Introduction
Activating point mutations in Kirsten rat sarcoma
viral oncogene homolog (KRAS) are present in ~30% of human cancer,
including the vast majority of types of pancreatic cancer and large
fractions of lung, colon and breast cancer (1). Furthermore, elevation of the activity of
KRAS and other ras family members due to upstream signaling is very
common in most types of cancer (2).
KRAS is a small guanosine triphosphatase (GTP-ase) that mediates
survival, proliferation and cytoskeletal organization of cancer
cells by transducing or enhancing down-stream signals of the dual
specificity mitogen-activated protein kinase kinase mek-2
(MEK)/extracellular signaling-regulated kinase (ERK) and of the
phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling
cascade (3). The presence of
constitutively active KRAS is associated with chemotherapy
resistance and poor patient outcome, but so far attempts to
directly target KRAS have failed (4).
KRAS exerts its functions at the inner cellular membrane and
requires continuous recycling of mislocalized protein through
endosomes. The microtubule network is critically involved in this
subcellular segregation and trafficking of endosomal recycling
(5,6).
Drugs that target microtubules include taxanes and vinca
alcaloids. These drugs are among the best studied, longest
standing cancer drugs and act as microtubule stabilizers that slow
or block mitosis at the metaphase-anaphase transition, eventually
resulting in apoptosis (7). Tissue
penetration of taxanes and vinca alcaloids is however
limited and the therapeutic index is low.
Plinabulin (NPI-2358) is a synthetic analogue of
halimide, an Aspergillus-derived natural product that
targets the tubulin network through a mechanism that is distinct
from that of taxanes and vinca alcaloids: Instead of
stabilization of polymerized microtubules, plinabulin inhibits
polymerization by interacting with the colchicine-binding domain of
β-tubulin (8,9). Drugs that target the colchicine-binding
domain were historically deemed too toxic to be effective
anticancer agents, but novel compounds including plinabulin are
more specific and potent inhibitors of tubulin polymerization
(7), and tissue penetration is more
favorable compared to classic tubulin stabilizing drugs (10). Furthermore, plinabulin demonstrated a
favorable safety profile and encouraging antitumor responses in two
early phase clinical trials (10,11). Based
on these results, plinabulin is currently in phase III clinical
development in combination with docetaxel and in phase I/II in
combination with nivolumab for advanced non-small cell lung cancer
(NCT02504489 and NCT02812667). In the present study the inhibition
of tubulin polymerization by plinabulin was described and the
preclinical results of the efficacy of plinabulin in a KRAS driven
murine tumor models and outline effects of plinabulin on
intracellular KRAS trafficking and down-stream signaling were
reported.
Materials and methods
Compounds and reagents.
Plinabulin was obtained from BeyondSpring
Pharmaceuticals, Inc. (New York, NY, USA), irinotecan was obtained
from Teva Pharmaceuticals (Irvine, CA, USA), docetaxel and
paclitaxel were from Selleckchem (Houston, TX, USA). For in
vivo studies, all compounds were diluted in 5% dextrose for
intraperitoneal (i.p.) or intravenous (i.v.) injection of indicated
doses. For in vitro studies, all drugs were diluted in
complete growth media with 1% dimethyl-sulfoxide (DMSO). In
vitro EGF treatment was performed at a concentration of 100
ng/ml.
Tubulin polymerization assays.
Microtubule protein (MTP) preparations consisting of
70% tubulin and 30% microtubule-associated proteins were isolated
from a bovine brain, polymerized into microtubules and monitored by
light scattering at 350 nm. Transmission electron microscopy was
used to determine the mean length distribution of microtubules in
the absence or presence of the drug. In brief, samples were fixed
at room temperature with 4% formaldehyde and 2.5% glutaraldehyde in
0.1 M PIPES buffer for 30 min and stained at room temperature for 7
min utilizing primary antibodies and for 2 min. with gold-labeled
secondary antibodies. Embeding was performed at 60˚C with epoxy
resin. Section thickness was 60 nm. Grids were viewed in a Jeol
electron microscope-1200 EX11 (JEOL, Ltd., Tokyo, Japan) at x2,000
and x30,000 magnification. The Zeiss MOPIII was used to determine
microtubule length distributions and mean lengths for at least 100
microtubules per sample.
Cell culture.
All cell lines were obtained from the American Type
Culture Collection (Manassas, VA, USA). HCT-15, A549 and MDA-MB-231
cells were maintained in RPMI-1640 medium (Lonza Group, Ltd.,
Basel, Switzerland). LoVo cells were maintained in F12-K medium
(Corning/Cellgro; Corning Inc., Corning, NY, USA). DF1 cells were
maintained in Gibco Dulbecco's modified Eagle's medium (DMEM;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). All cell culture
media were supplemented with 10% fetal bovine serum (FBS; Avantor,
Inc., Radnor, PA, USA) and housed in a 5% CO2 atmosphere
at 37˚C, except the DF1 cells which were cultured at 39˚C.
Immunocytology.
