Introduction
Albumin is the most abundant plasma protein (35-50
g/l human serum) with a molecular weight of 66.5 kDa. The functions
and binding properties of human serum albumin (HSA) are multifold
(1). HSA facilitates the transport
and disposition of various endogenous and exogenous components to
their specified targets. Albumin is emerging as a versatile protein
carrier for ligand targeting and also for peptide pharmacokinetic
profile improvement or for protein-based ligands (2). An X-ray crystallographic study reveal
that the heart shaped HSA consists of three structurally similar
domains (I, II and III), each of which contains two subdomains (A
and B) (3). It has been suggested
that the principal regions of the ligand which bind to HSA are
located in hydrophobic cavities in subdomains IIA and IIIA, which
respectively are designated as sites I and II (4,5). The
binding affinity offered by site I is mainly through hydrophobic
interactions, whilst site II involves a combination of hydrophobic,
hydrogen bonding and electrostatic interactions (6). The unique nature of the HSA ligand
binding properties reflect its multi domain organization, and it is
also one of the most important structure-function associations ever
reported for monomeric proteins (7).
HSA is known to be able to carry almost every small molecule; thus,
it may be a potential contender for being a cargo molecule/or
nano-vehicle for clinical, biophysical and industrial purposes
(8,9).
This protein serves an essential function as a transporter of
various unsaturated fatty acids including arachidonic acid (AA) and
hormones including L-thyroxin to the target sites (10). Polyunsaturated fatty acids (PUFAs) are
a group of fatty acids which contain more than one double bond in
their backbone. This class comprises a number of important
compounds, including essential fatty acids (11). AA is one of the most important
biological lipids which is present in most organic species, is a
physiologically significant omega-6 fatty acid and also the
precursor of prostaglandins and other active molecules (12). Human are able to easily metabolize
linoleic acid to form n-6 PUFA. It has been suggested that AA
functions as a protective agent against melanoma cancer and blood
coagulation (13). Furthermore, lipid
mediators generated from long-chain PUFA (AA in the n-6 series and
eicosapentaenoic acid and docosahexaenoic acid in the n-3 series)
have important functions in immune regulation and inflammation
(14,15). As mentioned above, HSA binds to a
large variety of ligands and delivers them to their target organs,
functioning as protein carriers. Thus, the present study aimed to
identify the effects of the physiochemical properties of AA on HSA
stability and structure. Their interaction was assessed based on
thermodynamic, structural and molecular dynamics (MD).
Materials and methods
Materials
AA was purchased from Sigma-Aldrich; Merck KGaA. The
stock solution was prepared in buffer
(KH2PO4=0.925 gr, KHPO4=0.825 gr,
169 ml with double-distilled water, pH=6.8). HSA was purchased from
Sigma-Aldrich; Merck KGaA. The HSA solutions were prepared 30 min
prior to the experiments. In the present study, 40 µM HSA and 10 µM
AA concentrations were used. For denaturation experiments, 6 M urea
was prepared.
HSA chemical and thermal
denaturation
To evaluate the effects of AA on protein chemical
stability, the HSA chemical denaturation profiles were recorded by
the titration of 40 µM protein solution with aliquots from a 6 M
urea stock solution. These experiments were performed in the
absence and presence of a 10 µM AA concentration. The protein
conformational changes were obtained at wavelength of 280 nm by
spectrophotometric technique.
Fluorescence quenching
experiments
HSA thermal denaturation fluorescence measurements
were performed using a spectrofluorometer (Cary Eclipse model 100,
equipped with a thermostatically controlled cuvette compartment).
Variable temperatures were utilized (10-90˚C) and the emission
spectra were recorded at 357 nm with excitation wavelength of 290
nm with an increase of 1 nm. The HSA (40 µM) intrinsic fluorescence
was evaluated using thermal scanning, in the absence and presence
of 10 µM AA.
Intrinsic fluorescence
The HSA intrinsic fluorescence evaluations were
performed in the presence and absence of AA via a
spectrofluorometer (Cary model Eclipse) using a 1 cm quartz cell
and a thermostat bath. A 40 µM HSA solution was prepared and
titrated with increasing concentrations of AA (0, 40, 80, 120, 160,
200, 240, 280, 320 and 360 µM) in 50 mM buffer solution with 298 K
temperature. An appropriate blank which corresponded to the buffer
was subtracted in order to correct for the background fluorescence.
The excitation wavelength was 290 nm, and the emission spectra were
recorded from 300 to 420 nm. The maximum emission intensities were
used to calculate the binding constants, binding sites occupation
and thermodynamic parameters evaluation (16). The quenching process is described by
the Stern-Volmer equation: F0/F = 1 +
KSV[Q] = 1 +
kqτ0[Q]. Where F0 and F are
the steady-state fluorescence intensities in the absence and in the
presence of quencher (AA), respectively, KSV the
Stern-Volmer quenching constant, and [Q] the concentration of the
quencher. kq is the quenching rate constant of the biomolecule,
τ0 is the average lifetime of the biomolecule without
quencher (17).
The number of binding sites and the binding constant
of the AA and HSA interaction were obtained by the following
equation: log[(F0-F)/F] = log(Kb) + n
log([Q]), where K is the binding constant for a site and n
is the number of bindings per albumin, respectively. The plot of
log [(F0-F)/F0] versus log [Q] yields
log(Kb) as the intercept and n as the slope. The
values for K and n were obtained from the intercept and the
slope (18). In order to map the HSA
and AA interactions, the thermodynamic parameters were calculated
using the Vant Hoff equation (19,20):
ln(Kb) = -(ΔΗ/RT) + (ΔS/R). Where the
Kb is the binding constant, R is the universal
gas constant (8.314 kJ/mol), ΔH and ΔS are enthalpy and entropy
changes during the quenching process. The free energy changes (ΔG)
associated with the interaction of AA and HSA were calculated from
the following equation: ΔG = ΔH - TΔS = -RT ln(K).
Lifetime measurements
Fluorescence lifetime measurements were performed
using an Edinburgh Instruments (FLS 920) spectrofluorimeter in
laser mode with excitation and emission wavelengths of 255 and 320
nm, respectively. The spectrofluorimeter in photon counting mode
equipped with a temperature-controlled cell connected to a
circulating water bath was used. The fluorescence lifetime of 40 µM
HSA in buffer phosphate (pH 7) was measured in the presence and
absence of 100-fold concentrations of 0, 20 and 80 µM AA. Using a
double-exponential decay function of the decay profiles with F900
analysis software, the lifetime values were determined from the
reconvolution fit analysis. Applying the reduced χ2
value, the goodness of fit was assessed (close to 1 in all
cases).
Furthermore, according to the following equation and
using the amplitude βi with the Euler angle in the
ith frame, component lifetime τFi, the mean fluorescence
lifetime (τF) was calculated by taking into account the amplitude
αi (21): τF =
Σi (βi
τFi2)/Σi (αi
τFi).
Circular dichroism (CD)
measurements
CD measurements were performed using a JASCO
spectropolarimeter (model J-800; JASCO Applied Sciences) equipped
with a thermoelectrically controlled cell holder under a constant
nitrogen flow. Cuvettes with path lengths of 1 mm were utilized.
Far-ultraviolet (UV)-CD spectra were recorded in the range of
190-260 nm. Near-UV-CD of the adjacent chain of aromatic amino
acids was absorbed in the range of 250-290 nm. A concentration of
40 µM HSA and AA concentrations of 0, 20, 80 and 100 µM were used.
The slit width was set at 5 nm, and the speed of scanning was 30
nm/min-1.
