Introduction
Characterization of novel biological activities of
chemical compounds is closely associated with identification of
potential drug prototypes. Phenanthridine derivatives are a group
of low-molecular-weight (179-600 Da) non-peptide organic compounds,
which are being actively studied, primarily due to their ability to
bind efficiently with DNA and RNA (intercalators) (1) and also used as dyes (2). Representative compounds of this group
have been shown to exhibit significant inhibitory activity against
parasitic Leishmania protists (3), as well as activity against MCF-7, PC3
and HeLa tumor cell lines (4).
Moreover, phenanthridine derivatives bind to specific target
proteins both in vitro and in vivo, such as Bcl-XL
(5), topoisomerase I (6) and poly (ADP-ribose) polymerases
(7), causing anti-tumor and
anti-apoptotic effects. Thus, phenanthridines present a class of
compounds with a wide (but not fully studied) spectrum of
biological and pharmacological properties.
The search for novel antiviral drug prototypes,
including compounds against HIV-1, is one of the top priorities of
pharmacological screening programs during the initial stages of
drug discovery. In the present study, it was hypothesized that
phenanthridine derivatives may exhibit biological activity against
HIV-1, since one of the derivatives,
2,3,6,8-tetrachlorophenanthridine, has been characterized as a
HIV-1 protease (HIVp) dimerization inhibitor, as shown in our
previous studies (8,9).
The aim of the present study was to assess the
antiviral activity of eight new phenanthridine derivatives with
homologous structures to 2,3,6,8-tetrachlorophenanthridine on HIVp.
This viral enzyme is an obligatory dimer serving a key role in the
HIV-1 life cycle, however a high rate of mutation during HIV-1
replication reduces the efficacy of chemotherapy (10-12).
Bioinformatics (structure-activity relationship
predictions and molecular docking simulations), surface plasmon
resonance (SPR) and enzyme inhibition assays were used to assess
the activities of the derivatives. At least one of the new
phenanthridine derivatives,
3,3,9,9-tetramethyl-3,4,9,10-tetrahydro-2H,8H-phenanthridine-1,7-dione
(compound 2a), showed different affinities for the monomeric and
dimeric forms of HIVp. Docking estimations with the lowest binding
energy corresponded to the positioning of compound 2a in two
regions of HIVp, the active site cavities and the flaps domain.
Thus, it was suggested that binding of compound 2a restricted
substrate access to the active site of HIVp due to the inhibitory
effect of this compound.
Materials and methods
Recombinant proteins and chemical compounds.
The purity of the recombinant HIVp (Baсhem Holding AG) was >95%
as determined by SDS-PAGE. A micrOTOF-Q II (Bruker Corporation)
mass-spectrometer was used as an additional quality control check
of the protein preparation according to standard protocols of
protein identification (data not shown) (13,14).
Chromogenic substrate VII, acetyl-pepstatin (a known competitive
inhibitor of HIVp) (15) and the
‘interfacial’ hexapeptide Palmitoyl-Thr-Val-Ser-Tyr-Glu-Leu were
obtained from Bachem Holding AG. A set of phenanthridine
derivatives (Table I) was obtained
from Asinex and Sigma-Aldrich; Merck KGaA. Protease inhibitors and
phenanthridine derivatives were dissolved in 100% DMSO. Stock
solutions were stored at -20˚C.
1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide HCl,
N-hydroxysuccinimide and 1M ethanolamine HCl (pH 8.5) were obtained
from GE Healthcare. Maleic acid was obtained from Sigma-Aldrich;
Merck KGaA.
 | Table IPhenanthridine derivatives and their
binding properties. |
Table I
Phenanthridine derivatives and their
binding properties.
