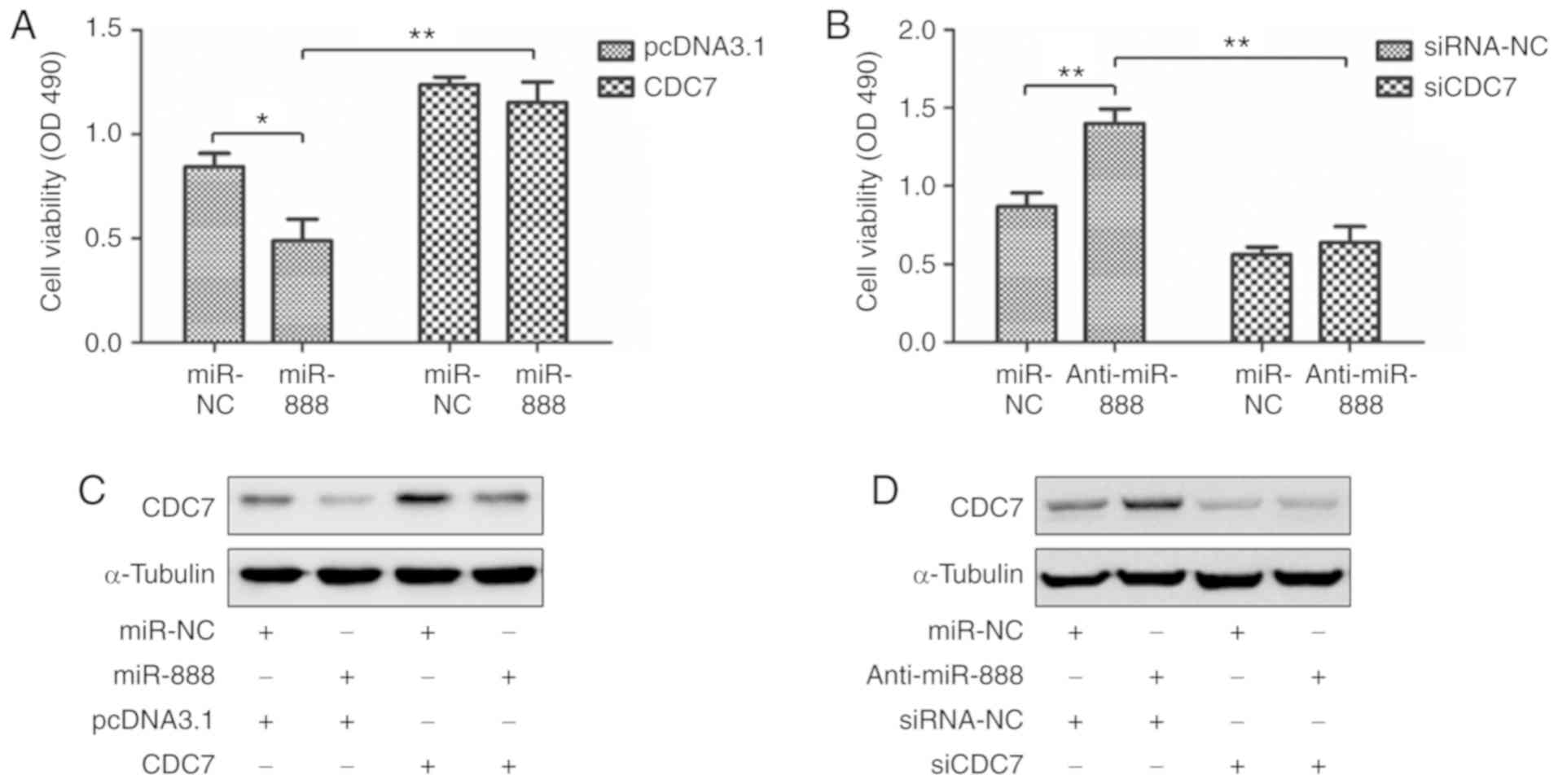
To clarify whether miR-888 inhibited A549 cell
proliferation by targeting CDC7, rescue experiments were performed.
