Introduction
The incidence of tumors is rising due to population
growth, aging and advancements in tumor diagnostics (1), making malignant tumors a significant
threat to human health and life (2,3).
Cancer treatment options include surgery, radiotherapy,
chemotherapy, targeted therapy, hormone therapy and immunotherapy
(1), with chemotherapy being the
most widely used across various cancer types. However,
chemoresistance remains a complex clinical challenge, leading to
the majority of cancer-related deaths (4,5).
Understanding the mechanisms of chemoresistance is therefore
essential for enhancing therapeutic efficacy, and the present
article reviewed the latest developments in tumor chemoresistance.
Notably, since numerous studies have already detailed the drug
resistance mechanisms of molecular targeted therapies, the present
review did not delve into those aspects (6–8).
Anatomical barriers that limit or obstruct the entry
of foreign substances into target tissues or cells play a
significant role in tumor chemoresistance. For example, in mammals,
the blood-brain barrier, composed of tightly connected endothelial
cells, basement membranes and astrocytes, restricts the passage of
most chemotherapeutic drugs, thereby reducing their efficacy
against central nervous system cancers. Mechanosensitive
Sox2+ tumor cells, for instance, develop chemoresistance
by constructing a blood-tumor barrier around capillaries (9). Similarly, the blood-testis barrier,
formed by testicular supporting cells, protects germ cells from
toxic substances. Sertoli cells, which are resistant to apoptosis
despite strong inflammatory stimuli (for example, LPS and IL-18),
play a pivotal role in maintaining the homeostasis of the
testicular environment, and this anti-apoptotic and
anti-inflammatory capability further enhances their resistance to
chemotherapy (10).
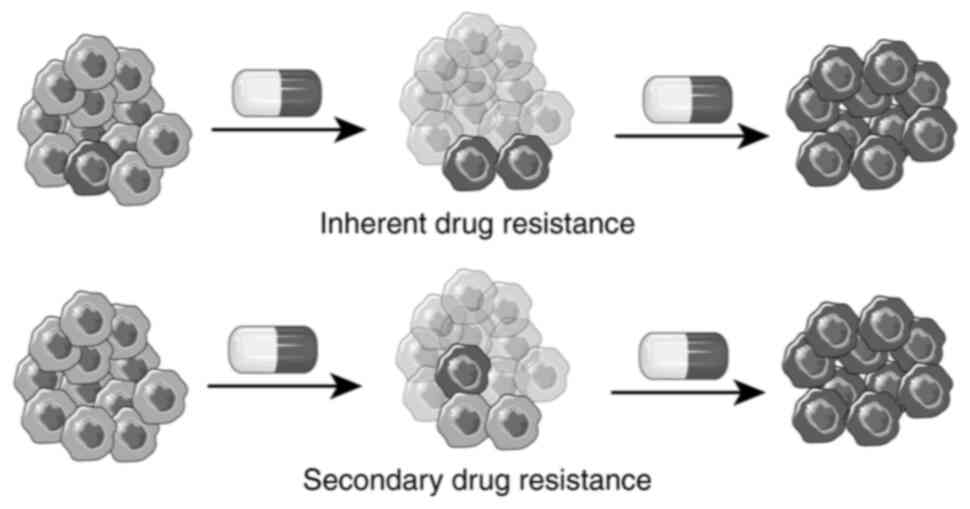
Chemotherapy resistance in cancer can be categorized
into inherent and secondary resistance based on its origin.
Inherent resistance refers to the presence of drug-resistant cells
within the tumor tissue before treatment begins (11). This concept suggests that drug
resistance is a ‘fait accompli’, with relapse occurring once
preexisting resistant cells repopulate (12). By contrast, secondary drug
resistance arises when tumor cells develop resistance to
chemotherapeutic drugs due to intracellular changes induced by the
drugs themselves (Fig. 1).
Chemotherapy resistance can also be classified into
primary drug resistance and multidrug resistance, depending on the
response of tumor cells to chemotherapy agents (13). Primary drug resistance occurs when
tumor cells develop resistance to a therapeutic drug after
treatment, commonly due to decreased drug uptake or increased drug
efflux. Multidrug resistance, on the other hand, involves tumor
cells becoming resistant not only to the therapeutic agent but also
to other chemotherapeutic drugs to which they have not been
previously exposed. This phenomenon is driven by mechanisms such as
enhanced drug efflux, increased DNA repair, neutralization of
chemotherapeutic agents, closure of nuclear pores and tumor cell
dormancy.
Mechanisms of action of chemotherapy
drugs
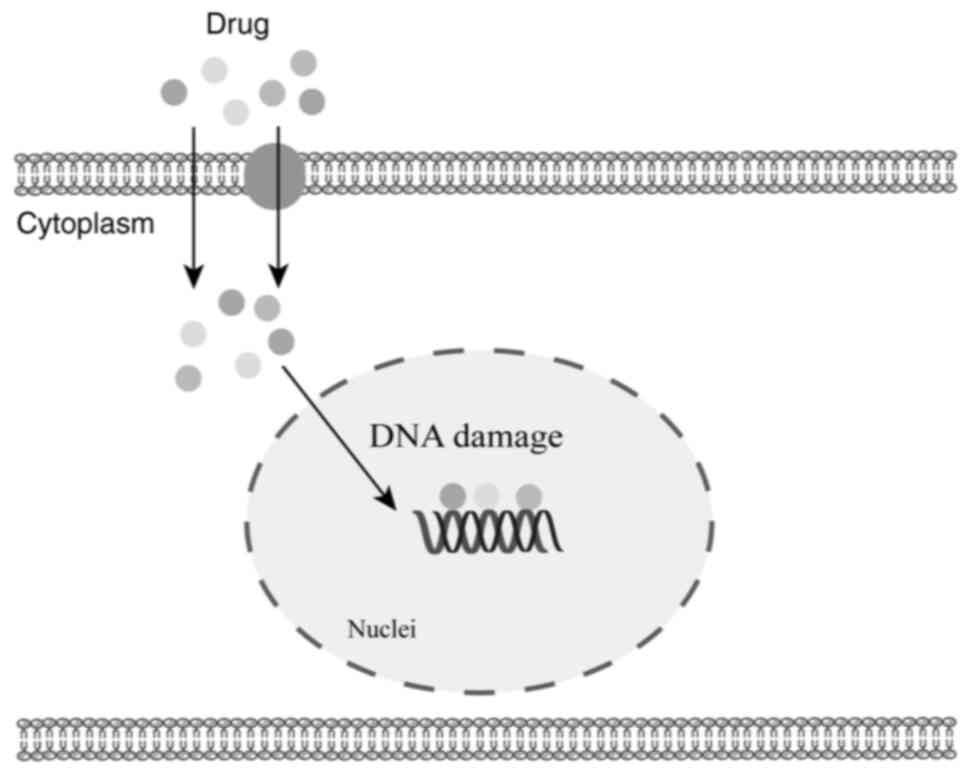
Chemotherapeutic agents encompass a wide range of
categories, including alkylating agents, antimetabolites,
antibiotics and antitumor phytopharmaceuticals, among others.
Although these agents employ diverse mechanisms of action, the
majority must penetrate the cell nucleus to exert their therapeutic
effects (Fig. 2).
Alkylating agents, such as cyclophosphamide and
cisplatin, are capable of targeting both proliferative and
non-proliferative cells. These agents possess active alkylating
groups that generate electrophilic groups with positive carbon
ions. These ions form covalent bonds with various nucleophilic
groups within cells, leading to cross-linking between DNA molecules
or between DNA and proteins, ultimately causing disruptions in DNA
replication and transcription, culminating in cell death (14,15).
Furthermore, alkylating agents have been shown to induce protein
oxidation and acetylation, which can impair the lipid homeostasis
of the nuclear membrane and destabilize the cellular genome
(15). Cisplatin, a cis-alkylating
agent, specifically induces cross-links within DNA double strands,
resulting in the cessation of DNA replication and transcription, or
even causing DNA strand breaks and apoptosis (16). As the first metal-based
chemotherapeutic drug, cisplatin has been extensively used, though
its side effects, including ototoxicity and nephrotoxicity, have
posed significant challenges in its clinical application (17,18).
Antimetabolites, structurally similar to natural
metabolites but lacking their corresponding biological functions,
competitively inhibit and disrupt nucleic acid and protein
synthesis, ultimately leading to cell death (19). The most notable example is
5-fluorouracil, which inhibits thymidine synthetase, thereby
impairing DNA synthesis within the body (20).
Antitumor antibiotics, derived from microorganisms,
exert their effects by directly damaging DNA or intercalating
within it, thereby disrupting transcription (21). A key example is doxorubicin, which
is extensively employed in the treatment of hematological
malignancies, breast cancer and lung cancer (22).
Plant-derived antitumor agents, such as paclitaxel
and vincristine, inhibit tumor cell proliferation by targeting
microtubule proteins. Paclitaxel primarily prevents the
depolymerization of microtubules during mitosis, whereas
vincristine inhibits microtubule polymerization (23). Despite the clinical success of these
plant-derived anticancer drugs, tumor resistance to them remains
prevalent, particularly in some gastrointestinal cancers that
exhibit resistance from the onset of chemotherapy.
Molecular mechanisms of chemotherapy
resistance
Decreased drug entry into cells
Plant-derived chemotherapeutic agents, such as
paclitaxel and vincristine, possess favorable lipid solubility,
enabling them to permeate cells via passive diffusion through the
lipid bilayer of the cell membrane (24,25).
By contrast, cisplatin, a metal-based anticancer drug, increases
its water solubility by replacing chloride ions with
Pt(NH3)2(H2O)2.
However, this substitution process occurs slowly, necessitating
that cisplatin primarily enters cells through active transport
mechanisms (26).
Research has identified that defective expression of
copper ion transporter protein 1 (CTR1) correlates with reduced
platinum accumulation in tissues, leading to decreased therapeutic
efficacy of platinum-based drugs in various tumors (27). Under basal conditions, CTR1 is
predominantly localized in the perinuclear region of cultured
cells, but in the presence of copper complexes, CTR1 is highly
expressed on the cell membrane, facilitating cisplatin's active
transport. The uptake of cisplatin mediated by CTR1 depends on its
metal-bound extracellular region, which is connected to the
N-glycan chain at Asn15 and the O-glycan chain at Thr27 (28). Although cisplatin does not alter the
subcellular localization of CTR1, co-localization studies using
fluorescent cisplatin derivatives revealed their presence in
CTR1-associated vesicular structures, indicating that cisplatin is
endocytosed concurrently with CTR1 and transported into the cell
(29).