| | | | | | | | | Predicted
IC50, µM/inhibition, % |
|---|
| Compound no. | Cat. no. | Source | Name | Formula | Mw, Da | Structural
formula | ADME prediction, Drug
likeness/medicinal chemistry | HIV-integrase | HIV-1 protease | HIV-1 reverse
transcriptase |
|---|
| 1a | BAS 01025496 | ASINEX | 6-morpholino
4-yl-phenanthridine |
C17H16N2O | 264 |  | Lipinski Yes Ghose
Yes Veber Yes Egan Yes Muegge Yes/PAINS 0 alert Brenk 1 alert | 95/47 | 175/53 | 18/41 |
| 2a | BAS 13558724 | ASINEX |
3,3,9,9-tetramethyl-3,4,9,10-tetrahydro-2H,
8H-phenanthridine-1,7 dione |
C17H21NO2 | 271 |  | Lipinski Yes Ghose
Yes Veber Yes Egan Yes Muegge Yes/PAINS 0 alert Brenk 0 alert | 376/44 | 86/52 | 82/53 |
| 3a | BAS 17325280 | ASINEX | 3-methyl-
7.8,9,10,-tetrahydro- phenanthridine-6-thiol |
C14H15NS | 229 |  | Lipinski Yes Ghose
Yes Veber Yes Egan Yes Muegge Yes PAINS 0 alert Brenk 1 alert | 67/53 | 6/50 | 28/39 |
| 4a | S292982 | Sigma | 6-chloro-benzo(J)
phenenthridine | C17H10ClN | 263 |  | Lipinski No Ghose
Yes Veber Yes Egan Yes Muegge No/PAINS 0 alert Brenk 3 alert | 11/56 | 195/39 | - |
| 5a | S771163 | Sigma | 2-(2-nitro-
6-phenanthridinilamino)- 1-butanol | C17H17N3O3 | 311 |  | Lipinski Yes Ghose
Yes Veber Yes Egan Yes Muegge Yes/PAINS 0 alert Brenk 2 alert | 84/44 | 113/31 | 19/32 |
| 6a | R247324 | Sigma | 5-methyl-
phenanthridinium, iodide | C14H12IN | 321 |  | Lipinski Yes Ghose
Yes Veber Yes Egan Yes Muegge No/PAINS 1 alert Brenk 3 alert | - | 191/41 | 33/33 |
| 7a | S811238 | Sigma | 2-bromo-
6-methylphenanthridine nitrate | C14H11BrN | 336 |  | Lipinski Yes Ghose
Yes Veber Yes Egan Yes Muegge Yes/PAINS 0 alert Brenk 2 alert | 86/50 | 142/47 | - |
| 8a | 262692 | Sigma | Phenanthridine | C13H9N | 179 |  | Lipinski Yes Ghose
Yes Veber Yes Egan Yes Muegge No/PAINS 0 alert Brenk 1 alert | - | 234/41 | 33/31 |
Absorption, distribution, metabolism
and excretion (ADME) and activity prediction for compounds
For prediction of ADME properties, the online
resource SwissADME (swissadme.ch)
(16) was used. Prediction of the
activity spectrum for the phenanthridine derivatives against key
HIV-1 enzymes (integrase, protease and reverse transcriptase) was
performed using HIVprotI (bioinfo.imtech.res.in/manojk/hivproti/) (17).
SPR
Biacore 3000 and 8K biosensors (GE Healthcare) were
used for registration of intermolecular interactions. HIVp protein
was covalently immobilized on the surface of a standard CM5 sensor
chip (GE Healthcare) in sodium maleate buffer (pH 6.0) using a
standard amine coupling protocol according to the manufacturer's
protocol. The procedures for obtaining monomeric and dimeric forms
of HIVp on the optical chip were performed as described previously
(8,9). Sodium acetate buffer (0.1 M, pH 5.0)
containing 1 mM EDTA and 3% DMSO (v/v) was used as the running
buffer. A biosensor channel without immobilized HIVp protein was
used as a reference channel for subtraction of non-specific binding
to the chip surface. The biosensor signal was recorded in resonance
units (RU); one RU approximately corresponded to the binding of 1
pg material per 1 mm2 chip surface area. Sensograms of
association and dissociation of compound/HIVp complexes were
processed separately with the calculation of association
(kon)and dissociation rate (koff) constant
values using BIAevaluation version 4.1 (GE Healthcare). Apparent
dissociation constant values (Kd) were calculated as the
koff/kon ratio.