miR-888 mimics, and CDC7 overexpression plasmid without the 3′-UTR
were co-transfected into A549 cells, and miR-888 inhibitor and
small interfering RNAs targeting CDC7 (siCDC7) were co-transfected
into A549 cells. MTS assays and western blotting revealed that
overexpression of CDC7 abolished the miR-888-mediated inhibition of
proliferation in A549 cells (Fig. 5A
and C; P=0.0260, P=0.0035, respectively), while knockdown of
CDC7 markedly attenuated miR-888 inhibitor-mediated promotion of
proliferation in A549 cells (Fig. 5B
and D; P=0.0057, P=0.0015, respectively). These results
indicated that CDC7 was required for miR-888-mediated inhibition of
proliferation in lung adenocarcinoma A549 cells.
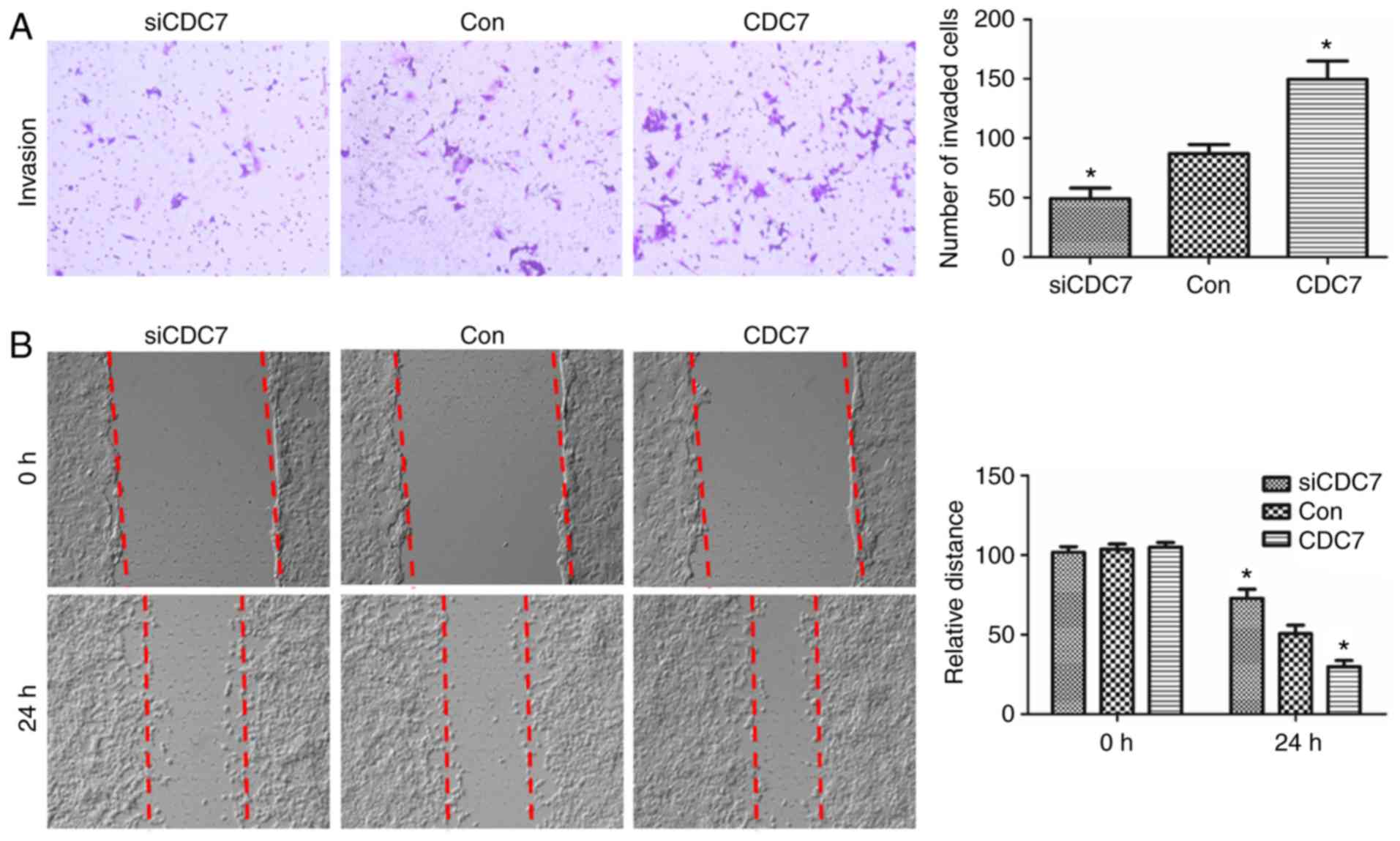
Effect of CDC7 on invasion and
migration of lung adenocarcinoma cells
To clarify whether miR-888 promoted A549 cell
invasion and migration by targeting CDC7, the effect of CDC7 on
cell invasion and migration was first assessed through gain- and
loss-of-function experiments. CDC7 was overexpressed or silenced in
A549 cells by transfection with pcDNA3.1-CDC7 and CDC7 siRNA.