Organic cation transporters (OCTs) are essential for
the cellular uptake of endogenous cationic substrates, hydrophilic
exogenous compounds, and platinum-based anticancer drugs. OCTs are
highly expressed in the renal basement membranes of humans and
mice, playing a significant role in mediating cisplatin uptake
(30). Elevated levels of OCT2 in
pre-chemotherapy biopsy samples have been associated with improved
prognosis when treated with cisplatin-inclusive regimens (31). Conversely, in OCT2 knockout (−/-)
mouse models, both nephrotoxicity and ototoxicity were
significantly reduced following cisplatin treatment (32). Although overexpressing transport
proteins exogenously is not yet feasible in clinical practice,
current strategies focus on developing novel carriers, such as
nanomaterial conjugates, to enhance the intracellular delivery of
chemotherapeutic drugs. This approach aims to increase the
intracellular concentration of these drugs, thereby enhancing their
efficacy in tumor cell eradication.
Increased chemotherapy drug
excretion
Members of the ABC protein transporter family,
primarily located in cell membranes, are responsible for expelling
cytotoxic substances, including chemotherapeutic drugs, out of the
cell by utilizing ATP hydrolysis for energy (33). This process impedes the accumulation
of therapeutic drug concentrations in target cells or organs,
thereby contributing to chemoresistance (34). Moreover, ABC transporter proteins
have been implicated in promoting tumor cell migration and invasion
(35). The ABC transporter family
is categorized into seven subgroups (ABCA-ABCG) comprising 48
proteins, based on sequence homology (36). All subfamilies, except the ABCF
family, have been linked to tumor chemoresistance, with key members
including ABCB1, ABCC1 and ABCG2.
The ABCB1 protein, also known as P-glycoprotein
(P-gp), serves a physiological role in safeguarding sensitive
tissues and the fetus from endogenous and exogenous toxins. It
primarily facilitates the transport of lipid-soluble drugs, such as
paclitaxel, vincristine and adriamycin (34,36–38).
Although P-gp binding substrates like everolimus have shown
potential in reversing chemoresistance in vitro and in
animal models by interacting with extracellular binding epitopes,
clinical trials have not yielded significant results, leaving the
underlying mechanisms unclear. Developing inhibitors that induce
conformational changes in ATP transport proteins remains a
promising avenue for future research (34,39–41).
The ABCC1 protein, also known as multidrug
resistance-associated protein 1 (MRP1), was the second drug
resistance transporter identified after P-gp. MRP1 transports a
wide array of pathophysiological substrates, including folic acid,
bilirubin, anthracyclines, glutathione (GSH) and glucuronide
conjugates, though it is less effective at transporting
paclitaxel-like drugs compared with other ABC transporters
(36,41–43).
Tumor cells overexpressing MRP1 exhibit lower intracellular GSH
levels and higher GSH efflux, leading to its designation as a GSH
transporter protein. After GSH binds to chemotherapeutic drugs to
form a complex, MRP1-mediated co-extrusion reduces the
intracellular concentration of these drugs, contributing to tumor
cell drug resistance (41,44).
ABCG2, also known as breast cancer resistance
protein, was first identified in breast cancer-resistant tumors and
functions as a hemi-transport protein, requiring homo- or
heterodimerization to act as an effective transporter (45). Cryo-electron microscopy has revealed
that ABCG2 can bind and transport chemotherapeutic agents such as
topotecan and mitoxantrone, as well as P-gp inhibitors such as
tariquidar (46).
Currently, the primary strategy for modulating the
ABC transporter protein family involves targeting overexpressed
proteins, but clinical benefits have been limited. Future research
may focus on developing drugs that induce conformational changes in
ABC transporter proteins, potentially offering a new direction for
overcoming chemoresistance.
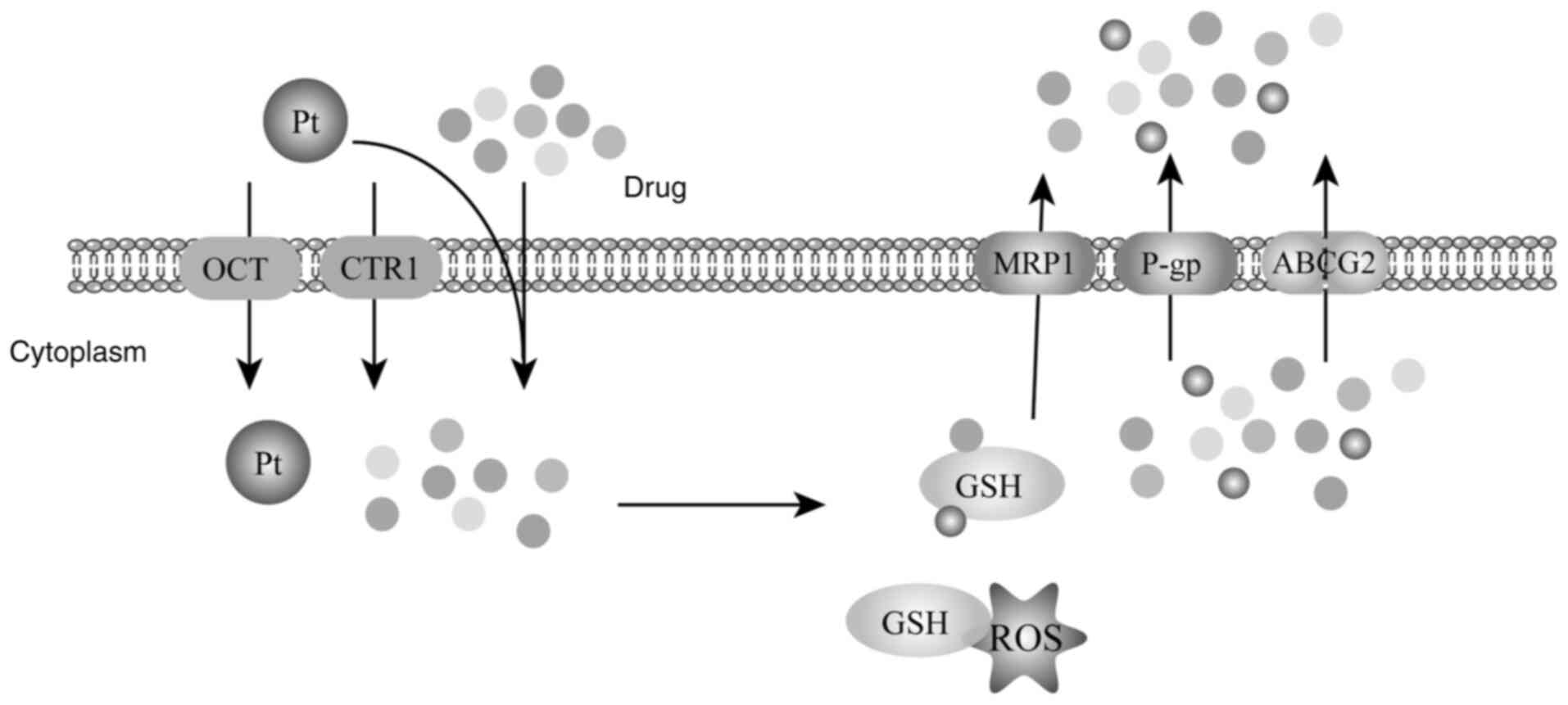
GSH and drug binding
GSH, a tripeptide consisting of glutamate, cysteine
and glycine, is a critical component of the cellular antioxidant
system, essential for tumor cells in scavenging reactive oxygen
species (ROS) and neutralizing chemotherapeutic agents (47). Predominantly existing in its reduced
form within the cell, GSH reacts with oxidizing agents such as ROS,
converting to GSH disulfide (GSSG). Excess GSSG can be expelled
from the cell or reduced back to GSH via GSH reductase. The
intracellular GSH concentration typically ranges from 2–10 mM,
~1,000-fold higher than in the extracellular environment (2–10 µM)
(47).
GSH also plays a critical role in ROS scavenging as
part of the cellular antioxidant system. Elevated GSH levels in
tumor cells safeguard them from ROS-induced DNA damage and
apoptosis by neutralizing ROS during tumor growth. Research
indicates that reducing GSH levels in tumor cells can elevate ROS
levels, thereby enhancing the cytotoxic effects of therapeutic
agents on these cells (48,49) (Fig.
3).
Moreover, GSH is involved in the neutralization of
cytotoxic substances under physiological conditions, with numerous
chemotherapeutic drugs, such as cisplatin and adriamycin, acting as
binding substrates for GSH (50).
Catalyzed by GSH-S-transferase (GST), GSH directly binds to
electrophilic xenobiotics in the cytoplasm, preventing these
chemotherapeutic drugs from entering the nucleus and exerting their
inhibitory effects. The resulting GSH-drug conjugates are then
exported from the cell via MRP1, thereby reducing the intracellular
concentration of these drugs and contributing to the development of
chemoresistance in tumor cells (51). Experimental evidence suggests that
decreasing intracellular GSH levels can reverse cisplatin
resistance and enhance its therapeutic efficacy (52).
Intracellular GSH levels are modulated by various
factors. Transforming growth factor β (TGF-β) is a well-known
inhibitor of normal epithelial cell proliferation, and its
suppression can increase the susceptibility of epithelial tissues
to cancer. Interestingly, while high TGF-β signaling expression in
the skin prevents benign papillary tumors from progressing to
malignancy, it paradoxically facilitates the malignant
transformation of squamous cell carcinoma stem cells into squamous
carcinoma, thereby promoting metastasis (53). Upon activation, TGF-β ligands bind
to TbRII, which phosphorylates TbRI. The activated TbRI then
transmits signals by phosphorylating intracellular effectors SMAD2
and SMAD3 (SMAD2/3). These phosphorylated SMAD proteins form a
complex with SMAD4, which interacts with other transcriptional
regulators to activate downstream target genes. Notably, TGF-β
reporter gene-positive basal cells exhibit high expression of genes
involved in GSH metabolism and significantly lower ROS levels
(53). In RNA-seq analysis of
tyrosine kinase receptor inhibitor (TKI)-resistant non-small cell
lung cancer, aldo-keto reductase 1B1 (AKR1B1) was found to be
highly expressed in multiple drug-resistant cell lines, promoting
GSH biosynthesis through STAT3-mediated upregulation of
SLC7A11-dependent cystine uptake, leading to TKI resistance
(54).
Both GSH and GST levels are elevated in tumor cells
compared with normal cells, closely correlating with drug
resistance. GSH levels can be measured using gold nanoparticles
detected by a lateral flow plasma biosensor or even visually, with
quantification achieved through automated analysis software. This
method has shown a strong positive correlation between GSH levels
and temozolomide resistance in GBM cells (55).