Fluorescence anisotropy
measurements
Fluorescence anisotropy is a phenomenon where the
light emitted by a fluorophore has unequal intensities along
different axes of polarization. The steady state anisotropy (r) is
given by:
where IVV and IVH are the intensities measured with
vertically polarized excitation, as indicated by the first
subscript, and detected through vertically or horizontally oriented
emission polarizers, respectively, as indicated by the second
subscript. The factor
which is measured using horizontally polarized
excitation, corrects for instrument polarization bias. Fluorescence
spectra and steady-state emission anisotropies of all HSA samples
were measured using a Cary Eclipse fluorimeter (Agilent
Technologies, Inc.). Anisotropy was measured with a manual
polarizer and the G factor correction was performed manually, with
a reference fluorophore. The labelled protein was then excited at
start (635 nm) and stop (640 nm) points. Furthermore, the parallel
and perpendicular emissions were monitored at maximum emission of
580 nm with a manual polarizer. The anisotropy measurement of the
protein was then repeated with AA (21,22).
Auto Dock technique (Visual MD)
To study the interactions between HSA and AA,
molecular docking was performed by the Auto Dock Vina 1.5.6
program). The docking process was performed to predict the
preferred orientation and binding affinity of the ligands. The HSA
crystal structure was obtained from a protein data bank (4L9Q PDB
code). The graphical images obtained represent the best pose which
were prepared using LigPlot v.4.5.3 software (23).
Protein-ligand docking
The AA molecular docking process to HSA was
performed using AutoDock Vina. The docking method in which receptor
side chains and the ligand are flexible was used for the
aforementioned process. To assess the permissible torsions for the
ligand, characterizing the search space coordinates and adding
polar hydrogen atoms to the protein, the graphical AutoDock tool
was applied (24). Following this,
the docking process of domain I and II was performed via a grid
size of 126 x 126 x 126 along the X, Y and Z axes with 1 Å spacing.
The lowermost binding energy for the docking conformation of
AA2-HSA and AA3-HSA complexes was created using AutoDock Vina,
which were assumed as the primary conformation for the MD
simulation process.
MD of AA and HSA
The conformational modifications of ligand-enzyme
complexes were estimated using a MD method via the GROMACS 4.5.4
(University of Groningen Royal Institute of Technology) package
software and the GROMOS96 43a1 force field. A periodic box full of
water molecules was designated for the complexes which was achieved
by the MD method (25). The HSA
topology characteristics were created using the aforementioned
GROMACS program. The AA topology parameters were created by means
of the Dundee PRODRG server (26).
The complex was immersed in a cubic box of extended simple point
charge water molecules. The steepest descent method of 10,000 steps
followed by the conjugation gradient method for 10,000 steps was
utilized to minimize the energy in order to release incompatible
contacts. The system equilibration phase constant substance amount,
pressure and temperature (NPT) and constant substance amount,
