Introduction
Preoperative chemoradiotherapy (CRT) is considered a
standard therapy for local control in patients with locally
advanced rectal cancer. Pathological complete response (pCR) in the
patients treated with CRT implies the absence of residual cancer in
the resected specimen. Since the recurrence rate after curative
surgery in the patients with pCR has been reported to be lower than
that in non-responders (1),
increasing the pCR rate is necessary to improve patient prognosis.
However, 5-FU-based conventional CRT has been revealed to result in
pCR for only 13–17 % of patients (2), and these results are insufficient.
Hence, chemotherapeutic regimens for increasing tumor
radiosensitivity are being investigated worldwide.
Preclinical studies on solid cancer cells revealed
that ionizing radiation (IR) activated the PI3K/AKT/mTOR signaling
pathway, which was considered to be one of the radioresistance
mechanisms (3,4). However, in a phase-II clinical trial
for patients with locally advanced rectal cancer, CRT combined with
rapamycin, an mTOR inhibitor, resulted in pCR in 3% of patients and
the radiosensitizing effect of mTOR inhibitor was not observed
(5).
An mTOR inhibitor is known to induce autophagy by
dephosphorylating UNC-51-like kinase (ULK1). Autophagy is
considered as a cause for chemo/radiotherapy resistance in cancer
cells due to its function of self-protection and self-degradation
(6,7). This cytoprotective function of
autophagy was reported to play a role in resistance to the
antitumor effect of mTOR inhibitor (8). In addition, in a previous study, it
was demonstrated that the combination of temsirolimus (TEM), an
mTOR inhibitor, and chloroquine (CQ), an autophagy inhibitor,
improved the antitumor effect in colorectal cancer (CRC) cells
(9). Based on this result, we
hypothesized that the simultaneous inhibition of mTOR signaling and
autophagy is essential to enhance radiosensitivity. Therefore, in
the present study, we aimed to determine whether the co-inhibition
of mTOR and autophagy using TEM and CQ enhanced radiosensitivity in
CRC cells.
Materials and methods
Cell culture and reagents
The human CRC cell lines, HT-29 and SW480, were
purchased from the Japanese Cancer Research Resource Bank. Cells
were cultured in RPMI-1640 medium (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) supplemented with 5% fetal bovine serum (FBS)
and 1% antibiotic/antimycotic solution in a 5% CO2
incubator at 37°C. The mTOR inhibitor TEM and the autophagy
inhibitor chloroquine diphosphate (CQ) were purchased from
Sigma-Aldrich; Merck KGaA. Rabbit antibodies against phospho-S6
ribosomal protein (Ser235/236), S6 ribosomal protein (5G10),
phospho-eukaryotic translation initiation factor 4E-binding protein
1 (phospho-4E-BP1; Thr37/46), and 4E-BP1 were purchased from Cell
Signaling Technology (Tokyo, Japan); those against
microtubule-associated protein light chain 3 beta (LC3B), protein
62 [p62; also known as sequestosome-1 (SQSTM1)], and β-actin were
obtained from Medical and Biological Laboratories (MBL, Nagoya,
Japan); and alkaline phosphatase-labeled goat anti-rabbit antibody
was purchased from Abcam (Cambridge, UK).
Preparation of radiosensitizers and
exposure to IR
The experimental doses of TEM and CQ were determined
to be 80 nM and 20 µM, respectively based on a previous study
(9). The cells were pretreated with
TEM (TEM group), CQ (CQ group), both (TEM+CQ group), or no
treatment (the control group) for 1 h before exposure to IR. Cells
were irradiated at doses of ~6 Gy (1.0 Gy/sec) with an X-ray
generator (Pantac HF350; Kyoto, Japan). Irradiated cells were used
in subsequent experiments. The experimental radiation dose was
determined according to several preceding studies (10–12),
including our previous study using colorectal cell lines (13).
Clonogenic assay
The SW480 and HT-29 cells from each group were
seeded at densities of 200-1,200 cells in a 6-well plate. Cells
were pretreated with or without TEM and/or CQ for 1 h followed by
exposure to IR at doses of 0, 2, 4 or 6 Gy. After incubation for 24
h, the medium was changed to a fresh one without drugs. The medium
was then changed to a fresh one every 48 h for 14 days. Then, the
cells were fixed with 70% ethanol at room temperature for 30 min,
and stained with 0.5% crystal violet solution at room temperature
for 30 min, and the colonies were counted. The surviving fraction
(SF) in each group was calculated as follows: SF=no. of colonies
with IR/no. of colonies without IR. For each experimental
condition, colonies from three different wells were counted. All SF
values were expressed as the mean ± standard deviation (SD).
Cell proliferation assay
SW480 and HT-29 cells (5×104 cells/well)
were seeded in 96-well flat-bottomed plates and incubated for 72 h
with or without TEM and/or CQ after exposure to IR at a dose of 4
Gy. Then, MTS reagent (Promega Corp., Madison, WI, USA) was added
to each well, cells were incubated for 3 h, and spectrophotometric
assessment was performed with a 490-nm filter using Varioskan
Flash, a microplate reader (Life Technologies; Thermo Fisher
Scientific, Inc.). All experiments were performed in triplicate
wells, and the data for cell proliferation were expressed as the
mean ± SD.
Western blot analysis
SW480 and HT-29 cells were incubated for 24 h with
or without TEM and/or CQ after exposure to IR at a dose of 4 Gy.
Whole cell protein extracts were obtained by lysing cells in Bolt
LDS sample buffer (Life Technologies; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with protease and phosphatase
inhibitors (Roche Diagnostics, Basel, Switzerland). Solubilized
proteins were quantified using the Qubit protein assay kit (Life
Technologies; Thermo Fisher Scientific, Inc.) and 40 µg of protein
samples were separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) with 4–12 % Bis-Tris Gel (Life
Technologies; Thermo Fisher Scientific, Inc.) and transferred to
polyvinylidene difluoride (PVDF) membranes (pore size 0.2 µm) using
the Bolt system (Life Technologies; Thermo Fisher Scientific,
Inc.). After blocking with the iBind solution (Life Technologies;
Thermo Fisher Scientific, Inc.) at room temperature for 5 min,
western blotting was performed using the iBind Western System (Life
Technologies; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions; the membranes were incubated with
rabbit antibodies against phospho-S6 ribosomal protein (Ser235/236;
dilution 1:1,000; cat. no. 4857), S6 ribosomal protein (5G10;
dilution 1:1,000; cat. no. 2217), phospho-4E-BP1 (Thr37/46;
dilution 1:1,000; cat. no. 2855), 4E-BP1 (dilution 1:1,000; cat.