Beyond the GSH antioxidant system, eukaryotes
possess another critical antioxidant mechanism-the thioredoxin
system-which manages intracellular oxidative stress and supports
cell proliferation when GSH is depleted (56). Cytosolic flavin adenine dinucleotide
oxidoreductase 1 (TXNRD1), a selenoprotein with extensive
antioxidant and redox regulatory functions, is overexpressed in
numerous cancers and plays a key role in their growth and survival.
Inhibiting TXNRD1, especially in the context of GSH depletion,
increases oxidative stress within tumor cells, leading to their
death (57). Given the protective
role of the GSH system in shielding tumor cells from
chemotherapeutic agents and ROS, exploring the regulatory
mechanisms of this system and developing strategies to deplete GSH
in tumor cells using molecularly targeted drugs or nanomaterials
holds significant therapeutic promise.
Closure of nuclear pores and
exocytosis of drugs in the nucleus
The nuclear pore complex is a basket-like structure
embedded within the inner and outer nuclear membranes, featuring
apertures of ~70-80 nm and a channel diameter of ~9 nm,
facilitating the exchange of substances between the nucleus and the
cytoplasm. Numerous chemotherapeutic agents, such as platinum and
fluoropyrimidines, which inhibit DNA replication, must traverse the
nuclear pore complex to reach their targets and exert their
cytotoxic effects (58). Vault
particles, barrel-shaped structures primarily composed of major
vault protein (MVP), vADP-ribose polymerase, and
telomerase-associated protein 1, are considered to interact with
the nuclear pore complex. MVP, which constitutes 70% of the vault's
mass, is considered to mediate the translocation of macromolecules,
potentially rerouting chemotherapeutic drugs away from their
subcellular targets and contributing to multidrug resistance in
tumor cells (59,60). However, this hypothesis requires
further validation.
Lung resistance-related protein (LRP), also known as
major vault protein, is a key component of vault particles and
nuclear pore complexes in humans (61). Studies have demonstrated that LRP is
highly expressed in numerous tumor cells that lack P-gp expression,
and its elevated levels are associated with reduced chemotherapy
sensitivity (62). In a lung
adenocarcinoma cell model, gefitinib-resistant cells exhibited
significant upregulation of YB-1, which promotes downstream AKT
signaling and activates epithelial-mesenchymal transition (EMT),
thereby increasing resistance to gefitinib through direct
activation of MVP (63). CD73, a
glycosylphosphatidylinositol-anchored plasma membrane protein,
physiologically hydrolyzes extracellular adenosine monophosphate
into adenosine and inorganic phosphate. Evidence suggests that CD73
interacts with MVP, activating the SRC-AKT pathway and affecting
gemcitabine chemosensitivity in pancreatic ductal adenocarcinoma
(64).
While LRP is known to enhance tumor cell resistance
to chemotherapeutic agents, MVP deficiency has been associated with
a reduced likelihood of tumor progression. In HBV-encoded protein X
(HBx)-transgenic (TG) mice crossed with LRP-deficient mice,
significant reductions in tumor number, size, liver-to-body weight
ratio, alanine aminotransferase levels and alpha-fetoprotein levels
were observed compared with HBx-TG mice carrying the HBx,
indicating that LRP loss greatly diminishes tumor progression and
extends survival time (65).
Vault RNAs (vtRNAs), non-coding RNAs comprising ~5%
of vault particles, have been found to show high expression in cell
lines with elevated EBV protein levels, particularly those
expressing high levels of LMP1. Overexpression of vtRNAs has been
associated with increased EBV expression and the inhibition of
apoptosis through the overexpression of NOL3 and BCL-xl (59). Although most chemotherapeutic
agents, including platinum compounds, antibiotics and
phytochemicals, require nuclear entry to function effectively, the
mechanisms governing their translocation across the nuclear
membrane remain underexplored. Further investigation into the
nuclear transport mechanisms of chemotherapeutic drugs and other
macromolecules is essential for advancing the understanding of
tumor chemoresistance.
Increased DNA repair
The DNA damage response (DDR) pathway plays a
pivotal role in recognizing, signaling and repairing DNA damage
caused by endogenous or exogenous factors, including
chemotherapeutic agents. It also regulates cell cycle progression
through DNA repair mechanisms, mitigating damage and preventing
apoptosis in the case of unrepaired lesions. As such, the DDR
pathway is a critical target in cancer therapy (66–68).
Numerous chemotherapeutic drugs, such as platinum compounds,
alkylating agents and antibiotics, induce DNA double-strand breaks
by directly binding to DNA. These breaks are among the most lethal
forms of DNA damage; failure to repair them triggers cell death.
Enhanced DNA repair mechanisms are a key factor in the development
of multidrug resistance in tumor cells (58,69–71).
Two primary pathways are responsible for repairing
DNA double-strand breaks (DSBs): non-homologous end-joining (NHEJ)
and homologous recombination (HR). HR typically requires sister
chromatids as templates, limiting its activity to the S and G2
phases of the cell cycle. The efficiency of DNA repair largely
depends on HR-promoting factors including BRCA1 and NHEJ-promoting
proteins such as TP53BP1 (72).
BRCA1 and BRCA2 are essential for repairing DSBs, playing a key
role in inhibiting tumor cell proliferation and contributing to
drug resistance (73–75). Cells with defective BRCA1 or BRCA2
genes exhibit reduced homologous recombination repair capacity,
making them more sensitive to DNA-damaging agents such as cisplatin
and poly (ADP-ribose) polymerase (PARP) inhibitors (75).
DNA topoisomerase-2 (TOP2) is an enzyme essential
for DNA replication, transcription and recombination, with a known
role in promoting cancer across various tumors (76). In hepatocellular carcinoma cells,
continuous exposure to regorafenib leads to upregulation of TOP2A
expression, while silencing TOP2A increases the sensitivity of
these cells to regorafenib (77).
PARP, a poly ADP-ribose polymerase, is involved in
chromatin modification, DNA replication, transcription and DNA
repair, particularly through base excision repair of single-strand
DNA (ssDNA) damage. In the absence of functional PARP, ssDNA breaks
remain unrepaired, leading to DSBs during subsequent cell cycles as
DNA replication forks encounter ssDNA regions. Cells with intact
DSB repair pathways can repair such breaks and survive. PARP
inhibitors (PARPi) specifically target BRCA1/2-deficient tumors,
inducing apoptosis (73). BRCA1/2
mutant cells are particularly vulnerable to PARPi due to their
inability to repair DSBs effectively (73). Treatment with the PARP inhibitor
olaparib has been shown to restore sensitivity to conventional
chemotherapy in patients with prostate cancer resistant to standard
treatments and carrying defective DNA repair genes, such as BRCA1/2
(78).
The phosphatase and tensin homolog (PTEN) gene is
critical in regulating DNA damage repair, chromosome stability and
cell cycle progression through phosphatase-independent mechanisms.
Phosphorylation at tyrosine 240 (pY240-PTEN) is frequently observed
in patients with tumors undergoing radiotherapy. This
phosphorylated form of PTEN is highly expressed, associates with
chromatin via interaction with Ki-67, facilitates RAD51
recruitment, and enhances DNA repair processes, contributing to
chemoresistance and poor prognosis (70).
Schlafen family member 11 (SLFN11) is another key
regulator of DNA damage repair and is linked to the cellular
response to DNA-damaging agents in vitro. Replication
protein A (RPA), a heterotrimeric complex, is the primary
eukaryotic single-stranded DNA-binding protein, crucial for various
DNA metabolic pathways, including DNA replication, recombination,
damage checkpoints and repair. SLFN11 is recruited to sites of DNA
damage in an RPA-dependent manner, destabilizing the RPA-ssDNA
complex. Elevated levels of SLFN11 result in defects in checkpoint
maintenance and homologous recombination repair, thereby
sensitizing cells to DNA-damaging agents (79). Enhancer of zeste homolog 2 (EZH2)
induces the H3K27me3 histone modification, leading to the silencing
of SLFN11 in vivo. The use of EZH2 inhibitors in combination
with standard cytotoxic therapies has been identified to prevent
secondary resistance and improve the efficacy of chemotherapy in
both chemo-sensitive and chemoresistant small-cell lung cancer
models (80).
Mutations in DNA methyltransferase 3A (DNMT3A),
particularly at arginine 882 (DNMT3Amut), are commonly associated
with poor response to erythromycin chemotherapy. DNMT3Amut cells
exhibit impaired nucleosome expulsion and chromatin remodeling in
response to anthracyclines, which in turn hinders the recruitment
of the histone chaperone SPT-16. This defect impairs the cell's
ability to detect and repair DNA damage, exacerbating the DNMT3Amut
phenotype. Additionally, DNMT3Amut cells display reduced CHK1
phosphorylation, along with diminished downstream p53
phosphorylation/stabilization and apoptotic signaling, further
contributing to chemoresistance (81).
DNA mismatch repair (MMR) is a conserved process
that identifies and corrects spontaneously misincorporated bases
during DNA replication, ensuring genomic integrity (82). Impairment in MMR leads to
microsatellite instability (MSI), a hallmark found in >20
different tumor types (82). Werner
helicase (WRN), part of the RecQ family of DNA helicases, is
critical in maintaining genomic stability, DNA repair, replication,
transcription and telomere maintenance. WRN has been identified as
a synthetic lethal target in deficient (d)MMR/MSI-H cancers, being
essential for the survival of dMMR/MSI-H cells both in vitro
and in vivo. Knockdown of WRN in these cells induces
double-stranded DNA breaks and significant genomic instability,
leading to apoptosis. WRN inhibition has proven effective in dMMR
colorectal cancer models that develop secondary resistance to
broad-spectrum chemotherapeutic agents such as irinotecan,
oxaliplatin, or 5-FU, as well as combinations involving epidermal
growth factor receptor (EGFR) monoclonal antibodies and BRAF or
NTRK inhibitors (82).
While enhanced DNA repair can contribute to
chemotherapy resistance, molecular alterations in MMR-related genes
can also drive tumorigenesis and progression (83). Studies have revealed that cells
exposed to targeted therapies (for example, EGFR inhibitors or BRAF
inhibitors) temporarily downregulate the expression of DDR-related
genes, including those involved in MMR and HR. This transient
suppression of DDR capacity is reversible, with gene expression
returning to normal levels after the cessation of targeted therapy
(84). The presence of DNA damage
and repair mechanisms within tumor cells-and whether these
mechanisms are actively engaged to prevent apoptosis-presents a
strategic opportunity for combination therapies. Exploiting the
transient DDR deficiencies that tumors experience during treatment
could enhance the effectiveness of anticancer therapies.