HIVp enzyme inhibition assay
To measure the enzymatic activity of HIVp,
chromogenic substrate VII, with a maximum absorption spectra in the
UV region (wavelength 300 nm), was used. All measurements were
performed at 25˚C as described previously (8,9).
Molecular docking simulations
DockPrep tools of UCSF Chimera version 1.14
(18-20)
were used to prepare the PDB files of the crystallographic
structure of HIVp 1Z1R (21) and
phenanthridine derivatives for docking. Hydrogens as well as
charges were added and water molecules were removed. Minimization
procedure consisted of 300 steps with the steepest descent and
conjugate gradient algorithms used (22). Molecular docking was performed using
iGEMDOCK version 2.1(23) with
stable (slow) docking default settings (population size, 300;
generations, 80; number of solutions, 10).
Results
Activity prediction for compounds and
molecular docking simulations
The antiviral activity for the set of phenanthridine
derivatives was determined for three main HIV-1 enzymes: Integrase,
HIV-1 protease and reverse transcriptase. IC50 values
and percent inhibition values were predicted for all compounds
using HIVprotI (Table I). The
phenanthridine derivatives were predicted to exhibit activity
against HIV-1p (IC50 value range, 6-234 µM with 31-53%
inhibition), as well as against the HIV-1 integrase and reverse
transcriptase, although to a lesser extent.
Further molecular docking analysis of the
phenanthridine derivatives over the entire surface of both
monomeric and dimeric form of HIVp was performed using iGEMDOCK.
Docking of HIVp dimerization inhibitor
2,3,6,8-tetrachlorophenanthridine (8,9) was used
as reference for analysis of binding to the HIVp monomer/monomer
interface.
Together, the molecular simulations indicated that
all phenanthridine derivatives tested docked in both the dimeric
and monomeric forms of HIVp (Table
II). Amino acid residues Ile50 of the dimeric, as
well as Val32, Ile47, Gly48,
Gly49, Ile84 and Ile54 of the
monomeric form of HIVp were involved in van der Waals interactions
with the phenanthridine derivatives (Table II). Subsequently, experimental
validation of activities and binding models, predicted using
bioinformatics tools, was performed using SPR and enzyme activity
assays.
| Table IIDocking array of predicted HIVp amino
acid residues interacting with phenanthridine derivatives. |
Table II
Docking array of predicted HIVp amino
acid residues interacting with phenanthridine derivatives.
| HIV protease | Compound no. | Docking
posesa | Energy | Ile3cb | Leu24 | Thr25 | Ala28 | Val32 | Ile47 | Gly48 | Gly49 | Ile50 | Ile54 | Ile84 | Leu90 | Ile93 | Thr96 |
|---|
| Monomeric form | 5ac | 5 | -89.9 | -7.1 | -5.7 | -10.7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | -9.2 | -9.6 | -12.7 |
| 1a | 0 | -89.9 | 0 | 0 | 0 | -4.5 | -13.5 | -6.8 | -5.7 | -3.8 | 0 | -4.5 | -15.9 | 0 | 0 | 0 |
| 4ac | 3 | -85 | 0 | 0 | 0 | -3.3 | -11.7 | -10.3 | -5.8 | -3.2 | 0 | -0.6 | -14.8 | 0 | 0 | 0 |
| 2ac | 7 | -80 | 0 | 0 | 0 | -4.8 | -13.2 | -3.8 | -2.8 | -5 | 0 | -6.4 | -10.8 | 0 | 0 | 0 |
| 3a | 3 | -73.2 | 0 | 0 | 0 | -3.7 | -12.1 | -7.8 | -7.2 | -5.9 | 0 | -6.1 | -5.1 | 0 | 0 | 0 |