no. 9452) all from CST; LC3B (dilution 1:1,000; cat. no. PM036),
p62, also known as sequestosome-1 (SQSTM1) (dilution 1:1,000; cat.
no. PM045), or β-actin (dilution 1:1,000; cat. no. PM053) all from
MBL at room temperature for 3 h, followed by alkaline
phosphatase-labeled goat anti-rabbit antibody (dilution 1:2,000;
cat. no. ab97048; Abcam) at room temperature for 2 h. The membranes
were incubated in CDP-Star solution (Life Technologies; Thermo
Fisher Scientific, Inc.) for 5 min, and chemiluminescence was
detected using ChemiDoc XRS system (Bio-Rad Laboratories, Inc.,
Tokyo, Japan). The experiment was performed three times. The
density of each band was measured using ImageJ software (version
1.4.3; National Institutes of Health, Bethesda, MD, USA) and
expressed as the mean ± SD. The phosphorylated isoform fraction of
S6 and 4E-BP1 were normalized by the respective total protein
levels.
Acidic vesicular organelle
(autophagosome) detection
SW480 and HT-29 cells were prepared and treated as
aforementioned. The cells were collected in FACS tubes and stained
with 5 µg/ml acridine orange (Sigma-Aldrich; Merck, Tokyo, Japan)
for 15 min at room temperature. Then, the samples were washed twice
with Ca2+/Mg2+-free phosphate-buffered saline
[PBS(−)] and immediately analyzed using a BD FACSCalibur flow
cytometer (BD Biosciences, San Jose, CA, USA), or examined under a
fluorescence microscope (BZ-8100; Keyence Corp., Tokyo, Japan). The
experiments were performed three times. All numerical data were
expressed as the mean ± SD.
Apoptosis and cell death analysis
SW480 and HT-29 cells were prepared and treated as
aforementioned, and the harvested cells were stained with a
combination of Annexin V-fluorescein isothiocyanate (FITC) and
propidium iodide (PI) (Annexin V-FITC apoptosis detection kit; BD
Biosciences), according to the manufacturer's instructions. The
samples were immediately analyzed by flow cytometer. This method
allows the distinction between cells in early (Annexin
V-FITC+/PI−) and late (Annexin
V-FITC+/PI+) apoptosis; however, in the
present study, these subpopulations were counted together and
expressed as a fraction of the apoptotic cells. Next, for the
quantification of caspase-3/7 activity, cells were labeled with
CellEvent caspase-3/7 green detection reagent (Life Technologies;
Thermo Fisher Scientific, Inc.) and analyzed by flow cytometry.
Alternatively, for calculating the proportion of dead cells, the
cells were stained with trypan blue (Sigma-Aldrich; Merck KGaA),
and dead cells (stained with trypan blue) and alive cells
(unstained cells) were counted immediately. All experiments were
performed three times, and all data were expressed as the mean ±
SD.
Statistical analysis
Statistical significance of the differences was
determined using the unpaired, two-tailed Student's t-test for
single comparisons and one-way ANOVA followed by Tukey's post hoc
test for multiple comparisons. In addition, the interactions of
radiation levels and different drug therapies were analyzed by
two-way ANOVA. All analyses were performed using the JMP Pro 13.0
software (SAS Institute, Inc., Cary, NC, USA). Differences with
P<0.05 were considered statistically significant.
Results
IR activates the mTOR signaling
pathway and induces autophagy in CRC cells
To investigate whether IR activates the mTOR
signaling pathway and induces autophagy in human CRC cell lines,
the levels of mTOR pathway-related proteins and autophagy-related
proteins in SW480 and HT-29 cells before and after IR exposure were
determined by western blotting (Fig. 1A
and B). The levels of the phosphorylated isoforms of mTOR
pathway-related proteins, S6 ribosomal protein and 4E-BP1,
normalized by the respective total proteins, were significantly
increased after IR exposure in both cell lines, indicating that IR
activated the mTOR signaling pathway in CRC cells. Similarly,
conversion of LC3-I to LC3-II, which plays an important role in
autophagosome formation, was observed, and the LC3-II/LC3-I ratios
increased after IR exposure, whereas the levels of p62, which is
consumed during autophagy, decreased after IR exposure, in both
cell lines. These results indicate that IR induced autophagy in CRC
cells. The time-dependent levels of p-S6 normalized by total S6
protein (p-S6/total S6 ratios) and LC3-II/LC3-I ratios in cells
after the IR exposure are displayed in Fig. 1C and D. In both the cell lines,
p-S6/total S6 ratios increased immediately after IR exposure and
then decreased at 48 h after exposure, whereas LC3-II/LC3-I ratios
increased slowly and remained high at 48 h after IR exposure.
Similarly, the number of acidic autophagosomes stained with
acridine orange significantly increased after IR exposure in a
time-dependent manner, in both cell lines (Fig. 1E and F).