Effect of TME on chemotherapy
resistance
The tumor microenvironment (TME) is characterized by
several distinct features that differentiate it from normal
tissues, including acidic pH, hypoxia, elevated ROS, upregulated
antioxidant systems and overexpression of specific enzymes
(47). Hypoxia, a hallmark of
tumors, arises from the imbalance between oxygen consumption and
vascular oxygen supply in tumor tissues and is a critical factor in
promoting tumorigenesis and progression. This condition drives
metabolic reprogramming in tumor cells, allowing them to adapt to
the hypoxic TME (85). While
platinum-based chemotherapeutic drugs target rapidly proliferating
cancer cells, the quiescent cell population, often associated with
hypoxia, remains largely unaffected by such treatments.
Hypoxia-inducible factor 1 (HIF-1), composed of alpha and beta
subunits, is a key regulator of cellular hypoxic responses. HIF-1α
specifically modulates the cellular response to hypoxia,
influencing processes such as apoptosis, proliferation,
vasodilation, energy metabolism and angiogenesis. Salidroside has
been identified to promote the degradation of HIF-1α, and its
administration in combination with platinum drugs can reverse
platinum resistance and inhibit metastasis induced by the hypoxic
TME (86).
Hypoxia also contributes to tumor growth by
affecting exosome secretion. Exosomes, nanoscale extracellular
vesicles (30–150 nm in diameter), facilitate the transfer of
proteins, RNA and other molecules between cells within the TME,
thereby influencing the behavior of surrounding cells. In the
context of EGFR-mutant lung cancer, a common target in clinical
therapy, drug resistance remains a significant challenge. It has
been demonstrated that cells with wild-type EGFR can be
internalized by EGFR-mutant cancer cells via clathrin-dependent
endocytosis, leading to the acquisition of a wild-type EGFR
phenotype. This phenotypic change activates downstream PI3K/AKT and
MAPK signaling pathways, thereby triggering drug resistance
(87). Additionally, the
EGFR-targeted drug oxitinib has been shown to promote exosome
release by upregulating Rab GTPase, further contributing to drug
resistance (87).
In acute myeloid leukemia (AML), the apoptosis
repressor with caspase recruitment domain (ARC) protein serves as a
potent independent marker of poor prognosis. ARC activates NF-κB,
leading to increased IL1β expression in AML cells, which in turn
elevates the expression of CCL2, CCL4 and CXCL12 in mesenchymal
stromal cells (MSCs) (88). When
AML cells are co-cultured with MSCs, IL1β expression is further
elevated, driving AML cell migration towards CCL2, CCL4 and CXCL12.
Inhibition of IL1β has been revealed to reduce AML cell migration
(88). Moreover, co-cultures of AML
and MSCs have been found to increase Cox-2 expression in MSCs
through PGE2-mediated signaling in an ARC/IL1β-dependent manner,
thereby modulating ARC expression and enhancing the chemoresistance
of AML cells (89).
Microorganisms within the TME are significant
contributors to cancer progression and treatment resistance. A
study on ovarian epithelial cancer revealed that antibiotic use
during chemotherapy was linked to poorer overall survival,
primarily due to bacterial imbalance. Stool analysis indicated that
co-treatment with antibiotics and cisplatin disrupted 49
non-resistant intestinal microbes in mice, leading to accelerated
tumor growth and cisplatin resistance compared with mice treated
with chemotherapy alone. This resistance was characterized by
reduced apoptosis, increased DNA damage repair and enhanced
angiogenesis. However, transplanting cecal microbes from control
mice into the co-treated mice restored cisplatin sensitivity
(90). Similarly, L-lactic acid
produced by Lactobacillus iners in tumors reprograms
cervical tumor metabolism, enhancing resistance to gemcitabine
combined with 5-FU chemotherapy (91). Fusobacterium nucleatum and
its metabolite succinic acid induce resistance to anti-PD-1
monoclonal antibody immune checkpoint blockade therapy in
colorectal cancer by inhibiting key immune pathways and reducing
CD8+ T cell migration into the TME (92). In addition, telomelysin OBP-301, a
telomerase-specific, replication-competent oncolytic adenovirus
with an hTERT promoter upstream of the E1 gene, has been
demonstrated to enhance Akt phosphorylation in hepatocellular
carcinoma cells. However, the histone deacetylase inhibitor AR42
reduces telomerase-induced Akt phosphorylation and enhances
telomerase-induced apoptosis. Combined treatment with telomelysin
and AR42 demonstrated synergistic anti-hepatocellular carcinoma
effects (93).
Tumor cells do not exist in isolation; their
characteristics are intricately linked to the TME. The interactions
between tumor cells and other cells, including bacteria within the
microenvironment, as well as the unique pH and hypoxic conditions,
profoundly influence tumor cell characteristics and drug
sensitivity. Inflammation within the TME promotes drug resistance
by activating pro-inflammatory cytokines and signaling pathways.
This chronic inflammatory response not only drives tumor growth and
metastasis but also significantly reduces cancer cell sensitivity
to chemotherapy through the regulation of resistance protein
expression (94). Investigating how
the microenvironment contributes to chemotherapy resistance may
provide new therapeutic strategies that focus on targeting the TME,
rather than the tumor cells alone.
Tumor cells escape from the pressure of
chemotherapy through dormancy
What is tumor cell dormancy?
Tumor recurrence years after chemotherapy or surgery
is often attributed to the presence of dormant cancer cells within
the tumor mass. Dormancy refers to a reversible state in which
cells cease division, exhibit low metabolic activity, and reduce
mRNA synthesis, yet remain responsive to external stimuli (95–97).
Dormancy can occur in early metastatic sites as well as in residual
lesions following chemotherapy (98–100).
Traditionally, cellular dormancy was viewed as a quiescent state
associated with the G0 phase of the cell cycle (101). However, it was recently suggested
that these non-dividing cells exist in a slow-cycling survival
mode, akin to embryonic stasis, and are therefore also known as
slow-cycling or drug-resistant persister cells (100).
Dormant tumor cells differ from tumor stem-like
cells, though there is overlap between the two. Tumor stem-like
cells are generally considered a subset of dormant tumor cells,
possessing the ability to remain dormant, yet not all dormant cells
possess stem cell properties such as self-renewal and
differentiation.
Most chemotherapeutic agents target proliferating
cells, allowing tumor cells to evade therapeutic stress by entering
a dormant state, thereby contributing to drug resistance (102,103). Although some reactivated cells may
remain sensitive to chemotherapy, the prognosis for patients with
recurrent tumors is often poor (100,104). The induction of dormancy in
response to drug treatment underscores the importance of accurately
identifying dormant tumor cells to improve guidance of clinical
drug use (105).
How to determine tumor cell
dormancy
Next-generation sequencing and lentiviral barcoding
experiments have revealed that xenograft tumors arising after
chemotherapy-induced dormancy do not exhibit significant reductions
in genetic or barcode complexity, indicating that dormancy is a
non-genetic state (100). Tumor
cell dormancy functions as a survival strategy, enabling cells to
adapt to external stress rather than representing a distinct
subpopulation; all tumor cells have the potential to enter a
dormant state (100). Currently,
there are no specific markers for identifying dormant tumor cells.
Detection primarily relies on immunohistochemistry,
immunofluorescence and western blot analysis of relevant
indicators, typically associated with tumor stem-like cells, such
as CK, Sox-2 and CD133, as well as dormancy activation markers
including Ki-67, cyclin D1, C-myc, VEGF and proliferating cell
nuclear antigen. TUNEL staining is also commonly used (106). Additionally, tracking the mitotic
kinetics of dormant tumor cells with lipophilic fluorescent dyes
serves as another method for determining cellular dormancy
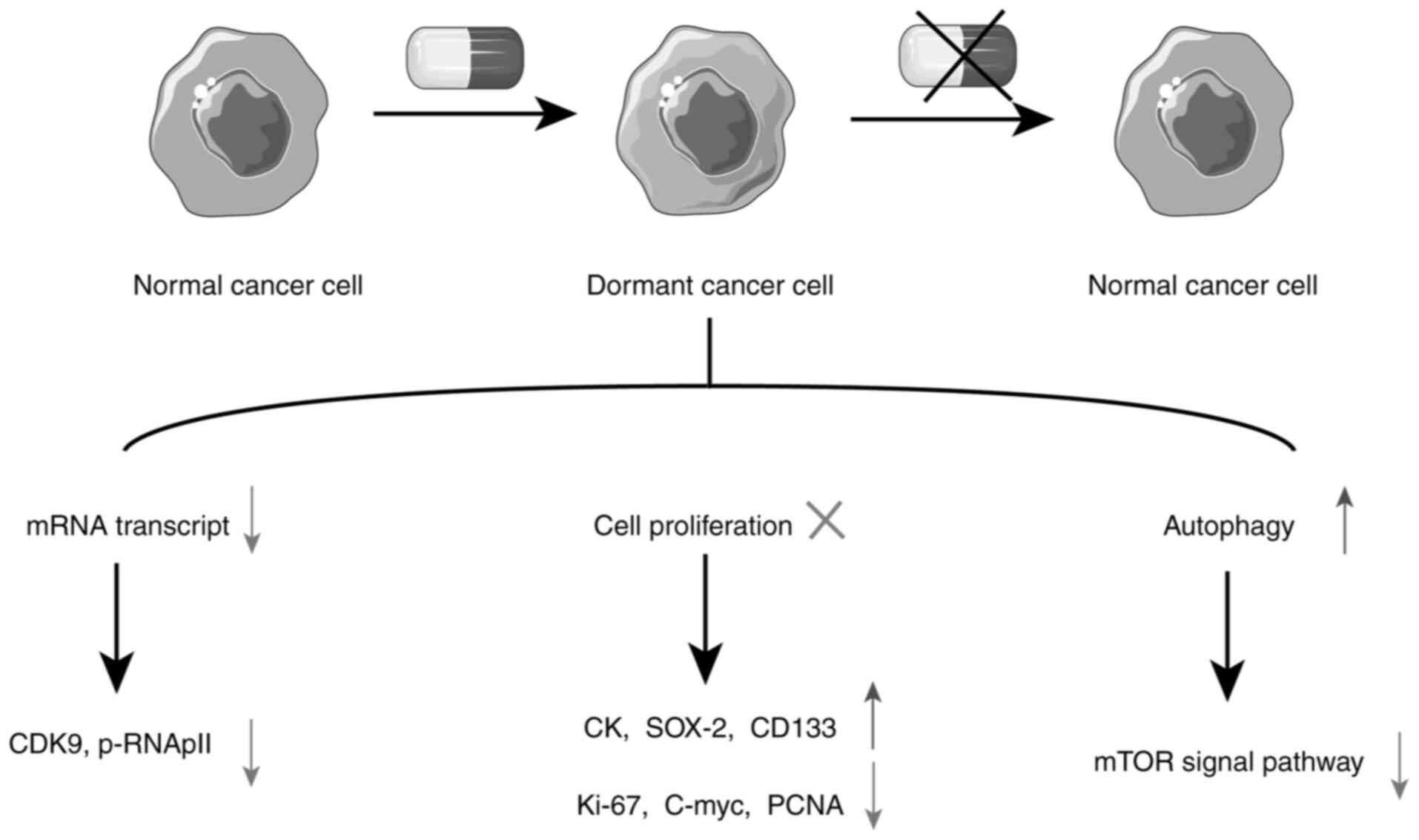
(107). One of the key features of
dormant cells is reduced mRNA synthesis activity. Since mRNA is
transcribed by RNA polymerase II (RNApII), low RNApII
phosphorylation is highly specific to dormant cells, with
cyclin-dependent kinase 9 (CDK9) being critical for
RNApII-dependent gene transcription. The Optical Stem Cell Activity
Reporter system distinguishes dormant cells from proliferating
cells by detecting intracellular transcriptional status (95). In vitro immobilization
techniques can also be employed to isolate and recover dormant
cancer cells. This can be achieved using a microfluidic
flow-focusing device that allows individual cells coated with
agarose to be immobilized and survive on silica gel wells.