| 7a | 4 | -69.8 | 0 | 0 | 0 | -3.1 | -14.2 | -9.9 | -4.6 | -3.8 | 0 | -4.3 | -10.1 | 0 | 0 | 0 |
| 6a | 9 | -64.8 | 0 | 0 | 0 | -4 | -13.2 | -9.2 | -6.8 | -5 | 0 | -4 | -7.3 | 0 | 0 | 0 |
| 8a | 0 | -47.6 | 0 | 0 | 0 | -3.6 | -13.8 | -9.1 | -6.5 | -4.6 | 0 | -4.2 | -6.3 | 0 | 0 | 0 |
| Dimeric form | 1a | 7 | -31.3 | 0 | 0 | 0 | -6.7 | -4.3 | -5.9 | -6.2 | -7.2 | -7.4 | 0 | -1.6 | 0 | 0 | 0 |
| 3a | 3 | -28.9 | 0 | 0 | 0 | -5.5 | -4 | -4.4 | -4.8 | -6.5 | -5.1 | 0 | -1.6 | 0 | 0 | 0 |
| 2ac | 5 | -31.3 | 0 | 0 | 0 | 0 | 0 | 0 | -0.4 | -2.7 | -14.4 | 0 | -3.7 | 0 | 0 | 0 |
| 5ac | 0 | -28.9 | 0 | 0 | 0 | -0.7 | 0 | 0 | -0.3 | -3.3 | -19.4 | 0 | -1.2 | 0 | 0 | 0 |
| 4ac | 7 | -27.5 | 0 | 0 | 0 | 0 | 0 | 0 | -0.2 | -2.4 | -11.9 | 0 | -2.1 | 0 | 0 | 0 |
| 7a | 5 | -13.5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | -3.2 | 0 | -1.5 | 0 | 0 | 0 |
| 6a | 3 | -12.7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | -4.7 | 0 | -1.4 | 0 | 0 | 0 |
| 8a | 3 | -8.3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | -5.8 | 0 | -0.1 | 0 | 0 | 0 |
SPR
An optical biosensor test system, consisting of the
immobilized monomeric or dimeric form of HIVp in the different flow
cells (8,9), was used to screen the binding of
phenanthridine derivatives at a concentration of 100 µM. Only 3
compounds, compounds 2a, 4a and 5a, showed positive binding signals
with HIVp monomer and dimer exceeding 10 RU (cut-off level with
correction for the baseline drift upon analyte injection).
Furthermore, each of these compounds were injected at different
concentrations to calculate the Kd values of the
compound/HIVp complexes (Table
III). SPR binding sensograms of compound 2a, which formed the
most affine complexes with HIVp, are shown in Fig. S1.
| Table IIISummary of molecular docking
simulations, surface plasmon resonance analysis and HIV protease
enzyme inhibition assay for phenanthridine derivatives. |
Table III
Summary of molecular docking
simulations, surface plasmon resonance analysis and HIV protease
enzyme inhibition assay for phenanthridine derivatives.
| Parameters | HIVp | 1a | 2a | 3a | 4a | 5a | 6a | 7a | 8a |
|---|
| Surface plasmon
resonance Kd, µM | Dimer | No | ~7 | No | ~1,000 | ~500 | No | No | No |
| | Monomer | No | ~2 | No | ~100 | ~300 | No | No | No |
| Enzyme inhibition
assay IC50, µM | | No | ~36 | No | ~45 | No | No | No | No |
| Binding regions of
HIVpa | Dimer | II | II | II | II | I | II | II | II |
| | Monomer | I, II | I, II | I, II | I, II | III | I, II | I, II | I, II |
Inhibition of HIVp activity by the
phenanthridine derivatives
HIVp enzymatic activity was sensitive to the known
inhibitors acetyl-pepstatin and the ‘interfacial’ hexapeptide. HIVp
activity decreased by 60 and 35% compared to control (without any
inhibitor) (data not shown), respectively, which indicated that
biochemical testing provided relevant results. Among the eight
phenanthridine derivatives tested, only compounds 2a and 4a
demonstrated HIVp inhibitory activity (Table III). Fig. S2 shows HIVp activity data in the
presence of different concentrations of compound 2a.