A549 cells (1,000 cells/cm2) were seeded
overnight on coverslips. Cells were fixed in 4% paraformaldehyde
for 10 min at room temperature, followed by permeabilization using
0.1% Triton in PBS for 5 min and blocking of unspecific binding
with 5% donkey serum (Jackson ImmunoResearch Laboratories, Inc.,
West Grove, PA, USA) in PBS. These steps were done at room
temperature and washing with PBS was done between any steps.
Primary antibodies were rabbit anti-early endosomal antigen (EEA)1
(1:300; cat. no. 610456; BD Bioscences, San Jose, CA, USA) and
mouse anti-KRAS (1:100; cat. no. ab172949; Abcam, Cambridge, MA,
USA) and were applied overnight at 4˚C. After additional washing
steps, cells were incubated with secondary donkey anti-rabbit-488
(1:200; cat. no. 711-545-152l; Jackson ImmunoResearch Laboratories,
Inc.) and anti-mouse-Cy3 (1:200; cat. no. 715-165-150; Jackson
ImmunoResearch Laboratories, Inc.) antibodies in PBS with 5% donkey
serum for 30 min at room temperature, followed by washing and
addition of Hoechst 33342 Solution (1 µg/ml; Thermo Fisher
Scientific, Inc.) for 3 min, followed by washing, H2O
for 3 min, 100% ethanol for 3 min and air-drying prior to embedding
(all steps at room temperature). Images were captured with a Zeiss
LSM 780 NLO confocal microscope and quantification was performed
using ImageJ, version 1.51 (National Institute of Health, Bethesda,
MD, USA).
In vitro cell viability assays.
Indicated cell lines were grown in standard
serum-containing media (RPMI with 10% FBS from American Type
Culture Collection). Cell viability was measured in triplicate
utilizing the adenosine triphosphate-based
CellTiter-Glo® assay (Promega Corporation, Madison, WI,
USA).
Murine xenograft models.
Animal studies were carried out in accordance with
the Guide for the Care and Use of Laboratory Animals, as adopted by
the U.S. National Institutes of Health. Animal research was
prospectively approved by the Institutional Animal Care and Use
Committee (Animal assurance institutional #A3226-01; Protocol
#50842, Fred Hutchinson Cancer Research Center, Seattle, WA, USA).
Female athymic Foxn1nu (BALB/c nude) mice between 4 to 6 weeks of
age were obtained from Harlan Laboratories, Inc. (Madison, WI,
USA). A total of 84 mice weighing 18-24 g and kept under germ-free
conditions with a 12-h light/dark cycle at 20-24˚C with 40-60%
humidity. For the HCT-15 and LoVo models, mice were anesthetized by
isoflurane and inoculated with 0.1 ml of a 50% media/50% Matrigel
mixture containing a suspension of cultured tumor cells
(1x107 cells/mouse for LoVo, 5x106
cells/mouse for HCT-15). As the tumors grew, the length (largest
diameter) and width (smallest diameter) of tumors were measured
using a digital caliper to determine the mean radius for tumor
volume estimates assuming a spherical geometry and calculation per
Vtumor = 4/3 x π x
r3. Automated calculation and documentation of
tumor volume estimates was done using the animal study management
software Study Director V.2.1.1 (Studylog Systems, Inc., San
Francisco, CA, USA). When tumor volume estimates exceeded 50-60
mm3, mice were randomized into control and treatment
groups, being pair-matched by tumor size, and dosing was initiated
(Day 1). Mice were euthanized as described below.
RCAS/t-va glioma models.
A total of 40 male (n=23) or female (n=17)
N/t-va;Ink4a/Arf-/- agouti mice (Jackson Laboratory, Bar
Harbor, ME, USA) were kept under germ-free conditions in a 12 h
light/12 h dark cycle at 20-24˚C with 40-60% humidity. Mice were
aged 6-8 weeks and weighed 18-24 g. These mice expressed the avian
t-va receptor under control of the nestin promoter were injected
intracranially with DF-1 cells (American Type Culture Collection)
as hosts for the replicative avian retroviral gene transfer vector
RCAS (Addgene, Inc., Cambridge, MA, USA). Tumors were generated
utilizing two RCAS vectors for the expression of platelet-derived
growth factor B (PDGFB) and either KRAS encoding the activating
mutation KRASG12A, or green fluorescent protein
(GFP). DF-1 cells infected with these RCAS strains were injected
into the brains of adult mice (aged 4 to 6 weeks) under the
anesthetic isoflurane. A total of 1 µl of a 1:1 mixture for a total
combined injection of 4x104 transfected DF-1 cell
suspension was delivered using a 30-gauge needle attached to a
Hamilton syringe. Coordinates were bregma 0 mm, lateral -0.5 mm
(right of midline) and depth -1.5 mm from the dural surface. Mice
were put on the study when they lost >0.3 g total over 2
consecutive days or displayed outward signs of a tumor. Mice (n=89)
were treated with vehicle or plinabulin at 7.5 mg/kg twice per week
for up to 10 weeks. The mice were euthanized in a carbon dioxide
chamber (flow rate of CO2 displacing 25% of the chamber
volume per minute for 5 min total in accordance with American
Veterinary Medical Association Euthanasia Guidelines of 2013) when
they lost >20% of their body weight, displayed a lack of
mobility, inability to feed or weighed <14 g for a male/12 g for
a female. Animal death was confirmed following at least 1 min of
immobility within the chamber, which was followed immediately by
decapitation and harvesting of the brain tissue.