Typically, actively proliferating cells do not survive the
immobilization process, but dormant cells can be re-awakened by
in situ digestion of the agarose gel and effectively
recovered through magnetic separation of the silica gel (108) (Fig.
4).
Mechanisms of tumor cell dormancy
In breast cancer, the tyrosine kinase receptor TIE2
induces cell dormancy and resistance to 5-FU and adriamycin by
activating the expression of cyclin-dependent kinase inhibitors
CDKN1A and CDKN1B (109). The
treatment of diabetes with metformin or a high-fat diet can promote
the survival of dormant ER+ breast cancer cells through
the upregulation of the AMPK signaling pathway and the activation
of fatty acid oxidation (105).
FBX8, a member of the F-box protein family, plays a role in
maintaining tumor cell dormancy under the pressure of chemotherapy
with drugs such as oxaliplatin and 5-FU. Overexpression of FBX8 in
dormant cells enhances the degradation of HIF-1α, CDK4 and C-Myc
via the ubiquitin-proteasome pathway, thereby prolonging the
dormancy period, whereas FBX8 knockdown shortens this period
(106). In paclitaxel-treated
non-small cell lung cancer, regulator of G protein signaling 2
(RGS2) mediates translational arrest and dormancy in tumor cells
through the proteasomal degradation of activating transcription
factor 4, while antagonizing RGS2 in the endoplasmic reticulum
pathway induces apoptosis in dormant cells (110). CXCL10 has been identified as a
factor that can reactivate the proliferation of dormant tumor cells
within micro-metastases, making it a potential therapeutic target
for addressing tumor dormancy (111). Elevated intracellular copper
levels are generally associated with tumor progression; however,
copper carrier drugs such as elesclomol can induce cell death in a
copper-ion-dependent manner. Consequently, blocking copper uptake
can inhibit tumor cell proliferation and induce dormancy, reducing
overall cell mortality (112,113).
The MAPK/ERK signaling pathway, a classical
proliferation-related pathway, also plays a role in the induction
of cell dormancy. In EGFR-mutated non-small cell lung cancer, the
combination of EGFR inhibitors and TKIs drives tumor cells into a
senescence-like dormant state characterized by high YAP/TEAD
activity, owing to ERK1/2 blockade. Inhibiting YAP and TEAD in
conjunction with EGFR/MEK inhibition has been shown to enhance
apoptosis, effectively depleting dormant cells (114).
Myc is a critical gene that regulates tumor cell
growth and proliferation. Inhibition of Myc or
bromodomain-containing protein 4 prompts tumor cells to enter
dormancy by reducing the initiation of apoptosis. Conversely,
inducing Myc expression increases chemotherapy sensitivity,
suggesting that maintaining dormancy through Myc inhibition
post-chemotherapy or disrupting dormancy by inhibiting CDK9 could
be promising therapeutic strategies against dormant tumor cells
(115).
Autophagy plays a pivotal role in cellular stress
response by eliminating damaged organelles, misfolded proteins and
abnormal protein aggregates, regulating mitochondrial mass, and
preventing the accumulation of ROS, thereby aiding cell survival
(102). Studies have revealed that
when tumor cells enter a dormant state, the activity of the
autophagy-related mTOR pathway decreases, while
autophagy-associated phenotypes increase (102,116). Conversely, inhibiting autophagy
with drugs such as chloroquine can reawaken dormant cells,
prompting them to re-enter the proliferative state (117). The combination of autophagy
inhibitors with chemotherapeutic agents has been found to induce
the death of dormant cells (100,118). Thus, autophagy is integral not
only to the initiation and maintenance of dormancy but also to the
transition from dormancy to proliferation.
The TME significantly contributes to inducing tumor
cell dormancy. In a mouse model of breast cancer metastasis, Ki-67
immunofluorescence revealed that tumor cells metastasizing to the
lungs entered a dormant state. This metastatic dormancy was
dependent on the presence of T cells, particularly
CD39+PD-1+CD8+ T cells, which
induced cell cycle arrest by secreting IFNγ and TNF-α, thereby
promoting a dormant phenotype (119). Hypoxia, another key feature of the
TME, also influences dormancy. Under hypoxic conditions, the
expression of CSN8, a subunit of the COP9 signalosome, is elevated.
This upregulation is accompanied by increased levels of dormancy
markers (NR2F1, DEC2 and p27) and hypoxia markers (HIF-1α and
GLUT1), along with decreased expression of the proliferation marker
Ki-67, suggesting that CSN8 plays a role in regulating
hypoxia-induced dormancy (120).
While most chemotherapeutic agents target
proliferating cells and are ineffective against dormant cells,
nimustine has shown efficacy in targeting dormant cells in
BRCA1-deficient mice. Platinum-induced intra-strand crosslinks can
be repaired by nucleotide excision during the G0-G1 phase; however,
in BRCA1-deficient tumor cells, nimustine-induced inter-strand
crosslink repair is impaired, leading to cell death (98).
The study of tumor cell dormancy and its role in
chemotherapy resistance remains an emerging field. The definitions,
markers and specific mechanisms of tumor cell dormancy remain
incompletely understood. However, continued research is essential
to elucidate how tumor cells evade chemotherapy and survive as
micro-metastases, ultimately improving therapeutic strategies.
Conclusion and future directions
In recent decades, the molecular mechanisms
underlying chemoresistance have become increasingly
well-understood. These mechanisms include reduced drug uptake into
cells, increased drug efflux, GSH-mediated drug neutralization,
nuclear pore closure, enhanced DNA repair, promotion of tumor cell
dormancy, and the complex interactions between tumor cells and the
TME. Although the mechanisms underlying chemotherapy resistance in
tumor cells are increasingly well understood, their complexity in
clinical practice presents significant challenges. For example, the
use of inhibitors targeting multidrug resistance transporter
proteins such as MDR1 is complicated by their role in immune
function. Literature indicates that CD8+ T cells secrete
various cytokines to mediate immune responses and exert cytotoxic
effects on viruses and tumor cells, processes in which MDR1 plays a
critical role. MDR1 is essential for the development of naive
CD8+ T cells, regulated primarily by Runt-Related
transcription factors, which help inhibit oxidative stress, enhance
cell survival, and protect the mitochondrial function of nascent
CD8+ cytotoxic T lymphocytes. Therefore, inhibiting MDR1
in patients with cancers could impair immune function, potentially
leading to treatment failure (121).
The development of nanomaterials designed to deliver
chemotherapeutic agents directly to specific tumor sites or
intracellular targets is a rapidly expanding area of research aimed
at reversing chemoresistance. For example, a nanomaterial
containing an iron oxide core has been engineered to deliver the
cytotoxic agent adriamycin and the TLR3 agonist polyinosinic:
Polycytidylic acid (Poly IC) to both breast cancer and dendritic
cells. The Endoglin-binding peptide on the nanomaterial targets
triple-negative breast cancer cells, inducing apoptosis through
multiple mechanisms, thereby inhibiting tumor growth and
metastasis. This approach has shown significant success in
prolonging the survival of mouse models with aggressive and
resistant triple-negative breast cancer metastases (122). Current strategies to combat
GSH-mediated cellular resistance focus on GSH depletion or
targeting GST. For instance, ethacraplatin, a platinum prodrug,
inhibits GST by releasing ethacrynic acid, and encapsulating this
compound in nanomicelles has been identified to enhance the
intracellular accumulation of cisplatin (123).
Some polysulfides have been developed to target high
GSH levels in tumor cells, serving as a prodrug backbone that also
reverses multidrug resistance (5,124).
Additionally, nanocarriers such as dendritic mesoporous silica
nanoparticles can integrate components such as ultrasmall
Fe3O4 nanoparticles, Mn2+ ions and
the glutaminase inhibitor Telaglenastat (CB-839) into their large
mesopores to form nanodrugs. These nanodrugs exhibit
peroxidase-mimetic activity under acidic conditions, catalyzing the
decomposition of hydrogen peroxide (H2O2)
into hydroxyl radicals (−OH) and depleting existing GSH while
blocking endogenous GSH synthesis. This process enhances
ROS-mediated tumor catalytic therapy (125). Given the ongoing challenges with
traditional inhibitors in combating chemotherapy resistance, the
development of new nanomaterials represents a promising strategy
for overcoming multidrug resistance in tumors, and it is expected
to be a key focus of future research.
Acknowledgements
Not applicable.
Funding
The present study was partially supported by the Natural Science
Foundation of Hunan Province (2023JJ50135, 2023JJ30521), the
Projects of the Health Commission of Hunan Province (202201043124),
the Aid Program for Science and Technology Innovative Research Team
in Higher Educational Institutions of Hunan Province [No.
(2023)233] and the Research Foundation of Education Bureau of Hunan
(grant no. 21B0444).
Availability of data and materials
Not applicable.
Authors' contributions
XW and WHZ conceived and designed the analysis,
conducted the research, and drafted the manuscript. LHX created the
figures. LYZ and PL contributed to the revision and polishing of
the manuscript. ZL, WJG, QQ, DLC and XZ were involved in the
conception and design of the analysis. XZ made significant
contributions to the discussion of the content and reviewed, edited
and finalized the manuscript. All authors read and approved the
final version of the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Miller KD, Nogueira L, Devasia T, Mariotto
AB, Yabroff KR, Jemal A, Kramer J and Siegel RL: Cancer treatment
and survivorship statistics, 2022. CA Cancer J Clin. 72:409–436.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2022. CA Cancer J Clin. 72:7–33. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zheng R, Zhang S, Zeng H, Wang S, Sun K,
Chen R, Li L, Wei W and He J: Cancer incidence and mortality in
China, 2016. J Natl Cancer Cent. 2:1–9. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bukowski K, Kciuk M and Kontek R:
Mechanisms of multidrug resistance in cancer chemotherapy. Int J
Mol Sci. 21:32332020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xiao X, Wang K, Zong Q, Tu Y, Dong Y and
Yuan Y: Polyprodrug with glutathione depletion and cascade drug
activation for multi-drug resistance reversal. Biomaterials.