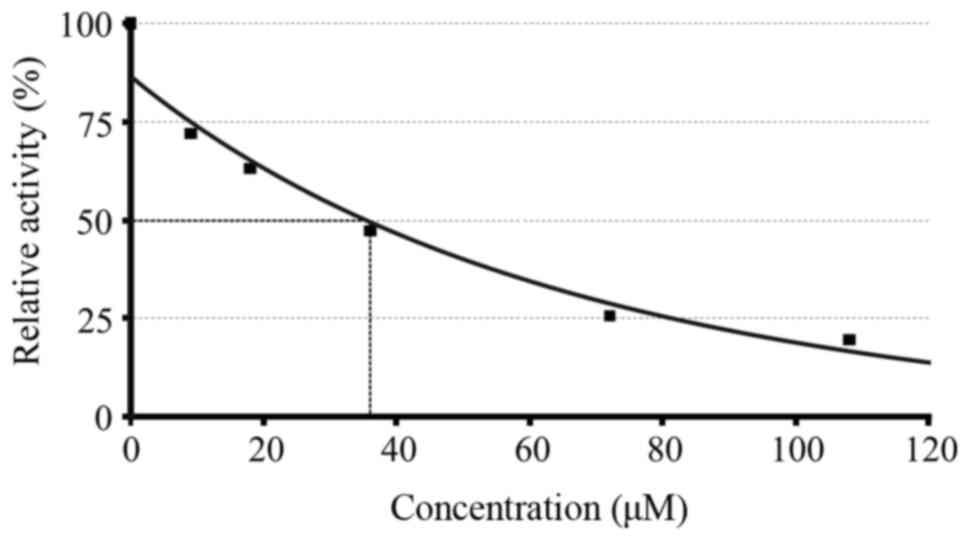
IC50 calculation for compound 2a is shown in Fig. 1.
Discussion
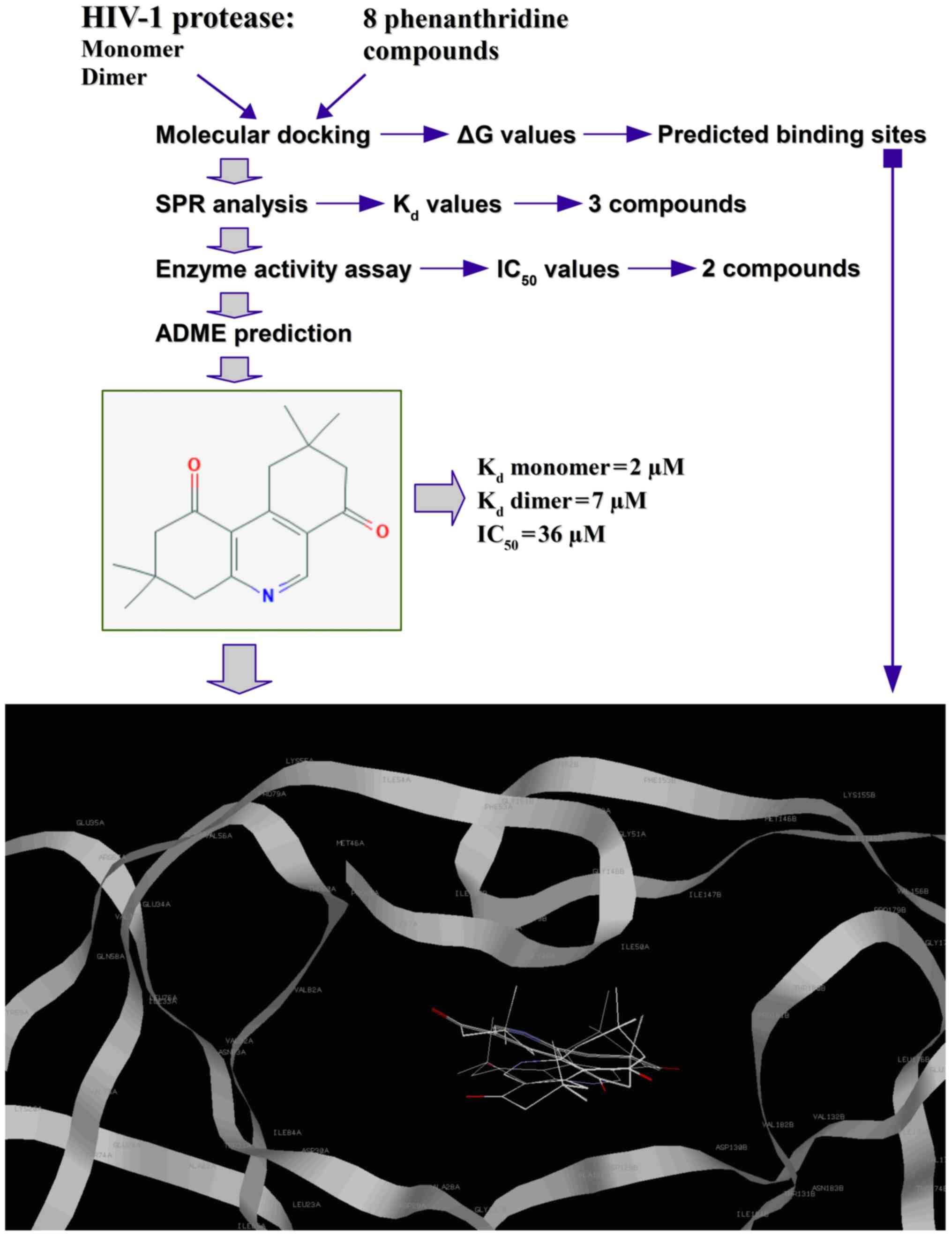
A flow chart of the experimental procedure used in
the present study is shown in Fig.