Transwell and wound-healing assays were performed to explore the
effects of CDC7 on invasion and migration in A549 cells. The
results revealed that overexpression of CDC7 promoted the invasion
and migration of A549 cells (Fig. 6A
and B; P=0.0319, P=0.0219, respectively), and downregulation of
CDC7 revealed the opposite effects (Fig. 6A and B; P=0.0463, P=0.0355,
respectively). These results indicated that the miR-888/CDC7 axis
was not the dominant pathway driving the regulation of cell
migration and invasion via miR-888 in A549 cells.
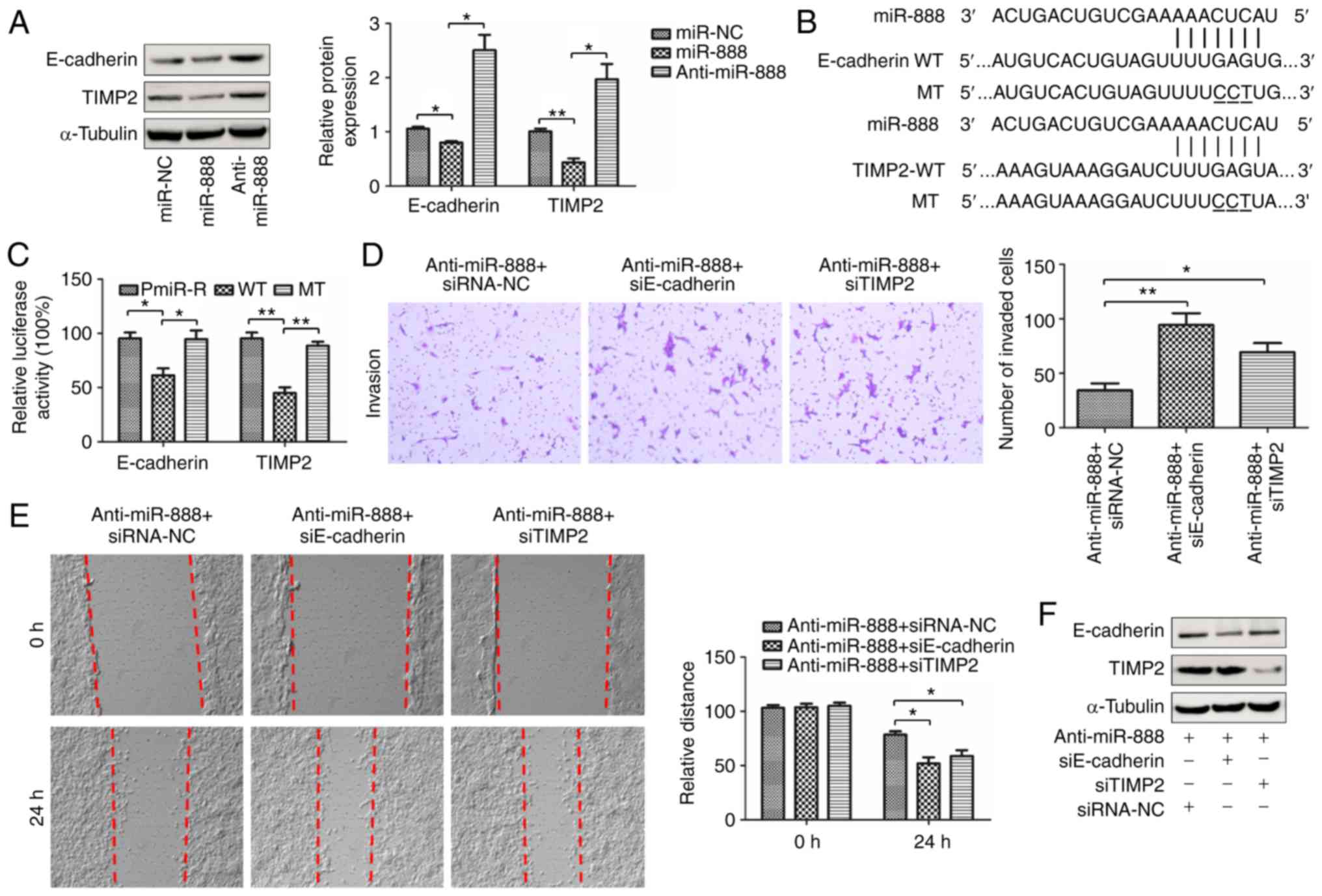
miR-888 promotes invasion and
migration of lung adenocarcinoma cells by targeting E-cadherin and
TIMP2
E-cadherin and TIMP2 have been reported to be
targets of miR-888 to promote invasion and migration in breast
cancer cells and prostate cancer cells respectively (15,16).
