Introduction
Alzheimer's disease (AD) is a common
neurodegenerative disease worldwide in the elderly. The main
neuropathological characteristic of AD is the deposition of amyloid
β peptide (Aβ) in brain, which plays critical roles in promoting
cognitive impairments and neuronal injuries in the onset and
development of AD. Aβ is mainly produced by the sequential cleavage
of amyloid precursor protein (APP) through β- and γ-secretase
(1). In progression of AD, the
imbalance between Aβ production and clearance contributes to
elevation of Aβ levels in the central nervous system (2), which leads to neuronal oxidative
stress and inflammation, ultimately resulting in neuronal
dysfunction and cognitive deficits (3). Although the specific mechanism of Aβ
accumulation remains unclear, increasing evidence suggested that
alterations in autophagy and inflammation are closely associated
with Aβ generation and clearance in AD (4,5).
However, there are still no effective drugs in improving neuronal
injuries and cognitive dysfunction through modulation of autophagy
and inflammation in AD.
Autophagy is a cell cycle process by which the cells
hydrolyze macromolecules in response to various stress signals
(6). Its primary function is to
remove aged or damaged organelles and maintaining essential energy
homeostasis (7). Additionally,
autophagy plays an important role in the occurrence of numerous
diseases, such as cancer, diabetes and neurodegenerative diseases
(8). It has been reported that Aβ
peptides can be produced by cleavage of APP in autophagosomes
during the autophagic renewal of APP-rich organelles (9,10).
The autophagy function can affect Aβ levels and thus modulates the
pathological changes in AD (11).
Moreover, autophagy also plays significant roles in metabolism of
Aβ and autophagy deficits may also cause the aggregation of Aβ
(12). Although how autophagy
exactly affects AD pathology remains undefined, regulating
autophagy may be an important target in developing new drugs for
AD.
Neuroinflammation has long been considered to be
associated with AD pathology. Neuroinflammation and Aβ production
are two critical early steps in the development of AD (13). Inflammasomes are large cytosolic
multiprotein complexes that can activate caspase-1-mediated
inflammatory responses (14).
Caspase-1 is an important protease that causes the maturation and
secretion of pro-inflammatory cytokines, such as IL-1β and IL-18
(15). The release of these
cytokines contributes to synaptic dysfunction, neuronal death and
inhibition of neurogenesis. Additionally, inflammatory cytokines
(such as IL-1β and TNF-α) are also reported to involve in the
process of APP proteolytic cleavage to increase the production of
Aβ1-42 peptides (16). Nucleotide-binding and
oligomerization domain (NOD)-like receptors (NLRs) are important
subfamilies of inflammasomes. Previously, the NLR-family pyrin
domain-containing 1 (NLRP1) inflammasome, widely expressed in brain
particularly in neurons, has received extensive attention. An ~25-
to 30-fold increase of NLRP1 immunopositivity in neurons was
observed in AD brains compared with non-AD brains (17). By contrast, inhibition of NLRP1
inflammasome could downregulate the Aβ accumulation in AD model
mice (18). A previous study by
the authors indicated that NLRP1 activation is closely involved in
aging-related cognitive impairments and neuronal injuries (19). However, the specific link between
NLRP1 inflammasome and regulation of Aβ production remains
unclear.
Ginsenoside Rg1 (Rg1) is a saponin extracted from
ginseng. Numerous experiments have shown that Rg1 has multiple
neuroprotective effects both in vivo and in vitro
(20,21). A recent study by the authors
suggested that Rg1 can prevent H2O2-induced
neuronal injury in vitro by inhibiting the activation of
NLRP1 inflammasome in hippocampal neurons (22). A recent study by the authors also
suggested that Rg1 treatment significantly attenuates neuronal
injury and Aβ generation and deposition in APP/PS1 mice (23). In recent years, it has been
increasingly reported that autophagy can be regulated through the
modulation of inflammatory pathways (24,25), suggesting that associating NLRP1
inflammasome with autophagy may provide new ideas for delaying AD.
However, there is little study on whether Rg1 can improve AD
pathology through regulating NLRP1 inflammasome and autophagic
pathways. In the present study, it was hypothesized that Rg1 may
reduce Aβ deposition through inhibiting NLRP1 inflammasome and
autophagy dysfunction, resulting in improvement of cognitive
impairments and neuronal injuries. The effects of Rg1 treatment on
olfactory detection ability, learning and memory function and Aβ
deposition in APPPS1 mice were firstly explored. Meanwhile, the
regulation of Rg1 treatment on NLRP1 inflammasome and autophagy
dysfunction in APP/PS1 mice was further investigated. The present
study may provide new targets and drug options for inhibiting the
occurrence and development of AD.
Materials and methods
Animals and treatment
The APP/PS1 transgenic AD model mice were provided
by the Model Animal Research Center of Nanjing University. The mice
were bred in the animal center of Anhui Province. Male APP/PS1 mice
were used in the present study, and age- and gender-matched WT
littermates were utilized as control. Mice were housed in standard
laboratory conditions with free access to standard food and water
(temperature, 22–25°C; atmosphere, ~50–60%; 12-h light/dark cycle).
The mice were raised in a standard laboratory with free food and
water and were allowed to adapt to the laboratory conditions before
testing. The experiment was performed (approval no. LLSC20211172)
according to the protocol of Animal Ethics Committee of Anhui
Medical University (Hefei, China).
To study the protective effects of Rg1 on APP/PS1
male mice, six-month old (WT) or APP/PS1 male mice were divided
randomly into 6 groups (n=10 in each group; weight, 25–35 g): WT-9M
group, APP/PS1-6M group, APP/PS1-9M model group, APP/PS1 + apocynin
(50 mg/kg), APP/PS1 + Rg1 (5 mg/kg) and APP/PS1 + Rg1 (10 mg/kg)
groups. Rg1 (Chengdu Desite Biotechnology Co.; Content >98%) and
apocynin (Merck Millipore) were dissolved with water and were
administered orally (0.1 ml/10 g) for 12 weeks. The doses of Rg1
and apocynin were reported in previous studies (26,27). The WT-9M and APP/PS1-9M groups
were given equal volume of solvent for 12 weeks.
Buried food test (BFT)
Previously, olfactory dysfunction has been reported
in AD patients even in the early stages of AD (28). Therefore, the changes of olfactory
dysfunction were examined using a previously described method
(29). Briefly, mice were
subjected to restricted diet for 24 h prior to the test. All mice
were acclimated to the test chamber for 1 h prior to testing. A
small piece of food was randomly placed in a corner of a clean test
cage 1 cm below the bedding. The mouse was then placed in the test
cage at a constant distance from the hidden food. ANY-maze
behavioral tracking software was used to record the latency of the
mouse to find food. If the mouse failed to find the buried food
within 5 min, the latency period was recorded as 300 sec. A total
of 9 h after the aforementioned experiment, surface pellet tests
were performed using the same protocol, but with the pellets placed
on the surface to exclude possible motor impairment.
Morris water maze (MWM) test
MWM is an important method to evaluate learning and
memory impairments (30). The MWM
consists of a circular pool (120 cm in diameter and 60 cm in
height) filled with water and a video tracking system on top. MWM
test includes four training trials from first day (day 1) to day 4
and the exploration trials on day 5 as previously described
(31). During the training
trials, the animals were placed in the water from each of the 4
quadrants in turn and ensured that their heads were to the wall of
the pool. The animals were given 60 sec to find a platform hidden
underwater. If animals did not find the platform within 60 sec,
they were guided to the platform and were allowed to stay on the
platform for 30 sec to familiarize and memorize the location of the
platform. The mean escape latency (MEL, sec) of the four trials
each day was recorded to indicate the learning performance. On day
5, the platform was removed, and each mouse was detected a swimming
probe trial for 60 sec from the first quadrant. The latency first
to the platform (LFP, sec), the swimming time in the platform
quadrant (STP, sec), and the number crossing platform (NCP) were
recorded to indicate the memory results.
H&E staining
After behavior test, the mouse was euthanized by
cervical dislocation, and the brain tissues (n=4) were removed and
fixed with 4% paraformaldehyde at 4°C for 24 h. The tissues were
sectioned into 5-µm after paraffin-embedded using a slicer (Leica
Microsystems GmbH). The pathological changes were examined by using
intelligent tissue slice imaging system (3DHISTECH) after the slice
was stained with hematoxylin for 4 min and eosin for 40 sec at room
temperature (RT).
Thioflavin-S Staining
The sections (n=4) were deparaffinized by xylene and
were hydrated by graded alcohol. Then the slices were stained with
Thioflavin-S (1%, MedChemExpress USA, HY-D0972) at 37°C for 5 min,
then were hydrated with 70% ethanol. After being washed twice with
PBS, the slices were stained with Hoechst 33258 at RT for 5 min.
Then the slices were sealed and examined with an intelligent slice
imaging system (3DHISTECH). The green density was double-blindly
analyzed from 3 regions (magnification, ×400) respectively in the
cortex and hippocampal CA1 by using an Image-Pro Plus 6.0 analysis
system (Media Cybernetics, Inc.) to evaluate the deposition of
Aβ.
Immunofluorescence
Briefly, the slides (n=4) were deparaffinized and
hydrated as aforementioned. Then the slides were treated with 0.25%
Triton X-100 (cat. no. ST797-100 ml; Beyotime Institute of
Biotechnology) at RT for 30 min, and were blocked with 1%BSA (cat.
no. ST2249-5g; Beyotime Institute of Biotechnology) at RT for 60
min. Next the slides were incubated with polyclonal
Aβ1–42 (1:150; cat. no. bs-0076R; BIOSS) and
post-synaptic density scaffolding protein 95 (PSD95) (1:100; cat.
no. GB11277; Wuhan Servicebio Technology Co., Ltd.) antibodies
overnight at 4°C. The second day, the slides were treated with a
secondary antibody conjugated to Rhodamine (1:200; cat. no.
ZF-0317; OriGene Technologies, Inc.) at RT for 60 min. Lastly, the
slides were sealed with anti-fade medium and were imaged by an
intelligent slice imaging system (3DHISTECH). The red fluorescence
density was analyzed from 3 regions (magnification, ×400)
respectively in the cortex and hippocampal CA1 by using an
Image-Pro Plus 6.0 analysis system to evaluate the changes of PSD95
and Aβ1-42 expression.
Immunohistochemistry
The slides (n=4) were deparaffinized and were
hydrated as aforementioned. Then, the slides were treated with
H2O2 (3%) at RT for 10 min to eliminate
endogenous peroxidase. Afterward, the slides were treated with
boiling sodium citrate buffer (3.23 g/l, pH 6.0; OriGene
Technologies, Inc.) to restore antigen. Next, the slides were
blocked with 10% goat serum (cat. no. C0265; Beyotime Institute of
Biotechnology) for 30 min at RT and were then incubated with
specific primary antibodies against Beclin1 (1:100; cat. no.
