Introduction
The World Health Organization (2019) reported that
heart disease, stroke and other cardiovascular diseases are the
most frequent causes of death, with ischemic heart disease (IHD)
accounting for the largest number of cardiovascular
diseases-related deaths (1,2). The Global Burden of Disease (GBD)
study from 2016 stated that IHD and stroke are both types of
cardiovascular diseases, and cardiovascular diseases overall are
categorized as the leading cause of death in humans (2).
IHD refers to heart disease caused by insufficient
blood circulation due to coronary artery stenosis, which manifests
as decreased blood perfusion of the heart, resulting in myocardial
ischemia, decreased metabolism and abnormal cardiac function
(3,4). When the vascular diameter decreases or
occlusion occurs due to coronary artery endothelial cell
thickening, lipid deposition or thrombosis, an imbalance develops
between myocardial oxygen supply and demand, which further causes
cardiomyocyte apoptosis or necrosis (5). Therefore, cardiac blood flow should be
restored as soon as possible after myocardial ischemia to prevent
myocardial necrosis; however, myocardial ischemia/reperfusion (I/R)
also damages cardiomyocytes, which is termed myocardial I/R injury
(MIRI) (6). MIRI is known to be
associated with the incidence and mortality rate of cardiovascular
diseases (7), as the myocardium
develops biochemical and metabolic alterations during I/R. These
include rapid intracellular ATP exhaustion, calcium overload
leading to cell membrane disruption, synthesis of a large number of
reactive oxygen species (ROS) and a decreased mitochondrial
membrane potential, leading to caspase-3 activation and myocardial
hypercontracture (8-11),
thereby leading to cardiomyocyte death.
Ischemic preconditioning (IPC), one of the most
common methods used to decrease the severity of myocardial injury
in myocardial infarction (MI) studies, refers to administration of
transient non-lethal infarction stimulation prior to the occurrence
of fatal myocardial ischemia events. This stimulation induces
immune system mechanisms and releases endogenous protective
substances, such as adenosine, bradykinin and nitric oxide, to
protect the myocardium and preserve energy metabolism, thereby
decreasing the damage caused as well as the risk of fatal
infarction (12,13). IPC in vivo has predominantly
been performed in animal studies, and the earliest study by Murry
et al (14) found that IPC
could protect the myocardium against persistent ischemic injury and
reduce the infarct size (14-17).
Subsequently, several studies have employed IPC in a number of
animal experiments, such as rabbit, rat, murine and swine animal
models, and proved that IPC protected the heart after MIRI, and
also protected against abnormal systolic function and arrhythmia
after ischemia (18-21).
Exercise is key for promoting and maintaining human
health. Regular aerobic exercise can promote metabolism, improve
hormone equilibrium, improve cognitive function, and it is also an
important factor that controls cardiovascular disease (22,23).
It is well known that exercise can improve systemic circulation to
increase cardiopulmonary function, increase coronary artery
perfusion and improve cardiac tolerance (24-26).
It was previously demonstrated that exercise can be
beneficial to myocardial function, as it can increase the
ventricular dimensions, myocardial mass, cardiac output and stroke
volume (27,28). The effect of exercise training on MI
was first evidenced in the study by McElroy et al (29). They found that the formation of new
blood vessels in the hearts of rats after exercise reduced the
infarct size after MI. In animals, running on a treadmill and
swimming are primarily employed as interventional methods. For
example, rats that undergo treadmill training are shown to have
improved recovery in blood pumping capacity following myocardial
ischemia, and they also exhibit increased levels of cardiac
antioxidant enzymes, which decrease lipid peroxidation (30,31).
Swimming can increase cardiac antioxidant capacity in rats and
improve the systolic function of the aged myocardium. In addition,
swimming training prior to MI can increase cardiac arteriole
density and aid in improving cardiac function at the remodeling
phase (32-34).
Reducing cell death and myocardial injury caused by
IHD and MIRI has been an important focus of research. Mitochondrial
dysfunction following MIRI can induce apoptotic cell death, since
the mitochondrial apoptogenic factor, cytochrome c, is
released into the cytoplasm to activate caspase-3-mediated
apoptosis (35); however,
cytochrome c leakage to the cytosol can be inhibited by
Bcl-2/Bcl-xL (36). Furthermore,
myocardial I/R causes the release of TNF-α, which is a
proinflammatory cytokine that exacerbates MIRI at an early stage of
reperfusion by activating NF-κB, facilitating leukocyte
infiltration and inducing apoptosis (37,38).