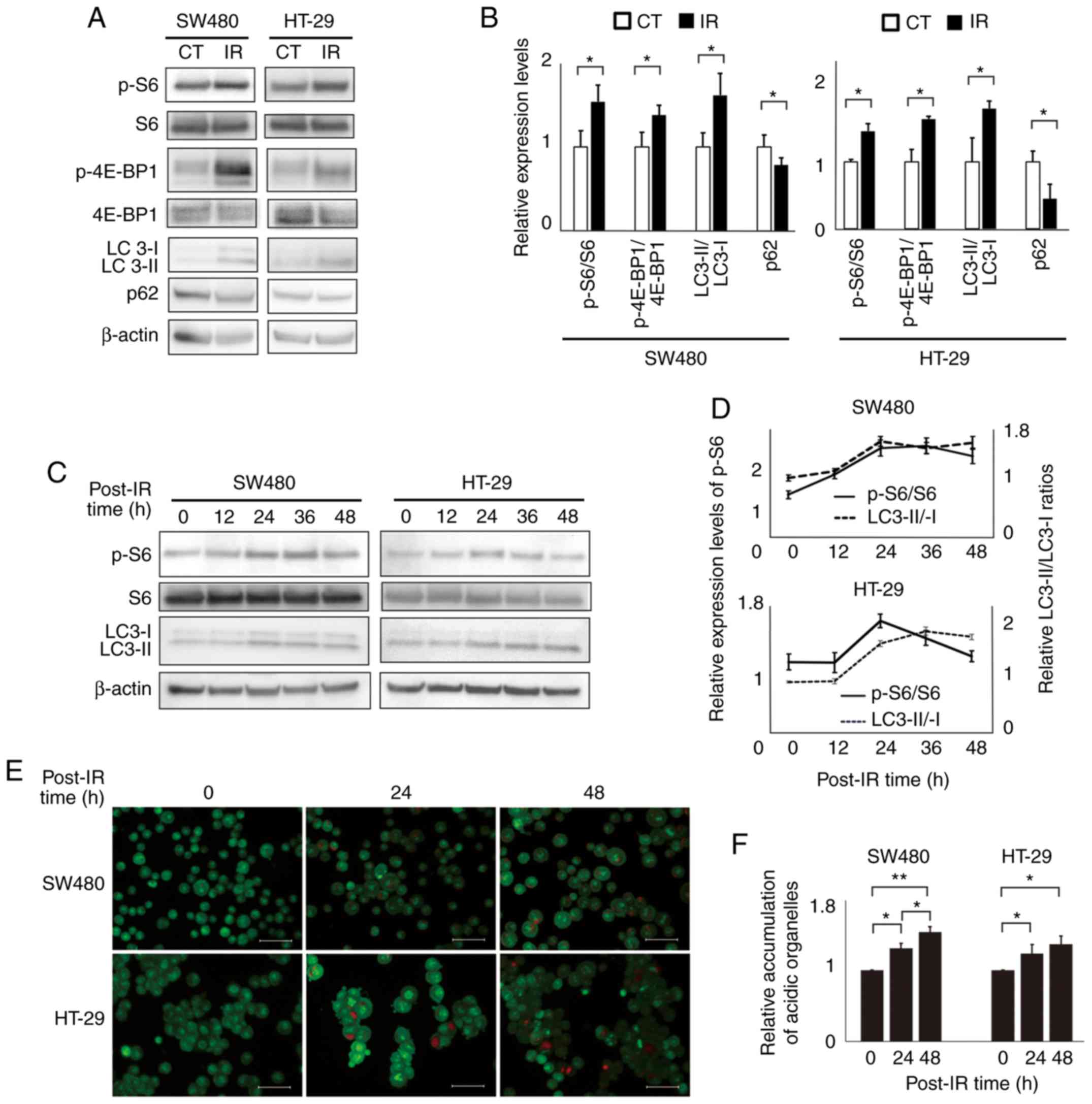 | Figure 1.Ionizing radiation activates the mTOR
pathway and induces autophagy in human colorectal cancer cells.
HT-29 and SW480 cells were treated with ionizing radiation at a
dose of 4 Gy and mTOR pathway activation and autophagy induction
were investigated. (A) Western blot analysis of the mTOR
pathway-related proteins, p-S6, total S6, p-4E-BP1, and total
4E-BP1; and autophagy-related proteins LC3B-II/I and SQSTM1/p62 in
cell lines 24 h after radiotherapy. β-actin was used as the
control. (B) Bar plots represent the relative expression levels of
each protein, as determined via densitometric analysis. The mTOR
pathway-related proteins were presented as the phosphorylated
isoform ratios normalized by the respective total proteins. The
autophagy-related proteins were presented as LC3-II/LC3-I ratios
and the p62 expression levels were normalized to β-actin levels.
The data are expressed as the mean ± SD from the results of three
independent experiments (n=3). *P<0.05. Unpaired Student's
t-tests were used. (C) p-S6, total S6, and LC3B-II/I levels at
different intervals after radiotherapy. (D) Line graphs represent
the changes in the expression levels of p-S6/total S6 and
LC3-II/LC3-I ratio, as determined via densitometric analysis. The
data are expressed as the mean ± SD (n=3). (E) Acridine orange
staining and fluorescence microscopy results for the accumulation
of acidic vesicular organelles (bright red fluorescence) in cell
lines 24 h after radiotherapy. Scale bar, 20 µm. (F) Bar plots
represent flow cytometric results for formation of acidic vesicular
organelles in cell lines 24 h after radiotherapy. The values of
each therapy group were standardized by the value of the control,
without any treatment. The data are expressed as the mean ± SD
(n=3). *P<0.05, **P<0.01. One-way ANOVA was used. p-S6,
phospho-S6 ribosomal protein; p-4E-BP1, phospho-eukaryotic
translation initiation factor 4E-binding protein 1; SQSTM1,
sequestosome-1. |
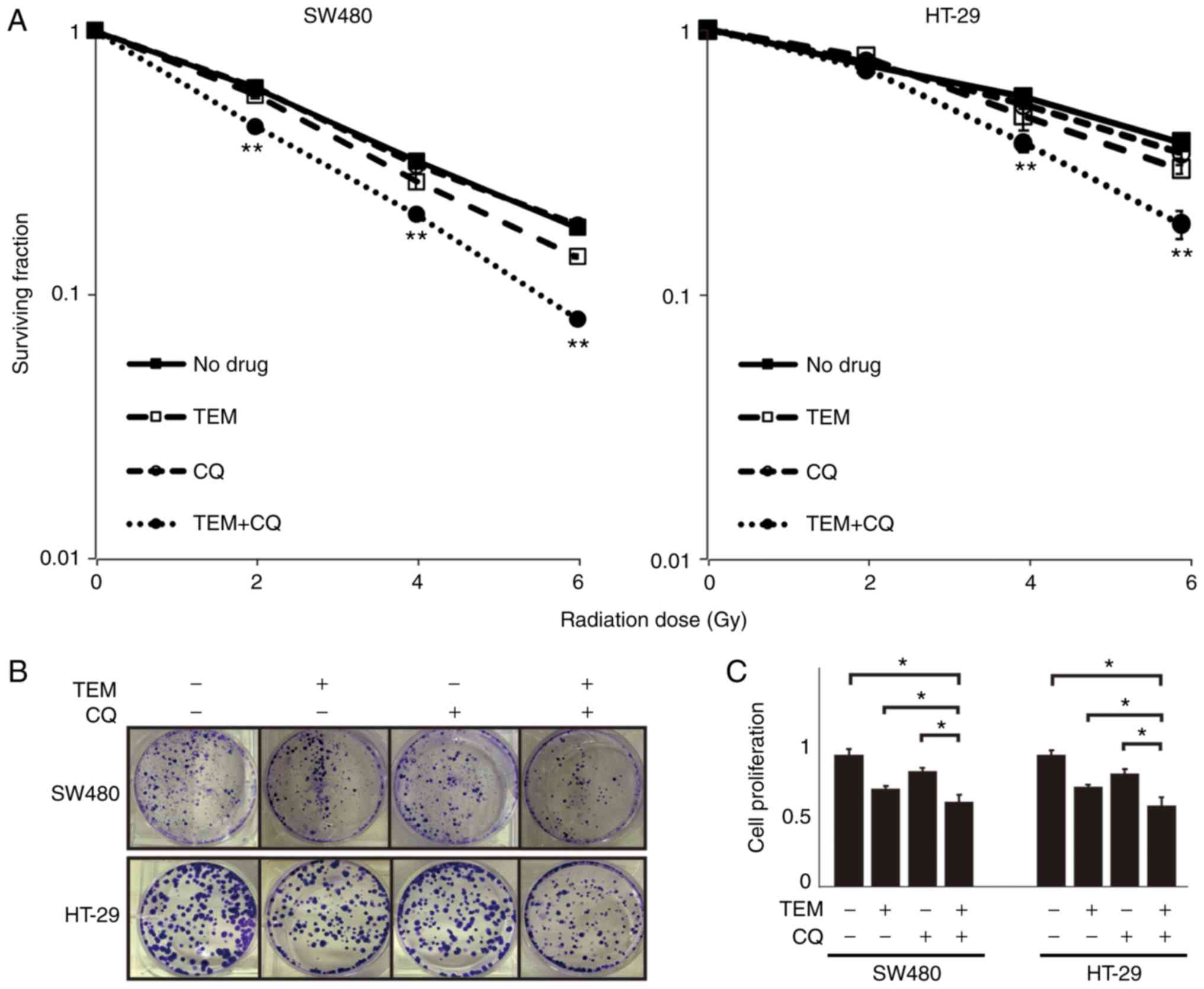
TEM and CQ combination therapy
increases radiosensitivity in CRC cells
Next, whether mTOR inhibition and/or autophagy
inhibition increases radiosensitivity in CRC cells was
investigated, by studying the dose-dependent efficacies of IR in
cells with or without TEM and/or CQ. In the clonogenic assay
(Fig. 2A and B), a two-way ANOVA
revealed the interaction of radiation levels and drug therapies in
SW480 cells (P<0.001) and HT-29 cells (P<0.001). Although CQ
or TEM monotherapy did not affect the suppressive effect of
irradiation, the combination of TEM and CQ significantly enhanced
the radiosensitivity at 2, 4 and 6 Gy in SW480 cells and at 4 and 6