11306-1-AP) and LC3 (1:100; cat. no. 14600-1-AP; both from
ProteinTech Group, Inc.) overnight at 4°C. The next day, the slides
were washed with PBS three times 5 min per time and were incubated
with a rabbit-general secondary antibody (1:500; cat. no. S0001;
Affinity Biosciences) at RT for 1 h. The slides were stained by a
DAB kit (OriGene Technologies, Inc.) to produce brownish staining.
Then the slides were mounted and examined by an intelligent tissue
slice imaging system (3DHISTECH). The quantification of Beclin1 and
LC3 was detected blindly by using Image-Pro Plus 6.0 software from
3 fields (magnification, ×400) respectively in the cortex and
hippocampal CA1.
Immunoblot analysis
Total protein in the hippocampus and cortex tissues
(n=4) were extracted with RIPA lysis buffer (Beyotime Institute of
Biotechnology) containing protease and phosphatase inhibitors by
using a cryogenic tissue grinder (Shanghai Jingxin Industrial
Development Co., Ltd.) 60 Hz for 50 sec, at 4°C. The protein
concentration was detected by using a BCA Protein Assay kit
(Beyotime Institute of Biotechnology). The proteins (20 µg) were
separated by 12% SDS-PAGE and were transferred onto a PVDF membrane
(MilliporeSigma). Then, the membrane was treated with 5% non-fat
milk dissolved in TBS-0.05% Tween-20 (TBST) buffer at RT for 1 h
and then incubated with corresponding primary antibodies overnight
at 4°C: PSD95 (1:1,000), NLRP1 (1:2,000; cat. no. ab3683; Abcam),
ASC (1:500; cat. no. bs-6741R; BIOSS), Caspase-1 (1:1,000; cat. no.
ab1872; Abcam), IL-1β (1:1,000; cat. no. ab9722; Abcam), IL-6
(1:1,000; cat. no. 21865-1-AP; ProteinTech Group, Inc.), TNF-α
(1:1,000; cat. no. WL01581; Wanleibio Co., Ltd.), AMPK (1:1,000;
cat. no. AF6423; Affinity Biosciences), p-AMPK (1:1,000; cat. no.
bs-4010; Bioworld Technology, Inc.), mTOR (1:1,000; cat. no.
66888-1-lg; ProteinTech Group, Inc.), p-mTOR (1:1,000; cat. no.
bs-4706; Bioworld Technology, Inc.), Beclin1 (1:1,000), LC3
(1:1,000), P62 (1:500; cat. no. WL02385; Wanleibio Co., Ltd.) and
β-actin (1:2,000; cat. no. GB12001; Wuhan Servicebio Technology
Co., Ltd.). The second day, the membrane was washed with TBST and
was incubated in secondary antibody conjugated horseradish
peroxidase (1:10,000; cat. no. ZF-2301; OriGene Technologies, Inc.)
at RT for 1 h. After washed with TBST, the bands were visualized by
an ECL reagent (Amersham; Cytiva) and were imaged with a Chemi mini
imaging system (Q4600 Mini; Shanghai Bioshine Technology; URL,
http://bioshine.bioon.com.cn/). The band
density of protein was examined using an ImageJ 6.0 software
(National Institutes of Health). The target band density was
normalized to β-actin.
Reverse transcription-quantitative
(RT-q) PCR analysis
The total RNAs were extracted from hippocampus and
cortex tissues (n=4) by using a TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) as previously
described (32). Firstly, the
cDNA was synthesized with PrimeScript™ Reverse Transcriptase
buffers with dNTP (cat. no. RR037A; Takara Bio, Inc.) from total
RNA. The thermocycling conditions were at 37°C for 15 min, then at
85°C for 5 sec followed by cooling to 4°C. The qPCR analyses for
mRNAs of ASC, NLRP1, IL-1β, Caspase-1 and β-actin were examined
with a SYBR®Premix Ex Taq II RTPCR kit (cat. no. RR820A;
Takara Bio). The level of β-actin mRNA was used as internal
control. The primer sequences (TsingKe Biological Technology) were
as follows: NLRP1 forward, 5′-TGGCACATCCTAGGGAAATC-3′ and reverse,
5′-TCCTCACGTGACAGCAGAAC-3′; ASC forward,
5′-GTCACAGAAGTGGACGGAGTG-3′ and reverse,
5′-CTCATCTTGTCTTGGCTGGTG-3′; Caspase-1 forward,
5′-CGTGGAGAGAAACAAGGAGTG-3′ and reverse,
5′-AATGAAAAGTGAGCCCCTGAC-3′; IL-1β forward,
5′-CTGCTTCCAAACCTTTGACC-3′ and reverse, 5′-AGCTTCTCCACAGCCACAAT-3′;
and β-actin forward, 5′-GATTACTGCTCTGGCTCCTAGC-3′ and reverse,
5′-GACTCATCGTACTCCTGCTTGC-3′. The PCR protocol was at 95°C for 30
sec, followed by amplification at 95°C for 5 sec (40 cycles), then
at 60°C for 30 sec. The qPCR was performed with a Real-time PCR
System (cat. no. CFX96; Bio-Rad Laboratories, Inc.). The CT values
of the samples were calculated, and the 2−ΔΔCq method
was used to analyze transcript levels of the target mRNAs as
previously described (33).
Drug-target molecular docking
To verify whether there is an association between
Rg1 and target protein, the molecular docking between ginsenoside
Rg1 and NLRP1 was monitored. Firstly, the 3D Protein Data Bank
(PDB) format file of NLRP1 (PDB ID: 6XKK) was obtained from the
RCSB PDB, the 2D SDF of Rg1 was downloaded (PubChem CID: 441923)
from the PubChem database, and the 2D structure was imported into
ChemBio3D14.0 software for structural optimization, revealing the
3D mol2 structure. Then, the drug molecules and proteins were
imported into AutoDock Tool to perform hydrogenation, charge
calculation, atom addition and ROOT docking. The lower binding
energy indicates the improvement of the docking result. Pymol 2.5.2
software (Schrödinger, Inc.) was used to analyze the docking
results.
Statistical analysis
All results were showed as the mean ± SD. The
GraphPad Prism 8.0 software (GraphPad Software, Inc.) was used for
statistical analysis. The results of four-day training in MWM were
analyzed by repeated-measure two-way analysis of variance (ANOVA)
and other data were analyzed by one-way ANOVA. Tukey's post hoc
test was performed to compare the significance between groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Rg1 treatment improves abnormal
behavior function in APP/PS1 mice
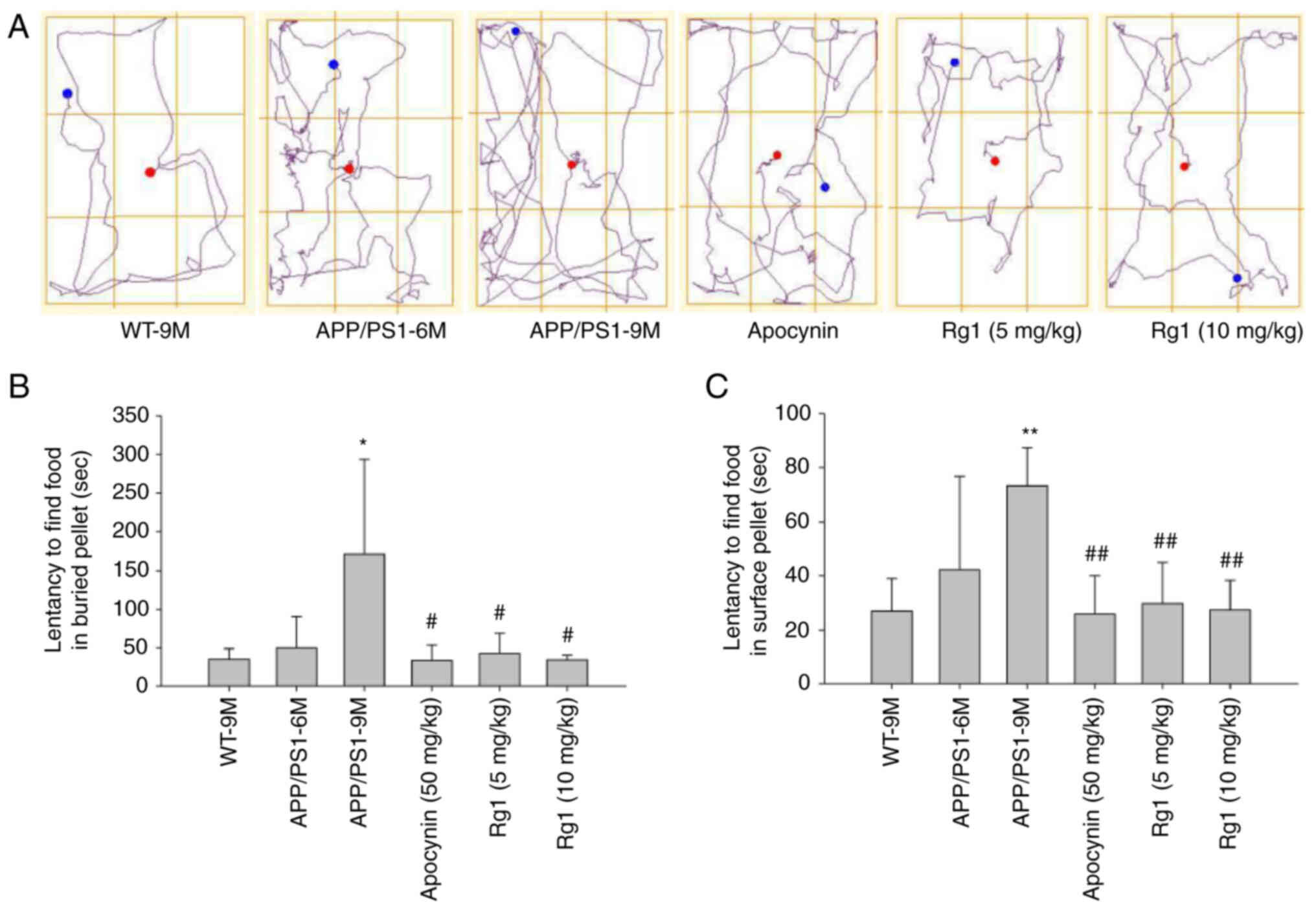
Firstly, BFT was used to observe the effects of Rg1
on olfactory function in APP/PS mice. The buried pellet results
showed that there were no differences in the latency to find the
food between the WT-9M and APP/PS1-6M group mice, while in the
APP/PS1-9M group mice the latency to find the food was
significantly increased compared with the WT-9M group mice.