Statistical analysis.
All statistical comparisons were performed using
GraphPad Prism 7 (GraphPad Software, Inc., San Diego, CA, USA).
Kaplan-Meier survival curves were prepared using and analyzed using
the log-rank (Mantel-Cox) test. Time series data were analyzed
using linear regression with the mean ± standard error calculated
for each time-point. Direct comparisons were performed using a
two-tailed t-test. P<0.05 was considered to indicate a
statistically significant difference. All experiments were
performed in triplicate unless stated otherwise.
Results
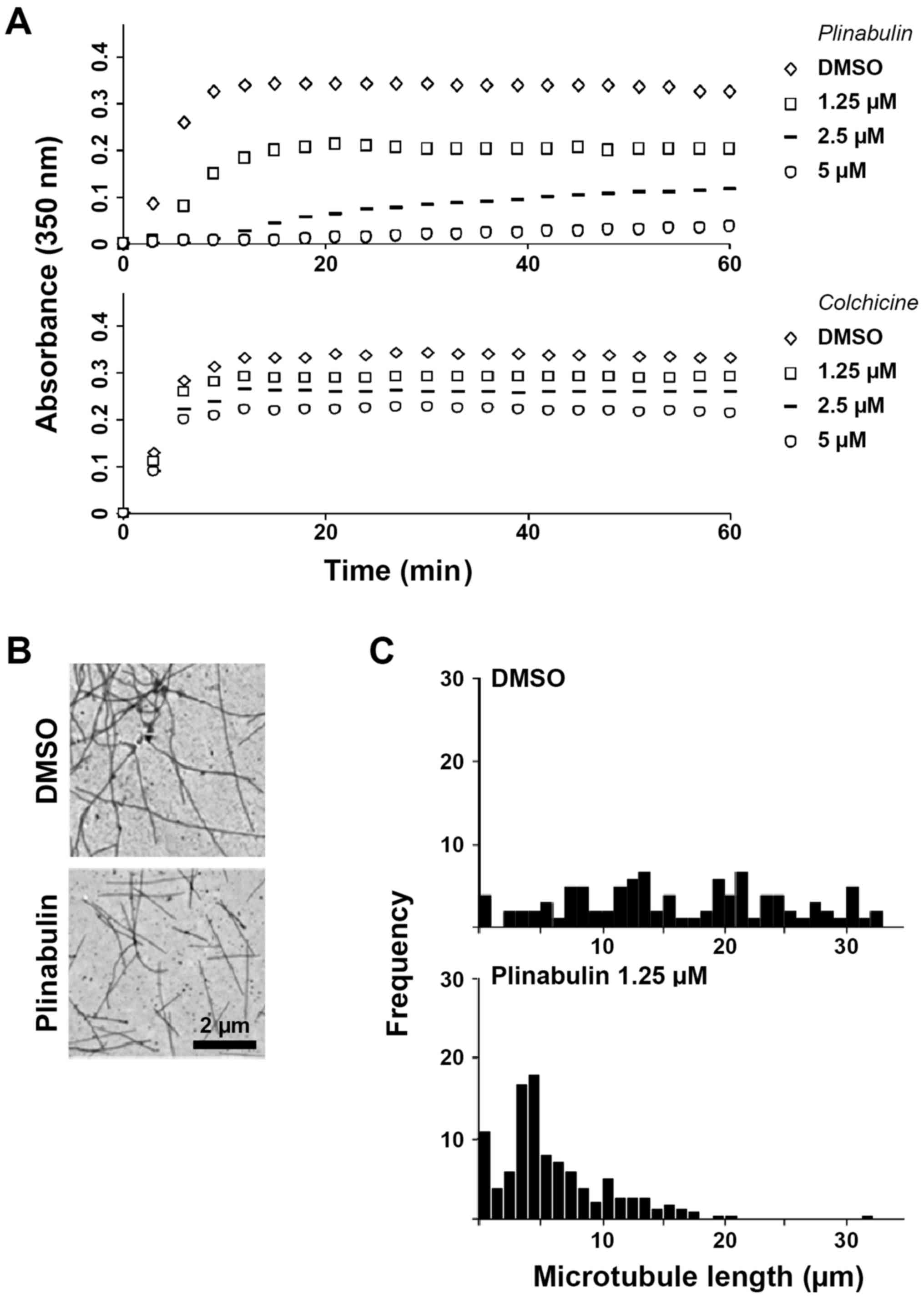
Plinabulin inhibits microtubule
polymerization.