Animal experiments were performed in the present study to examine
whether exercise intervention could protect against cardiovascular
disease and to explore the roles of apoptotic signaling elements,
such as TNF-α, activated caspase-3 and Bcl-2. The findings of the
present study may lead to optimization of exercise programming for
healthy individuals, athletes and patients with heart failure
seeking strategies to enhance cardiac function (39).
Materials and methods
Ethics approval
The present study was approved by the Animal
Experiment Committee of University of Taipei (Taipei, Taiwan; IACUC
approval no. UT110001). The animals used in the study were managed
in a humane manner in accordance with the Guide for the Care and
Use of Laboratory Animals (40).
Animal preparation
A total of 42 Sprague-Dawley male rats, which were
purchased from BioLASCO, and weighed 250-280 g, were used in the
present study. A total of 1 week prior to the experiment, the rats
were moved to the laboratory to allow them to acclimate. The rats
were given ad libitum access to food and water, and
maintained with a 12 h light/dark cycle from 6:00 a.m. to 6:00
p.m., and the room temperature was controlled at 25˚C and a
humidity of 55±5%.
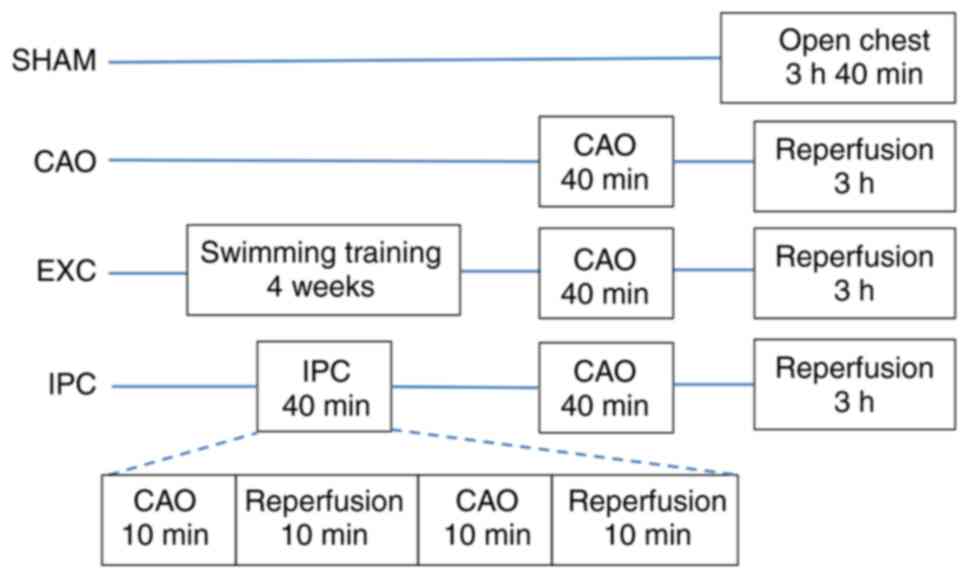
The rats were randomly assigned to one of four
groups as follows: Sham control group (Sham, n=10), myocardial
ischemia group (coronary artery occlusion; CAO, n=10), swimming
training group (EXC, n=10) and IPC group (IPC, n=10). Rats in the
EXC group were allowed to acclimate to swimming training at the age
of 3 weeks. Swimming started at 15 min per day and was increased to
180 min per day at day 6. At 4-7 weeks of age, the rats underwent 3
h of swimming training per day. Water temperature was controlled at
35-37˚C. The forced swimming test procedure was employed for
swimming training (41). Since the
forced swimming test procedure was employed in the swimming
training, the rats were kept in the status of swimming throughout
the training period in which they could not reach the bottom or
hang on the side with the continued supervision of the
experimenters. In other words, the time of immobility and climbing
while the rats in the water was completely excluded.
Experimental protocol
The rats were anesthetized by intraperitoneal
injection of 20 mg/kg BW Zoletil 50 [tiletamine + zolezepam; Virbac
(Taiwan) Co., Ltd.] and intraperitoneal injection of 10 mg/kg BW
Balanzine (xylazine 2% w/v) (42).