volume and temperature (NVT) as performed at 300 K for 200 psec
followed by MD production run for 20 nsec. As described in previous
studies (27,28), the atomic coordinates were registered
every 2.0 psec during the MD simulation process. In this section
parameters, including root mean square deviation (RMSD) and root
mean square fluctuation (RMSF), were measured.
Results
Chemical and thermal denaturation
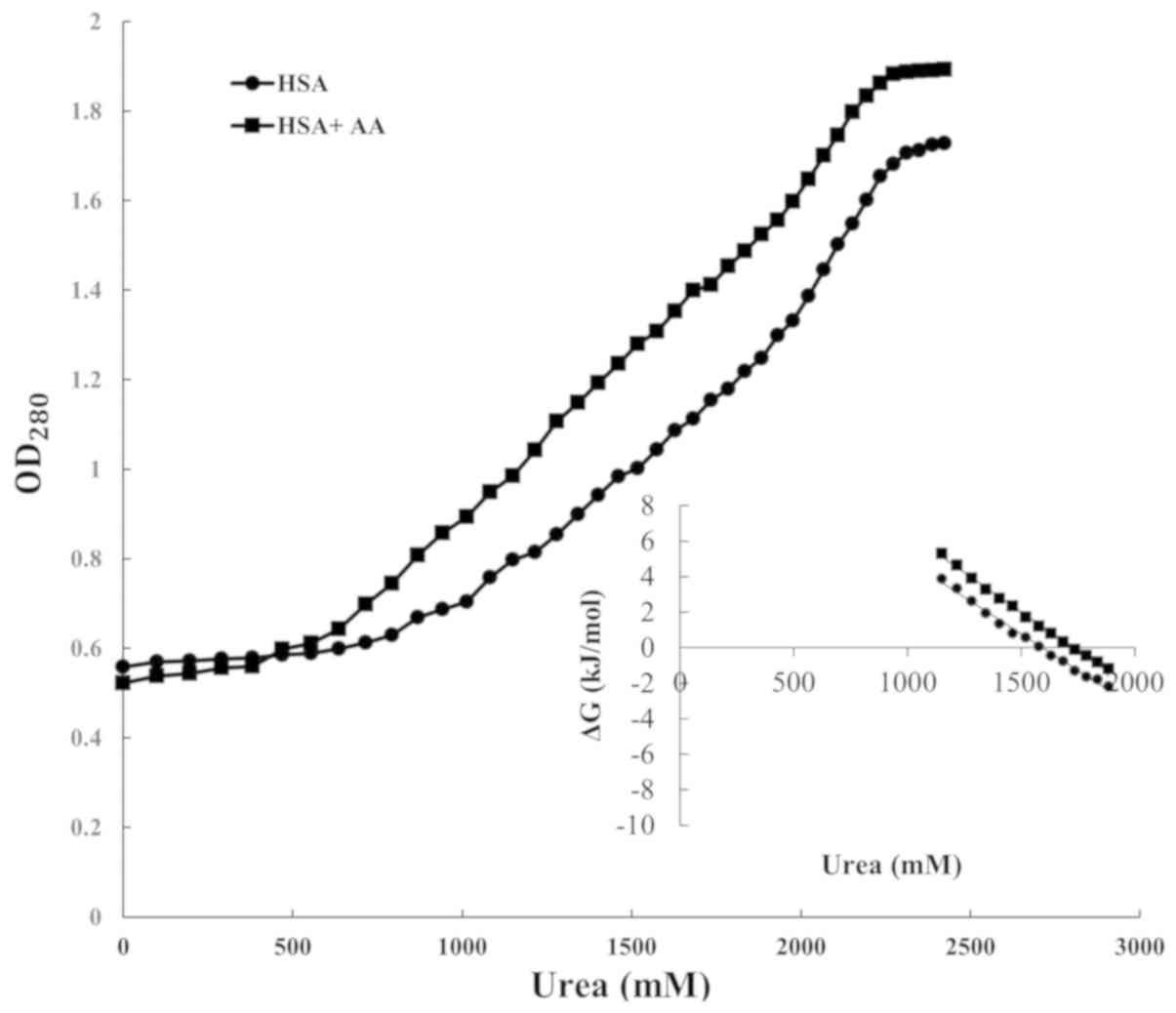
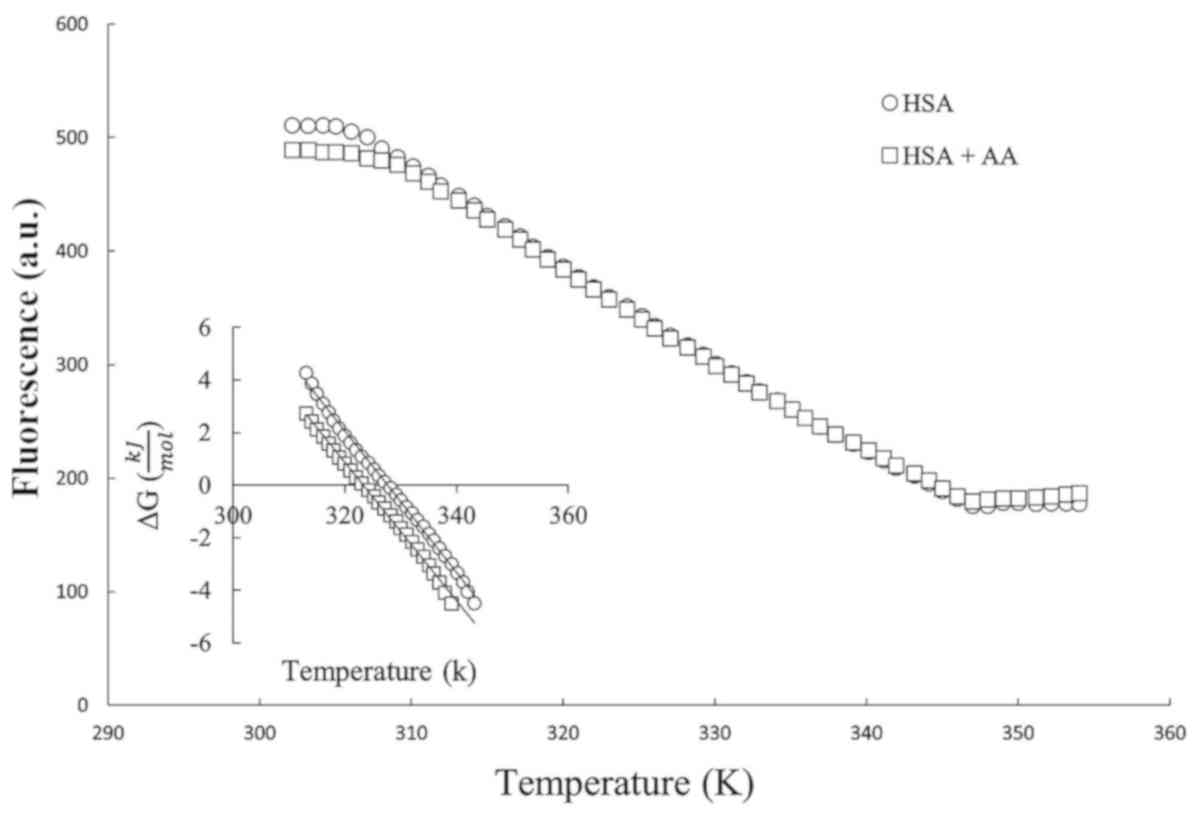
Chemical and thermal denaturation profiles were
obtained from urea titration and thermal scanning in the absence
and presence of AA in specific concentrations, which are depicted
in Figs. 1 and 2. Each profile is a sigmoidal curve; thus,
this process is described as a single denaturant-dependent step
based on the two-step theory. Determination of the standard Gibbs
free energy of denaturation (ΔG0), as a criterion of the
conformational stability of a globular protein, is based on two
state theory and the equation 1(29):
ΔG0 = -RT ln(K). The secondary plots of Figs. 1 and 2
are illustrated insets. From these linear plots, the ΔG0
varies linearly in a denaturation-dependent manner (urea
concentration and temperature) over a limited region. The equation
for determining the ΔG0 is as follows: ΔG0 =
ΔG0(H2O) - m[denaturant]. Where
ΔG0(H2O) is the free energy of conformational
stability in the absence of a denaturant, while m is the measure of
ΔG0 dependence in denaturant concentration.
In chemical denaturation,
[Urea]1/2 is the denaturant concentration
which the protein needs to reach to halve its two-state transition.
In thermal denaturation, the protein melting point (Tm)
is a temperature which the protein needs to reach to halve its
two-state transition. The magnitudes of
ΔG0(H2O), [Urea]1/2 and
Tm were determined from replots, and are summarized in
Table I.
 | Table IThermodynamic parameters obtained
from the chemical and thermal denaturation curves. |
Table I
Thermodynamic parameters obtained
from the chemical and thermal denaturation curves.
| | Chemical
denaturation | Thermal
denaturation |
|---|
| Sample | [Ligand]/2, M | ΔG(H2O),
kJ/mol | M, kJ/mol | Tm,
K | ΔG0(298
K), kJ/mol |
|---|
| HSA | 0.16 | 13.19 | 83 | 327.7 | 88 |
| HSA/AA | 0.13 | 9.6 | 72 | 323.4 | 85 |
Internal fluorescence experiments
Internal fluorescence spectroscopy is a method which
is widely used to investigate the microenvironment of amino acid
residues by measuring the emission wavelength shift (30). The HSA internal fluorescence spectra
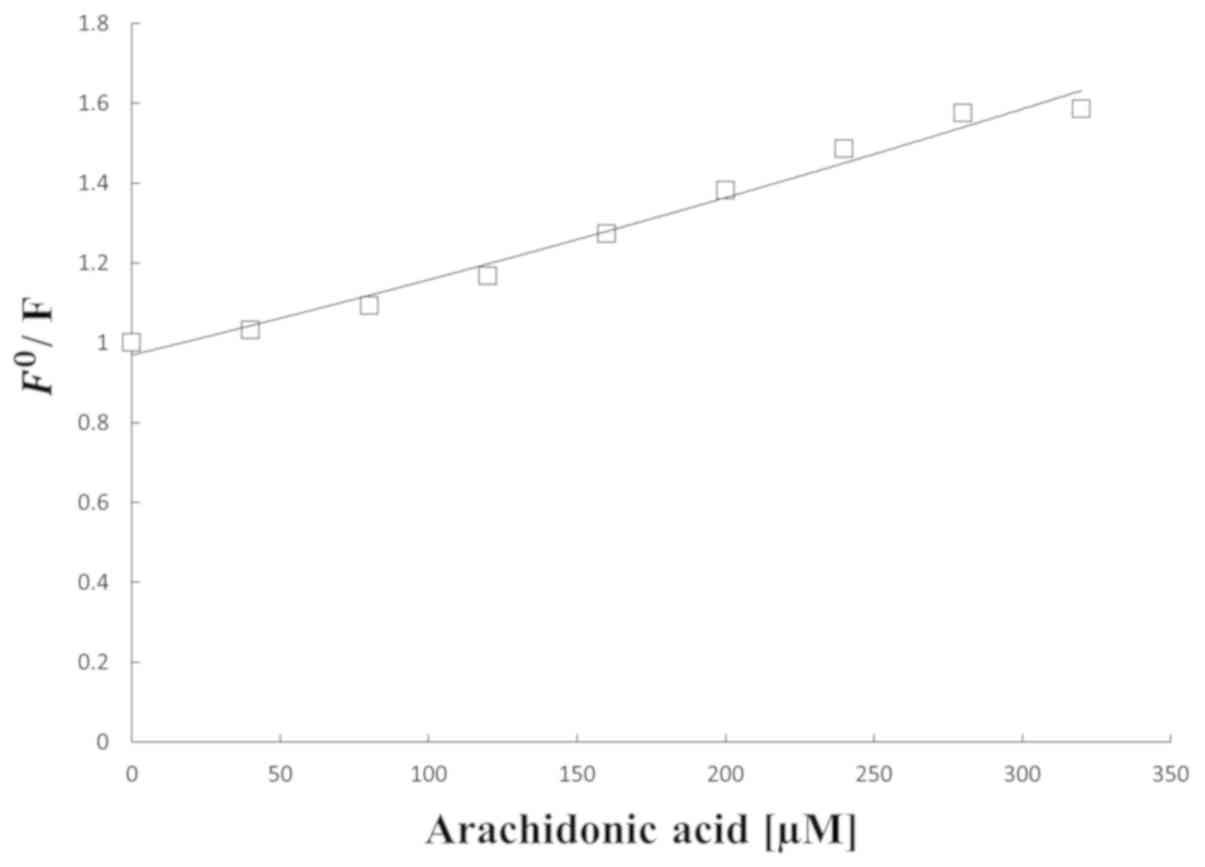
with various amounts of AA are presented in Fig. 3. The HSA fluorescence intensity
regularly decreased with the addition of AA. Furthermore, it was
demonstrated that fluorescence quenching occurred in the binding.
Additionally, it implies that the interaction between AA and HSA
may affect the Trp residue microenvironment. Thermodynamic
parameters of the binding reaction, obtained from the Stern-Volmer
curve, confirm the binding force. Therefore, the
temperature-dependent thermodynamic parameters were analyzed to
characterize the acting forces between AA and HSA (31). The results of the data sequential
analysis using the Vant Hoff equation are illustrated in Table II. These were confirmed parameters by
Fig. 4.