The overlapping binding sites of plinabulin and
colchicine at the α/β-tubulin interface led to characterizing and
comparing the effects of the two drugs on microtubule dynamics. In
a cell-free MTP-containing model system, plinabulin inhibited
microtubule formation more potently than colchicine, as measured by
turbidity spectra (Fig. 1A). The
concentrations of plinabulin and colchicine at which polymerization
was inhibited by 50% (IC50) were 2.4 µM [standard
deviation (SD) +/- 0.4, N=4 independent experiments] and 7.6 µM (SD
+/- 2.4, N=3), respectively. The tubulin polymerization profiles
obtained in the presence of different concentrations of plinabulin
or colchicine differed in two ways: First, the initial rate of
increase in absorbance over time decreased with increasing drug
concentrations of plinabulin, indicating that there is a lag period
for microtubule formation. In contrast, the initial rate of
polymerization is unchanged at all concentrations of colchicine.
Second, microtubule formation in the presence of plinabulin does
not reach steady state at high drug concentrations, as indicated by
the absorbance values that increase linearly with time, whereas in
the presence of high concentrations of colchicine steady state is
readily reached. These data are suggestive of a reduction of the
pool of soluble, assembly-competent tubulin in the presence of
plinabulin. In contrast, colchicine binds nearly irreversibly and
induces a rate-limiting conformational alterations in tubulin that,
once incorporated into the lattice, inhibits further microtubule
polymerization at substoichiometric concentrations (12). Transmission electron microscopy was
performed to further validate these results. No microtubule
aggregates were identified in the presence of plinabulin 1.25 µM
(Fig. 1B) and 2.5 µM (data not
shown). The mean microtubule length was however potently reduced to
34% of the DMSO-treatment group at 1.25 µM plinabulin, whereas
colchicine 1.25 µM reduced the mean microtubule length to 63%
(Fig. 1C; Fig. S1). Next, how these results translated
in a cellular system was evaluated. The IC50 of
plinabulin for inhibiting mitosis in MCF-7 breast cancer cells was
17 nM and mitosis was halted at the prometaphase (Fig. S2), suggesting that tubulin bound by
plinabulin may not be assembly competent to be incorporated into
microtubules.
Plinabulin synergizes with
chemotherapy in KRAS-driven xenograft cancer models.
In consideration to the limited activity of
chemotherapy in various cancer types with constitutive KRAS
activity, whether the distinct mechanism of action of plinabulin
yielded antitumor activity in these types of cancer was evaluated.
In a panel of 8 human cell lines derived from pancreatic,
colorectal and renal cancer, plinabulin inhibited growth in the
nanomolar range in all cell lines, except for the pancreatic cancer
cell line HPAC (Fig. 2A). This led
the present study to also investigate the putative anticancer
effects of plinabulin in vivo in a panel of five KRAS
mutated xenograft models. In contrast to the in vitro
results, plinabulin monotherapy failed to inhibit the growth of
LoVo and HCT-15 colorectal cancer (Fig.
2B; Fig. S3A) and exerted no
more than a moderate effect on the growth of MDA-MB-231 breast
cancer alone (Fig. 2C). The growth of