After tracheotomy, each rat was intubated and ventilated, and the
carotid artery was cannulated for the direct BP measurement.
Throughout the experiment, heart rate, systolic blood pressure
(SBP), diastolic blood pressure (DBP), and mean arterial pressure
(MAP) were recorded using a direct measurement method through the
carotid artery with a Biopac MP150 (Biopac Data Acquisition
System). The purpose of detecting hemodynamic changes was to
observe the heart condition of each group of experimental rats
before, during and after the experiment. Electrocardiography leads
were placed on limbs. After median sternotomy, a 4-0 silk suture
was passed around the proximal part of the left anterior descending
coronary artery. The ends of the silk suture were threaded through
a small vinyl tube to form a snare. The body temperature was
monitored using a rectal thermometer and maintained at 37˚C with
heating pads throughout the experiments.
After achieving hemodynamic stability for 20 min,
the rats were divided randomly into four groups. Group 1 rats
(Sham) underwent the same surgical procedures without any
pretreatment, CAO or reperfusion. Group 2 rats (CAO) did not
receive any pretreatment. Group 3 rats (EXC) underwent 4 weeks of
swimming training intervention prior to surgery. In group 4 rats
(IPC), IPC was induced via two 10-min episodes of CAO. The two
episodes of CAO were followed by 10 min of reperfusion. At 10 min
after the previously mentioned treatments, a 40-min CAO was induced
in the rats in groups 2, 3 and 4 by pulling the snare around the
left anterior descending coronary artery. Successful occlusion was
verified by observing the development of ST segment elevation and
changes in the QRS complex on the electrocardiogram and cyanotic
changes in the myocardium in the occluded area. After 40 min of
CAO, the snare was released for reperfusion for 3 h. Reperfusion
was confirmed by refilling of the coronary artery and visualizing a
reactive hyperemic response. Arterial pressure, heart rate and
electrocardiography were recorded simultaneously and continuously
throughout the experiment. The grouping flowchart is shown in
Fig. 1. After the operation, rats
were euthanized by exsanguination. Blood specimens were collected
from the carotid artery and serum separated after centrifugation
(2,000 x g at 4˚C for 20 min) for biochemical research.
Determination of the area at risk
(AAR) and MI size
The dyeing methods of Zhao et al (43) were followed. After the experiment
was completed, heparin (1,000 U) was injected intravenously.
Subsequently, the heart was resected and the previous MI site was
re-ligated. Next, 1% Evans blue dye (MilliporeSigma) was perfused
into the ascending aorta. After perfusion was completed, five
transverse sections were made through the left ventricle and
interventricular septum. The sections were immersed in 1% triphenyl
tetrazolium chloride solution and heated in a 37˚C thermostatic
water bath for 20 min. After heating, the sections were weighed,
and 10% formalin solution was added to fix the sections at room
temperature for 24 h. The colors of blue, pale and red respectively
represented the normal, infarcted and ischemic tissue. The total
weight of the AAR and MI were calculated and the sum was taken. AAR
is defined as the myocardial tissue within the vascular territory
that is distal to the culprit lesion of the infarct-related artery.
AAR is the percentage of the weight of the red area divided by the
weight of the left ventricle; MI is the percentage of the weight of
the white area divided by the red area (44).
Biochemical analysis of cardiac
function
Blood was collected from rats in each group after
the experiment was completed. The levels of troponin I, lactate
dehydrogenase (LDH) and creatine phosphokinase (CPK) were
subsequently measured. CPK and LDH activities were determined
according to standard methods using diagnostic kits from BioSystems
S.A. Assessment of serum troponin I was carried out via
enzyme-linked immunosorbent assay (ELISA) using a commercial kit
purchased from DRG International, Inc. The concentration of
Troponin-I, LDH and CPK in the blood is increased following
myocardial damage, such as acute myocardial infarction, shock,
hypoxia or acute coronary artery disease. Therefore, the
aforementioned factors can be used as the clinical diagnosis of
myocardial infarction and evaluation (45).