Meanwhile, it was revealed that Rg1 and apocynin treatment
significantly reduced the latency to find the food compared with
the APP/PS1-9M group mice (Fig. 1A
and B, P<0.05). Additionally, the surface pellet test showed
similar results to the buried pellet test (Fig. 1C, P<0.01), suggesting that
there was no motor dysfunction in the experimental mice. These
results suggested that Rg1 treatment can improve olfactory
dysfunction in APP/PS1 mice.
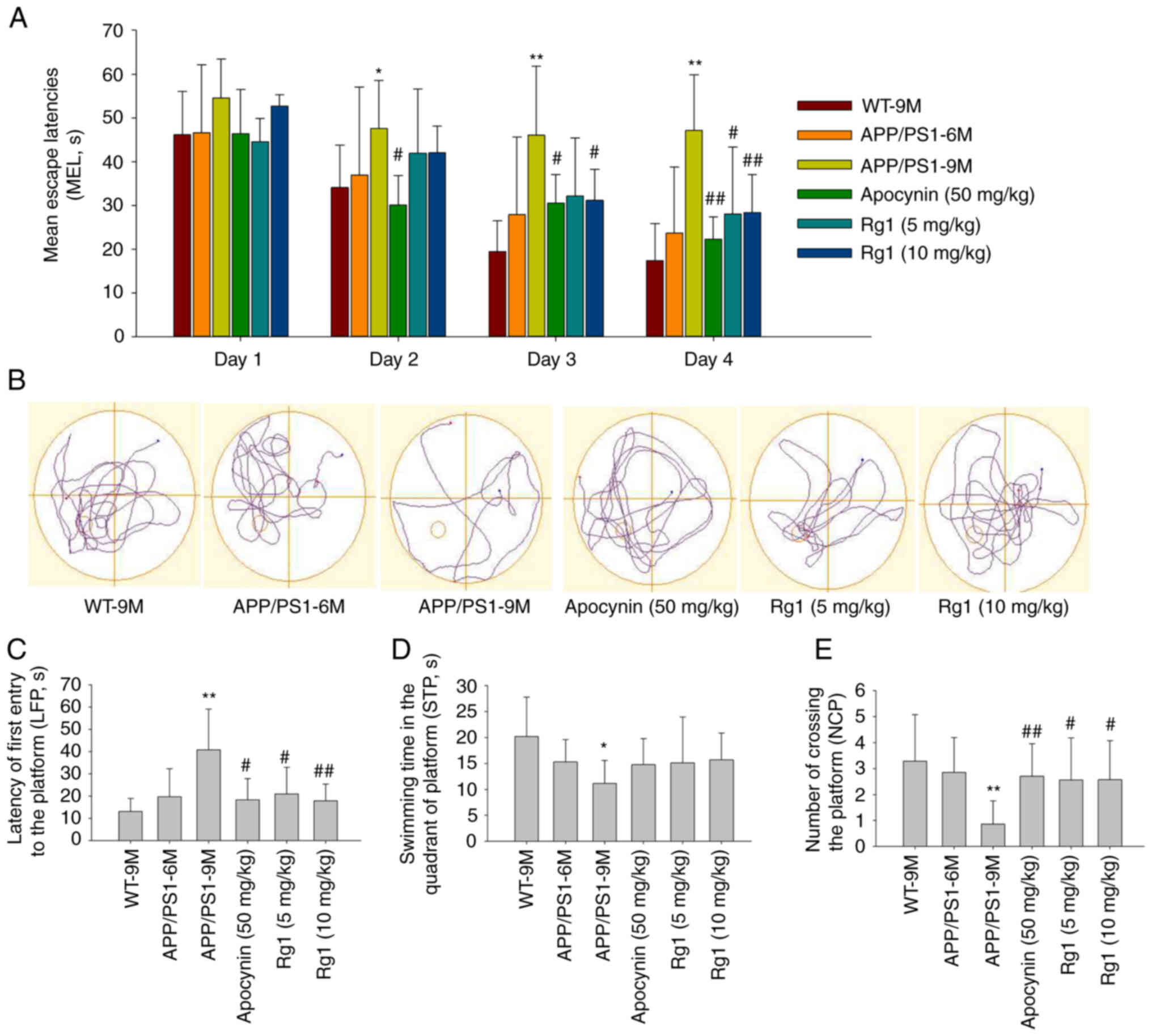
Then, MWM was conducted to examine the learning and
memory ability of mice. On the first day (day 1), there was no
significant difference in escape latency among the groups. With the
progress of the experiment, the escape latency became increasingly
shorter in the WT-9M and administration groups. While in APP/PS1-9M
group, the escape latency was significantly longer than that of the
WT-9M mice in days 2–4 (Fig. 2A,
P<0.05 or P<0.01). Meanwhile, it was identified that the
escape latency was significantly reduced by apocynin treatment in
days 2–4, by Rg1 (5 mg/kg) in day 4 and Rg1 (10 mg/kg) in days 3–4
compared with APP/PS1-9M group (Fig.
2A, P<0.05 or P<0.01). On the fifth day of the probe
test, the results showed that the LFP was significantly increased,
the NCP and the STP were significantly decreased in APP/PS1-9M mice
compared with the WT-9M group (Fig.
2B-E, P<0.05 or P<0.01). While compared with the
APP/PS1-9M group, treatment with apocynin and Rg1 (5 and 10 mg/kg)
could significantly reduce the LFP and increase the NCP (Fig. 2B-E, P<0.05 or P<0.01). The
results indicated that Rg1 treatment can significantly improve the
learning and memory impairments in APP/PS1 mice.
Rg1 treatment alleviates neuronal
damages and Aβ depositions in APP/PS1 mice
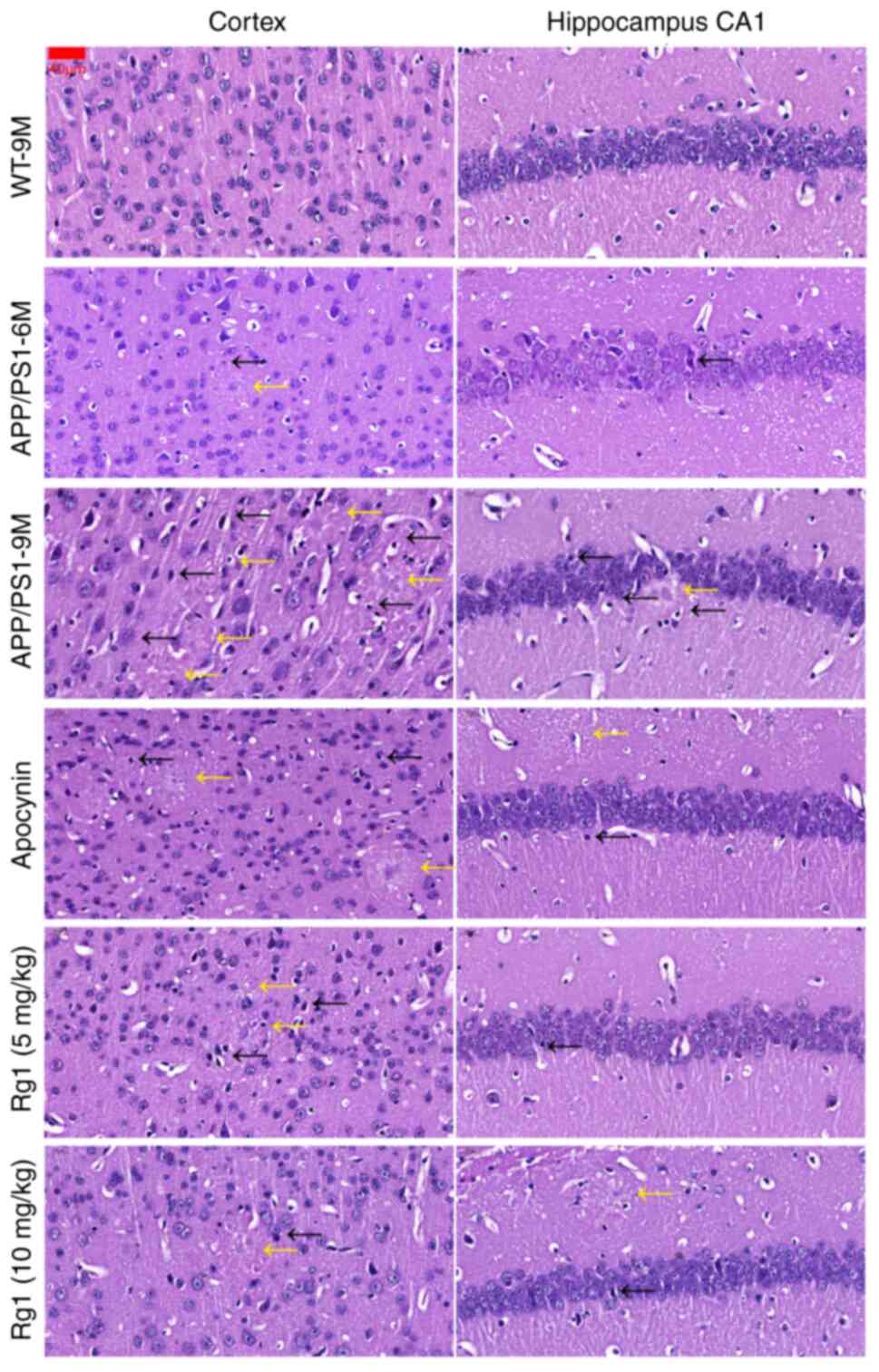
The H&E staining results revealed that, compared
with WT-9M, there were few pathological changes in the cortex and
hippocampal CA1 areas in APP/PS1-6M mice, while in APP/PS1-9M mice,
neuronal damages, such as pyknosis and nucleoli inconspicuous
(black arrows) and Aβ plaques (yellow arrows), were increased in
the cortex and hippocampal CA1 (Fig.
3). Compared with APP/PS1-9M, treatment with apocynin and Rg1
(5 and 10 mg/kg) could alleviate the neuronal damages in the cortex
and hippocampal CA1 (Fig. 3). The
results indicated that Rg1 treatment could improve neuronal damages
in APP/PS1 mice.