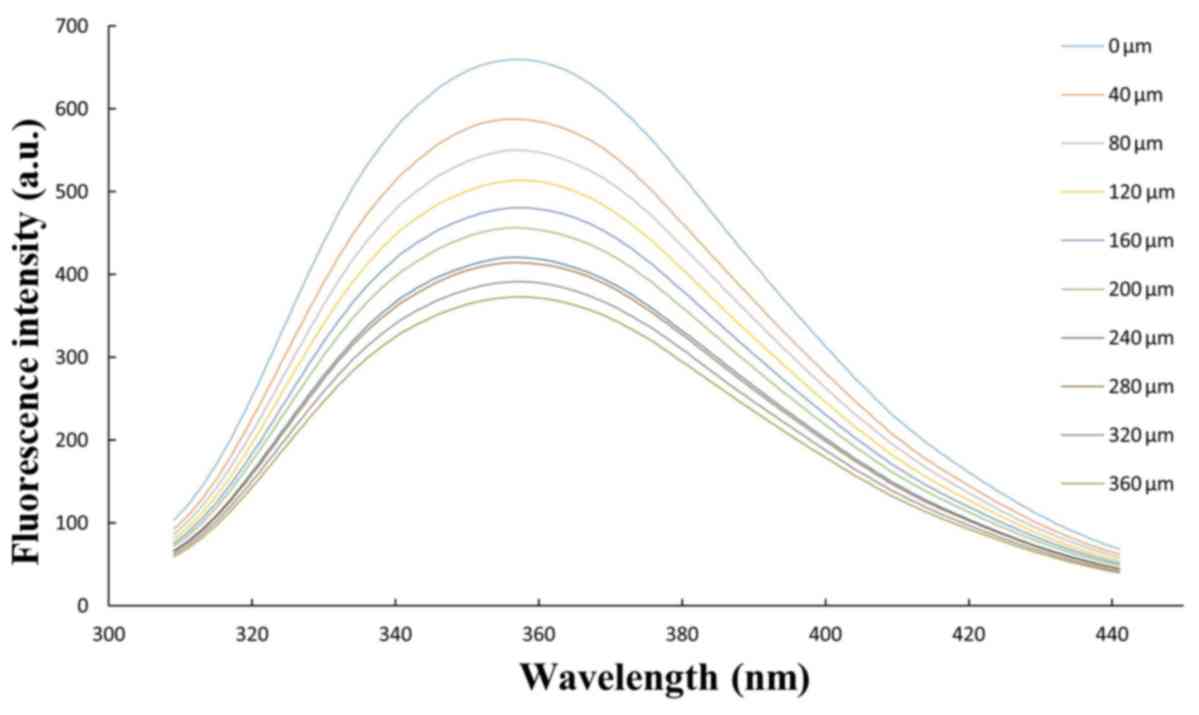 | Figure 3.Curve obtained from fluorescence
spectrum of HSA (T=300 K, excitation wavelength, 290 nm) in the
presence of a range of concentrations of arachidonic acid (0, 40,
80, 120, 160, 200, 240, 280, 320 and 360 µM), with HSA=40 µM. HSA,
human serum albumin. |
| Table IIBinding and thermodynamic parameters
obtained from the Stern-Volmer equation following the interaction
of HSA/AA. |
Table II
Binding and thermodynamic parameters
obtained from the Stern-Volmer equation following the interaction
of HSA/AA.
| Sample | T, K | n | R | KSV,
104l/mol | ∆G0,
kJ/mol | ∆H0,
kJ/mol | ∆S0,
J/(mol K) |
|---|
| HSA/AA | 298 | 2 | 0.98 | 5.3 | -31.13 | -22.66 | 28.42 |
Lifetime measurements
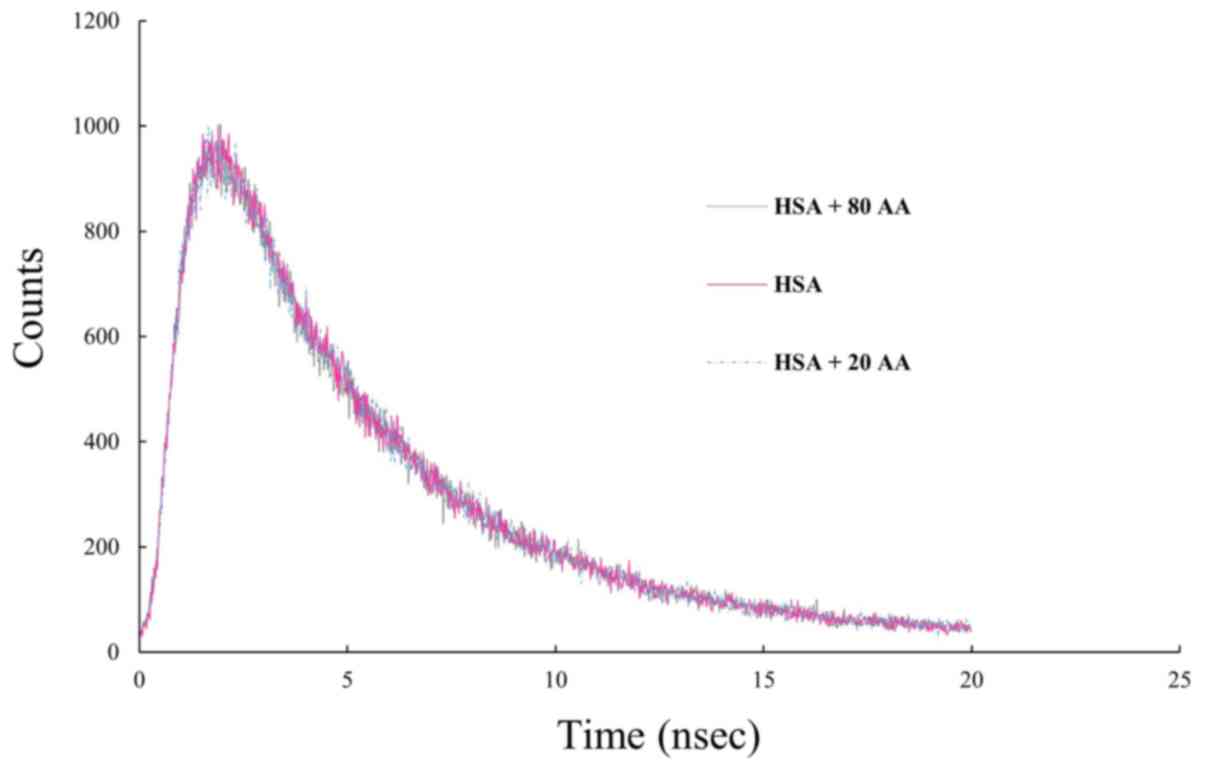
Based on the results of the fluorescence lifetime
measurements, adding 20 and 80 µM AA to the protein solution
resulted in no significant change in the fluorescence lifetime of
HSA (Fig. 5). Therefore, changing the
concentration AA over time does not change, which indicates that
the interaction is static.