270:1206492021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bedard PL, Hyman DM, Davids MS and Siu LL:
Small molecules, big impact: 20 Years of targeted therapy in
oncology. Lancet. 395:1078–1088. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li X, Li M, Huang M, Lin Q, Fang Q, Liu J,
Chen X, Liu L, Zhan X, Shan H, et al: The multi-molecular
mechanisms of tumor-targeted drug resistance in precision medicine.
Biomed Pharmacother. 150:1130642022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Phan TG and Croucher PI: The dormant
cancer cell life cycle. Nat Rev Cancer. 20:398–411. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen X, Momin A, Wanggou S, Wang X, Min
HK, Dou W, Gong Z, Chan J, Dong W, Fan JJ, et al: Mechanosensitive
brain tumor cells construct blood-tumor barrier to mask
chemosensitivity. Neuron. 111:30–48. e142023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Inoue T, Aoyama-Ishikawa M, Uemura M,
Kohama K, Fujisaki N, Murakami H, Yamada T and Hirata J: The role
of death receptor signaling pathways in mouse Sertoli cell
avoidance of apoptosis during LPS- and IL-18-induced inflammatory
conditions. J Reprod Immunol. 158:1039702023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Russo M, Crisafulli G, Sogari A, Reilly
NM, Arena S, Lamba S, Bartolini A, Amodio V, Magrì A, Novara L, et
al: Adaptive mutability of colorectal cancers in response to
targeted therapies. Science. 366:1473–1480. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Diaz LA Jr, Williams RT, Wu J, Kinde I,
Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al:
The molecular evolution of acquired resistance to targeted EGFR
blockade in colorectal cancers. Nature. 486:537–540. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zheng N, Fang J, Xue G, Wang Z, Li X, Zhou
M, Jin G, Rahman MM, McFadden G and Lu Y: Induction of tumor cell
autosis by myxoma virus-infected CAR-T and TCR-T cells to overcome
primary and acquired resistance. Cancer Cell. 40:973–985.e7. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hirota K, Ooka M, Shimizu N, Yamada K,
Tsuda M, Ibrahim MA, Yamada S, Sasanuma H, Masutani M and Takeda S:
XRCC1 counteracts poly(ADP ribose)polymerase (PARP) poisons,
olaparib and talazoparib, and a clinical alkylating agent,
temozolomide, by promoting the removal of trapped PARP1 from broken
DNA. Genes Cells. 27:331–344. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ovejero S, Soulet C and Moriel-Carretero
M: The alkylating agent Methyl methanesulfonate triggers lipid
alterations at the inner nuclear membrane that are independent from
its DNA-damaging ability. Int J Mol Sci. 22:74612021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ghosh S: Cisplatin: The first metal based
anticancer drug. Bioorg Chem. 88:1029252019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fetoni AR, Paciello F and Troiani D:
Cisplatin chemotherapy and cochlear damage: Otoprotective and
chemosensitization properties of polyphenols. Antioxid Redox
Signal. 36:1229–1245. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Curry JN and McCormick JA:
Cisplatin-induced kidney injury: Delivering the goods. J Am Soc
Nephrol. 33:255–256. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Balboni B, El Hassouni B, Honeywell RJ,
Sarkisjan D, Giovannetti E, Poore J, Heaton C, Peterson C, Benaim
E, Lee YB, et al: RX-3117 (fluorocyclopentenyl cytosine): A novel
specific antimetabolite for selective cancer treatment. Expert Opin
Investig Drugs. 28:311–322. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo J, Yu Z, Das M and Huang L: Nano
codelivery of oxaliplatin and folinic acid achieves synergistic
chemo-immunotherapy with 5-fluorouracil for colorectal cancer and
liver metastasis. Acs Nano. 14:5075–5089. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Z, Li C, Wu Q, Tu Y, Wang C, Yu X, Li
B, Wang Z and Sun S and Sun S: MEDAG enhances breast cancer
progression and reduces epirubicin sensitivity through the
AKT/AMPK/mTOR pathway. Cell Death Dis. 12:972021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li J, Yu K, Pang D, Wang C, Jiang J, Yang
S, Liu Y, Fu P, Sheng Y, Zhang G, et al: Adjuvant capecitabine with
docetaxel and cyclophosphamide plus epirubicin for triple-negative
breast cancer (CBCSG010): An open-label, randomized, multicenter,
phase III trial. J Clin Oncol. 38:1774–1784. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
You JH, Lee J and Roh JL: PGRMC1-dependent
lipophagy promotes ferroptosis in paclitaxel-tolerant persister
cancer cells. J Exp Clin Cancer Res. 40:3502021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen Y, Liu R, Li C, Song Y, Liu G, Huang
Q, Yu L, Zhu D, Lu C, Lu A, et al: Nab-paclitaxel promotes the
cancer-immunity cycle as a potential immunomodulator. Am J Cancer
Res. 11:3445–3460. 2021.PubMed/NCBI
|
|
25
|
Elshamy AM, Salem OM, Safa MAE, Barhoma
RAE, Eltabaa EF, Shalaby AM, Alabiad MA, Arakeeb HM and Mohamed HA:
Possible protective effects of CO Q10 against vincristine-induced
peripheral neuropathy: Targeting oxidative stress, inflammation,
and sarmoptosis. J Biochem Mol Toxicol. 36:e229762022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rajković S, Živković MD and Djuran MI:
Reactions of dinuclear Platinum(II) complexes with peptides. Curr
Protein Pept Sci. 17:95–105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim ES, Tang X, Peterson DR, Kilari D,
Chow CW, Fujimoto J, Kalhor N, Swisher SG, Stewart DJ, Wistuba II
and Siddik ZH: Copper transporter CTR1 expression and tissue
platinum concentration in non-small cell lung cancer. Lung Cancer.
85:88–93. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lv X, Song J, Xue K, Li Z, Li M, Zahid D,
Cao H, Wang L, Song W, Ma T, et al: Core fucosylation of copper
transporter 1 plays a crucial role in cisplatin-resistance of
epithelial ovarian cancer by regulating drug uptake. Mol Carcinog.
58:794–807. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kalayda GV, Wagner CH and Jaehde U:
Relevance of copper transporter 1 for cisplatin resistance in human
ovarian carcinoma cells. J Inorg Biochem. 116:1–10. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao H, Zhang S, Hu T, Qu X, Zhai J, Zhang
Y, Tao L, Yin J and Song Y: Omeprazole protects against
cisplatin-induced nephrotoxicity by alleviating oxidative stress,
inflammation, and transporter-mediated cisplatin accumulation in
rats and HK-2 cells. Chem Biol Interact. 297:130–140. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Naka A, Takeda R, Shintani M, Ogane N,
Kameda Y, Aoyama T, Yoshikawa T and Kamoshida S: Organic cation
transporter 2 for predicting cisplatin-based neoadjuvant
chemotherapy response in gastric cancer. Am J Cancer Res.
5:2285–2293. 2015.PubMed/NCBI
|
|
32
|
Hucke A, Rinschen MM, Bauer OB, Sperling
M, Karst U, Köppen C, Sommer K, Schröter R, Ceresa C, Chiorazzi A,
et al: An integrative approach to cisplatin chronic toxicities in
mice reveals importance of organic cation-transporter-dependent
protein networks for renoprotection. Arch Toxicol. 93:2835–2848.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Verhalen B, Dastvan R, Thangapandian S,
Peskova Y, Koteiche HA, Nakamoto RK, Tajkhorshid E and Mchaourab
HS: Energy transduction and alternating access of the mammalian ABC
transporter P-glycoprotein. Nature. 543:738–741. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Alam A, Kowal J, Broude E, Roninson I and
Locher KP: Structural insight into substrate and inhibitor
discrimination by human P-glycoprotein. Science. 363:753–756. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pasello M, Giudice AM and Scotlandi K: The
ABC subfamily A transporters: Multifaceted players with incipient
potentialities in cancer. Semin Cancer Biol. 60:57–71. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hanssen KM, Haber M and Fletcher JI:
Targeting multidrug resistance-associated protein 1
(MRP1)-expressing cancers: Beyond pharmacological inhibition. Drug
Resist Updat. 59:1007952021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim Y and Chen J: Molecular structure of
human P-glycoprotein in the ATP-bound, outward-facing conformation.
Science. 359:915–919. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Miyamoto S, Azuma K, Ishii H, Bessho A,
Hosokawa S, Fukamatsu N, Kunitoh H, Ishii M, Tanaka H, Aono H, et
al: Low-dose erlotinib treatment in elderly or frail patients with
EGFR mutation-positive non-small cell lung cancer: A multicenter
phase 2 trial. JAMA Oncol. 6:e2012502020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Robey RW, Pluchino KM, Hall MD, Fojo AT,
Bates SE and Gottesman MM: Revisiting the role of ABC transporters
in multidrug-resistant cancer. Nat Rev Cancer. 18:452–464. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Carvalho DM, Richardson PJ, Olaciregui N,
Stankunaite R, Lavarino C, Molinari V, Corley EA, Smith DP, Ruddle
R, Donovan A, et al: Repurposing vandetanib plus everolimus for the
treatment of ACVR1-mutant diffuse intrinsic pontine glioma. Cancer
Discov. 12:416–431. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cole SPC: Targeting multidrug resistance
protein 1 (MRP1, ABCC1): Past, present, and future. Annu Rev
Pharmacol Toxicol. 54:95–117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Johnson ZL and Chen J: ATP binding enables
substrate release from multidrug resistance protein 1. Cell.
172:81–89.e10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vulsteke C, Lambrechts D, Dieudonné A,
Hatse S, Brouwers B, van Brussel T, Neven P, Belmans A, Schöffski
P, Paridaens R and Wildiers H: Genetic variability in the multidrug
resistance associated protein-1 (ABCC1/MRP1) predicts hematological
toxicity in breast cancer patients receiving (neo-)adjuvant
chemotherapy with 5-fluorouracil, epirubicin and cyclophosphamide
(FEC). Ann Oncol. 24:1513–1525. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cole SPC: Multidrug resistance protein 1
(MRP1, ABCC1), a ‘multitasking’ ATP-binding cassette (ABC)
transporter. J Biol Chem. 289:30880–30888. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Taylor NMI, Manolaridis I, Jackson SM,
Kowal J, Stahlberg H and Locher KP: Structure of the human
multidrug transporter ABCG2. Nature. 546:504–509. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kowal J, Ni D, Jackson SM, Manolaridis I,
Stahlberg H and Locher KP: Structural basis of drug recognition by
the multidrug transporter ABCG2. J Mol Biol. 433:1669802021.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Niu B, Liao K, Zhou Y, Wen T, Quan G, Pan
X and Wu C: Application of glutathione depletion in cancer therapy:
Enhanced ROS-based therapy, ferroptosis, and chemotherapy.