the KRAS mutated DU-145 prostate cancer (Fig. 2D) and A549 lung adenocarcinoma
(Fig. S3B) models was not affected
by plinabulin monotreatment. However, in combination with standard
chemotherapeutic agents administered in these cancers, the additive
or synergistic effects of plinabulin were observed on the
inhibition of tumor growth, including combinations with the
topoisomerase inhibitor irinotecan and with the tubulin targeting
agents, paclitaxel and docetaxel (Fig.
2; Fig. S3). These data point
towards effects of plinabulin other than direct cytotoxicity and
therefore led to down-stream KRAS signaling being evaluated
next.
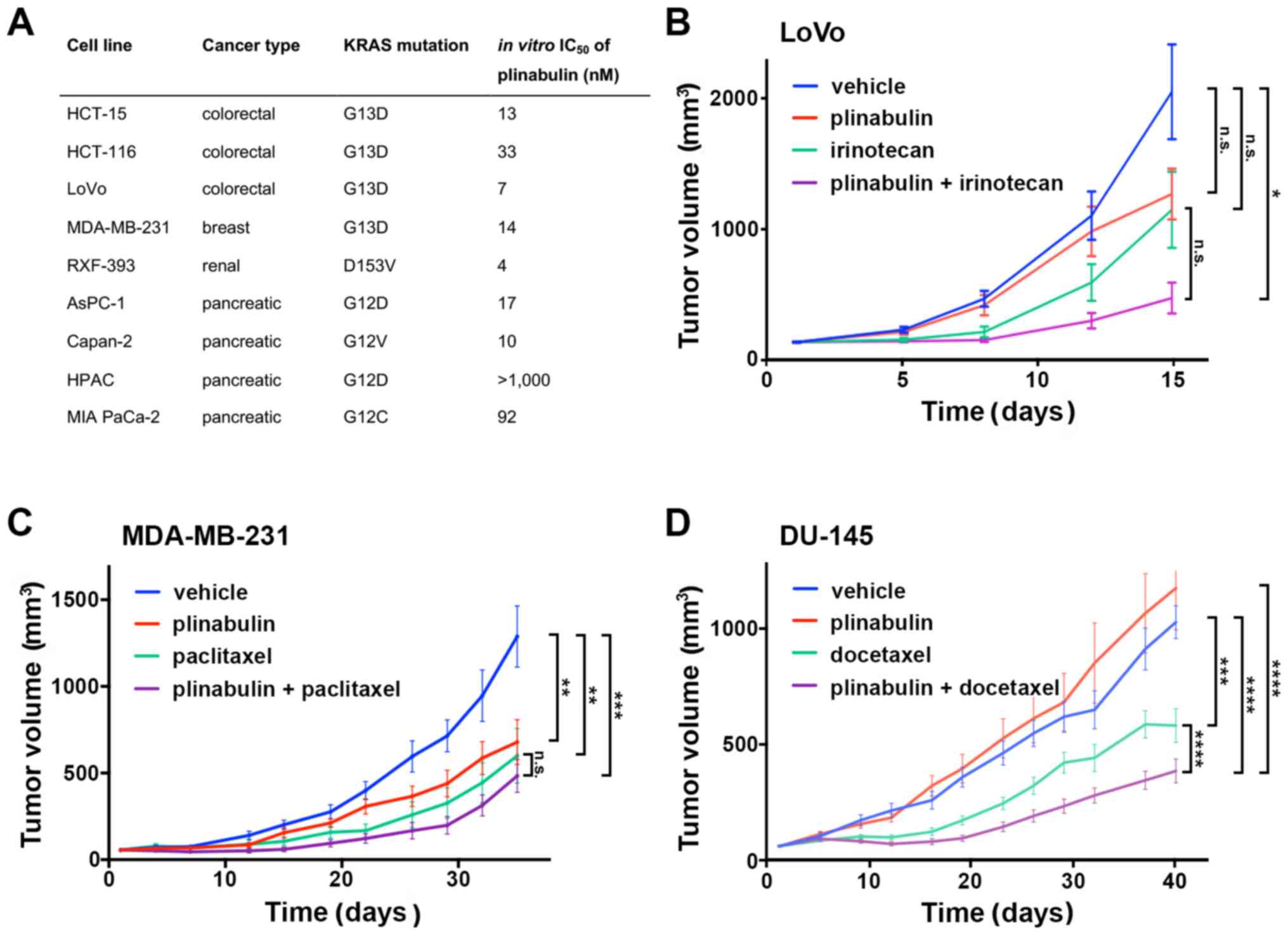 | Figure 2Plinabulin inhibits tumor growth in
KRAS mutated cancer models. (A) In vitro growth
inhibition of KRAS mutated cancer cell lines by plinabulin.
A total of 1,000 cells per well were seeded in 96-well plates in
triplicate overnight and treated in serum-free medium with
plinabulin at log2 increments of 1 nM-16.4 µM plinabulin
in 0.2% DMSO or DMSO 0.2% alone for 72 h prior to assessment of
adenosine triphosphate content. Human KRAS mutated (B) LoVo
colon cancer, (C) MDA-MB-231 breast cancer and (D) DU-145 prostate
cancer xenograft models were injected as indicated with 5% dextrose
solution (vehicle) i.p. or i.v., plinabulin 7.5 mg/kg i.p. on 5
subsequent days, (B) irinotecan 80 mg/kg i.p. once weekly for 3
weeks, (C) paclitaxel 16 mg/kg i.p. on 5 subsequent days, and (D)
docetaxel 12.5 mg/kg i.v. on days 1, 3 and 5. Tumor volumes were
measured twice weekly in N=10 mice per treatment group.
**P<0.001 and ***P<0.0001. i.v.,
intravenous; i.p., intraperitoneal; NS, not significant. |
Tubulin depolymerization yields
displacement of KRAS to endosomes.
In non-small cell lung and other types of cancer,
downstream effects of KRAS signaling include the activation of the
MEK/ERK and PI3K/Akt pathways (3).
Furthermore, ERK is regulated by a variety of converging pathways
that are activated under cellular stress including chemotherapy
with tubulin-targeting agents, whereas the key activator of Akt is
the KRAS target phosphoinositide 3-kinase (PI3K) (13). The proto-oncogene phosphatase and
tensin homolog (PTEN) is the key inhibitor of PI3K, therefore
rendering intact PTEN a pre-requisite for KRAS-dependency of cancer
cells (14). In PTEN proficient A549
lung cancer cells, disassembly of the microtubule network utilizing
plinabulin concentrations in the range of its IC50 for
in vitro tubulin depolymerization led to rapid accumulation
of KRAS in early endosomes (mean endosomal EEA1-colocalized KRAS in
DMSO vs. Plinabulin =2 vs. 31%, P<0.001, N=5) (Fig. 3A and B).
Furthermore, overall KRAS levels were decreased upon plinabulin
treatment, which may reflect lysosomal degradation, but this needs
to be investigated further. The two key down-stream effectors of
KRAS signaling, ERK and Akt, were affected differently by
plinabulin treatment and subsequent KRAS depletion: ERK
phosphorylation was increased, but epidermal growth factor
(EGF)-stimulated Ser473-phosphorylation of Akt was reduced
(Fig. 3C; 100 ng/ml EGF for indicated
times) (15). These findings indicate
that displacement of KRAS and potentially other tubulin-dependent
kinases may yield decreased downstream PI3K signaling, while
alternative signaling pathways act on the ERK signaling
cascade.