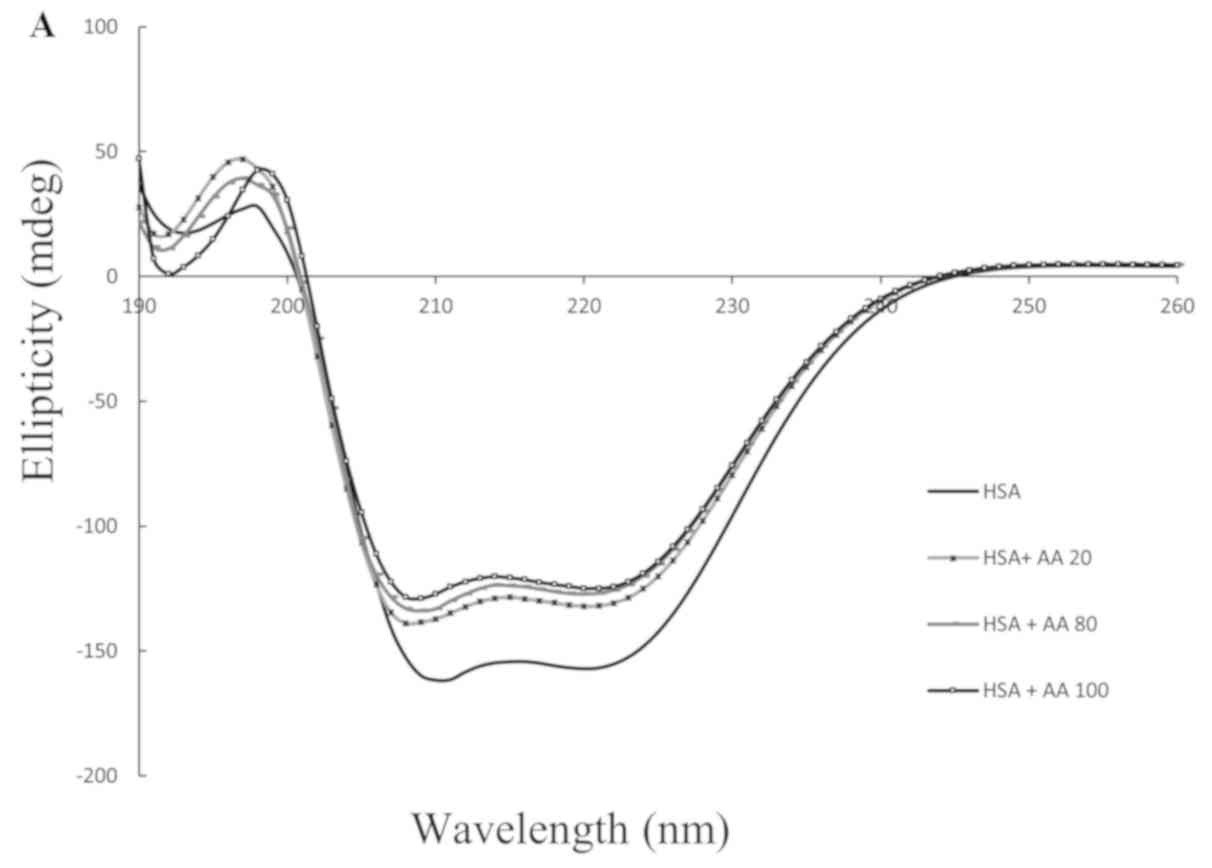
CD spectroscopic experiments
The far-UV-CD spectra of HSA exhibited two negative
bands in the 208 nm and 222 nm regions, which represent the β-sheet
and α-helical structure of the protein, respectively. The
near-UV-CD method obtained the HSA tertiary regular structure
values (32). When the AA
concentration was gradually increased from 20 to 100 µM, the CD
exhibited fundamental changes (Fig.
6A and B).
Anisotropy measurement
HSA was labeled with a fluorophore and the
anisotropy of the labeled protein was measured prior to and
following AA binding. The dye to protein ratio was low and the
anisotropy of unlabeled protein was 0.28. When AA was added to the
labeled protein, the anisotropy decreased to 0.26. Interactions
between HSA and AA and the third structure are not stable. This may
be attributed to conformational changes in HSA and the generation
of another binding site upon interaction with AA, which are in
agreement with our CD and anisotropy results (33).
Molecular docking and dynamics
To determine the interaction of AA with HSA specific
binding sites, a molecular docking process was performed. The main
result to note of the reaction is the binding position of AA with
HSA. One previous study demonstrated which knowledge about the
protein-ligand binding may be useful to understand the function and
effectiveness of AA and HSA as potential therapeutic agents
(34). The selective side-chain
residue flexibility is a great option which is available on
AutoDock Vina docking software (35).
The aim of the present study was to make the protein-ligand
interaction environment more pragmatic in order to decrease the
computer processing time. The conformational modification of the
receptor with regards to ligand selectivity demonstrate that
receptor flexibility may have an essential function in computer
drug design, which is evaluated in current study (36). To determine the structural changes,
which are induced by ligand binding (with AA), the MD simulation
for free HSA (subdomain II and III) and AA2-HSA/AA3-HSA complexes
was performed and the results were consequently compared. Based on
structural characteristics, the stability of the HSA protein was
able to be studied during the simulation time. The evolution time
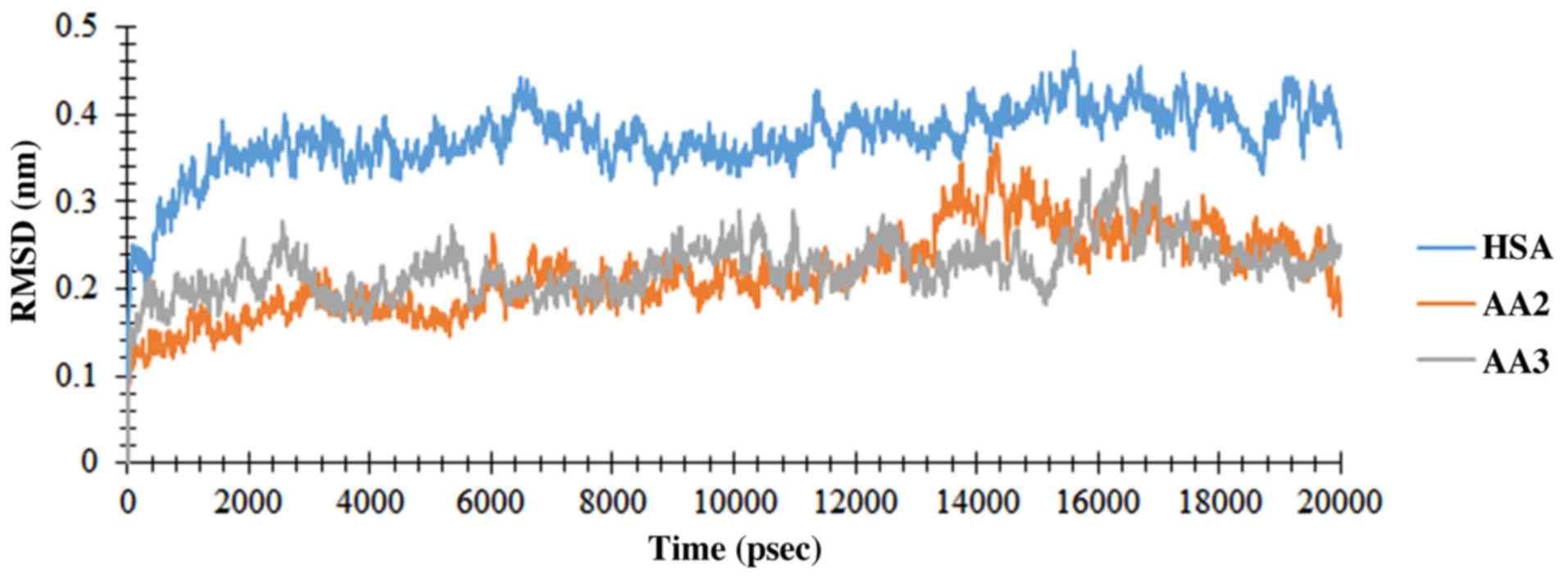
of RMSD from the preliminary structure was calculated for three
simulation runs. The RMSDs of Cα atoms, which are representative of
two complexes, are demonstrated in Fig.