Thus, it was speculated that miR-888-induced invasion and migration
may be linked to E-cadherin and TIMP2. To demonstrate this
hypothesis, a series of experiments were performed. The protein
expression of E-cadherin and TIMP2 in A549 cells transfected with
miR-888 mimics and inhibitor was first assessed using western
blotting assay. The results revealed that overexpression of miR-888
in A549 cells reduced the protein levels of E-cadherin and TIMP2
and knockdown of miR-888 increased the expression of E-cadherin and
TIMP2 in A549 cells (Fig. 7A;
P=0.0402, P=0.0320, P=0.0019, P=0.0182, respectively). To further
assess if miR-888 directly targeted E-cadherin and TIMP2, the
putative binding site of miR-888 was predicted and luciferase
reporter assays were performed to ascertain the prediction
(Fig. 7B). Luciferase reporter
assays revealed that miR-888 mimics significantly decreased
luciferase activity in cells transfected with the wild-type miR-888
binding site, but not in cells transfected with a construct with
the mutant miR-888 binding site, (Fig.
7C; P=0.0154, P=0.0309, P=0.0026, P=0.0024, respectively). It
was next assessed whether miR-888 induced invasion and migration in
A549 cells by targeting E-cadherin and TIMP2, by performing rescue
experiments. miR-888 inhibitor and small interfering RNAs targeting
E-cadherin (siE-cadherin) or TIMP2 (siTIMP2) were co-transfected
into A549 cells. Transwell and wound-healing assays revealed that
knockdown of E-cadherin markedly attenuated miR-888
inhibitor-mediated suppression of invasion and migration in A549
cells (Fig. 7D and E; P=0.0085,
P=0.0146, respectively). Similar results were observed in A549
cells co-transfected with miR-888 inhibitor and TIMP2 siRNA
(Fig. 7D and E, right panel;
P=0.0294, P=0.0341, respectively). In addition, western blotting
was performed to detect the expression of E-cadherin and TIMP2 in
A549 cells co-transfected with miR-888 inhibitor and small
interfering RNAs targeting E-cadherin or TIMP2 (Fig. 7F). The data indicated that miR-888
promoted invasion and migration of lung adenocarcinoma cells by
targeting E-cadherin and TIMP2.