2. Predicted using molecular docking, amino acid residues in
positions 44-57 of the HIVp monomer represent the so-called flaps
(or flaps domains) that are considered to be involved in regulation
of the substrate access to the active site of the enzyme due to the
different conformational states of the flaps (opened to closed)
(24,25). An exception to this was compound 5a,
which, according to the docking model (Table SI), interacted with the monomeric
form of HIVp within the monomer/monomer contact area
(Pro1Gln2Ile3Thr4 and
Thr96'Leu97'Asn98'Phe99')
(26,27). In addition, compound 5a was
stabilized by the hydrophobic interactions with Leu24',
Thr26' and Leu90, Ile93 (Table II). A similar binding model for
2,3,6,8-tetrachlorophenanthridine was predicted (Table SI). An association between
Kd values and predicted binding models was observed.
Interactions of compounds 2a, 4a and 5a with the monomeric form of
HIVp exhibited notably lower values of predicted binding energy in
the docking models compared with the dimeric form.
Compound 4a demonstrated a positive result in the
HIVp enzyme inhibition assay (IC50 value, 45 µM), had
low binding affinity to the dimeric and monomeric forms of HIVp
(Kd, ~1x10-4 and ~1x10-5 M,
respectively). Compound 2a showed a Kd and
IC50 value of 2-7 and 36 µМ, respectively. The binding
affinity of compound 2a to the HIVp monomer (Kd~2 µМ)
was 4x higher than 2,3,6,8-tetrachlorophenanthridine
(Kd~9 µМ) (8,9). It is worth noting that the predicted
binding sites for compound 2a were different from those for
2,3,6,8-tetrachlorophenanthridine which targeted the HIVp
interface. In general, compound 2a had the most favorable ADME
parameters among all the phenanthridine derivatives tested and thus
may be used as the basic structure in the search for more potent
drug prototypes aimed at inhibition of HIV-1p.
Additional SPR experiments showed no significant
effect of the phenanthridine derivatives on the re-association of
the HIVp dimeric form on the optical chip (Table SII). This indirectly indicated the
absence of the compound's interference with the HIVp dimerization
process. The optimal docking conformations with the lowest binding
energy values corresponded to the positioning of compound 2a in two
regions of HIV-1p, the active site cavities and the flaps domain
(Table III), and it was
hypothesized that the inhibitory effect of compound 2a on HIV-1p
protease activity may be due to the restriction of substrate access
to the HIVp active site.
In conclusion, through the use of complex
methodological approaches, including experimental and
bioinformatics methods, it was demonstrated that the phenanthridine
derivatives with different spatial arrangements of the substituent
groups exhibited differing efficiencies of HIVp inhibition as well
as different SPR binding profiles with the monomeric and dimeric
forms of HIVp. Furthermore, according to the molecular docking
simulations, phenanthridine/HIVp binding models varied
significantly, indicating a difference in their potential
mechanisms of action on HIVp. These results confirmed our
hypothesis that phenanthridines, as a class of low-molecular-weight
compounds, in addition to their already known beneficial biological
and pharmacological activities against several different types of
cancer, may additionally possess antiviral activity against
HIV-1.