Histological examination of the
heart
At the end of the experiment, the hearts from four
rats in each group were harvested. Part of the heart was fixed
using 10% paraformaldehyde at room temperature for 24 h,
dehydrated, embedded in paraffin, cut into 4-µm sections and
mounted on glass slides. The sections were then deparaffinized
using xylene at room temperature for 10 min, and counterstained
using hematoxylin and eosin (H&E) at room temperature for 7
min. Finally, the samples were analyzed using an inverted light
microscope (CKX53; Olympus Corporation) at x400 magnification. The
severity of myocardial injury was determined using a morphological
scoring system as follows: 0, no damage; 1, interstitial edema and
focal necrosis; 2, diffuse myocardial cell swelling and necrosis;
3, necrosis with contraction bands and neutrophil infiltrate; and
4, widespread necrosis with contraction bands, neutrophil
infiltrate and hemorrhage. The evaluation of the myocardial damages
based on the HE staining was the average of the scores from two
separate examiners (46). To make
comparisons between groups, the mean score and standard deviation
of each group was calculated.
TUNEL staining analysis of the
heart
Briefly, when the experiments were completed, the
hearts were cut into 4-µm sections and fixed in acetone at room
temperature for 24 h. Then, the samples were moved into a solution
containing terminal deoxynucleotidyl transferase and detection
buffer (Roche Diagnostics GmbH), conjugated with horseradish
peroxidase, and incubated at 37˚C for 60 min. A
diamino-benzidine-chromogen (Boehringer) was used. Several further
steps were introduced to obtain the end value of the number of
TUNEL-positive nuclei, including the samples were analyzed using an
inverted light microscope (CKX53; Olympus Corporation) at x400
magnification, then selecting an area randomly, counting the nuclei
in that area, and converting the value to a percentage by dividing
it by the total number of the cell nuclei.
Western blot analysis
Briefly, when the experiments were completed, the
hearts of four rats from each group were moved into the tissue
protein extraction reagent (T-PER; Thermo Fisher Scientific, Inc.)
at 4˚C for homogenization; the myocardial cell lysate was prepared
in cold lysis buffer [25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1%
NP-40, 1% sodium deoxycholate, 0.1% SDS]. Samples were centrifuged
at 10,000 x g at 4˚C for 10 min, and a modified Bradford assay
which compared with the conventional Bradford method was used to
determine protein concentrations. Notably, the characteristics are
stable, the measurement speed is fast, the sensitivity is high and
the error rate is small. A total of 60 µg of protein were loaded
and separated using 15% SDS-PAGE and then transferred to a
nitrocellulose membrane. After blocking with 5% non-fat dry milk in
Tris-buffered saline containing 0.1% Tween-20 (TBST) at 37˚C for 30
min, the membranes were incubated with anti-caspase-3 (cat. no.
IMG-144A; Imgenex), anti-TNF-α (cat. no. sc-52746; Santa Cruz
Biotechnology) or anti-Bcl-2 antibodies (cat. no. 633502;
BioLegend; all 1:1,000) in 5% non-fat dry milk at 4˚C for 24 h. The
membranes were then incubated in 5% non-fat dry milk in TBST
containing secondary antibody (m-IgGκ BP-HRP; cat. no. sc-516102;
Santa Cruz Biotechnology; 1:1,000) conjugated to horseradish
peroxidase. To visualize the peroxidase activity, an enhanced
chemiluminescence substrate system was used, followed by exposing
the membranes to hyperfilms. β-actin (cat. no. 643802; BioLegend;
1:1,000) was used at room temperature for 2 h as a loading control.
ImageJ version 1.6 (National Institutes of Health) was used for
densitometry analysis, and each gel was normalized against the
background density.
Capillary density measurements in
tissue sections
To visualize the capillaries in the myocardium,
endothelial cells were stained with CD31, often used as a
biological marker to represent the capillary vessels in the
myocardium. Angiogenesis was quantitatively assessed based on
CD31-positive capillary vessels for the determination of capillary
density. For immunostaining, a rabbit anti-rat CD31 antibody (cat.
no. sc-376764; Santa Cruz Biotechnology; 1:100) was used at room
temperature for 15 min. Staining was visualized by reaction with
DAB (MilliporeSigma; 1:20). Capillaries were visualized in the
myocardium as a brown precipitate, identified as having a diameter
<20 µm and a layer of endothelial cells without smooth muscle
cells. Computer-assisted morphometry was performed using Image Pro
Plus version 5.0 (Media Cybernetics, Inc).