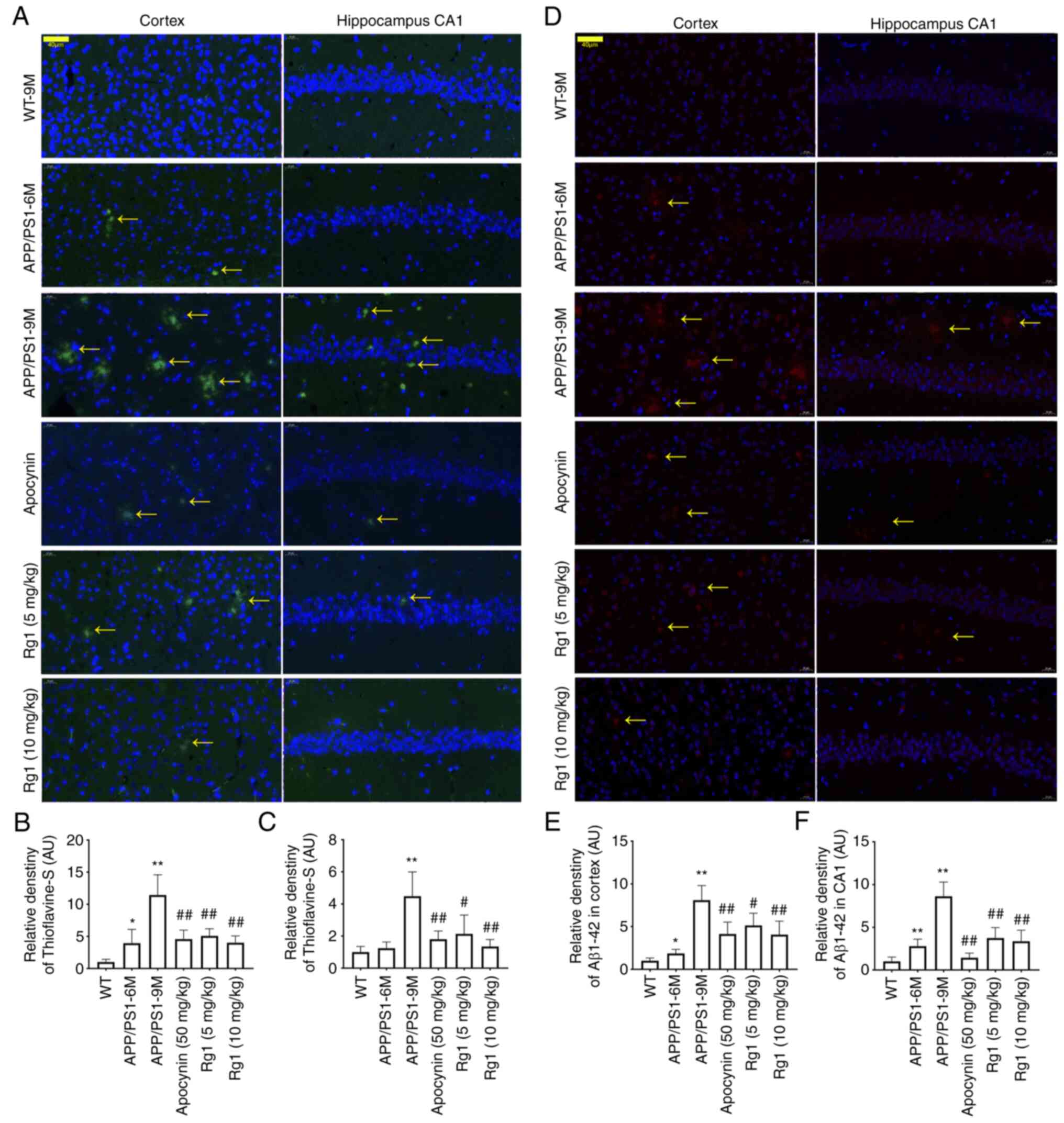
To confirm the effect of Aβ deposition, the
Thioflavin-S staining and immunofluorescence were conducted to
detect the Aβ deposition and Aβ1–42 level. The
Thioflavin-S staining results demonstrated that there was a small
amount of Aβ deposition in the cortex and hippocampal CA1 in
APP/PS1-6M mice, while in APP/PS1-9M mice, the Aβ depositions
(yellow arrows) were significantly increased compared with WT-9M
(Fig. 4A-C, P<0.05 or
P<0.01). Compared with APP/PS1-9M, treatment with apocynin and
Rg1 could significantly reduce Aβ depositions in the cortex and
hippocampal CA1 (Fig. 4A-C,
P<0.05 or P<0.01). The immunofluorescence results of
Aβ1–42 were similar to that of Thioflavin-S staining,
suggesting that Aβ1–42 expression was significantly
increased in APP/PS1-9M mice, and was significantly downregulated
by apocynin and Rg1 treatment in the cortex and hippocampal CA1
(Fig. 4D-F, P<0.05 or
P<0.01). The results indicated that Rg1 treatment decreases Aβ
generation and deposition in APP/PS1 mice.
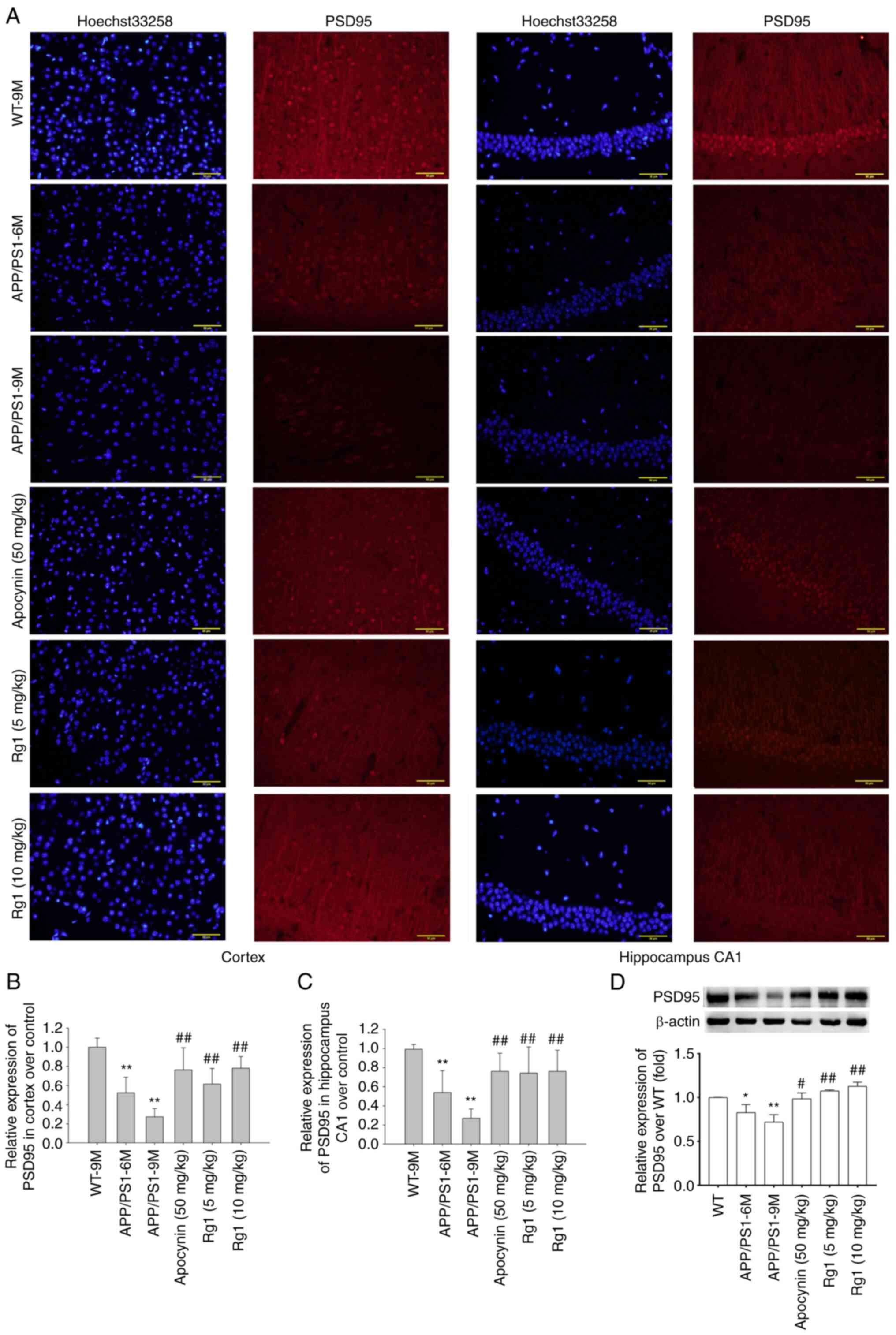
Rg1 treatment increases PSD95
expression in APP/PS1 mice
AD models not only have Aβ deposition, but also are
accompanied by synaptic dysfunction (34). Therefore, the expression of PSD95
was detected by immunofluorescence and immunoblot analysis. The
immunofluorescence results showed that the expression of PSD95 was
significantly decreased in the cortex and hippocampal CA1 in both
APP/PS-6M and APP/PS1-9M mice compared with WT-9M mice,
particularly in APP/PS1-9M mice (Fig.
5A-C, P<0.01). After Apocynin and Rg1 treatment, the
expression of PSD95 was significantly increased in the cortex and
hippocampal CA1 (Fig. 5A-C,
P<0.01). The immunoblot results of PSD95 were similar to that of
immunofluorescence results, suggesting that the expression of PSD95
was significantly decreased in APP/PS-6M and APP/PS1-9M mice, and
was significantly increased after Apocynin and Rg1 treatment
(Fig. 5D, P<0.05 or
P<0.01). These results indicated that Rg1 treatment can improve
synaptic dysfunction in APP/PS1 mice.