Gy in HT-29 cells. In the MTS assay, TEM or CQ treatment in cells
exposed to IR decreased cell proliferation, compared to that
observed with IR exposure alone (Fig.
2C). Moreover, cell proliferation after the combination therapy
of TEM and CQ was lower than that observed after the monotherapies
with TEM or CQ.
TEM and CQ combination therapy
inhibits the activation of mTOR pathway and autophagy in IR-exposed
cells
Next, the levels of mTOR pathway-related proteins
and autophagy-related proteins in each group were assessed
(Fig. 3A and B). The levels of
IR-induced phosphorylated isoforms of S6 and 4E-BP1 significantly
decreased after TEM monotherapy but were not affected by CQ
monotherapy. The IR-induced conversion of LC3-I to LC3-II
significantly increased after CQ or TEM treatment and the
LC3-II/LC3-I ratios were the highest after TEM and CQ combination
therapy. Furthermore, p62 consumed by IR decreased after TEM
monotherapy but increased after CQ monotherapy and was the highest
after the combination therapy. Furthermore, acridine orange
staining revealed that the number of IR-induced acidic
autophagosomes increased after treatment with a combination of TEM
and CQ, compared to that observed after monotherapy with TEM or CQ
(Fig. 3B). These results were
demonstrated in both SW480 and HT-29 cells.
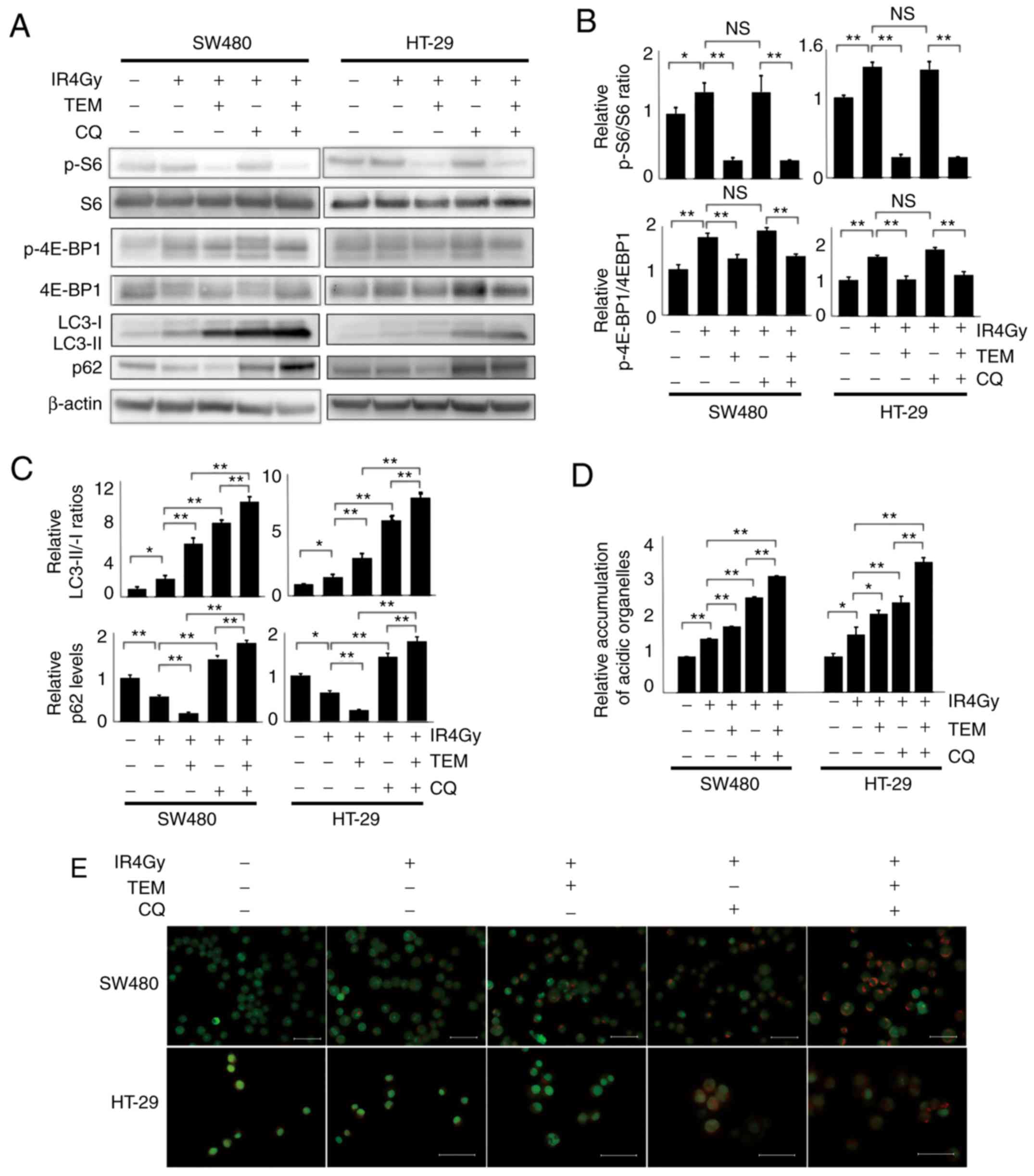 | Figure 3.Combination of TEM, CQ and IR
downregulates mTOR pathway-related proteins and autophagy-related
proteins in SW480 and HT-29 cells. The cells were treated with 80
nM TEM and/or 20 µM CQ followed by 4 Gy radiotherapy and examined
24 h after radiotherapy. (A) Western blot analysis of mTOR pathway-
and autophagy-related proteins. (B) Bar plots represent the
relative expression levels of mTOR-related proteins, p-S6/total S6,
and p-4E-BP1/total 4E-BP1, as determined via densitometric
analysis. The data are expressed as the mean ± SD (n=3).
*P<0.05, **P<0.01. One-way ANOVA was used. (C) Bar plots
represent the relative expression levels of autophagy-related
proteins, LC3-II/LC3-I ratios, and p62 levels. The data are
expressed as the mean ± SD (n=3). *P<0.05, **P<0.01. One-way
ANOVA was used. (D) Bar plots represent flow cytometric results
revealing the formation of acridine orange-stained acidic vesicular
organelles in cells 24 h after radiotherapy. The values of each
group were standardized by the value of the control without any
treatment. The data are expressed as the mean ± SD (n=3).
*P<0.05, **P<0.01. One-way ANOVA was used. (E) Fluorescence
microscopy images showing acridine orange-stained autophagic
vesicles in cells. Scale bar, 20 µm. NS, not significant; TEM,
temsirolimus; CQ, chloroquine; IR, ionizing radiation; p-S6,
phospho-S6 ribosomal protein; p-4E-BP1, phospho-eukaryotic