Statistical analysis
Data are presented as the mean ± standard deviation,
and statistical analysis was performed using SPSS 20.0 (IBM Corp.).
Hemodynamic variables were analyzed using a two-way repeated
measures ANOVA. Otherwise, data were analyzed using a one-way ANOVA
and a Bonferroni post hoc multiple comparisons test. The ordinal
values of the myocardial injury scores were analyzed using a
Mann-Whitney U test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Comparison of mortality rates amongst
the four groups
A total of 42 rats were used in the present study.
Of those 42 rats, 1 rat in the CAO group died from heart failure,
which was defined as a progressive decrease of the systolic
arterial pressure to <50 mmHg with global left ventricular
dilatation and poor contraction; and 1 rat in the IPC group
developed ventricular fibrillation and died. These two rats were
excluded from the study. No statistically significant difference
was observed amongst the four groups in terms of mortality rate
(P=0.591).
Comparison of changes in hemodynamic
variables during the experiments amongst the four groups
No significant difference was found amongst the four
groups in terms of mean arterial pressure and heart rate, at
baseline and throughout the experiments. In addition, the changes
in hemodynamic variables did not reach a level of statistical
significance during the experiments in the four groups (Table I).
 | Table IHemodynamic changes during the
experiments.a |
Table I
Hemodynamic changes during the
experiments.a
| | CAO | CAR |
|---|
| Factor | Group | n | Baseline 1 | Baseline 2 | 20 min | 40 min | 1 h | 2 h | 3 h |
|---|
| Mean arterial
pressure, mmHg | | | | | | | | | |
|
1 | Sham | 10 | 84±5 | 78±5 | 73±5 | 72±3 | 83±6 | 74±4 | 75±4 |
|
2 | CAO | 10 | 82±8 | 82±4 | 75±9 | 73±4 | 73±5 | 73±6 | 84±5 |