Biomaterials. 277:1211102021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang J, Bai J, Zhou Q, Hu Y, Wang Q, Yang
L, Chen H, An H, Zhou C, Wang Y, et al: Glutathione prevents high
glucose-induced pancreatic fibrosis by suppressing pancreatic
stellate cell activation via the ROS/TGFβ/SMAD pathway. Cell Death
Dis. 13:4402022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen M, Zhao S, Zhu J, Feng E, Lv F, Chen
W, Lv S, Wu Y, Peng X and Song F: Open-source and
reduced-expenditure nanosystem with ROS self-amplification and
glutathione depletion for simultaneous augmented
chemodynamic/photodynamic therapy. ACS Appl Mater Interfaces.
14:20682–20692. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cheng X, Xu HD, Ran HH, Liang G and Wu FG:
Glutathione-depleting nanomedicines for synergistic cancer therapy.
ACS Nano. 15:8039–8068. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gao P, Yang X, Xue YW, Zhang XF, Wang Y,
Liu WJ and Wu XJ: Promoter methylation of glutathione S-transferase
pi1 and multidrug resistance gene 1 in bronchioloalveolar carcinoma
and its correlation with DNA methyltransferase 1 expression.
Cancer. 115:3222–3232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xu Y, Han X, Li Y, Min H, Zhao X, Zhang Y,
Qi Y, Shi J, Qi S, Bao Y and Nie G: Sulforaphane mediates
glutathione depletion via polymeric nanoparticles to restore
cisplatin chemosensitivity. ACS Nano. 13:13445–13455. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Oshimori N, Oristian D and Fuchs E: TGF-β
promotes heterogeneity and drug resistance in squamous cell
carcinoma. Cell. 160:963–976. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang KR, Zhang YF, Lei HM, Tang YB, Ma
CS, Lv QM, Wang SY, Lu LM, Shen Y, Chen HZ and Zhu L: Targeting
AKR1B1 inhibits glutathione de novo synthesis to overcome acquired
resistance to EGFR-targeted therapy in lung cancer. Sci Transl Med.
13:eabg64282021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pang HH, Ke YC, Li NS, Chen YT, Huang CY,
Wei KC and Yang HW: A new lateral flow plasmonic biosensor based on
gold-viral biomineralized nanozyme for on-site intracellular
glutathione detection to evaluate drug-resistance level. Biosens
Bioelectron. 165:1123252020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Harris IS, Endress JE, Coloff JL, Selfors
LM, McBrayer SK, Rosenbluth JM, Takahashi N, Dhakal S, Koduri V,
Oser MG, et al: Deubiquitinases maintain protein homeostasis and
survival of cancer cells upon glutathione depletion. Cell Metab.
29:1166–1181.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Stafford WC, Peng X, Olofsson MH, Zhang X,
Luci DK, Lu L, Cheng Q, Trésaugues L, Dexheimer TS, Coussens NP, et
al: Irreversible inhibition of cytosolic thioredoxin reductase 1 as
a mechanistic basis for anticancer therapy. Sci Transl Med.
10:eaaf74442018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang X, Wang M, Feng J, Qin B, Zhang C,
Zhu C, Liu W, Wang Y, Liu W, Huang L, et al: Multifunctional
nanoparticles co-loaded with Adriamycin and MDR-targeting siRNAs
for treatment of chemotherapy-resistant esophageal cancer. J
Nanobiotechnology. 20:1662022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Amort M, Nachbauer B, Tuzlak S, Kieser A,
Schepers A, Villunger A and Polacek N: Expression of the vault RNA
protects cells from undergoing apoptosis. Nat Commun. 6:70302015.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu S, Hao Q, Peng N, Yue X, Wang Y, Chen
Y, Wu J and Zhu Y: Major vault protein: A virus-induced host factor
against viral replication through the induction of type-I
interferon. Hepatology. 56:57–66. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bai H, Wang C, Qi Y, Xu J, Li N, Chen L,
Jiang B, Zhu X, Zhang H, Li X, et al: Major vault protein
suppresses lung cancer cell proliferation by inhibiting STAT3
signaling pathway. BMC Cancer. 19:4542019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Shen W, Qiu Y, Li J, Wu C, Liu Z, Zhang X,
Hu X, Liao Y and Wang H: IL-25 promotes cisplatin resistance of
lung cancer cells by activating NF-κB signaling pathway to increase
of major vault protein. Cancer Med. 8:3491–3501. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lou L, Wang J, Lv F, Wang G, Li Y, Xing L,
Shen H and Zhang X: Y-box binding protein 1 (YB-1) promotes
gefitinib resistance in lung adenocarcinoma cells by activating AKT
signaling and epithelial-mesenchymal transition through targeting
major vault protein (MVP). Cell Oncol (Dordr). 44:109–133. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yu X, Liu W, Wang Z, Wang H, Liu J, Huang
C, Zhao T, Wang X, Gao S, Ma Y, et al: CD73 induces gemcitabine
resistance in pancreatic ductal adenocarcinoma: A promising target
with non-canonical mechanisms. Cancer Lett. 519:289–303. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yu H, Li M, He R, Fang P, Wang Q, Yi Y,
Wang F, Zhou L, Zhang Y, Chen A, et al: Major vault protein
promotes hepatocellular carcinoma through targeting interferon
regulatory factor 2 and decreasing p53 activity. Hepatology.
72:518–534. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Pilié PG, Tang C, Mills GB and Yap TA:
State-of-the-art strategies for targeting the DNA damage response
in cancer. Nat Rev Clin Oncol. 16:81–104. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Gourley C, Balmaña J, Ledermann JA, Serra
V, Dent R, Loibl S, Pujade-Lauraine E and Boulton SJ: Moving from
poly (ADP-Ribose) polymerase inhibition to targeting DNA repair and
DNA damage response in cancer therapy. J Clin Oncol. 37:2257–2269.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Pettitt SJ, Frankum JR, Punta M, Lise S,
Alexander J, Chen Y, Yap TA, Haider S, Tutt ANJ and Lord CJ:
Clinical BRCA1/2 reversion analysis identifies hotspot mutations
and predicted neoantigens associated with therapy resistance.
Cancer Discov. 10:1475–1488. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ma J, Benitez JA, Li J, Miki S, Ponte de
Albuquerque C, Galatro T, Orellana L, Zanca C, Reed R, Boyer A, et
al: Inhibition of nuclear PTEN tyrosine phosphorylation enhances
glioma radiation sensitivity through attenuated DNA repair. Cancer
Cell. 35:504–518.e7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Goodall J, Mateo J, Yuan W, Mossop H,
Porta N, Miranda S, Perez-Lopez R, Dolling D, Robinson DR, Sandhu
S, et al: Circulating cell-free DNA to guide prostate cancer
treatment with PARP inhibition. Cancer Discov. 7:1006–1017. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Dev H, Chiang TW, Lescale C, de Krijger I,
Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei W,
Ostermaier M, et al: Shieldin complex promotes DNA end-joining and
counters homologous recombination in BRCA1-null cells. Nat Cell
Biol. 20:954–965. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Schlacher K: PARPi focus the spotlight on
replication fork protection in cancer. Nat Cell Biol. 19:1309–1310.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lord CJ and Ashworth A: Mechanisms of
resistance to therapies targeting BRCA-mutant cancers. Nat Med.
19:1381–1388. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ray Chaudhuri A, Callen E, Ding X, Gogola
E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et
al: Replication fork stability confers chemoresistance in
BRCA-deficient cells. Nature. 535:382–387. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Awah CU, Chen L, Bansal M, Mahajan A,
Winter J, Lad M, Warnke L, Gonzalez-Buendia E, Park C, Zhang D, et
al: Ribosomal protein S11 influences glioma response to TOP2
poisons. Oncogene. 39:5068–5081. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wang Z, Zhu Q, Li X, Ren X, Li J, Zhang Y,
Zeng S, Xu L, Dong X and Zhai B: TOP2A inhibition reverses drug
resistance of hepatocellular carcinoma to regorafenib. Am J Cancer
Res. 12:4343–4360. 2022.PubMed/NCBI
|
|
78
|
Mateo J, Carreira S, Sandhu S, Miranda S,
Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A,
Tunariu N, et al: DNA-repair defects and olaparib in metastatic
prostate cancer. N Engl J Med. 373:1697–1708. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Mu Y, Lou J, Srivastava M, Zhao B, Feng
XH, Liu T, Chen J and Huang J: SLFN11 inhibits checkpoint
maintenance and homologous recombination repair. EMBO Rep.
17:94–109. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Gardner EE, Lok BH, Schneeberger VE,
Desmeules P, Miles LA, Arnold PK, Ni A, Khodos I, de Stanchina E,
Nguyen T, et al: Chemosensitive relapse in small cell lung cancer
proceeds through an EZH2-SLFN11 axis. Cancer Cell. 31:286–299.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Guryanova OA, Shank K, Spitzer B, Luciani
L, Koche RP, Garrett-Bakelman FE, Ganzel C, Durham BH, Mohanty A,
Hoermann G, et al: DNMT3A mutations promote anthracycline
resistance in acute myeloid leukemia via impaired nucleosome
remodeling. Nat Med. 22:1488–1495. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Picco G, Cattaneo CM, van Vliet EJ,
Crisafulli G, Rospo G, Consonni S, Vieira SF, Rodríguez IS,
Cancelliere C, Banerjee R, et al: Werner helicase is a
synthetic-lethal vulnerability in mismatch repair-deficient
colorectal cancer refractory to targeted therapies, chemotherapy,
and immunotherapy. Cancer Discov. 11:1923–1937. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Germano G, Lamba S, Rospo G, Barault L,
Magrì A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G, et
al: Inactivation of DNA repair triggers neoantigen generation and
impairs tumour growth. Nature. 552:116–120. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gay CM, Parseghian CM and Byers LA: This
is our cells under pressure: Decreased DNA damage repair in
response to targeted therapies facilitates the emergence of
drug-resistant clones. Cancer Cell. 37:5–7. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Guo W, Qiao T, Dong B, Li T, Liu Q and Xu
X: The effect of hypoxia-induced exosomes on anti-tumor immunity
and its implication for immunotherapy. Front Immunol.