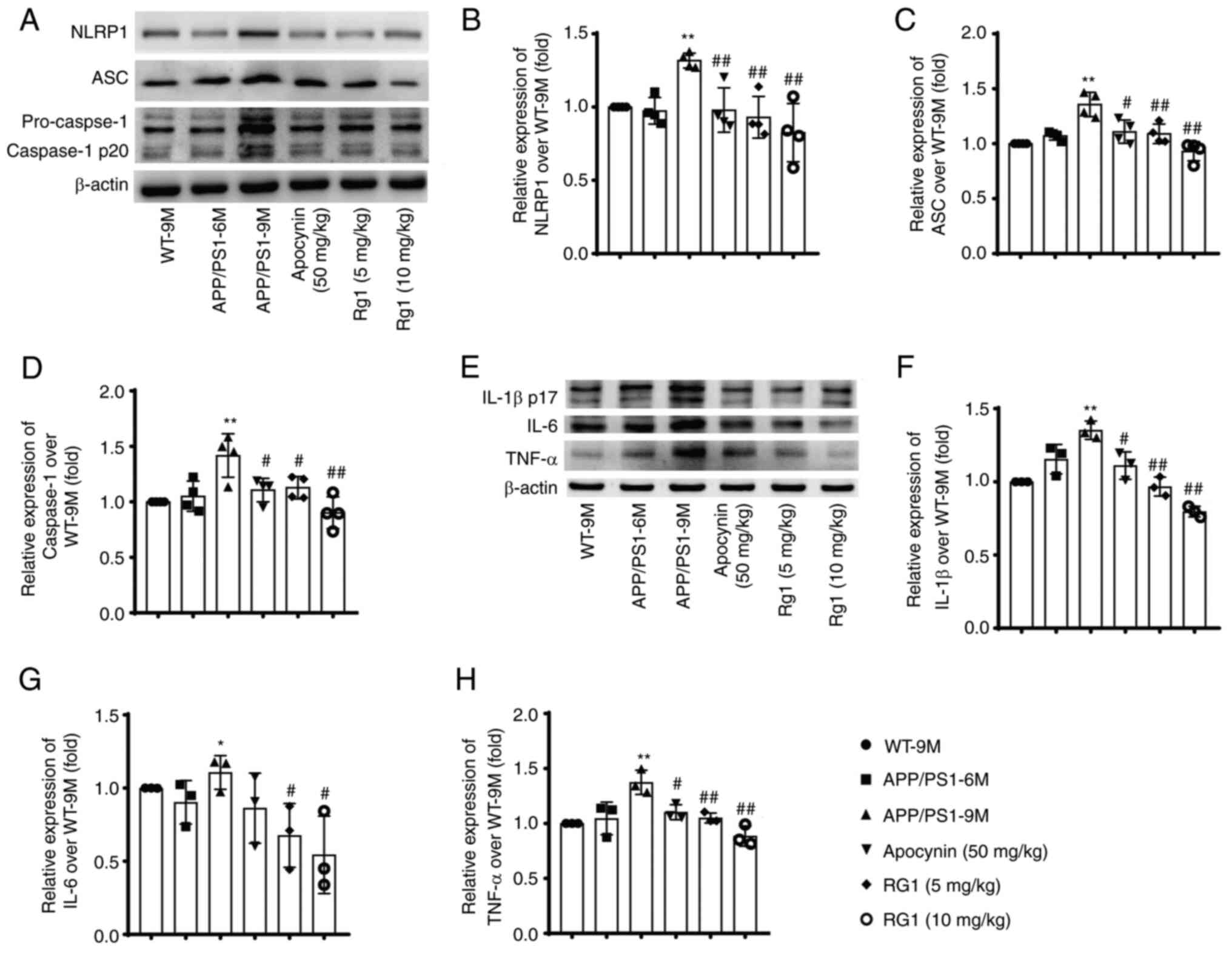
Rg1 treatment decreases the expression
of NLRP1 inflammasome in APP/PS1 mice
It has been reported that NLRP1 inflammasome is
involved in learning and memory impairments and neuronal damages
during the aging process in mice (19). Therefore, the effect of Rg1
treatment on the expression of NLRP1 inflammasome proteins was
further examined through immunoblot and RT-qPCR. The results showed
that, compared with WT-9M, there was no significant increase in the
expression of NLRP1 inflammasome-related proteins in the APP/PS1-6M
group. While in the APP/PS1-9M group, the expression levels of
NLRP1, ASC, caspase-1, IL-1β, IL-6 and TNF-α were significantly
increased. Compared with the APP/PS1-9M group, after apocynin and
Rg1 treatment, the expression levels of these proteins were
significantly downregulated (Fig.
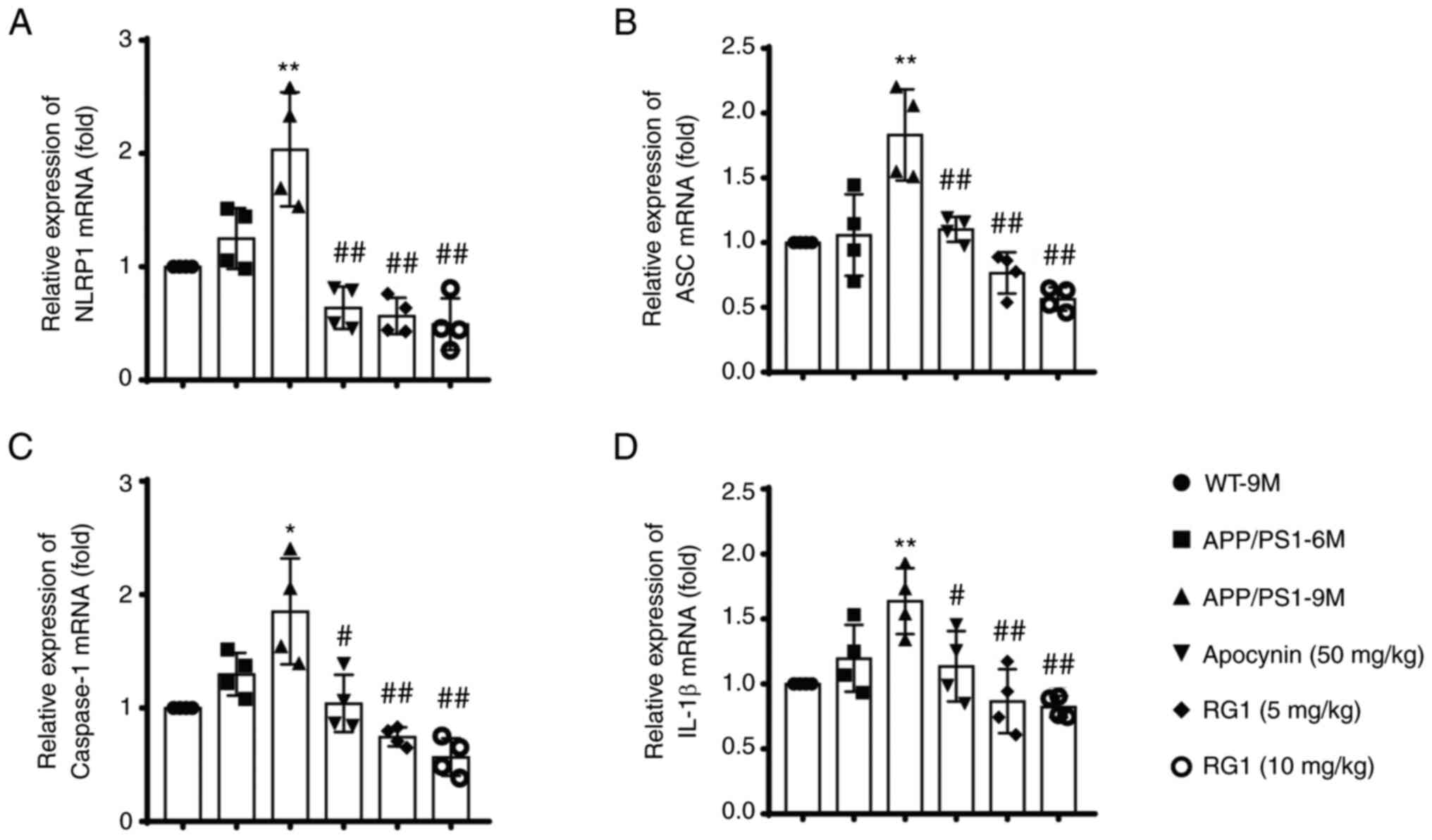
6A-H, P<0.05 or P<0.01). Similar results were observed
using qPCR; the levels of NLRP1, ASC, caspase-1 and IL-1β mRNA were
significantly increased in the APP/PS1-9M mice and were
significantly decreased after apocynin and Rg1 treatment (Fig. 7A-D, P<0.05 or P<0.01). These
results suggested that Rg1 treatment can inhibit NLRP1 inflammasome
in APP/PS1 mice.
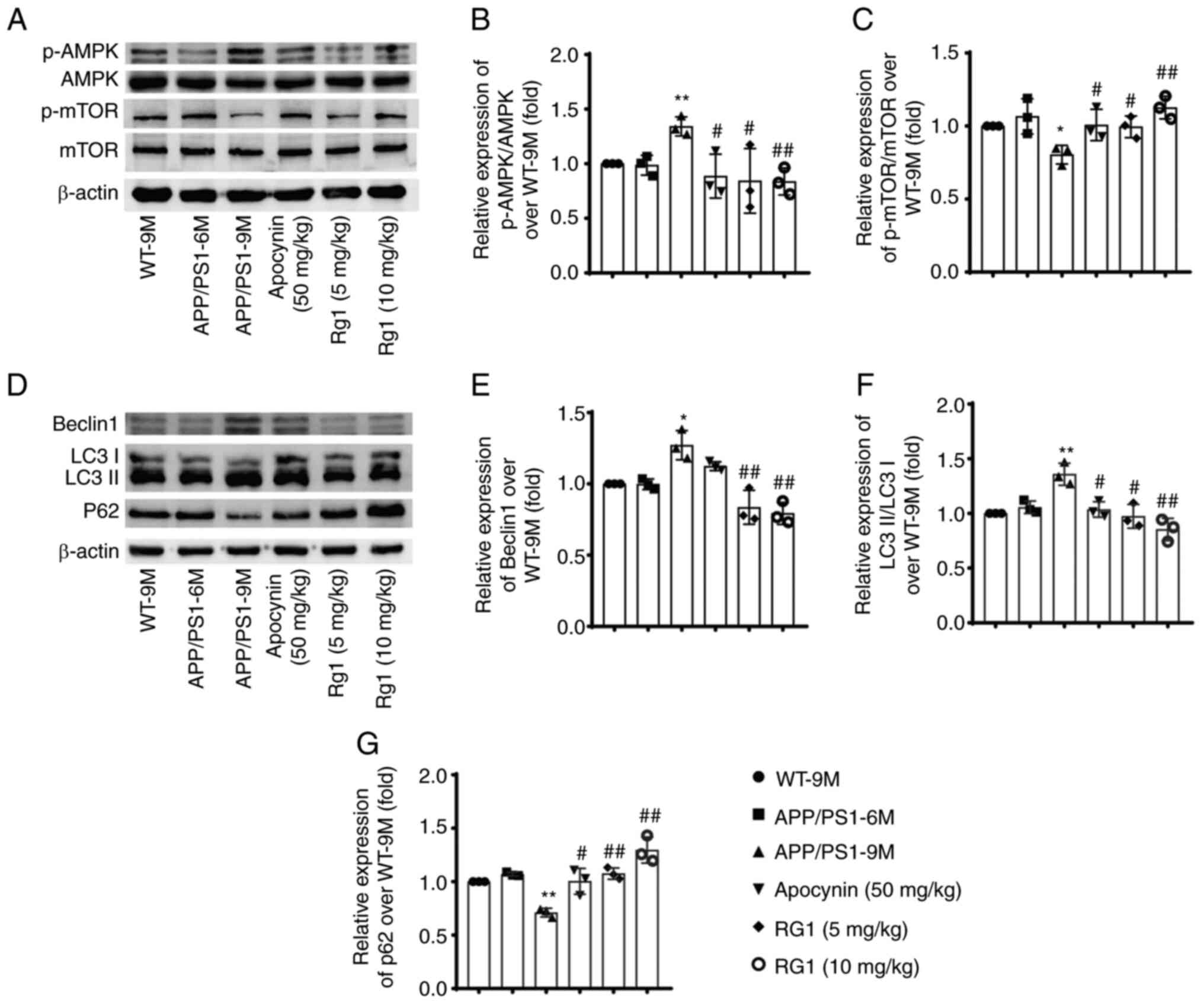
Rg1 treatment improves autophagy
dysfunction in APP/PS1 mice
Autophagy also plays important roles in Aβ
generation and metabolism, and its malfunction is involved in the
progress of AD (35). Therefore,
the effect of Rg1 on autophagy function in APP/PS mice was further
studied. The results showed that there were no significant
differences in the expression levels of p-AMPK/AMPK, p-mTOR/mTOR,
Beclin1, LC3 II/LC3 I and P62 in APP/PS1-6M mice compared with
WT-9M. While in APP/PS1-9M mice, the levels of p-AMPK/AMPK, Beclin1
and LC3 II/LC3 I were significantly increased, and the levels of
p-mTOR/mTOR and P62 were significantly decreased (Fig. 8A-G, P<0.05 or P<0.01).