translation initiation factor 4E-binding protein 1. |
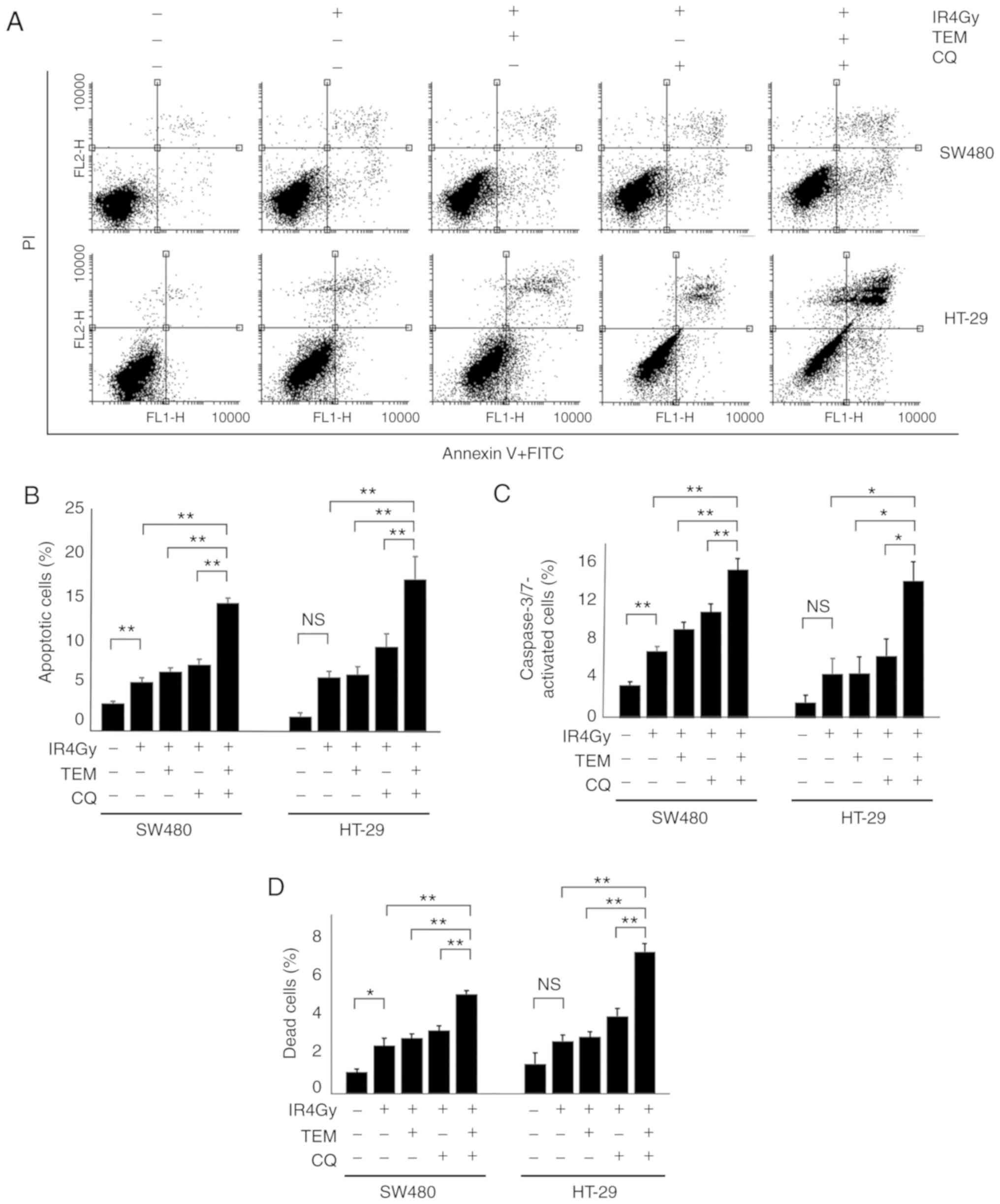
TEM and CQ combination therapy induces
apoptosis in IR-exposed cells
To analyze cell death in each group, apoptosis was
analyzed via double staining with Αnnexin V-FITC and PI (Fig. 4A). The proportion of apoptotic
cells, as Annexin V-positive cells, in each group are displayed in
Fig. 4B. The apoptosis rate after
IR exposure alone (5.5%) was significantly higher than that of the
control (3.1%), which indicated that IR induced apoptosis in SW480
cells. TEM or CQ treatment with IR exposure in SW480 cells
increased the apoptotic rate to 6.6 and 7.4%, respectively, but
these differences were not significant; however, treatment with a
combination of TEM and CQ with IR exposure significantly increased
the apoptotic rate up to 14.3%, compared to that observed after IR
exposure with or without TEM or CQ single agents. Similarly, the
proportion of dead cells stained with trypan blue in SW480 cells
treated with IR and a combination of TEM and CQ (5.2%) were
significantly higher than those in the control cells (1.1%) and
cells treated with IR alone (2.5%), TEM alone (2.9%), and CQ alone
(3.3%; Fig. 4D). The proportion of
activated caspase-3/7 in SW480 cells treated with IR and a
combination of TEM and CQ (15.5%) were significantly higher than
those in control cells (3.5%) and cells treated with IR alone
(7.0%), TEM alone (9.3%), and CQ alone (11.0%; Fig. 4C). Furthermore, similar results were
obtained from HT-29 cells.
Discussion
The PI3K/AKT/mTOR pathway is an intracellular
signaling pathway involving the phosphorylation of proteins
downstream of tyrosine kinase receptors, resulting in
phosphorylation of ribosomal protein S6 and 4E-BP1 (14). In cancer cells, the activation of
mTOR signaling promotes tumor growth and angiogenesis and inhibits
apoptosis (14,15). Furthermore, the PI3K/AKT/mTOR
pathway and autophagy are known to play important roles in cancer
radioresistance. In the present study, the effect of TEM (mTOR
inhibitor) and CQ (autophagy inhibitor) combination therapy was
investigated on the radiosensitivity of CRC cells.
In the present study, S6 and 4E-BP1 were
phosphorylated after IR exposure with a peak at 24–36 h after IR
exposure, indicating that mTOR signaling was activated by IR. The
activation of mTOR signaling by IR has been reported in endometrial
and breast cancer cells as well as human umbilical vein endothelial
cells (3,16,17)
but not in CRC cells. Since mTOR inhibitor has been reported to
inhibit the repair of IR-induced DNA damage in breast cancer cells,
mTOR signaling activated by IR is considered to play an important
role in radioresistance (4).