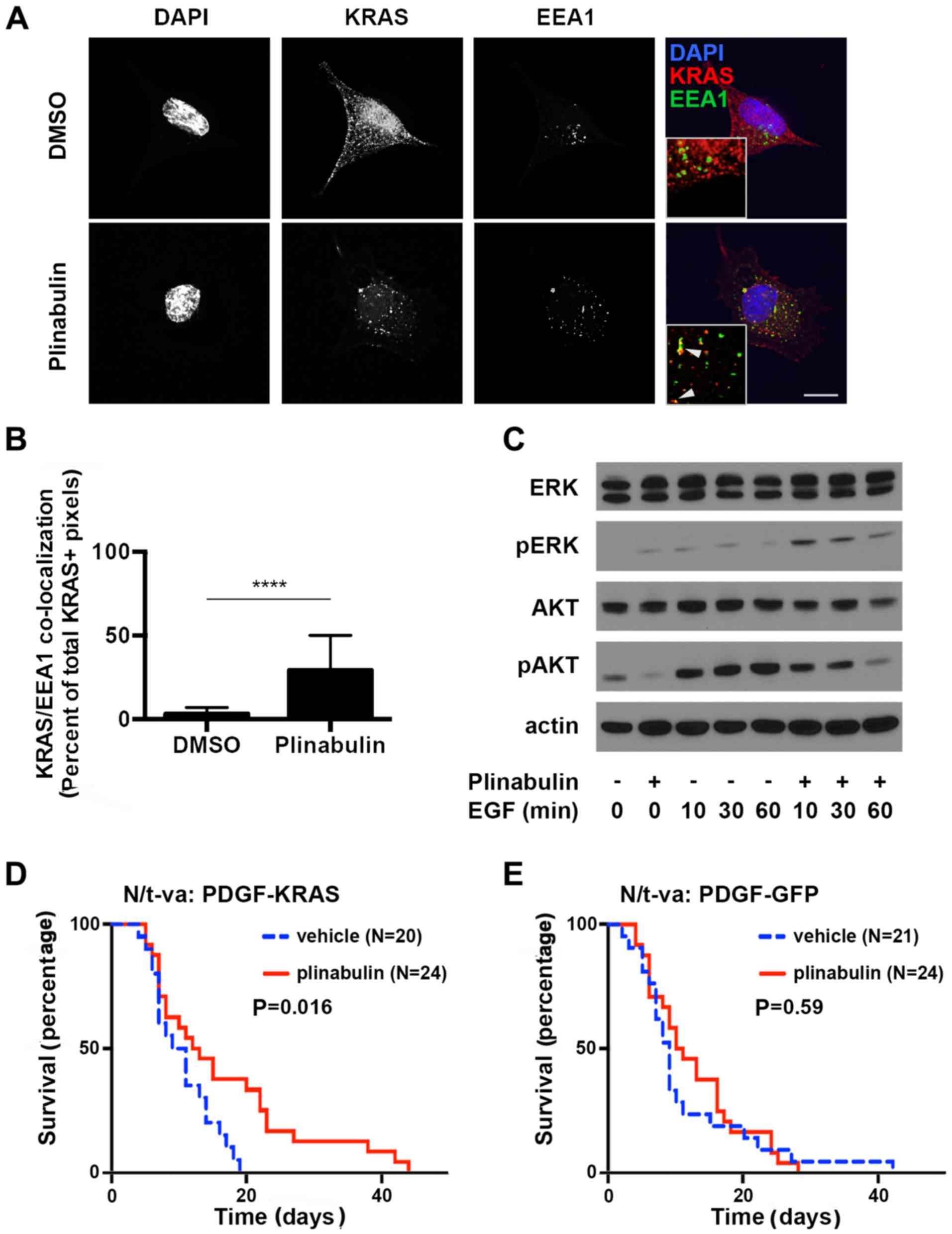 | Figure 3Plinabulin yields endosomal
accumulation of KRAS. (A) A549 cells were treated with 0.2% DMSO
(upper panel) or 1.25 µM plinabulin (lower panel) for 2 h prior to
immunofluorescence staining of DNA utilizing DAPI, (blue), EEA-1
(green) and KRAS (red). Scale bar, 10 µm. Inlays: 5x magnification;
arrowheads indicate co-localization of EEA-1 and KRAS. (B)
Quantification of the percentage EEA1/KRAS double-positive pixels
of total KRAS-positive pixels in five high power fields
(magnification, x100) from two independent experiments. (C)
Immunoblot analysis of A549 cells serum-starved overnight and
treated with 1% DMSO or 1.25 µM plinabulin for 2 h prior to
treatment with 100 ng/ml EGF for indicated times. Mouse gliomas
were generated in N/t-va;Ink4a/Arf-/- mice by
transduction with RCAS-PDGF and (D)
RCAS-KRASG12A, or (E) RCAS-GFP. Mice were treated
with 5% dextrose solution i.p. (vehicle) or plinabulin 7.5 mg/kg
i.p. for 5 subsequent days. Weight loss was utilized as an
indicator of tumor formation to determine the time-point of
treatment initialization.***P<0.0001. EGF, epithelial
growth factor; KRAS, Kirsten rat sarcoma viral oncogene homolog;
DAPI4, 6-Diamidino-2-phenylindole; ERK, extracellular signal
regulated kinase; GFP, green fluorescent protein; PDGF, platelet
derived growth factor; DMSO, dimethyl sulfoxide; EEA1, early
endosomal antigen 1; pAKT, phosphorylated protein kinase B. |
Plinabulin prolongs survival in a
RAS-driven glioma model.