|
3 | EXC | 10 | 85±6 | 88±3 | 81±5 | 74±5 | 79±6 | 83±4 | 83±8 |
|
4 | IPC | 10 | 80±4 | 81±5 | 76±5 | 79±4 | 80±4 | 84±6 | 85±6 |
| Heart rate,
beats/min | | | | | | | | | |
|
1 | Sham | 10 | 420±21 | 481±33 | 417±26 | 404±15 | 423±34 | 456±18 | 420±32 |
|
2 | CAO | 10 | 454±31 | 418±36 | 417±25 | 421±29 | 437±24 | 431±30 | 431±24 |
|
3 | EXC | 10 | 483±21 | 453±28 | 452±37 | 431±33 | 433±29 | 441±21 | 417±19 |
|
4 | IPC | 10 | 486±30 | 481±56 | 487±31 | 468±39 | 475±41 | 475±45 | 482±34 |
| Mean arterial
pressure-heart rate product/1,000, mmHg*beats/min | | | | | | | | | |
|
1 | Sham | 10 | 36.08±2.56 | 37.02±3.66 | 31.46±2.67 | 28.60±1.74 | 35.39±2.72 | 33.48±2.61 | 31.24±2.70 |
|
2 | CAO | 10 | 40.27±4.73 | 39.16±6.42 | 37.79±6.63 | 34.60±6.25 | 36.51±3.96 | 35.53±3.97 | 36.73±4.84 |
|
3 | EXC | 10 | 40.63±3.97 | 38.51±3.93 | 41.83±4.65 | 39.96±5.77 | 42.37±5.66 | 37.46±6.28 | 38.32±4.82 |
|
4 | IPC | 10 | 31.49±5.16 | 35.39±3.09 | 32.27±3.75 | 32.03±3.48 | 38.69±5.57 | 39.46±6.40 | 39.53±2.45 |
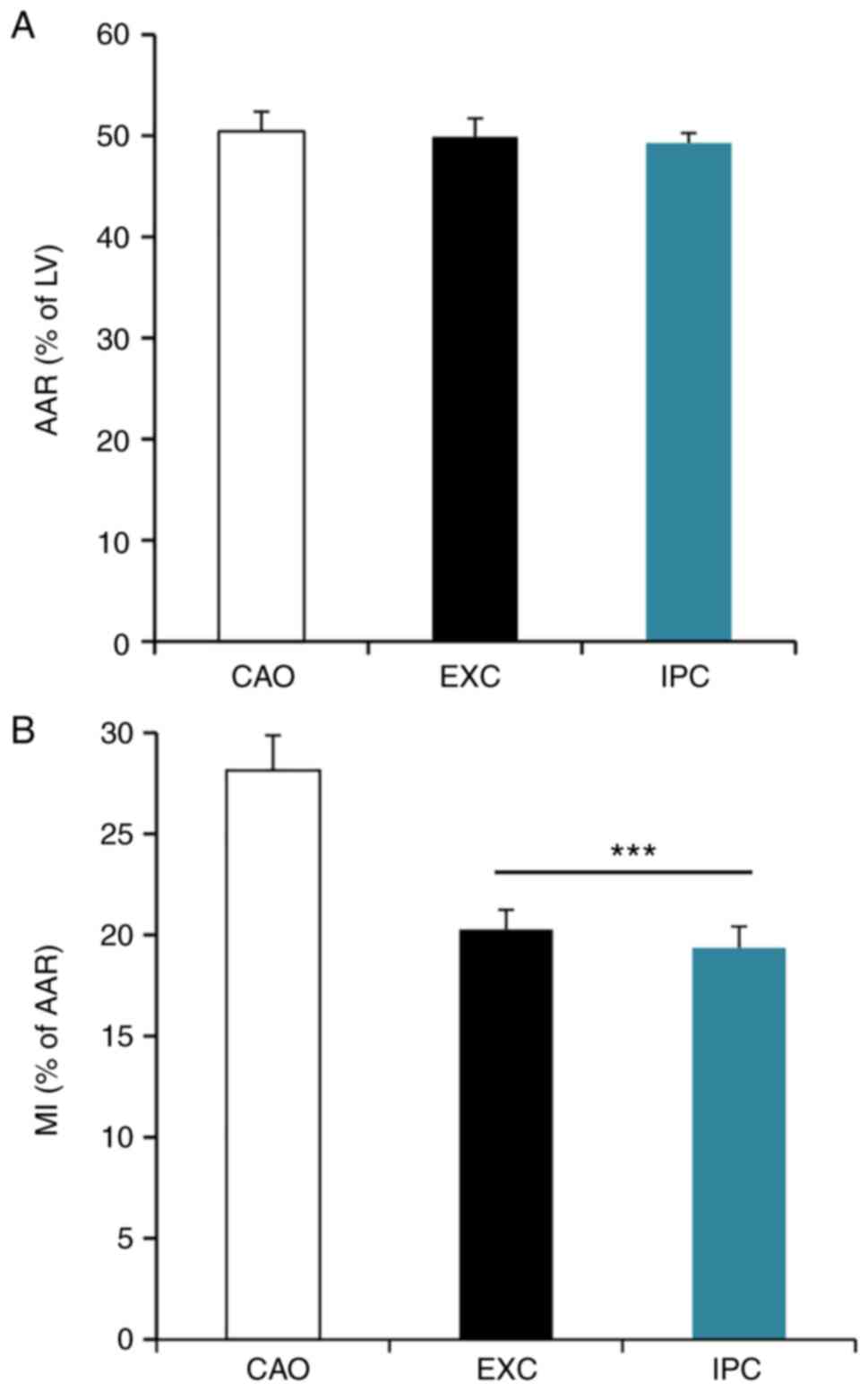
Comparison of AAR and MI size amongst
the CAO, EXC and IPC groups
When the experiments were completed, six rats from
each group were randomly selected to collect cardiac tissues for
AAR and MI size analysis. The AAR of the left ventricular infarct
area is shown in Fig. 2A. The MI
size, expressed as a percentage of AAR, is shown in Fig. 2B. The results demonstrated that
there was no statistically significant difference in AAR amongst
the three groups (50.44±1.75% in CAO vs. 49.90±1.62% in EXC and
49.29±0.88% in IPC; P>0.05), while the MI size was significantly
decreased in the EXC and IPC experimental groups compared with the
CAO group (20.27±0.88% in EXC and 19.37±0.95% in IPC vs.
28.15±1.54% in CAO; P<0.001).