Moreover, apocynin and Rg1 treatment could significantly reverse
the changes of these proteins in APP/PS1 mice (Fig. 8A-G, P<0.05 or P<0.01).
Furthermore, the expression levels of Beclin1 and LC3 were detected
by immunohistochemistry. There were similar results that the
expression levels of Beclin1 and LC3 were increased in APP/PS1-9M
mice and were significantly decreased after Rg1 treatment in cortex
and hippocampus CA1 (Figs. S1
and S2, P<0.05 or P<0.01).
These results indicated that Rg1 treatment can improve autophagy
dysfunction in APP/PS1 mice.
Molecular docking shows potential
interaction of Rg1 with NLRP1
Molecular docking was used to further verify the
interaction between Rg1 and NLRP1. The results showed that several
hydrogen bonds may form between Rg1 and NLRP1 (Fig. S3A and B). Meanwhile, the binding
energy (affinity: −5.31 kcal/mol) in the docking analysis showed
favorable binding results between Rg1 and NLRP1 (Fig. S3C).
Discussion
Although it is well known that the pathological
features of AD are obvious, the pathogenesis remains not completely
clear and there is currently little effective treatment for AD. The
Aβ cascade hypothesis is dominated in the field of AD research and
provides an intellectual framework for therapeutic interventions
(36). Based on this, various
mechanisms, and pathways of Aβ generation and deposition have been
established, such as ROS oxidative stress, mitochondrial
dysfunction and inflammatory process (37). In recent years, increasing
evidence has focused on the changes in neuroinflammation and
autophagy, which has brought new ideas to explore the pathogenesis
of AD. Ginsenoside Rg1 has been reported to possess neuroprotective
effects in numerous neurodegenerative diseases (20,21). Particularly, Rg1 treatment could
protect against lipopolysaccharide (LPS)-induced neuroinflammation
and neuronal apoptosis (38,39). A recent study by the authors
indicated that Rg1 treatment could significantly ameliorate
LPS-induced cognitive impairments and neuroinflammation through
inhibiting NLRP1 inflammasome in mice (40). Additionally, another recent study
indicated that Rg1 treatment could promote autophagy, resulting in
the inhibition of NLRP3 inflammasome in acute liver injuries
(41). However, whether Rg1
protects against AD through regulating autophagy remains unknown.
Therefore, it was hypothesized that Rg1 treatment may alleviate
neuronal damages and Aβ depositions by inhibiting NLRP1
inflammasome and autophagy dysfunction in APP/PS1 mice, which may
provide a new therapeutic strategy for AD. In the present study,
the results showed that Rg1 treatment significantly alleviated the
olfactory dysfunction, learning and memory impairments, Aβ
deposition and neuronal damage in APP/PS1 mice. The results also
demonstrated that Rg1 treatment significantly reduced the
expression levels of NLRP1 inflammasome, as well as reversed the
AMPK/mTOR pathway and inhibited the autophagy function in APP/PS1
mice. The present results suggested that Rg1 ameliorates Aβ
depositions and neuronal damages by regulating NLRP1 inflammasome
as well as the AMPK/mTOR-mediated autophagic pathway.
The APP/PS1 double transgenic mouse is a common AD
model, which shows significant memory dysfunction, Aβ deposition
and neuronal damages similarity to pathological features of AD at 8
months of age or even earlier (42). Therefore, the protective effects
of Rg1 in APP/PS1 mice from 6–9 months old were studied.
Additionally, Apocynin, as a NOX inhibitor by reducing ROS
generation, has been reported to possess protective effect against
neurodegenerative diseases (43,44) and has anti-inflammatory effects
(45). Apocynin has also been
reported to ameliorate neuronal damage by regulating autophagy
(46,47). Therefore, apocynin was chosen as a
positive drug in the current study. It was found that the
expression levels of NLRP1 inflammasome and autophagy-related
proteins were significantly decreased in the apocynin-treated mice,
suggesting that apocynin has a regulatory effect on inflammation
and autophagy in AD. Furthermore, previous studies have suggested
that olfactory dysfunctions (OD) may be an early biomarker for
neurodegeneration (48). It has
been reported that OD is observed in 85% of early-stage AD patients
(48). The present study
confirmed that there was no significant OD in APP/PS1-6M mice, but
the APP/PS1-9M mice had a significant OD. The result of MWM was
similar to the BFT that the APP/PS1-9M mice showed significant
learning and memory impairments but not in APP/PS1-6M mice.
Meanwhile, it was found that Rg1 and apocynin treatment could
significantly improve the OD and cognitive impairments in
APP/PS1-9M mice, suggesting that Rg1 could attenuate the
progression of AD. The structural and functional dysfunctions in
cortex and hippocampus, which are also vulnerable to Aβ
accumulation and are closely correlated to AD progression (49). The results of the present study
indicated that the APP/PS1-9M mice exhibited significant neuronal
injuries and Aβ depositions in the cortex and hippocampus CA1
regions, and Rg1 treatment significantly alleviated the neuronal
damages and Aβ accumulation. Synapse loss also correlates with
cognitive impairments in AD. PSD95, an important protein regulating
synapse function, is significantly reduced in the cortex and
hippocampus regions of patients with AD and is correlated
negatively with cognitive dysfunction of AD (50). The present results revealed that
the expression of PSD95 was significantly reduced in APP/PS1-9M
mice but was increased by Rg1 treatment. These data suggested that
Rg1 treatment could ameliorate neuronal injuries and Aβ deposition
in APP/PS1 mice.
Inflammation is considerably reported as an
essential factor of neuronal damage in the progression of AD
(51). It has been reported that
a number of proinflammatory cytokines are elevated in brain tissues
of AD, such as TNF-α, IL-6 and IL-1β (52,53). The TNF-α secreted by activated
microglia is involved in cognitive impairment (54). The IL-1β suppresses the long-term
potentiation (LTP), and induces learning and memory impairments by
activating p38-MAPK and GSK3 signaling (55). Furthermore, overexpression of
these cytokines contributes to synaptic plasticity dysfunction and
neuronal damages in 5×FAD mice (56). Furthermore, a handful of studies
suggested that the proinflammatory cytokines, such as TNF-α and
IL-1β, also play crucial roles in Aβ accumulation of AD. IL-1β and
TNF-α overexpression could increase Aβ generation from APP through
enhancement of β- and γ-secretase (57). In the present study, it was
identified that the expression levels of IL-1β, IL-6 and TNF-α were
significantly increased accompanied significant elevation of
Aβ1–42 generation and Aβ deposition in APP/PS1-9M mice.
Rg1 treatment could significantly inhibit the expression levels of
IL-1β, IL-6 and TNF-α and attenuate learning and memory impairments
and Aβ deposition. These data demonstrated that Rg1 may alleviate
Aβ deposition and learning and memory impairments through
inhibiting neuroinflammation. A recent study by the authors
suggested that Rg1 treatment could improve LPS-induced neuronal
injuries by downregulating the expression levels of NLRP1
inflammasomes in HT22 cells (58). Therefore, it was hypothesized that
Rg1 may delay the progression of AD by inhibiting NLRP1
inflammasome in APP/PS1 mice. The NLRP1 inflammasome is extensively
expressed in central nervous system, particularly in neurons and
serves as a platform for the recruitment of the
apoptosis-associated speck-like protein containing a CARD (ASC) and
procaspase-1 protease. Once activated, ASC activates caspase-1 and
then induces the processing and maturation of the IL-1β, IL-6 and
IL-18 (59). Previous studies
have shown that NLRP1 inflammasome is upregulated and accompanied
by neuronal damage and cognitive decline in mice model of AD
(15). The present results showed
that the expression levels of NLRP1, ASC, Caspase-1, IL-1β, IL-6
and TNF-α were all significantly increased in APP/PS1-9M mice, and
Rg1 treatment significantly decreased their expression levels. In
addition, the molecular docking analysis showed that there was
favorable binding result between Rg1 and NLRP1. These results
confirmed that activation of NLRP1 inflammasome is involved in AD
progression and Rg1 may alleviate learning and memory impairments
and Aβ disposition through inhibiting the NLRP1 inflammasome.
Autophagy is a lysosomal degradation pathway
essential for survival, differentiation, development and
homeostasis in cells (60). There
are three stages in autophagy biogenesis, including phagosome
membrane separation, phagosome elongation and phagocytosis of
random cytoplasmic contents, and autophagosome maturation and
fusion with lysosomes (61).
Increasing evidence has implicated autophagy in numerous major
neurodegenerative disorders, such as frontotemporal dementia, AD,
Parkinson's and Huntington's disease (62–65). Under normal conditions, autophagy
can remove accumulated intracellular toxicants or damaged
organelles from neuronal cells (66). For example, autophagy can
facilitate the degradation and clearance of APP as well as APP
cleavage products including Aβ (67,68). However, autophagy dysfunction is
also involved in the accretion of noxious proteins in the AD brain
(35). Therefore, autophagy may
be a double-edged sword in the progression of AD, and its function
is controversial (11,69). On the one hand, autophagy can
protect cells against apoptosis and necrosis by degrading harmful
substances (70). On the other
hand, its excessive activation can lead to autophagic stress, which
is defined as a relatively sustained imbalance in which rates of
autophagosome vacuoles formation exceed rates of autophagosome
vacuoles degradation (71). The
accumulated autophagic vacuoles were found to be increased in AD
brains and in APP/PS1 mice, suggesting a source of Aβ generation
(72). Therefore, excessive
autophagy may contribute to autophagic stress, and its normal
function will be blocked, thus aggravating the severity of AD
(73). Beclin1 is an
autophagy-associated protein that regulates the formation of
autophagosomes. During the process of autophagy, a cytosolic form
of LC3 (LC3 I) is conjugated to phosphatidylethanolamine to form
LC3-phosphatidylethanolamine (LC3 II), which is recruited to
autophagosome membranes (74).