Next, the specificity of plinabulin effects on KRAS
driven types of cancer in a simplified model system that was not
biased by co-mutations was determined. For this purpose, two
PTEN-proficient glioma gene transfer models that differed solely in
the absence or presence of constitutively active KRAS were
employed. Gliomas were generated in the brains of
Ink4a/Arf-deficient mice expressing the avian t-va receptor under
control of the nestin promoter (N/t-va:Ink4a/Arf-/-) to
allow gene transfer utilizing the avian retrovirus RCAS for
overexpression of PDGFB and GFP or KRASG12A.
Although activating KRAS mutations are rare in gliomas,
hyperactivation of RAS signaling is a key component of
gliomagenesis and histologically, these gliomas resemble the human
disease (16). Furthermore, given
that brain metastases are an important cause of cancer-associated
death (17-19),
the present study aimed to evaluate whether plinabulin was capable
of penetrating the blood brain barrier to a relevant extent.
Penetration of the blood-brain barrier by plinabulin is supported
by the observation that in a Quantitative Hole Body
Autoradioluminogrphy study, plinabulin rapidly entered the brain
parenchyma (Fig. S4). Treatment with
plinabulin prolonged survival in KRAS-expressing gliomas (Fig. 3D), but not in tumors that were solely
driven by PDGFB and Ink4a/Arf loss (Fig.
3E), therefore confirming penetration of the blood brain
barrier to a relevant extent and supporting the notion of efficacy
of plinabulin specifically in KRAS-mutated tumors.
Discussion
KRAS-driven cancer poses a major therapeutic
challenge. To date, precision medicine approaches have failed to
directly target KRAS (4) and standard
chemotherapy has only limited efficacy in KRAS-mutated cancer
(20). KRAS exerts its oncogenic
function primarily by activating the MEK/ERK and PI3K/Akt signaling
cascades, but dual inhibition of the two pathways is deemed too
toxic to be clinically applicable. However, targeting microtubules
has been suggested as a means to compromise endosomal recycling of
KRA (21) and combination of
docetaxel with the MEK-inhibitor selumetinib dramatically improved
the survival of patients with KRAS-driven non-small cell lung
cancer compared with docetaxel alone in an exploratory, randomized
phase II trial (22).
The authors' preclinical study supports the concept
of combining tubulin-targeting agents with inhibitors of the
MEK/ERK signaling axis: Upon plinabulin treatment, rapid
accumulation of KRAS in endosomal vesicles and a potent inhibitory
effect on Akt phosphorylation were observed, along with a
simultaneous increase in ERK activation that may compensate for Akt
inhibition. The underlying mechanism of this ERK activation likely
reflects a stress response through alternative kinases including
the stress-activated protein kinases (23).
Compensatory ERK activation may also explain the
lack of growth inhibition of most xenograft models tested here by
plinabulin monotherapy. Furthermore, effective targeting of KRAS
signaling through combined treatment with tubulin-targeting agents
and MEK/ERK inhibitors may require intact PTEN, as disinhibition of
PI3K signaling upon loss of PTEN activity renders Akt signaling
independent of KRAS.
Plinabulin may be well suited for such potentially
toxic combination treatments, given its favorable safety profile
and tolerability in clinical trials. Of note, tubulin
polymerization assays reported in the present study indicate that
plinabulin exerts its effects by a mechanism that is distinct and
potentially less toxic than that of colchicine and established
anticancer tubulin-targeting drugs: Plinabulin binds less avidly to
tubulin and its binding may be more readily reversible.
Furthermore, activity in a mutant KRAS-driven brain tumor model
underscores the favorable pharmacokinetics profile of plinabulin,
including crossing of the blood brain barrier.
In conclusion, the present preclinical study
supports the inclusion of tubulin-targeting agents in future
developments of combination treatments against KRAS-driven
cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by BeyondSpring
Pharmaceuticals Inc.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
HGW and GL designed the study. Data collection was
performed by LH, LD, YW, JB, JDH, KK, PG, JC, SN, MAJ, LW and HGW.
LH, LD, YW, JB, JDH, KK, PG, JC, SN, MAJ, LW, PJC and HGW analyzed
and interpreted data. PJC and HGW drafted and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All applicable international, national and/or
institutional guidelines for the care and use of animals were
followed. This article does not contain any studies with human
participants performed by any of the authors.