7. The free protein and the protein involved in the complex
achieved the appropriate stability after 1 psec, which proved the
equilibration of the system. The RMSD values of the HSA Cα-atoms
and the complexes indicated a near mean equilibration and
oscillation value for the whole system with maintained stability up
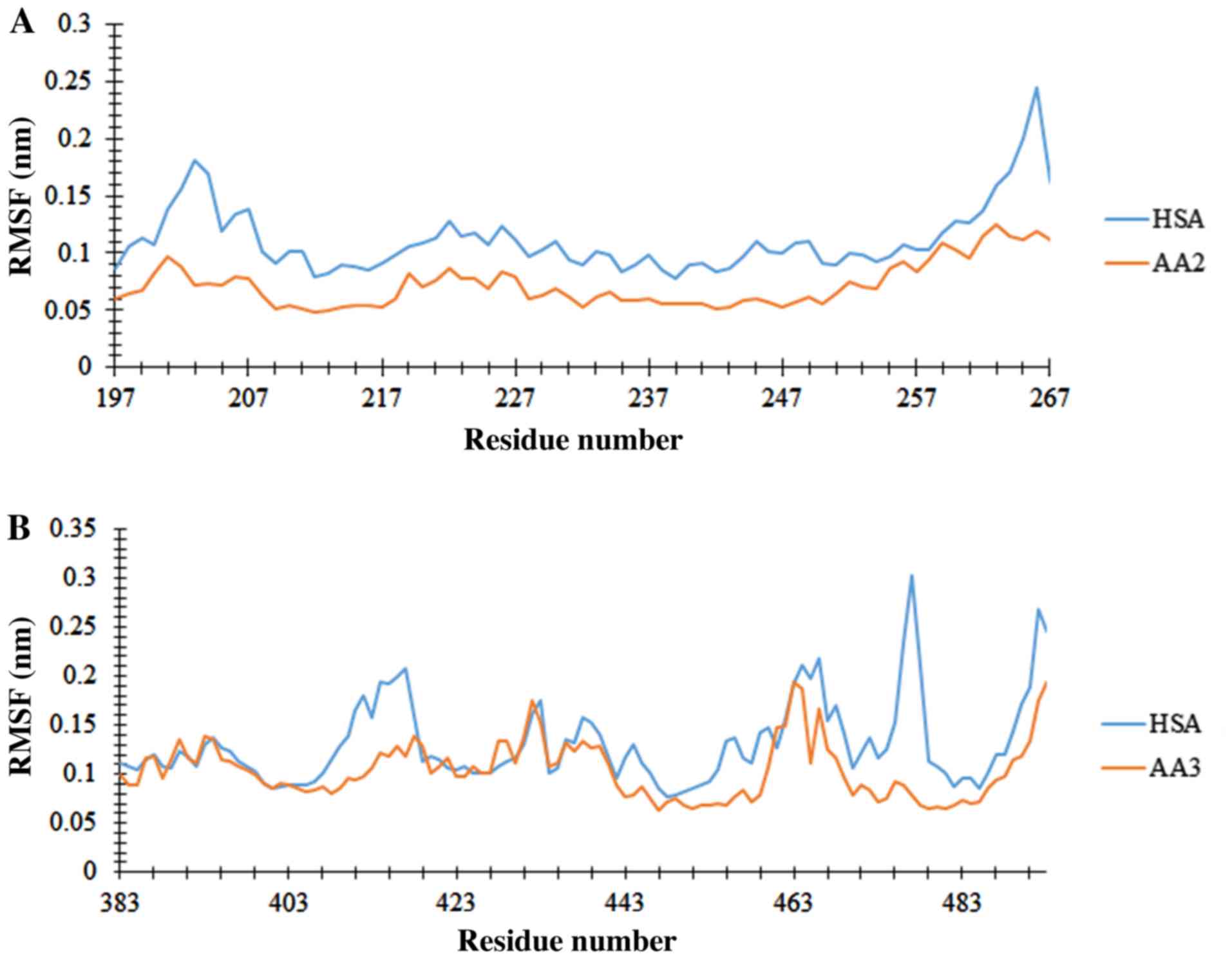
to the end of the simulation. To obtain the flexibility data, RMSF
was measured. To identify the flexible regions of the molecule,
RMSFs of Cα atoms were produced and are illustrated in Fig. 8. The RMSF value for the free protein
and the protein involved in the complex have similar trends as
illustrated in Fig. 8, while residues
262-267 and 474-480 in subdomain IIA and IIIA have the highest RMSF
values. Due to molecular movement restriction which was induced by
the ligand, the free protein demonstrates a higher RMSF value
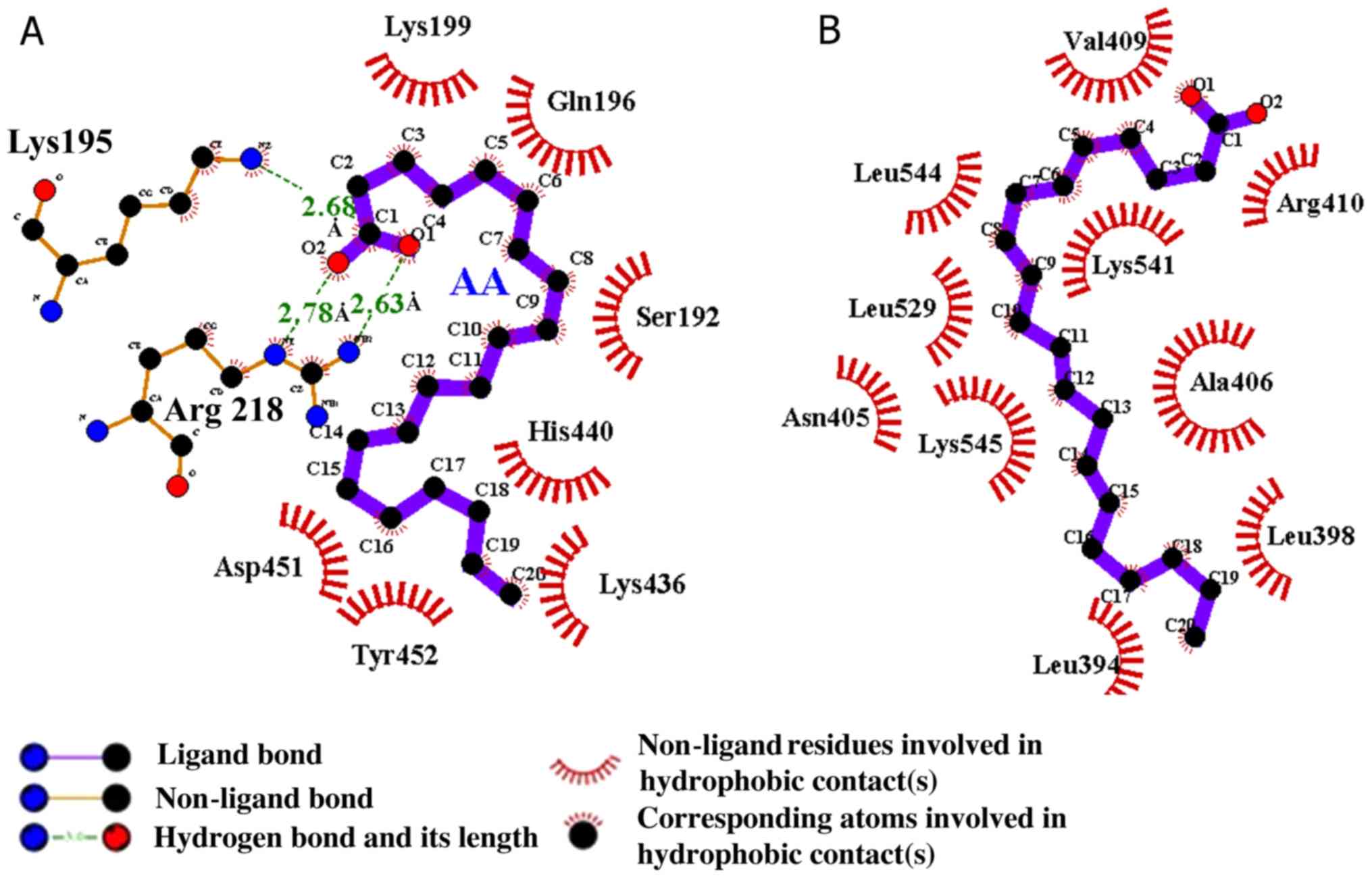
compared with the protein involved in the complex. Following MD
analysis, the AA-HSA complex in site I was formed by the hydrogen
bonds of Lys195 and Arg218 and hydrophobic interactions, as
indicated in Fig. 9A. Additionally,
the AA-HSA complex was created by the hydrophobic interaction in
site II, as illustrated in Fig. 9B.