P62 is an autophagic receptor that recognizes ubiquitinylated
proteins and interacts with LC3-II in the autophagosomes (75). Furthermore, the lysosome is an
essential organelle for Aβ generation in autophagic vacuoles after
autophagy activation (76). The
results of the present study indicated that the expression levels
of Beclin1 and LC3-II/LC3-I were decreased and p62 expression was
increased in parallel with Aβ deposition in APP/PS1-9M mice.
Meanwhile, it was found that treatment with Rg1 can significantly
reverse these changes in APP/PS1 mice. These data suggested that
there may be autophagic stress in APP/PS1-9M mice, and Rg1 may
alleviate Aβ deposition by inhibiting autophagy dysfunction in
APP/PS1 mice. To confirm the effect of Rg1 on regulation of
autophagy in AD, the change of the AMPK/mTOR pathway, which plays
an important role in the modulation of autophagy, was further
explored (77). The AMP-activated
protein kinase (AMPK) and mTOR are two main nutrient-sensing
pathways in response to stress. The AMPK/mTOR pathway can activate
autophagy by decreasing the activity of mTOR. In the present study,
it was found that the p-AMPK/AMPK was significantly upregulated and
the p-mTOR/mTOR was significantly downregulated in APP/PS1-9M mice.
In addition, Rg1 significantly decreased p-AMPK/AMPK and increased
mTOR/mTOR in APP/PS1-9M mice. The results further revealed that Rg1
may regulate autophagy function through regulation of AMPK/mTOR
pathway in APP/PS1 mice.
Additionally, it has been reported that excessive
IL-1β can induce autophagy dysfunction in primary cultures of
neurons, astrocytes and microglia (78). Moreover, the Beclin1 assembly rate
is positively correlated with IL-1β and TNF-α levels, and
conversely, TNF-α levels were negatively correlated with mTOR
levels in APP/PS1 mouse brain tissue (79). These data suggested that
interaction of inflammation and autophagy may be closely correlated
with AD progression. The present study suggested that both NLRP1
inflammasome and autophagy dysfunction coexist in APP/PS mice. Rg1
treatment may delay the progression of AD by inhibiting NLRP1
inflammasome and autophagy dysfunction in APP/PS1 mice.
In general, the present study indicated that Rg1
treatment improves olfactory dysfunction, learning and memory
impairments, neuronal damages and Aβ depositions in APP/PS1 mice.
Meanwhile, the present results revealed that Rg1 can regulate NLRP1
inflammasome as well as AMPK/mTOR-mediated autophagic function in
APP/PS1 mice. These data suggested that Rg1 may alleviate Aβ
deposition and AD progression by inhibiting NLRP1 inflammasome and
autophagy dysfunction. However, the present study only provided the
basic experimental results that Rg1 treatment ameliorated cognitive
dysfunction due to the inhibition of NLRP1 inflammasome and
autophagy in APP/PS1 mice and its exact mechanism needs to be
confirmed in vitro. Additionally, as the interrelationship
between NLRP1 inflammasome and autophagy are complex, more research
is required to fully elucidate the possible mechanisms in
progression of AD.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Mr. Dake Huang
(Synthetic Laboratory, Basic Medicine College) for the technical
assistance.
Funding
The present study was supported by the Major projects of Anhui
Provincial Department of Education (grant no. KJ2020ZD14) and the
National Natural Science Foundation of China (grant nos. 81671384
and 81970630).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
WZL and WPL conceived and designed the study and
critically revised the manuscript for intellectually important
content. LK, LH and XL performed the experiments and statistical
analysis and wrote the manuscript. YS collated the data. HZ and PJ
were mainly responsible for the histological experiments. YS and HZ
confirm the authenticity of all the raw data. PJ, RS and CW helped
to perform the experiments and wrote part of the manuscript. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
All experiments involving animals were approved
(approval no. LLSC20211172) by the Ethics Committee of Laboratory
Animals of Anhui Medical University (Hefei, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gouras GK, Olsson TT and Hansson O:
β-Amyloid peptides and amyloid plaques in Alzheimer's disease.
Neurotherapeutics. 12:3–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zolezzi JM, Bastias-Candia S, Santos MJ
and Inestrosa NC: Alzheimer's disease: Relevant molecular and
physiopathological events affecting amyloid-β brain balance and the
putative role of PPARs. Front Aging Neurosci. 6:1762014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer's disease: Progress and problems on the
road to therapeutics. Science. 297:353–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kuang H, Tan CY, Tian HZ, Liu LH, Yang MW,
Hong FF and Yang SL: Exploring the bi-directional relationship
between autophagy and Alzheimer's disease. CNS Neurosci Ther.
26:155–166. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Newcombe EA, Camats-Perna J, Silva ML,
Valmas N, Huat TJ and Medeiros R: Inflammation: The link between
comorbidities, genetics, and Alzheimer's disease. J
Neuroinflammation. 15:2762018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Parzych KR and Klionsky DJ: An overview of
autophagy: Morphology, mechanism, and regulation. Antioxid Redox
Signal. 20:460–473. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim KH and Lee MS: Autophagy-a key player
in cellular and body metabolism. Nat Rev Endocrinol. 10:322–337.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Saha S, Panigrahi DP, Patil S and Bhutia
SK: Autophagy in health and disease: A comprehensive review. Biomed
Pharmacother. 104:485–495. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nixon RA: Autophagy, amyloidogenesis and
Alzheimer disease. J Cell Sci. 120:4081–4091. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Steele JW, Fan E, Kelahmetoglu Y, Tian Y
and Bustos V: Modulation of autophagy as a therapeutic target for
Alzheimer's disease. Postdoc J. 1:21–34. 2013.PubMed/NCBI
|
|
11
|
Nilsson P, Loganathan K, Sekiguchi M,
Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T and Saido
TC: Aβ secretion and plaque formation depend on autophagy. Cell
Rep. 5:61–69. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Q, Liu Y and Sun M: Autophagy and
Alzheimer's disease. Cell Mol Neurobiol. 37:377–388. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Forloni G and Balducci C: Alzheimer's
disease, oligomers, and inflammation. J Alzheimers Dis.
62:1261–1276. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chavarria-Smith J and Vance RE: The NLRP1
inflammasomes. Immunol Rev. 265:22–34. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yap JKY, Pickard BS, Chan EWL and Gan SY:
The role of neuronal NLRP1 inflammasome in Alzheimer's disease:
Bringing neurons into the neuroinflammation game. Mol Neurobiol.
56:7741–7753. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang F and Jiang L: Neuroinflammation in
Alzheimer's disease. Neuropsychiatr Dis Treat. 11:243–256. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kaushal V, Dye R, Pakavathkumar P, Foveau
B, Flores J, Hyman B, Ghetti B, Koller BH and LeBlanc AC: Neuronal
NLRP1 inflammasome activation of caspase-1 coordinately regulates
inflammatory interleukin-1-beta production and axonal
degeneration-associated caspase-6 activation. Cell Death Differ.
22:1676–1686. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tan MS, Tan L, Jiang T, Zhu XC, Wang HF,
Jia CD and Yu JT: Amyloid-β induces NLRP1-dependent neuronal
pyroptosis in models of Alzheimer's disease. Cell Death Dis.
5:e13822014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun D, Gao G, Zhong B, Zhang H, Ding S,
Sun Z, Zhang Y and Li W: NLRP1 inflammasome involves in learning
and memory impairments and neuronal damages during aging process in
mice. Behav Brain Funct. 17:112021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chu SF, Zhang Z, Zhou X, He WB, Chen C,
Luo P, Liu DD, Ai QD, Gong HF, Wang ZZ, et al: Ginsenoside Rg1
protects against ischemic/reperfusion-induced neuronal injury
through miR-144/Nrf2/ARE pathway. Acta Pharmacol Sin. 40:13–25.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Z, Song Z, Shen F, Xie P, Wang J,
Zhu AS and Zhu G: Ginsenoside Rg1 prevents PTSD-like behaviors in
mice through promoting synaptic proteins, reducing Kir4.1 and TNF-α
in the hippocampus. Mol Neurobiol. 58:1550–1563. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu TZ, Shen XY, Sun LL, Chen YL, Zhang BQ,
Huang DK and Li WZ: Ginsenoside Rg1 protects against H2O2-induced
neuronal damage due to inhibition of the NLRP1 inflammasome
signalling pathway in hippocampal neurons in vitro. Int J Mol Med.
43:717–726. 2019.PubMed/NCBI
|
|
23
|
Zhang H, Su Y, Sun Z, Chen M, Han Y, Li Y,
Dong X, Ding S, Fang Z and Li W and Li W: Ginsenoside Rg1
alleviates Aβ deposition by inhibiting NADPH oxidase 2 activation
in APP/PS1 mice. J Ginseng Res. 45:665–675. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xue Z, Zhang Z, Liu H, Li W, Guo X, Zhang
Z, Liu Y, Jia L, Li Y, Ren Y, et al: lincRNA-Cox2 regulates NLRP3
inflammasome and autophagy mediated neuroinflammation. Cell Death
Differ. 26:130–145. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang Y, Li J, Rao T, Fang Z and Zhang J:
The role and mechanism of hyperoside against myocardial infarction
in mice by regulating autophagy via NLRP1 inflammation pathway. J
Ethnopharmacol. 276:1141872021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han Y, Li X, Yang L, Zhang D, Li L, Dong
X, Li Y, Qun S and Li W: Ginsenoside Rg1 attenuates cerebral
ischemia-reperfusion injury due to inhibition of NOX2-mediated
calcium homeostasis dysregulation in mice. J Ginseng Res.
46:515–525. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen Y, Ding S, Zhang H, Sun Z, Shen X,
Sun L, Yin Y, Qun S and Li W: Protective effects of ginsenoside Rg1
on neuronal senescence due to inhibition of NOX2 and NLRP1
inflammasome activation in SAMP8 mice. J Funct Foods.