Patient consent for publication
Not applicable.
Competing interests
LH, LD, YW and GKL are employed by BeyondSpring
Pharmaceuticals Inc. JDH, KK, and PG are employed by Translational
Drug Development Inc.
References
|
1
|
Prior IA, Lewis PD and Mattos C: A
comprehensive survey of Ras mutations in cancer. Cancer Res.
72:2457–2467. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Fernández-Medarde A and Santos E: Ras in
cancer and developmental diseases. Genes Cancer. 2:344–358.
2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22.
2003.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Ostrem JM and Shokat KM: Direct
small-molecule inhibitors of KRAS: From structural insights to
mechanism-based design. Nat Rev Drug Discov. 15:771–785.
2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Schmick M, Vartak N, Papke B, Kovacevic M,
Truxius DC, Rossmannek L and Bastiaens PIH: KRas localizes to the
plasma membrane by spatial cycles of solubilization, trapping and
vesicular transport. Cell. 157:459–471. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Thissen JA, Gross JM, Subramanian K, Meyer
T and Casey PJ: Prenylation-dependent association of Ki-Ras with
microtubules. Evidence for a role in subcellular trafficking. J
Biol Chem. 272:30362–30370. 1997.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Dumontet C and Jordan MA:
Microtubule-binding agents: A dynamic field of cancer therapeutics.
Nat Rev Drug Discov. 9:790–803. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Wang Y, Zhang H, Gigant B, Yu Y, Wu Y,
Chen X, Lai Q, Yang Z, Chen Q and Yang J: Structures of a diverse
set of colchicine binding site inhibitors in complex with tubulin
provide a rationale for drug discovery. FEBS J. 283:102–111.
2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Prota AE, Danel F, Bachmann F, Bargsten K,
Buey RM, Pohlmann J, Reinelt S, Lane H and Steinmetz MO: The novel
microtubule-destabilizing drug BAL27862 binds to the colchicine
site of tubulin with distinct effects on microtubule organization.
J Mol Biol. 426:1848–1860. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Mita MM, Spear MA, Yee LK, Mita AC, Heath
EI, Papadopoulos KP, Federico KC, Reich SD, Romero O, Malburg L, et
al: Phase 1 first-in-human trial of the vascular disrupting agent
plinabulin (NPI-2358) in patients with solid tumors or lymphomas.
Clin Cancer Res. 16:5892–5899. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Millward M, Mainwaring P, Mita A, Federico
K, Lloyd GK, Reddinger N, Nawrocki S, Mita M and Spear MA: Phase 1
study of the novel vascular disrupting agent plinabulin (NPI-2358)
and docetaxel. Invest New Drugs. 30:1065–1073. 2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ravelli RB, Gigant B, Curmi PA, Jourdain
I, Lachkar S, Sobel A and Knossow M: Insight into tubulin
regulation from a complex with colchicine and a stathmin-like
domain. Nature. 428:198–202. 2004.PubMed/NCBI View Article : Google Scholar
|
|
13
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Singh A, Greninger P, Rhodes D, Koopman L,
Violette S, Bardeesy N and Settleman J: A gene expression signature
associated with ‘K-Ras addiction’ reveals regulators of EMT and
tumor cell survival. Cancer Cell. 15:489–500. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Pennock S, Billing S, Wang Z and Wang Y:
Two-pulse endosomal stimulation of receptor tyrosine kinases
induces cell proliferation. Methods Mol Biol. 1652:127–133.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Uhrbom L, Dai C, Celestino JC, Rosenblum
MK, Fuller GN and Holland EC: Ink4a-Arf loss cooperates with KRas
activation in astrocytes and neural progenitors to generate
glioblastomas of various morphologies depending on activated Akt.
Cancer Res. 62:5551–5558. 2002.PubMed/NCBI
|
|
17
|
Langer CJ and Mehta MP: Current management
of brain metastases, with a focus on systemic options. J Clin
Oncol. 23:6207–6219. 2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Khuntia D, Brown P, Li J and Mehta MP:
Whole-brain radiotherapy in the management of brain metastasis. J
Clin Oncol. 24:1295–1304. 2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Basseville A, Bates S and Fojo T:
Pancreatic cancer: Targeting KRAS and the vitamin D receptor via
microtubules. Nat Rev Clin Oncol. 12:442–444. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Jänne PA, Shaw AT, Pereira JR, Jeannin G,
Vansteenkiste J, Barrios C, Franke FA, Grinsted L, Zazulina V,
Smith P, et al: Selumetinib plus docetaxel for KRAS-mutant advanced
non-small-cell lung cancer: A randomised, multicentre,
placebo-controlled, phase 2 study. Lancet Oncol. 14:38–47.
2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sánchez I, Hughes RT, Mayer BJ, Yee K,
Woodgett JR, Avruch J, Kyriakis JM and Zon LI: Role of SAPK/ERK
kinase-1 in the stress-activated pathway regulating transcription
factor c-Jun. Nature. 372:794–798. 1994.PubMed/NCBI
|