The optimal probable conformation was obtained with AA and HSA
following docking and MD analysis. The molecular affinity of AA2
and AA3 with HSA was reported as -5.8 and -5.0 kcal/mol,
respectively. These results demonstrated which AA binds forcefully
to the large hydrophobic cavity available on HSA at subdomain ΙΙA
of site Ι. Studies have confirmed that IIA and IIIA subdomains are
suitable as target and initial drug binding sites for the docking
process. Each experiment which evaluates the ligand-binding site
next to drug-binding sites I/II may be useful in selecting the best
docking simulation position. To measure the structural
modifications caused by the ligand (AA) binding the MD simulation
process for free HSA and IIA/IIIA subdomains, experiments were
performed and the results consequently compared. At present, the
results demonstrated that AA has the ability to bind effectively to
the large hydrophobic cavity, which is available on HSA at
subdomain ΙΙA of site Ι. The HSA binding site was evaluated to
recognize the site containing residues.
Discussion
A large number of previous studies indicated that
IIA/IIIA subdomains may be regarded as target sites for docking due
to their early drug binding site potential. Appraising other
binding sites may be more useful for a suitable site selection for
the MD simulation process. To evaluate the structural
modifications, which are provided by ligand (AA) binding, in the
present study, the MD simulation for HSA and IIA/IIIA subdomains
was performed and the results compared (37,38).
Thermodynamic parameters, which were obtained from the chemical and
thermal denaturation plots of HSA and AA, revealed that the protein
Gibbs free energy and Tm decreased following incubation
with AA. This instability was proven by structural analysis from
fluorescence and CD studies. The effects of AA binding on HSA
stability was investigated by urea-induced denaturation using
optical density 280 measurements via spectrophotometry and its
thermal denaturation following protein excitation in 290 nm
wavelength via spectrofluorometer techniques (Figs. 1 and 2).
The Gibbs free energy of HSA
(ΔGH2O) in the absence and present
of AA was revealed to be 13.19 and 9.6 kJ/mol, respectively, as
determined by the linear fittings of intrinsic spectrophotometric
data. As presented in Table I, all
the thermodynamic parameters including
ΔGH2O, Cm,
ΔG298K and Tm which are achieved from
chemical and thermal denaturation, revealed a relative HSA
instability following its interaction with AA (39). In the chemical denaturation, the urea
interacts with HSA by electrostatic forces, yielding a randomly
coiled conformation in its unfolded state, while the thermal
denaturation produces a molten globule state and the protein
aggregation; therefore, each method yield different structurally
unfolded states of HSA. From the efonidipine interaction with BSA,
it may be seen that ΔH = 68.04 kJ/mol, ΔS = 319.42 kJ/mol and ΔG =
-27.08 kJ/mol (40). These positive
values of ΔH and ΔS are associated with the hydrophobic nature of
the ligand-protein interaction. The negative value of
ΔG0, which is obtained from Stern-Volmer data analysis
as presented in Table II, identifies
that the HSA-AA binding is a spontaneous process. The positive
ΔH0 and ΔS0 values for AA indicate that the
force acting between these compound and AA is mainly a hydrophobic
interaction. Thus, the HSA non-polar hydrophobic groups may be
responsible for the main effect determination of AA-protein binding
(41). As demonstrated in Table II, the KSV values
may be obtained from the modified linear Stern-Volmer equation
slope. Additionally, the KSV of the AA
interaction with HSA suggests that the overall quenching is
dominated by static quenching (42,43). This
additionally reflects the lifetime measurements, in that the
interaction between HSA and AA is of a static nature. Hence, AA may
strongly quench the intrinsic fluorescence of HSA by static
quenching (44). HSA structural
experiments, which were evaluated by CD spectra in absence and
presence of B12 and 4-thio-5-methyluridine, exhibited two negative
bands in the UV region at 209 and 228 nm, which are the protein
characteristics of a β-sheet and a-helical structures. The results
revealed that B12 and 4-thio-5-methyluridine had fundamental
effects on HSA secondary structure (45,46), and
the results are in agreement with the results of the present study
obtained by CD from a HSA regular secondary structure subsequent to
its interaction with different concentrations of AA (Fig. 6A). The near-UV-CD results of the
aforementioned previous study are consistent with our data which
was provided by CD from a regular tertiary structure of HSA
following its interaction with different concentrations of AA
(Fig. 6B). The docking and MD results
indicated the effective binding of AA to IIA/IIIA subdomains (site
I and II) in HSA.
To conclude, based on the structure-function
association experiments and the function of HSA as a carrier
protein, it is necessary to change the structure and accept AA as a
ligand. These changes may be explained by the nature of
non-covalent physical interactions, which are induced by the ligand
minor instability and flexibility.
Acknowledgements
Not applicable.
Funding
The authors acknowledged the facilitative support of
the educational department of Azad University of Science and
Research, the Qazvin University of Medical Sciences and the
Biochemistry-Biophysics Institute of Tehran University.
Availability of data and materials
All the datasets generated and analyzed during the
present study are included in the published manuscript.
Authors' contributions
FMV drafted the manuscript, and analyzed and
interpreted the data. AF and HS conducted the study and analyses,
and collected background information. NG participated in the design
of the present study and performed the statistical analysis. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kratz F: Albumin as a drug carrier: Design
of prodrugs, drug conjugates and nanoparticles. J Control Release.
132:171–183. 2008.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Sarkar D, Mahata A, Das P, Girigoswami A,
Ghosh D and Chattopadhyay N: Deciphering the perturbation of serum
albumins by a ketocyanine dye: A spectroscopic approach. J
Photochem Photobiol B. 96:136–143. 2009.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Chatterjee T, Pal A, Dey S, Chatterjee BK
and Chakrabarti P: Interaction of virstatin with human serum
albumin: Spectroscopic analysis and molecular modeling. PLoS One.
7(e37468)2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Elsadek B and Kratz F: Impact of albumin
on drug delivery--new applications on the horizon. J Control
Release. 157:4–28. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lhiaubet-Vallet V, Sarabia Z, Boscá F and
Miranda MA: Human serum albumin-mediated stereodifferentiation in
the triplet state behavior of (S)- and (R)-carprofen. J Am Chem
Soc. 126:9538–9539. 2004.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jiménez MC, Miranda MA and Vayá I: Triplet
excited States as chiral reporters for the binding of drugs to
transport proteins. J Am Chem Soc. 127:10134–10135. 2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Fasano M, Curry S, Terreno E, Galliano M,
Fanali G, Narciso P, Notari S and Ascenzi P: The extraordinary
ligand binding properties of human serum albumin. IUBMB Life.
57:787–796. 2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ahmed-Ouameur A, Diamantoglou S,
Sedaghat-Herati MR, Nafisi Sh, Carpentier R and Tajmir-Riahi HA:
The effects of drug complexation on the stability and conformation
of human serum albumin: Protein unfolding. Cell Biochem Biophys.
45:203–213. 2006.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Varshney A, Sen P, Ahmad E, Rehan M,
Subbarao N and Khan RH: Ligand binding strategies of human serum
albumin: How can the cargo be utilized? Chirality. 22:77–87.
2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Anand U and Mukherjee S: Binding,
unfolding and refolding dynamics of serum albumins. Biochim Biophys
Acta. 1830:5394–5404. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Petitpas I, Petersen CE, Ha C-E,
Bhattacharya AA, Zunszain PA, Ghuman J, Bhagavan NV and Curry S:
Structural basis of albumin-thyroxine interactions and familial
dysalbuminemic hyperthyroxinemia. Proc Natl Acad Sci USA.
100:6440–6445. 2003.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zajdel A, Wilczok A, Chodurek E, Gruchlik
A and Dzierzewicz Z: Polyunsaturated fatty acids inhibit melanoma
cell growth in vitro. Acta Pol Pharm. 70:365–369. 2013.PubMed/NCBI
|
|
13
|
Nelson GJ, Schmidt PC, Bartolini G, Kelley
DS and Kyle D: The effect of dietary arachidonic acid on platelet
function, platelet fatty acid composition, and blood coagulation in
humans. Lipids. 32:421–425. 1997.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang C, Li A, Gao S, Zhang X and Xiao H:
The TIP30 protein complex, arachidonic acid and coenzyme A are
required for vesicle membrane fusion. PLoS One.