Supplementary Material
Surface plasmon resonance binding
sensograms of different concentrations of compound 2a to the (A)
monomeric and (B) dimeric form of HIV protease covalently
immobilized on a CM5 optical chip. The resulting sensograms are the
biosensor signal difference between the working (with immobilized
HIV protease protein) and control flow cell (without protein
immobilization). 1, 5 μM; 2, 15 μM; 3, 50 μM;
RU, resonance units.
Inhibition curves of the enzymatic
activity of HIV protease in the presence of different
concentrations of compound 2a. The experiments were performed in
duplicate. 1, control (without compound); 2, 9 μM; 3, 18
μM; 4, 36 μM; 5, 72 μM; 6, 108 μM.
The optimal docking conformations for
the predicted complexes of phenanthridine derivatives with dimeric
or monomeric forms of HIVp.
Surface plasmon resonance assessment
of the effect of the phenanthridine derivatives on the dimerization
of HIV1 protease.
Acknowledgements
Not applicable.
Funding
This research was funded by the Program of Basic
Scientific Research of National Science Academies for 2013-2020.
Biosensor 8K analysis and mass spectrometry quality control of
protein preparation were funded by MINOBRNAUKI (agreement no.
075-15-2019-1502).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request. Supplementary material are available at FigShare
repository 10.6084/m9.figshare.12837563.
Authors' contributions
PVE conceived the study. PVE and YVM performed the
experiments and wrote the manuscript. LAK and ASI analyzed and
interpreted the data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Azad I, Ahmad R, Khan T, Saquib M, Hassan
F, Akhter Y, Khan AR and Nasibullah M: Phenanthridine derivatives
as promising new anticancer agents: Synthesis, biological
evaluation and binding studies. Future Med Chem. 12:709–739.
2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tumir LM, Radić Stojković M and Piantanida
I: Come-back of phenanthridine and phenanthridinium derivatives in
the 21st century. Beilstein J Org Chem. 10:2930–2954.
2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Fuchino H, Kawano M, Mori-Yasumoto K,
Sekita S, Satake M, Ishikawa T, Kiuchi F and Kawahara N: In vitro
leishmanicidal activity of benzophenanthridine alkaloids from
Bocconia pearcei and related compounds. Chem Pharm Bull (Tokyo).
58:1047–1050. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wan M, Zhang L, Chen Y, Li Q, Fan W, Xue
Q, Yan F and Song W: Synthesis and anticancer activity evaluation
of novel phenanthridine derivatives. Front Oncol.
9(274)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bernardo PH, Wan KF, Sivaraman T, Xu J,
Moore FK, Hung AW, Mok HY, Yu VC and Chai CL: Structure-activity
relationship studies of Phenanthridine-based Bcl-XL inhibitors. J
Med Chem. 51:6699–6710. 2008.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Thai KM, Bui QH, Tran TD and Huynh TN:
QSAR modeling on benzo[c]phenanthridine analogues as topoisomerase
I inhibitors and anti-cancer agents. Molecules. 17:5690–5712.
2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Inbar-Rozensal D, Castiel A, Visochek L,
Castel D, Dantzer F, Izraeli S and Cohen-Armon M: A selective
eradication of human nonhereditary breast cancer cells by
phenanthridine-derived polyADP-ribose polymerase inhibitors. Breast
Cancer Res. 11(R78)2009.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Ershov PV, Gnedenko OV, Molnar AA, Lisitsa
AV, Ivanov AS and Archakov AI: Biosensor analysis of the
interaction of potential dimerization inhibitors with HIV-1
protease. Biomed Khim. 55:462–478. 2009.PubMed/NCBI(In Russian).
|
|
9
|
Ershov PV, Gnedenko OV, Molnar AA, Lisitsa
AV, Ivanov AS and Archakov AI: Kinetic and thermodynamic analysis
of dimerization inhibitors binding to HIV protease monomers by
surface plasmon resonance. Biomeditsinskaya Khimiya. 58:43–49.