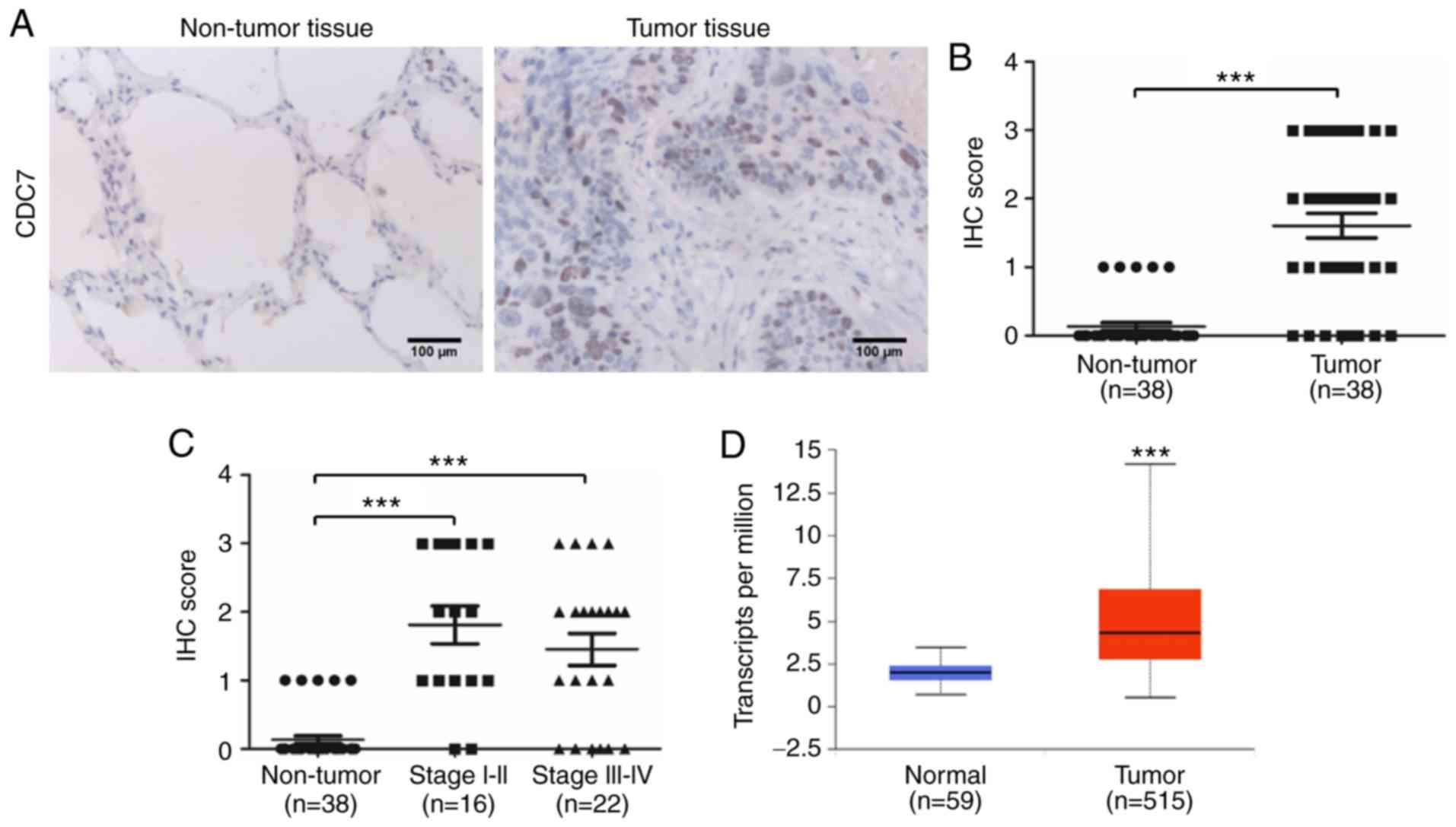
CDC7 is upregulated in the tumors of
patients with lung adenocarcinoma
Our data revealed that ectopic expression of miR-888
in A549 cells exhibited a tumor suppressive function by targeting
CDC7, while ectopic expression of miR-888 in A549 cells exhibited
an oncogenic function by targeting E-cadherin and TIMP2.
Furthermore, our data revealed that miR-888 was upregulated in the
tumors of patients with lung adenocarcinoma and markedly associated
with clinical stage progression in patients. Given that CDC7 plays
a critical role in ‘overexpression of miR-888 as a double-edged
sword in the progression of A549 cells’, the expression of CDC7 in
38 pairs of lung adenocarcinoma tissues was detected to investigate
the role of miR-888 in the tumors of patients with lung
adenocarcinoma. IHC results revealed that the protein level of CDC7
was high in lung adenocarcinoma tissues but was low or undetectable
in normal lung tissues (Fig. 8A).