Generally during the autophagic process, the
autophagy substrate SQSTM1/p62 is consumed, LC3 is converted from
its isotype I to II, and acidic organelles are accumulated in the
cell (18,19). In particular, the LC3-II/LC3-I ratio
is considered an important parameter to evaluate the degree of
autophagy. In the present study, p62 levels decreased and the
LC3-II/LC3-I ratios and accumulation of acidic organelles increased
with a peak at 36 h after IR exposure, indicating that autophagy
was induced by IR. These results are consistent with previous
studies on IR-induced autophagy in many solid cancer cells,
including CRC cells, indicating that IR-induced autophagy may play
a cytoprotective role (10–12).
Several studies have reported the
irradiation-induced activation of the mTOR pathway (3,16,17)
and autophagy pathway (10–12) in different cell lines, despite the
suppressive effect of the activated mTOR pathway on autophagy. It
has been reported that autophagy can be induced via several
pathways after radiation, such as those involving accumulation of
reactive oxygen (ROS), calcium ions and mitochondrial damage
(7). The present results indicated
that irradiation induced autophagy activation independently of the
mTOR pathway and that this activation overcame the suppressive
effect of the mTOR pathway; however, further studies are warranted
to confirm this. TEM treatment in combination with irradiation
increased the LC3-II/LC3-I ratio and p62 consumption (Fig. 3), indicating the suppressive effect
of the mTOR pathway on autophagy and enhancement of autophagy by
mTOR inhibition.
Next, a clonogenic assay was performed using TEM
and/or CQ, to determine whether the inhibition of mTOR signaling
and/or autophagy increases radiosensitivity in CRC cells. In both
SW480 and HT-29 cells, neither TEM nor CQ monotherapy influenced
the effect of radiation, but the interaction of the combination of
TEM and CQ and radiation levels was demonstrated by two-way ANOVA.
Western blotting revealed that TEM alone inhibited IR-induced mTOR
signaling but induced autophagy, as indicated by decreased p62 and
increased LC3-II/LC3-I ratios. mTOR is known to be a strong factor
for inhibiting autophagy; mTOR C1 phosphorylates ULK1 and
suppresses its function, which in turn suppresses the formation of
autophagosomes (7). An mTOR
inhibitor has been reported to induce autophagy by
dephosphorylating ULK1 and promoting the formation of
autophagosomes. In contrast, CQ alone was revealed to increase the
level of p62, indicating autophagy inhibition since CQ inhibits
autophagy in its late phase (9,20). As
anticipated, CQ monotherapy did not affect the levels of pS6 and
p4E-BP1, suggesting that CQ does not influence mTOR signaling.