65:1037132020. View Article : Google Scholar
|
|
28
|
Yu Q, Guo P, Li D, Zuo L, Lian T, Yu S, Hu
Y, Liu L, Jin Z, Wang R, et al: Olfactory dysfunction and its
relationship with clinical symptoms of Alzheimer disease. Aging
Dis. 9:1084–1095. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li W, Li S, Shen L, Wang J, Wu X, Li J, Tu
C, Ye X and Ling S: Impairment of dendrodendritic inhibition in the
olfactory bulb of APP/PS1 mice. Front Aging Neurosci. 11:22019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu Y, Zhang Y, Zheng X, Fang T, Yang X,
Luo X, Guo A, Newell KA, Huang XF and Yu Y: Galantamine improves
cognition, hippocampal inflammation, and synaptic plasticity
impairments induced by lipopolysaccharide in mice. J
Neuroinflammation. 15:1122018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hu YD, Pang W, He CC, Lu H, Liu W, Wang
ZY, Liu YQ, Huang CY and Jiang YG: The cognitive impairment induced
by zinc deficiency in rats aged 0~2 months related to BDNF DNA
methylation changes in the hippocampus. Nutr Neurosci. 20:519–525.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang B, Zhang Y, Wu W, Xu T, Yin Y, Zhang
J, Huang D and Li W: Chronic glucocorticoid exposure activates
BK-NLRP1 signal involving in hippocampal neuron damage. J
Neuroinflammation. 14:1392017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Morton H, Kshirsagar S, Orlov E, Bunquin
LE, Sawant N, Boleng L, George M, Basu T, Ramasubramanian B,
Pradeepkiran JA, et al: Defective mitophagy and synaptic
degeneration in Alzheimer's disease: Focus on aging, mitochondria
and synapse. Free Radic Biol Med. 172:652–667. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Uddin MS, Stachowiak A, Mamun AA, Tzvetkov
NT, Takeda S, Atanasov AG, Bergantin LB, Abdel-Daim MM and
Stankiewicz AM: Autophagy and Alzheimer's disease: From molecular
mechanisms to therapeutic implications. Front Aging Neurosci.
10:042018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Barage SH and Sonawane KD: Amyloid cascade
hypothesis: Pathogenesis and therapeutic strategies in Alzheimer's
disease. Neuropeptides. 52:1–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tonnies E and Trushina E: Oxidative
stress, synaptic dysfunction, and Alzheimer's disease. J Alzheimers
Dis. 57:1105–1121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mei X, Feng H and Shao B: Alleviation of
sepsis-associated encephalopathy by ginsenoside via inhibition of
oxidative stress and cell apoptosis: An experimental study. Pak J
Pharm Sci. 33:2567–2577. 2020.PubMed/NCBI
|
|
39
|
Jin Y, Peng J, Wang X, Zhang D and Wang T:
Ameliorative effect of ginsenoside Rg1 on
lipopolysaccharide-induced cognitive impairment: Role of
cholinergic system. Neurochem Res. 42:1299–1307. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dong X, Li L, Zhang D, Su Y, Yang L, Li X,
Han Y and Li W and Li W: Ginsenoside Rg1 attenuates LPS-induced
cognitive impairments and neuroinflammation by inhibiting NOX2 and
Ca2+-CN-NFAT1 signaling in mice. J Funct Foods.
87:1047912021. View Article : Google Scholar
|
|
41
|
Zhao J, He B, Zhang S, Huang W and Li X:
Ginsenoside Rg1 alleviates acute liver injury through the induction
of autophagy and suppressing NF-κB/NLRP3 inflammasome signaling
pathway. Int J Med Sci. 18:1382–1389. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Radde R, Bolmont T, Kaeser SA,
Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg
H, Gengler S, et al: Abeta42-driven cerebral amyloidosis in
transgenic mice reveals early and robust pathology. EMBO Rep.
7:940–946. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li J, Yang JY, Yao XC, Xue X, Zhang QC,
Wang XX, Ding LL and Wu CF: Oligomeric Aβ-induced microglial
activation is possibly mediated by NADPH oxidase. Neurochem Res.
38:443–452. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Qiu LL, Luo D, Zhang H, Shi YS, Li YJ, Wu
D, Chen J, Ji MH and Yang JJ: Nox-2-mediated phenotype loss of
hippocampal parvalbumin interneurons might contribute to
postoperative cognitive decline in aging mice. Front Aging
Neurosci. 8:2342016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dos-Santos-Pereira M, Guimarães FS,
Del-Bel E, Raisman-Vozari R and Michel PP: Cannabidiol prevents
LPS-induced microglial inflammation by inhibiting
ROS/NF-κB-dependent signaling and glucose consumption. Glia.
68:561–573. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ding Z, Liu S, Wang X, Khaidakov M, Dai Y
and Mehta JL: Oxidant stress in mitochondrial DNA damage, autophagy
and inflammation in atherosclerosis. Sci Rep. 3:10772013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lu Q, Harris VA, Kumar S, Mansour HM and
Black SM: Autophagy in neonatal hypoxia ischemic brain is
associated with oxidative stress. Redox Biol. 6:516–523. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dan X, Wechter N, Gray S, Mohanty JG,
Croteau DL and Bohr VA: Olfactory dysfunction in aging and
neurodegenerative diseases. Ageing Res Rev. 70:1014162021.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Harris JA, Devidze N, Verret L, Ho K,
Halabisky B, Thwin MT, Kim D, Hamto P, Lo I, Yu GQ, et al:
Transsynaptic progression of amyloid-β-induced neuronal dysfunction
within the entorhinal-hippocampal network. Neuron. 68:428–441.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cui YQ, Wang Q, Zhang DM, Wang JY, Xiao B,
Zheng Y and Wang XM: Triptolide rescues spatial memory deficits and
amyloid-β aggregation accompanied by inhibition of inflammatory
responses and MAPKs activity in APP/PS1 transgenic mice. Curr
Alzheimer Res. 13:288–296. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Minter MR, Taylor JM and Crack PJ: The
contribution of neuroinflammation to amyloid toxicity in
Alzheimer's disease. J Neurochem. 136:457–474. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Rubio-Perez JM and Morillas-Ruiz JM: A
review: Inflammatory process in Alzheimer's disease, role of
cytokines. ScientificWorldJournal. 2012:7563572012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Alam Q, Alam MZ, Mushtaq G, Damanhouri GA,
Rasool M, Kamal MA and Haque A: Inflammatory Process in Alzheimer's
and Parkinson's diseases: Central role of cytokines. Curr Pharm
Des. 22:541–548. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Torres-Acosta N, O'Keefe JH, O'Keefe EL,
Isaacson R and Small G: Therapeutic potential of TNF-α inhibition
for Alzheimer's disease prevention. J Alzheimers Dis. 78:619–626.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pickering M and O'Connor JJ:
Pro-inflammatory cytokines and their effects in the dentate gyrus.
Progress in Brain Research. Scharfman HE: Elsevier; pp. 339–354.
2007, View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jiang Y, Li K, Li X, Xu L and Yang Z:
Sodium butyrate ameliorates the impairment of synaptic plasticity
by inhibiting the neuroinflammation in 5XFAD mice. Chem Biol
Interact. 341:1094522021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liao YF, Wang BJ, Cheng HT, Kuo LH and
Wolfe MS: Tumor necrosis factor-alpha, interleukin-1beta, and
interferon-gamma stimulate gamma-secretase-mediated cleavage of
amyloid precursor protein through a JNK-dependent MAPK pathway. J
Biol Chem. 279:49523–49532. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang Y, Ding S, Chen Y, Sun Z, Zhang J,
Han Y, Dong X, Fang Z and Li W: Ginsenoside Rg1 alleviates
lipopolysaccharide-induced neuronal damage by inhibiting NLRP1
inflammasomes in HT22 cells. Exp Ther Med. 22:7822021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Fan JJ, Gao B, Song AQ, Zhu YJ, Zhou J, Li
WZ, Yin YY and Wu WN: Spinal cord NLRP1 inflammasome contributes to
dry skin induced chronic itch in mice. J Neuroinflammation.
17:1222020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Doherty J and Baehrecke EH: Life, death
and autophagy. Nat Cell Biol. 20:1110–1117. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lee KM, Hwang SK and Lee JA: Neuronal
autophagy and neurodevelopmental disorders. Exp Neurobiol.
22:133–142. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Watanabe Y, Taguchi K and Tanaka M:
Ubiquitin, autophagy and neurodegenerative diseases. Cells.
9:20222020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Reddy PH and Oliver DM: Amyloid beta and
phosphorylated tau-induced defective autophagy and mitophagy in
Alzheimer's disease. Cells. 8:4882019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lu J, Wu M and Yue Z: Autophagy and
Parkinson's disease. Adv Exp Med Biol. 1207:21–51. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Croce KR and Yamamoto A: A role for
autophagy in Huntington's disease. Neurobiol Dis. 122:16–22. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Marino G, Madeo F and Kroemer G: Autophagy
for tissue homeostasis and neuroprotection. Curr Opin Cell Biol.
23:198–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhou F, van Laar T, Huang H and Zhang L:
APP and APLP1 are degraded through autophagy in response to
proteasome inhibition in neuronal cells. Protein Cell. 2:377–383.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Son SM, Jung ES, Shin HJ, Byun J and
Mook-Jung I: Aβ-induced formation of autophagosomes is mediated by
RAGE-CaMKKβ-AMPK signaling. Neurobiol Aging. 33:1006.e11–e23. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Spilman P, Podlutskaya N, Hart MJ, Debnath
J, Gorostiza O, Bredesen D, Richardson A, Strong R and Galvan V:
Inhibition of mTOR by rapamycin abolishes cognitive deficits and
reduces amyloid-beta levels in a mouse model of Alzheimer's
disease. PLoS One. 5:e99792010. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Li ZY, Chen LH, Zhao XY, Chen H, Sun YY,
Lu MH, Wang ZT, Chen M, Lu L, Huang W, et al: Clemastine attenuates
AD-like pathology in an AD model mouse via enhancing mTOR-mediated
autophagy. Exp Neurol. 342:1137422021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chu CT: Autophagic stress in neuronal
injury and disease. J Neuropathol Exp Neurol. 65:423–432. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Nixon RA, Wegiel J, Kumar A, Yu WH,
Peterhoff C, Cataldo A and Cuervo AM: Extensive involvement of
autophagy in Alzheimer disease: An immuno-electron microscopy
study. J Neuropathol Exp Neurol. 64:113–122. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang C, Zhang X, Teng Z, Zhang T and Li Y:
Downregulation of PI3K/Akt/mTOR signaling pathway in
curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur J
Pharmacol. 740:312–320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang R and Hu W: Asprosin promotes β-cell
apoptosis by inhibiting the autophagy of β-cell via AMPK-mTOR
pathway. J Cell Physiol. 236:215–221. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Mizushima N: A(beta) generation in
autophagic vacuoles. J Cell Biol. 171:15–17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lenoir O, Tharaux PL and Huber TB:
Autophagy in kidney disease and aging: Lessons from rodent models.
Kidney Int. 90:950–964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
François A, Terro F, Janet T, Rioux Bilan
A, Paccalin M and Page G: Involvement of interleukin-1β in the
autophagic process of microglia: Relevance to Alzheimer's disease.
J Neuroinflammation. 10:1512013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
François A, Rioux Bilan A, Quellard N,
Fernandez B, Janet T, Chassaing D, Paccalin M, Terro F and Page G:
Longitudinal follow-up of autophagy and inflammation in brain of
APPswePS1dE9 transgenic mice. J Neuroinflammation. 11:1392014.
View Article : Google Scholar : PubMed/NCBI
|