13:9159852022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Qin Y, Liu HJ, Li M, Zhai DH, Tang YH,
Yang L, Qiao KL, Yang JH, Zhong WL, Zhang Q, et al: Salidroside
improves the hypoxic tumor microenvironment and reverses the drug
resistance of platinum drugs via HIF-1α signaling pathway.
EBioMedicine. 38:25–36. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wu S, Luo M, To KKW, Zhang J, Su C, Zhang
H, An S, Wang F, Chen D and Fu L: Intercellular transfer of
exosomal wild type EGFR triggers osimertinib resistance in
non-small cell lung cancer. Mol Cancer. 20:172021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Carter BZ, Mak PY, Chen Y, Mak DH, Mu H,
Jacamo R, Ruvolo V, Arold ST, Ladbury JE, Burks JK, et al:
Anti-apoptotic ARC protein confers chemoresistance by controlling
leukemia-microenvironment interactions through a NFκB/IL1β
signaling network. Oncotarget. 7:20054–20067. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Carter BZ, Mak PY, Wang X, Tao W, Ruvolo
V, Mak D, Mu H, Burks JK and Andreeff M: An ARC-regulated
IL1β/Cox-2/PGE2/β-catenin/ARC circuit controls
leukemia-microenvironment interactions and confers drug resistance
in AML. Cancer Res. 79:1165–1177. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Chambers LM, Esakov Rhoades EL, Bharti R,
Braley C, Tewari S, Trestan L, Alali Z, Bayik D, Lathia JD, Sangwan
N, et al: Disruption of the gut microbiota confers cisplatin
resistance in epithelial ovarian cancer. Cancer Res. 82:4654–4669.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Johnston CD and Bullman S:
Bacteria-derived L-lactate fuels cervical cancer chemoradiotherapy
resistance. Trends Cancer. 10:97–99. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Jiang SS, Xie YL, Xiao XY, Kang ZR, Lin
XL, Zhang L, Li CS, Qian Y, Xu PP, Leng XX, et al: Fusobacterium
nucleatum-derived succinic acid induces tumor resistance to
immunotherapy in colorectal cancer. Cell Host Microbe.
31:781–797.e9. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Lin ZZ, Hu MC, Hsu C, Wu YM, Lu YS, Ho JA,
Yeh SH, Chen PJ and Cheng AL: Synergistic efficacy of
telomerase-specific oncolytic adenoviral therapy and histone
deacetylase inhibition in human hepatocellular carcinoma. Cancer
Lett. 556:2160632023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Vaidya FU, Sufiyan Chhipa A, Mishra V,
Gupta VK, Rawat SG, Kumar A and Pathak C: Molecular and cellular
paradigms of multidrug resistance in cancer. Cancer Rep (Hoboken).
5:e12912022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Freter R, Falletta P, Omrani O, Rasa M,
Herbert K, Annunziata F, Minetti A, Krepelova A, Adam L, Käppel S,
et al: Establishment of a fluorescent reporter of RNA-polymerase II
activity to identify dormant cells. Nat Commun. 12:33182021.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Summers MA, McDonald MM and Croucher PI:
Cancer cell dormancy in metastasis. Cold Spring Harb Perspect Med.
10:a0375562020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yeh AC and Ramaswamy S: Mechanisms of
cancer cell dormancy-another hallmark of cancer? Cancer Res.
75:5014–5022. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Pajic M, Blatter S, Guyader C, Gonggrijp
M, Kersbergen A, Küçükosmanoğlu A, Sol W, Drost R, Jonkers J, Borst
P and Rottenberg S: Selected alkylating agents can overcome drug
tolerance of G0-like tumor cells and eradicate
BRCA1-deficient mammary tumors in mice. Clin Cancer Res.
23:7020–7033. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Badia-Ramentol J, Linares J, Gómez-Llonin
A and Calon A: Minimal residual disease, metastasis and immunity.
Biomolecules. 11:1302021. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Rehman SK, Haynes J, Collignon E, Brown
KR, Wang Y, Nixon AML, Bruce JP, Wintersinger JA, Singh Mer A, Lo
EBL, et al: Colorectal cancer cells enter a diapause-like DTP state
to survive chemotherapy. Cell. 184:226–242.e21. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Cackowski FC and Heath EI: Prostate cancer
dormancy and recurrence. Cancer Lett. 524:103–108. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Akkoc Y, Peker N, Akcay A and Gozuacik D:
Autophagy and cancer dormancy. Front Oncol. 11:6270232021.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Damen MPF, van Rheenen J and Scheele CLGJ:
Targeting dormant tumor cells to prevent cancer recurrence. FEBS J.
288:6286–6303. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Lee J, Kim SH and Kang BJ: Prognostic
factors of disease recurrence in breast cancer using quantitative
and qualitative magnetic resonance imaging (MRI) parameters. Sci
Rep. 10:75982020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Hampsch RA, Wells JD, Traphagen NA,
McCleery CF, Fields JL, Shee K, Dillon LM, Pooler DB, Lewis LD,
Demidenko E, et al: AMPK activation by metformin promotes survival
of dormant ER+ breast cancer cells. Clin Cancer Res.
26:3707–3719. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhu X, Wang F, Wu X, Li Z, Wang Z, Ren X,
Zhou Y, Song F, Liang Y, Zeng Z, et al: FBX8 promotes metastatic
dormancy of colorectal cancer in liver. Cell Death Dis. 11:6222020.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Quayle LA, Spicer A, Ottewell PD and Holen
I: Transcriptomic profiling reveals novel candidate genes and
signalling programs in breast cancer quiescence and dormancy.
Cancers (Basel). 13:39222021. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Preciado J, Lam T, Azarin SM, Lou E and
Aksan A: Induction of dormancy by confinement: An agarose-silica
biomaterial for isolating and analyzing dormant cancer cells. J
Biomed Mater Res B Appl Biomater. 109:2117–2130. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Drescher F, Juárez P, Arellano DL,
Serafín-Higuera N, Olvera-Rodriguez F, Jiménez S, Licea-Navarro AF
and Fournier PG: TIE2 induces breast cancer cell dormancy and
inhibits the development of osteolytic bone metastases. Cancers
(Basel). 12:8682020. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Cho J, Min HY, Lee HJ, Hyun SY, Sim JY,
Noh M, Hwang SJ, Park SH, Boo HJ, Lee HJ, et al: RGS2-mediated
translational control mediates cancer cell dormancy and tumor
relapse. J Clin Invest. 131:e1719012021. View Article : Google Scholar
|
|
111
|
Clark AM, Heusey HL, Griffith LG,
Lauffenburger DA and Wells A: IP-10 (CXCL10) can trigger emergence
of dormant breast cancer cells in a metastatic liver
microenvironment. Front Oncol. 11:6761352021. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Yu Z, Zhou R, Zhao Y, Pan Y, Liang H,
Zhang JS, Tai S, Jin L and Teng CB: Blockage of SLC31A1-dependent
copper absorption increases pancreatic cancer cell autophagy to
resist cell death. Cell Prolif. 52:e125682019. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Tsvetkov P, Coy S, Petrova B, Dreishpoon
M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R,
Spangler RD, et al: Copper induces cell death by targeting
lipoylated TCA cycle proteins. Science. 375:1254–1261. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Kurppa KJ, Liu Y, To C, Zhang T, Fan M,
Vajdi A, Knelson EH, Xie Y, Lim K, Cejas P, et al:
Treatment-induced tumor dormancy through YAP-mediated
transcriptional reprogramming of the apoptotic pathway. Cancer
Cell. 37:104–122.e12. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Dhimolea E, de Matos Simoes R, Kansara D,
Al'Khafaji A, Bouyssou J, Weng X, Sharma S, Raja J, Awate P,
Shirasaki R, et al: An embryonic diapause-like adaptation with
suppressed Myc activity enables tumor treatment persistence. Cancer
Cell. 39:240–256.e11. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Hussein AM, Wang Y, Mathieu J, Margaretha
L, Song C, Jones DC, Cavanaugh C, Miklas JW, Mahen E, Showalter MR,
et al: Metabolic control over mTOR-dependent diapause-like state.
Dev Cell. 52:236–250. e72020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare
S, Kondo S, Kondo Y, Yu Y, Mills GB, et al: The tumor suppressor
gene ARHI regulates autophagy and tumor dormancy in human ovarian
cancer cells. J Clin Invest. 118:3917–3929. 2008.PubMed/NCBI
|
|
118
|
Anlaş AA and Nelson CM: Soft
microenvironments induce chemoresistance by increasing autophagy
downstream of integrin-linked kinase. Cancer Res. 80:4103–4113.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Tallón de Lara P, Castañón H, Vermeer M,
Núñez N, Silina K, Sobottka B, Urdinez J, Cecconi V, Yagita H,
Movahedian Attar F, et al:
CD39+PD-1+CD8+ T cells mediate
metastatic dormancy in breast cancer. Nat Commun. 12:7692021.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Ju S, Wang F, Wang Y and Ju S: CSN8 is a
key regulator in hypoxia-induced epithelial-mesenchymal transition
and dormancy of colorectal cancer cells. Mol Cancer. 19:1682020.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Chen ML, Sun A, Cao W, Eliason A, Mendez
KM, Getzler AJ, Tsuda S, Diao H, Mukori C, Bruno NE, et al:
Physiological expression and function of the MDR1 transporter in
cytotoxic T lymphocytes. J Exp Med. 217:e201913882020. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Mu QG, Lin G, Jeon M, Wang H, Chang FC,
Revia RA, Yu J and Zhang M: Iron oxide nanoparticle targeted
chemo-immunotherapy for triple negative breast cancer. Mater Today
(Kidlington). 50:149–169. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Li S, Li C, Jin S, Liu J, Xue X, Eltahan
AS, Sun J, Tan J, Dong J and Liang XJ: Overcoming resistance to
cisplatin by inhibition of glutathione S-transferases (GSTs) with
ethacraplatin micelles in vitro and in vivo. Biomaterials.
144:119–129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Ling X, Chen X, Riddell IA, Tao W, Wang J,
Hollett G, Lippard SJ, Farokhzad OC, Shi J and Wu J:
Glutathione-scavenging poly(disulfide amide) nanoparticles for the
effective delivery of Pt(IV) prodrugs and reversal of cisplatin
resistance. Nano Lett. 18:4618–4625. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wu F, Du Y, Yang J, Shao B, Mi Z, Yao Y,
Cui Y, He F, Zhang Y and Yang P: Peroxidase-like active
nanomedicine with dual glutathione depletion property to restore
oxaliplatin chemosensitivity and promote programmed cell death. ACS
Nano. 16:3647–3663. 2022. View Article : Google Scholar : PubMed/NCBI
|