Cells treated with a combination of TEM and CQ revealed the highest
levels of p62 and LC3-II/LC3-I ratios, confirming the inhibition of
IR-activated mTOR signaling by TEM and the late phase inhibition of
TEM-induced autophagy by CQ. Therefore, autophagy induction after
TEM treatment and active mTOR signaling after CQ treatment indicate
that TEM or CQ alone does not affect the radiosensitivity of cells
and that both inhibition of mTOR and autophagy collectively enhance
radiosensitivity. Although the radiosensitizing effect of the mTOR
inhibitor alone was reported in pancreatic, breast, and prostate
cancer cells via in vivo and in vitro studies
(3,21,22),
the radiosensitizing effect of rapamycin was not observed in a
clinical trial of rectal cancer (5). Similarly, the radiosensitizing effect
of autophagy inhibitor 3-MA was reported in breast cancer cells
(23), but CQ did not affect
radiosensitivity in CRC cells (24). According to these studies, the
radiosensitizing effects of an mTOR inhibitor alone or CQ alone
differed among the cell lines (25). In contrast, the combination of an
mTOR inhibitor and CQ was reported to improve the
anti-proliferative effect in several cells, including CRC cells
(26,27), however, the radiosensitizing effect
of this combination has not yet been reported. Hence, it was
considered that inhibition of mTOR signaling or autophagy alone was
insufficient for enhancing radiosensitivity, but co-inhibition of
both these processes is essential in CRC cells.
Cell death analysis revealed a significantly high
apoptosis rate after TEM and CQ combination therapy but not after
TEM or CQ monotherapy. Previous studies reported the preservation
of phosphorylated AKT, an anti-apoptotic protein after treatment
with an mTOR inhibitor, indicating that it does not induce
apoptosis (28). Autophagy
maintains the quality of mitochondria, thus preventing the
accumulation of intracellular ROS (7). Although autophagic inhibition has been
reported to induce apoptosis through aberrant ROS accumulation, in
the present study no significant increase was revealed in the
apoptosis rates after treatment with CQ alone. In contrast, the
combination of an mTOR inhibitor and CQ was reported induce
apoptosis in a synergistic manner, consistent with the results
obtained in our study (8,9,27). An
mTOR inhibitor induces autophagosome formation and CQ leads to the
permeabilization of the autophagosome membrane, resulting in the
induction of apoptosis (29–31).
In the present study, the accumulation of autophagosomes was
observed in IR-exposed cells, but the accumulation was enhanced in
cells treated with IR and TEM. Thus, it was hypothesized that
adding CQ in the presence of excessive autophagosome accumulation
may contribute to increased apoptosis via autophagosome membrane
permeabilization.
There are several limitations of the present study.
First, this is an in vitro study, and the findings from an
in vivo study may not be consistent with our findings.
Furthermore, although the safety of the combination of TEM and CQ
has been demonstrated in a phase-I clinical study for several solid
cancers (32), clinical studies
investigating the effect of this combination on radiosensitivity
have not yet been performed, and prospective clinical trials are
warranted to confirm our results. Second, the radiosensitizing
effect of the combination of TEM and CQ in this study may be
attributed to their unique effects, and a combination of other mTOR
and autophagy inhibitors may not yield results consistent with
those obtained in the present study. Therefore, future
investigations using various mTOR and autophagy inhibitors are
required. This study would be greatly strengthened by future
validating studies using clinical colorectal cancer tissues or
colorectal cancer xenograft in vivo models, which were
beyond the scope of the present study.
In conclusion, the radiosensitizing effect of the
combination of TEM and CQ was demonstrated in CRC cells. This
combination therapy may be considered as an effective chemotherapy
combined with radiotherapy in patients undergoing neoadjuvant CRT
for rectal cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by Grants-in-Aid for
Scientific Research (grant nos. 16K07143, 16K07161, 17K10620,
17K10621, 17K10623 and 18K07194) from the Japan Society for the
Promotion of Science. This research was also supported by the
Project for Cancer Research and Therapeutic Evolution (P-CREATE;
grant no. 18cm0106502h0003) from the Japan Agency for Medical
Research and Development (AMED).
Availability of data and materials
All datasets generated during this study are
included in this published article.
Authors' contributions
HSh, KK, KH and HN aided in the concept and design
of the project. HSh, TT, TN, KO, KS, MK, KM, SE and HSo contributed
to manuscript preparation and data collection. HSh and KK
interpreted the results, analyzed the data, and revised the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Martin ST, Heneghan HM and Winter DC:
Systematic review and meta-analysis of outcomes following
pathological complete response to neoadjuvant chemoradiotherapy for
rectal cancer. Br J Surg. 99:918–928. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rödel C, Liersch T, Becker H, Fietkau R,
Hohenberger W, Hothorn T, Graeven U, Arnold D, Lang-Welzenbach M,
Raab HR, et al: Preoperative chemoradiotherapy and postoperative
chemotherapy with fluorouracil and oxaliplatin versus fluorouracil
alone in locally advanced rectal cancer: Initial results of the
German CAO/ARO/AIO-04 randomised phase 3 trial trial. Lancet Oncol.
13:679–687. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Albert JM, Kim KW, Cao C and Lu B:
Targeting the Akt/mammalian target of rapamycin pathway for
radiosensitization of breast cancer. Mol Cancer Ther. 5:1183–1189.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen H, Ma Z, Vanderwaal RP, Feng Z,
Gonzalez-Suarez I, Wang S and Zhang J, Roti Roti JL, Gonzalo S and
Zhang J: The mTOR inhibitor rapamycin suppresses DNA double-strand
break repair. Radiat Res. 175:214–224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Buijsen J, van den Bogaard J, Jutten B,
Belgers E, Sosef M, Leijtens JW, Beets GL, Jansen RL, Riedl RG,
Clarijs R, et al: A phase I–II study on the combination of
rapamycin and short course radiotherapy in rectal cancer. Radiother
Oncol. 116:214–220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chaurasia M, Bhatt AN, Das A, Dwarakanath
BS and Sharma K: Radiation-induced autophagy: Mechanisms and
consequences. Free Radic Res. 50:273–290. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hu L, Wang H, Huang L, Zhao Y and Wang J:
Crosstalk between autophagy and intracellular radiation response
(Review). Int J Oncol. 49:2217–2226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rosich L, Xargay-Torrent S, López-Guerra
M, Campo E, Colomer D and Roué G: Counteracting autophagy overcomes
resistance to everolimus in mantle cell lymphoma. Clin Cancer Res.