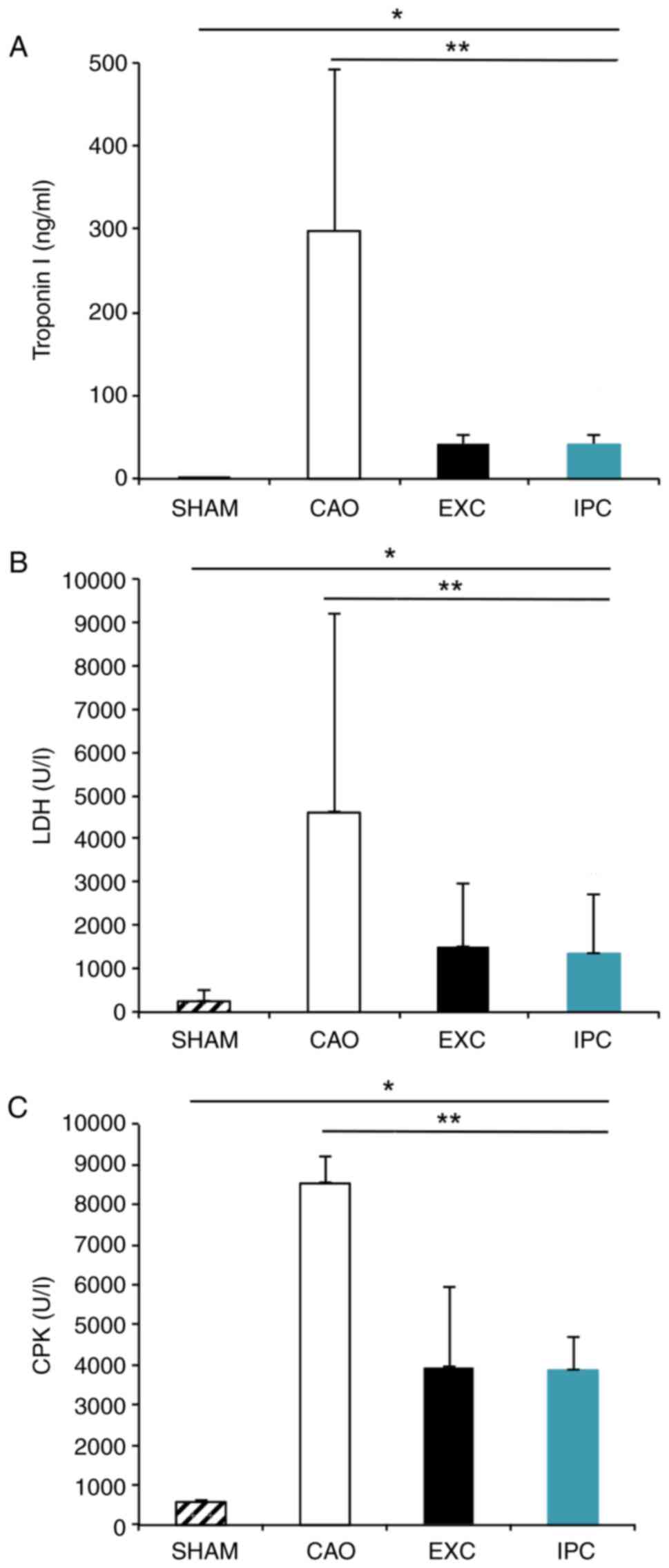
Intervention with EXC and IPC
significantly reduces the serum concentrations of troponin I, LDH
and CPK after myocardial I/R
When the experiments were completed, six rats from
each group were randomly selected to collect blood samples for
biochemical analysis of cardiac function. CAO group rats, compared
with Sham group rats, exhibited significantly increased serum
levels of troponin I (298.08±173.09 ng/ml vs. 0.68±0.47 ng/ml,
respectively; P<0.05; Fig. 3A),
LDH (4,606.17±1,057.12 U/l vs. 259.83±127.48 U/l, respectively;
P<0.001, Fig. 3B) and CPK
(8,531.33±591.00 U/l vs. 577.50±19.40 U/l, respectively;
P<0.001, Fig. 3C). The EXC and
IPC groups exhibited significantly less prominent increases in the
serum levels of troponin I (41.56±10.44 and 41.31±9.23 ng/ml,
respectively, vs. the CAO group; P<0.05; Fig. 3A), LDH (1,496.33±292.03 and
1,351.83±265.11 IU/l, respectively, vs. the CAO group; P<0.001;
Fig. 3B) and CPK (3,918.17±1,761.22
and 3,867.00±755.85 U/l, respectively, vs. the CAO group;
P<0.01; Fig. 3C). No significant
differences between the EXC and IPC groups were observed
(P>0.05).