Furthermore, quantitation data of the IHC score revealed that the
protein expression level of CDC7 was high in lung adenocarcinoma
tissues (Fig. 8B; P<0.001),
however, the expression of CDC7 was not associated with clinical
stage in patients (Fig. 8C).
Similar results were observed from UALCAN (http://ualcan.path.uab.edu/), an online database which
performs in-depth analyses of TCGA gene expression data (Fig. 8D; P<0.001). Collectively, our
data indicated that the miR-888/CDC7 axis did not work in the
tumors of patients with lung adenocarcinoma.
Discussion
Accumulating evidence has demonstrated that
dysregulation of miRNAs (microRNAs) plays essential roles in the
development and progression of cancers, such as lung cancer
(22). However, the molecular
mechanisms of miRNAs that contribute to lung cancer progression
have not been fully elucidated. miR-888 was revealed to be
upregulated in several tumors such as colorectal cancer,
endometrial cancers and an MCF-7 side population of human breast
cancer cells (10,11,15).
Consistent with previous studies, the present study study revealed
that the expression of miR-888 was significantly upregulated in
lung adenocarcinoma samples than in matched non-tumor tissues.
Moreover, its expression was asscoiated with the pathologic stage.
These findings indicated that miR-888 may function as an oncogene
in the progression of lung adenocarcinoma.
It has been reported that the targets of miRNA are
vital to the function of miRNA in cells, and one miRNA usually
targets multiple genes. Therefore, a miRNA may function as an
oncogene or tumor suppressor gene depending on the type of target
gene and the gene expression patterns on the tissue and the cell
type. For instance, Chen et al reported that miR-766
attenuated cell cycle progression of renal cell carcinoma (RCC)
cells by downregulating SF2 expression (23), while Yang et al reported that
miR-766 promoted the proliferation and metastasis of hepatocellular
carcinoma (HCC) cells by directly decreasing the NR3C2 expression
(24). miR-888 has been reported to
function as an oncogene in the progression of several cancers. For
example, Gao et al reported that miR-888 functioned as an
oncogene and predicted poor prognosis in colorectal cancer
(11). Huang et al reported
that miR-888 promoted cell migration and invasion in breast cancer
by targeting E-cadherin (15).
Lewis et al reported that miR-888 promoted cell
proliferation in prostate cancer by targeting RBL1 and SMAD4
(13). Hasegawa et al
reported that miR-888 promoted cell migration and invasion in
prostatic cancer by targeting tissue inhibitor of metalloproteinase
2 (TIMP2) (16). However, the role
of miR-888 has never been reported in lung adenocarcinoma. In this
study, the results revealed that ectopic expression of miR-888
functioned as a double-edged sword in the progression of lung
adenocarcinoma A549 cells. On the one hand, ectopic expression of
miR-888 not only promoted the migration and invasion of lung
adenocarcinoma cells by targeting E-cadherin or TIMP2, but also
inhibited the migration and invasion of lung adenocarcinoma cells
by targeting CDC7. Finally, the function of miR-888 as a promoter
in the migration and invasion of lung adenocarcinoma cells
suggested that the miR-888/CDC7 axis was not the dominant pathway
driving the regulation of cell migration and invasion via miR-888.
This result was consistent with the role of miR-888 in colorectal,
breast and prostate cancer (11,14,16).
On the other hand, ectopic expression of miR-888
significantly inhibited the proliferation of lung adenocarcinoma
cells by targeting CDC7, while previous studies in colorectal and
prostate cancer indicated that miR-888 promoted cellular
proliferation by targeting multiple targets (11,13).
These distinct results may indicate that miR-888 regulates cell
proliferation depending on the type of cancer cell or the gene
expression patterns on the tissue and the cell type. CDC7 is a
conserved serine-threonine kinase which plays a crucial role in the
initiation of DNA replication (25,26).
Thus, miR-888 may exert its inhibitory effect on cell proliferation
by blocking CDC7-mediated initiation of DNA synthesis and by
inducing G1 arrest. In addition, accumulating data has indicated
that CDC7 is overexpressed in many human cancers and tumor cell
lines, but has low or undetectable expression in normal tissues and
cell lines (19,27) and the overexpression of CDC7 was
revealed to be associated with advanced clinical tumor stages, poor
survival and chemoresistance (21,28).