2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Darke PL: Stability of dimeric retroviral
proteases. Meth Enzymol. 241:104–127. 1994.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Weber IT, Kneller DW and Wong-Sam A:
Highly resistant HIV-1 proteases and strategies for their
inhibition. Future Med Chem. 7:1023–1038. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Weber IT and Agniswamy J: HIV-1 Protease:
Structural perspectives on drug resistance. Viruses. 1:1110–1136.
2009.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Hao P, Ren Y and Xie Y: Label-free
relative quantification method for Low-abundance glycoproteins in
human serum by micrOTOF-Q. J Chromatogr B: Analyt Technol Biomed
Life Sci. 877:1657–1666. 2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Purushothaman AK and Pemiah B: Ultra high
performance liquid chromatography-ultraviolet-electrospray
ionization-micrOTOF-Q II analysis of flavonoid fractions from
Jatropha tanjorensis. Pharmacogn Mag. 10 (Suppl 3):S472–S479.
2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Richards AD, Roberts R, Dunn BM, Graves MC
and Kay J: Effective blocking of HIV-1 proteinase activity by
characteristic inhibitors of aspartic proteinases. FEBS Lett.
247:113–117. 1989.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Daina A, Michielin O and Zoete V:
SwissADME: A free web tool to evaluate pharmacokinetics,
drug-likeness and medicinal chemistry friendliness of small
molecules. Sci Rep. 7(42717)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Qureshi A, Rajput A, Kaur G and Kumar M:
HIVprotI: An integrated web based platform for prediction and
design of HIV proteins inhibitors. J Cheminform.
10(12)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Pettersen EF, Goddard TD, Huang CC, Couch
GS, Greenblatt DM, Meng EC and Ferrin TE: UCSF Chimera-a
visualization system for exploratory research and analysis. J
Comput Chem. 25:1605–1612. 2004.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wang J, Wang W, Kollman PA and Case DA:
Automatic atom type and bond type perception in molecular
mechanical calculations. J Mol Graph Model. 25:247–260.
2006.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Shapovalov MV and Dunbrack RL: A smoothed
Backbone-dependent rotamer library for proteins derived from
adaptive kernel density estimates and regressions. Structure.
19:844–858. 2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Martin JL, Begun J, Schindeler A,
Wickramasinghe WA, Alewood D, Alewood PF, Bergman DA, Brinkworth
RI, Abbenante G, March DR, et al: Molecular recognition of
macrocyclic peptidomimetic inhibitors by HIV-1 protease.
Biochemistry. 38:7978–7988. 1999.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ortiz D, delToro D, Ordyan M, Pajak J,
Sippy J, Catala A, Oh CS, Vu A, Arya G, Feiss M, et al: Evidence
that a catalytic glutamate and an ‘Arginine Toggle’ act in concert
to mediate ATP hydrolysis and mechanochemical coupling in a viral
DNA packaging motor. Nucl Acid Res. 47:1404–1415. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hsu KC, Chen YF, Lin SR and Yang JM:
iGEMDOCK: A graphical environment of enhancing GEMDOCK using
pharmacological interactions and Post-screening analysis. BMC
Bioinformatics. 12 (Suppl 1)(S33)2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Scott WR and Schiffer CA: Curling of flap
tips in HIV-1 protease as a mechanism for substrate entry and
tolerance of drug resistance. Structure. 8:1259–1265.
2000.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yu Y, Wang J, Chen Z, Wang G, Shao Q, Shi
J and Zhu W: Structural insights into HIV-1 protease flap opening
processes and key intermediates. RSC Adv. 7:45121–45128. 2017.
|
|
26
|
Babé LM, Pichuantes S and Craik CS:
Inhibition of HIV protease activity by heterodimer formation.
Biochemistry. 30:106–111. 1991.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Miller M, Schneider J, Sathyanarayana BK,
Toth MV, Marshall GR, Clawson L, Selk L, Kent SB and Wlodawer A:
Structure of complex of synthetic HIV-1 protease with a
substrate-based inhibitor at 2.3 A resolution. Science.
246:1149–1152. 1989.PubMed/NCBI View Article : Google Scholar
|