18:5278–5289. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaneko M, Nozawa H, Hiyoshi M, Tada N,
Murono K, Nirei T, Emoto S, Kishikawa J, Iida Y, Sunami E, et al:
Temsirolimus and chloroquine cooperatively exhibit a potent
antitumor effect against colorectal cancer cells. J Cancer Res Clin
Onco. 140:769–781. 2014. View Article : Google Scholar
|
|
10
|
Mo N, Lu YK, Xie WM, Liu Y, Zhou WX, Wang
HX, Nong L, Jia YX, Tan AH, Chen Y, et al: Inhibition of autophagy
enhances the radiosensitivity of nasopharyngeal carcinoma by
reducing Rad51 expression. Oncol Rep. 32:1905–1912. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun Q, Liu T, Yuan Y, Guo Z, Xie G, Du S,
Lin X, Xu Z, Liu M, Wang W, et al: MiR-200c inhibits autophagy and
enhances radiosensitivity in breast cancer cells by targeting
UBQLN1. Int J Cancer. 136:1003–1012. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Apel A, Herr I, Schwarz H, Rodemann HP and
Mayer A: Blocked autophagy sensitizes resistant carcinoma cells to
radiation therapy. Cancer Res. 68:1485–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okuno T, Kawai K, Hata K, Murono K, Emoto
S, Kaneko M, Sasaki K, Nishikawa T, Tanaka T and Nozawa H: SN-38
Acts as a radiosensitizer for colorectal cancer by inhibiting the
radiation-induced Up-regulation of HIF-1α. Anticancer Res.
38:3323–3331. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 168:960–976. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fingar DC, Richardson CJ, Tee AR, Cheatham
L, Tsou C and Blenis J: mTOR controls cell cycle progression
through its cell growth effectors S6K1 and 4E-BP1/eukaryotic
translation initiation factor 4E. Mol Cell Biol. 24:200–216. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Miyasaka A, Oda K, Ikeda Y, Sone K, Fukuda
T, Inaba K, Makii C, Enomoto A, Hosoya N, Tanikawa M, et al:
PI3K/mTOR pathway inhibition overcomes radioresistance via
suppression of the HIF1-α/VEGF pathway in endometrial cancer.
Gynecol Oncol. 138:174–180. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Edwards E, Geng L, Tan J, Onishko H,
Donnelly E and Hallahan DE: Phosphatidylinositol 3-kinase/Akt
signaling in the response of vascular endothelium to ionizing
radiation. Cancer Res. 62:4671–4677. 2002.PubMed/NCBI
|
|
18
|
Nishikawa T, Tsuno NH, Okaji Y, Shuno Y,
Sasaki K, Hongo K, Sunami E, Kitayama J, Takahashi K and Nagawa H:
Inhibition of autophagy potentiates sulforaphane-induced apoptosis
in human colon cancer cells. Ann Surg Oncol. 17:592–602. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sasaki K, Tsuno NH, Sunami E, Tsurita G,
Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, et al:
Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on
colon cancer cells. BMC Cancer. 10:3702010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Al-Bari MA: Chloroquine analogues in drug
discovery: New directions of uses, mechanisms of actions and toxic
manifestations from malaria to multifarious diseases. J Antimicrob
Chemother. 70:1608–1621. 2015.PubMed/NCBI
|
|
21
|
Cao C, Subhawong T, Albert JM, Kim KW,
Geng L, Sekhar KR, Gi YJ and Lu B: Inhibition of mammalian target
of rapamycin or apoptotic pathway induces autophagy and
radiosensitizes PTEN nulll prostate cancer cells. Cancer Res.
66:10040–10047. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Manegold PC, Paringer C, Kulka U, Krimmel
K, Eichhorn ME, Wilkowski R, Jauch KW, Guba M and Bruns CJ:
Antiangiogenic therapy with mammalian target of rapamycin inhibitor
RAD001 (Everolimus) increases radiosensitivity in solid cancer.
Clin Cancer Res. 14:892–900. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lomonaco SL, Finniss S, Xiang C,
Decarvalho A, Umansky F, Kalkanis SN, Mikkelsen T and Brodie C: The
induction of autophagy by gamma-radiation contributes to the
radioresistance of glioma stem cells. Int J Cancer. 125:717–722.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yan C, Luo L, Goto S, Urata Y, Guo CY, Doi
H, Kitazato K and Li TS: Enhanced autophagy in colorectal cancer
stem cells does not contribute to radio-resistance. Oncotarget.
7:45112–45121. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bristol ML, Emery SM, Maycotte P, Thorburn
A, Chakradeo S and Gewirtz DA: Autophagy inhibition for
chemosensitization and radiosensitization in cancer: Do the
preclinical data support this therapeutic strategy? J Pharmacol Exp
Ther. 344:544–552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Enzenmuller S, Gonzalez P, Debatin KM and
Fulda S: Chloroquine overcomes resistance of lung carcinoma cells
to the dual PI3K/mTOR inhibitor PI103 by lysosome-mediated
apoptosis. Anticancer Drugs. 24:14–19. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Seitz C, Hugle M, Cristofanon S,
Tchoghandjian A and Fulda S: The dual PI3K/mTOR inhibitor
NVP-BEZ235 and chloroquine synergize to trigger apoptosis via
mitochondrial-lysosomal cross-talk. Int J Cancer. 132:2682–2693.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Choo AY, Yoon SO, Kim SG, Roux PP and
Blenis J: Rapamycin differentially inhibits S6Ks and 4E-BP1 to
mediate cell-type-specific repression of mRNA translation. Proc
Natl Acad Sci USA. 105:17414–17419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shi TT, Yu XX, Yan LJ and Xiao HT:
Research progress of hydroxychloroquine and autophagy inhibitors on
cancer. Cancer Chemother Pharmacol. 79:287–294. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boya P, Gonzalez-Polo RA, Poncet D,
Andreau K, Vieira HL, Roumier T, Perfettini JL and Kroemer G:
Mitochondrial membrane permeabilization is a critical step of
lysosome-initiated apoptosis induced by hydroxychloroquine.
Oncogene. 22:3927–3936. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lakhter AJ, Sahu RP, Sun Y, Kaufmann WK,
Androphy EJ, Travers JB and Naidu SR: Chloroquine promotes
apoptosis in melanoma cells by inhibiting BH3 domain-mediated PUMA
degradation. J Invest Dermatol. 133:2247–2254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rangwala R, Chang YC, Hu J, Algazy KM,
Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel
AB, et al: Combined MTOR and autophagy inhibition: Phase I trial of
hydroxychloroquine and temsirolimus in patients with advanced solid
tumors and melanoma. Autophagy. 10:1391–1402. 2014. View Article : Google Scholar : PubMed/NCBI
|