6(e21233)2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Høstmark AT and Haug A: Percentage oleic
acid is inversely related to percentage arachidonic acid in total
lipids of rat serum. Lipids Health Dis. 12(40)2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Guo X-J, Sun X-D and Xu S-K: Spectroscopic
investigation of the interaction between riboflavin and bovine
serum albumin. J Mol Struct. 931:55–59. 2009. View Article : Google Scholar
|
|
17
|
Gao H, Lei L, Liu J, Kong Q, Chen X and Hu
Z: The study on the interaction between human serum albumin and a
new reagent with antitumour activity by spectrophotometric methods.
J Photochem Photobiol Chem. 167:213–221. 2004. View Article : Google Scholar
|
|
18
|
Ahmad E, Sen P and Khan RH: Structural
stability as a probe for molecular evolution of homologous albumins
studied by spectroscopy and bioinformatics. Cell Biochem Biophys.
61:313–325. 2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ghosh KS, Sahoo BK and Dasgupta S:
Spectrophotometric studies on the interaction between
(-)-epigallocatechin gallate and lysozyme. Chem Phys Lett.
452:193–197. 2008. View Article : Google Scholar
|
|
20
|
Ross PD and Subramanian S: Thermodynamics
of protein association reactions: Forces contributing to stability.
Biochemistry. 20:3096–3102. 1981.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Nigen M, Le Tilly V, Croguennec T,
Drouin-Kucma D and Bouhallab S: Molecular interaction between apo
or holo α-lactalbumin and lysozyme: Formation of heterodimers as
assessed by fluorescence measurements. Biochim Biophys Acta.
1794:709–715. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Rusinova E, Tretyachenko-Ladokhina V, Vele
OE, Senear DF and Alexander Ross JB: Alexa and Oregon Green dyes as
fluorescence anisotropy probes for measuring protein-protein and
protein-nucleic acid interactions. Anal Biochem. 308:18–25.
2002.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Laskowski RA and Swindells MB: LigPlot+:
multiple ligand-protein interaction diagrams for drug discovery. J
Chem Inf Model. 51:2778–2786. 2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Trott O and Olson AJ: AutoDock Vina:
Improving the speed and accuracy of docking with a new scoring
function, efficient optimization, and multithreading. J Comput
Chem. 31:455–461. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Morris GM, Goodsell DS, Halliday RS, Huey
R, Hart WE, Belew RK and Olson AJ: Automated docking using a
Lamarckian genetic algorithm and an empirical binding free energy
function. J Comput Chem. 19:1639–1662. 1998.
|
|
26
|
Lindahl E, Hess B and Van Der Spoel D:
GROMACS 3.0: A package for molecular simulation and trajectory
analysis. Mol Model Annu. 7:306–317. 2001. View Article : Google Scholar
|
|
27
|
Farasat A, Rahbarizadeh F, Hosseinzadeh G,
Sajjadi S, Kamali M and Keihan AH: Affinity enhancement of nanobody
binding to EGFR: In silico site-directed mutagenesis and molecular
dynamics simulation approaches. J Biomol Struct Dyn. 35:1710–1728.
2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hansson T, Oostenbrink C and van Gunsteren
W: Molecular dynamics simulations. Curr Opin Struct Biol.
12:190–196. 2002.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Schmittschmitt JP and Scholtz JM: The role
of protein stability, solubility, and net charge in amyloid fibril
formation. Protein Sci. 12:2374–2378. 2003.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ashoka S, Seetharamappa J, Kandagal P and
Shaikh S: Investigation of the interaction between trazodone
hydrochloride and bovine serum albumin. J Lumin. 121:179–186. 2006.
View Article : Google Scholar
|
|
31
|
Vekshin IL: Separation of the tyrosine and
tryptophan components of fluorescence using synchronous scanning
method. Biofizika. 41:1176–1179. 1996.(In Russian). PubMed/NCBI
|
|
32
|
Gokara M, Sudhamalla B, Amooru DG and
Subramanyam R: Molecular interaction studies of trimethoxy flavone
with human serum albumin. PLoS One. 5(e8834)2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhong D, Douhal A and Zewail AH:
Femtosecond studies of protein-ligand hydrophobic binding and
dynamics: Human serum albumin. Proc Natl Acad Sci USA.
97:14056–14061. 2000.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Gokara M, Narayana VV, Sadarangani V,
Chowdhury SR, Varkala S, Ramachary DB and Subramanyam R:
Unravelling the binding mechanism and protein stability of human
serum albumin while interacting with nefopam analogues: A
biophysical and insilico approach. J Biomol Struct Dyn.
35:2280–2292. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Abreu RM, Froufe HJ, Queiroz MJR and
Ferreira IC: Selective flexibility of side-chain residues improves
VEGFR-2 docking score using AutoDock Vina. Chem Biol Drug Des.
79:530–534. 2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Mohan V, Gibbs AC, Cummings MD, Jaeger EP
and DesJarlais RL: Docking: Successes and challenges. Curr Pharm
Des. 11:323–333. 2005.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Gou Y, Zhang Z, Li D, Zhao L, Cai M, Sun
Z, Li Y, Zhang Y, Khan H, Sun H, et al: HSA-based multi-target
combination therapy: Regulating drugs' release from HSA and
overcoming single drug resistance in a breast cancer model. Drug
Deliv. 25:321–329. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Shahabadi N, Fili SM and Kashanian S:
Human serum albumin interaction studies of a new copper (II)
complex containing ceftobiprole drug using molecular modeling and
multispectroscopic methods. J Coord Chem. 71:329–341. 2018.
View Article : Google Scholar
|
|
39
|
Shahabadi N, Bazvandi B and Taherpour A:
Synthesis, structural determination and HSA interaction studies of
a new water-soluble Cu (II) complex derived from 1,
10-phenanthroline and ranitidine drug. J Coord Chem. 70:3186–3198.
2017. View Article : Google Scholar
|
|
40
|
Wang N, Ye L, Zhao BQ and Yu JX:
Spectroscopic studies on the interaction of efonidipine with bovine
serum albumin. Braz J Med Biol Res. 41:589–595. 2008.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Lori C, Lantella A, Pasquo A, Alexander
LT, Knapp S, Chiaraluce R and Consalvi V: Effect of single amino
acid substitution observed in cancer on Pim-1 kinase thermodynamic
stability and structure. PLoS One. 8(e64824)2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Yuan T, Weljie AM and Vogel HJ: Tryptophan
fluorescence quenching by methionine and selenomethionine residues
of calmodulin: Orientation of peptide and protein binding.
Biochemistry. 37:3187–3195. 1998.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Li S, Huang K, Zhong M, Guo J, Wang WZ and
Zhu R: Comparative studies on the interaction of caffeic acid,
chlorogenic acid and ferulic acid with bovine serum albumin.
Spectrochim Acta A Mol Biomol Spectrosc. 77:680–686.
2010.PubMed/NCBI View Article : Google Scholar
|
|
44
|
OAbou-Zied OK and Al-Shihi OI:
Characterization of Subdomain IIA Binding Site of Human Serum
Albumin in its Native, Unfolded, and Refolded States Using Small
Molecular Probes. J Am Chem Soc. 130:10793–10801. 2008.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Hou HN, Qi ZD, Ouyang YW, Liao FL, Zhang Y
and Liu Y: Studies on interaction between Vitamin B12 and human
serum albumin. J Pharm Biomed Anal. 47:134–139. 2008.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhang HM, Chen TT, Zhou QH and Wang YQ:
Binding of caffeine, theophylline, and theobromine with human serum
albumin: A spectroscopic study. J Mol Struct. 938:221–228. 2009.
View Article : Google Scholar
|