Therefore, ectopic expression of miR-888 revealed a tumor
suppressive function by targeting CDC7 in A549 cells. However, our
results revealed that both miR-888 and CDC7 were significantly
upregulated in lung adenocarcinoma samples indicating that other
potential pathways may involve regulation of CDC7 in lung
adenocarcinoma patients, and that the miR-888/CDC7 axis was not the
dominant pathway for CDC7 regulation in patients with lung
adenocarcinoma. Furthermore, rescue experiments demonstrated that
overexpression of CDC7 abolished tumor suppressive function of
miR-888 in A549 cells (Fig. 5A).
Thus, the aforementioned results provided evidence that miR-888 may
function as an oncogene in the progression of lung adenocarcinoma
patients.
There are several limitations in the present study
when interpreting these results. First, our sample size was small,
in order to increase the accuracy of the conclusion, increased
sample sizes are required. Second, the mechanism underlying the
regulation of CDC7 in lung adenocarcinoma patients may be complex
and may involve cross-interactions of other signaling pathways,
which requires further investigation. Thirdly, further studies are
warranted to identify the roles miR-888 in the progression of lung
cancer through animal model studies.
In conclusion, our results revealed that ectopic
expression of miR-888 in A549 cells functioned as a double-edged
sword in the progression of lung adenocarcinoma cells by targeting
multiple targets. In addition, ectopic expression of miR-888
significantly inhibited the proliferation of lung adenocarcinoma
cells by targeting CDC7; on the other hand, ectopic expression of
miR-888 promoted the migration and invasion of lung adenocarcinoma
cells by targeting E-cadherin and TIMP2. Furthermore, both miR-888
and CDC7 were upregulated in tumor tissues of patients with lung
adenocarcinoma. Thus, our data demonstrated that miR-888 functioned
as an oncogene in the progression of lung adenocarcinoma and may
shed light on the therapeutic strategies for lung adenocarcinoma
treatment.
Acknowledgements
Specimens and diagnosis data were provided by the
Department of Pathology, Zhongshan Hospital Xiamen University
(Xiamen, China). We thank Dr Peter Cherepanov (Imperial College
London) for providing the pcDNA3.1-CDC7 constructs.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81502393).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
JXC conceived and designed the study, performed the
experiments and wrote the manuscript and agrees to be accountable
for all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Xiamen University (Xiamen, China), and written informed consents
were obtained from the patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CDC7
|
cell division cycle 7
|
|
miRNA
|
microRNA
|
|
TIMP2
|
tissue inhibitor of metalloproteinase
2
|
|
TCGA
|
The Cancer Genome Atlas
|
|
NSCLC
|
non-small cell lung carcinoma
|
|
IHC
|
immunohistochemistry
|
|
3′-UTR
|
3′-untranslated region
|
|
OS
|
overall survival
|
|
WT
|
wild-type
|
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Torre LA, Siegel RL, Ward EM and Jemal A:
Global cancer incidence and mortality rates and trends-An update.
Cancer Epidemiol Biomarkers Prev. 25:16–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Esquela-Kerscher A and Slack FJ:
Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer.
6:259–269. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hwang HW and Mendell JT: MicroRNAs in cell
proliferation, cell death, and tumorigenesis. Br J Cancer.
94:776–780. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Uddin A and Chakraborty S: Role of miRNAs
in lung cancer. J Cell Physiol. 2018. View Article : Google Scholar
|
|
8
|
Ma YS, Yu F, Zhong XM, Lu GX, Cong XL, Xue
SB, Xie WT, Hou LK, Pang LJ, Wu W, et al: miR-30 family reduction
maintains self-renewal and promotes tumorigenesis in
NSCLC-initiating cells by targeting oncogene TM4SF1. Mol Ther.
26:2751–2765. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Song Q, Ji Q, Xiao J, Wang L, Chen Y, Xu Y
and Jiao S: miR-409 inhibits human non-small-cell lung cancer
progression by directly targeting SPIN1. Mol Ther Nucleic Acids.
13:154–163. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hovey AM, Devor EJ, Breheny PJ, Mott SL,
Dai D, Thiel KW and Leslie KK: miR-888: A novel cancer-testis
antigen that targets the progesterone receptor in endometrial
cancer. transl Oncol. 8:85–96. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao SJ, Chen L, Lu W, Zhang L, Wang L and
Zhu HH: miR-888 functions as an oncogene and predicts poor
prognosis in colorectal cancer. Oncol Lett. 15:9101–9109.
2018.PubMed/NCBI
|
|
12
|
Bobowicz M, Skrzypski M, Czapiewski P,
Marczyk M, Maciejewska A, Jankowski M, Szulgo-Paczkowska A,
Zegarski W, Pawłowski R, Polańska J, et al: Prognostic value of
5-microRNA based signature in T2-T3N0 colon cancer. Clin Exp
Metastasis. 33:765–773. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lewis H, Lance R, Troyer D, Beydoun H,
Hadley M, Orians J, Benzine T, Madric K, Semmes OJ, Drake R, et al:
miR-888 is an expressed prostatic secretions-derived microRNA that
promotes prostate cell growth and migration. Cell Cycle.
13:227–239. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang S and Chen L: MiR-888 regulates side
population properties and cancer metastasis in breast cancer cells.
Biochem Biophys Res Commun. 450:1234–1240. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang S, Cai M, Zheng Y, Zhou L, Wang Q
and Chen L: miR-888 in MCF-7 side population sphere cells directly
targets E-cadherin. J Genet Genomics. 41:35–42. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hasegawa T, Glavich GJ, Pahuski M, Short
A, Semmes OJ, Yang L, Galkin V, Drake R and Esquela-Kerscher A:
Characterization and evidence of the miR-888 cluster as a novel
cancer network in prostate. Mol Cancer Res. 16:669–681. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang L, Liu Q, Lin D and Lai M: CD44v6
down-regulation is an independent prognostic factor for poor
outcome of colorectal carcinoma. Int J Clin Exp Patho.
8:14283–14293. 2015.
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bonte D, Lindvall C, Liu H, Dykema K,
Furge K and Weinreich M: Cdc7-Dbf4 kinase overexpression in
multiple cancers and tumor cell lines is correlated with p53
inactivation. Neoplasia. 10:920–931. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Q, Xie W, Wang N, Li C and Wang M:
CDC7-dependent transcriptional regulation of RAD54L is essential
for tumorigenicity and radio-resistance of glioblastoma. Transl
Oncol. 11:300–306. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sasi NK, Bhutkar A, Lanning NJ, MacKeigan
JP and Weinreich M: DDK promotes tumor chemoresistance and survival
via multiple pathways. Neoplasia. 19:439–450. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Del Vescovo V, Grasso M, Barbareschi M and
Denti MA: MicroRNAs as lung cancer biomarkers. World J Clin Oncol.
5:604–620. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen C, Xue S, Zhang J, Chen W, Gong D,
Zheng J, Ma J, Xue W, Chen Y, Zhai W, et al:
DNA-methylation-mediated repression of miR-766-3p promotes cell
proliferation via targeting SF2 expression in renal cell carcinoma.
Int J Cancer. 141:1867–1878. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang C, Ma X, Guan G, Liu H, Yang Y, Niu
Q, Wu Z, Jiang Y, Bian C, Zang Y and Zhuang L: MicroRNA-766
promotes cancer progression by targeting NR3C2 in hepatocellular
carcinoma. FASEB J. 33:1456–1467. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Masai H and Arai K: Cdc7 kinase complex: A
key regulator in the initiation of DNA replication. J Cell Physiol.
190:287–296. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Labib K: How do Cdc7 and cyclin-dependent
kinases trigger the initiation of chromosome replication in
eukaryotic cells? Genes Dev. 24:1208–1219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Datta A, Ghatak D, Das S, Banerjee T, Paul
A, Butti R, Gorain M, Ghuwalewala S, Roychowdhury A, Alam SK, et
al: p53 gain-of-function mutations increase Cdc7-dependent
replication initiation. EMBO Rep. 18:2030–2050. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Erbayraktar Z, Alural B, Erbayraktar RS
and Erkan EP: Cell division cycle 7-kinase inhibitor PHA-767491
hydrochloride suppresses glioblastoma growth and invasiveness.
Cancer Cell Int. 16:882016. View Article : Google Scholar : PubMed/NCBI
|