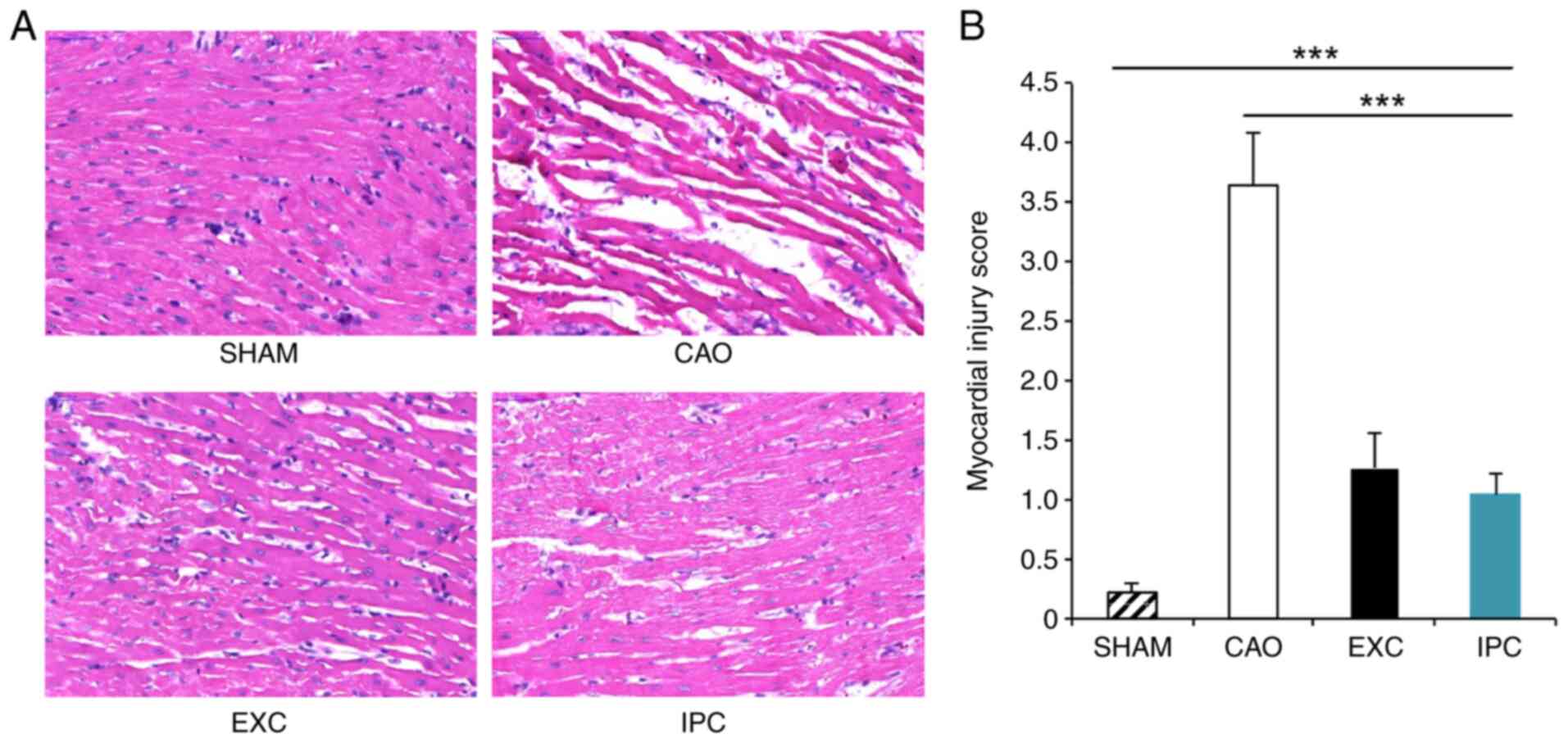
EXC and IPC interventions
significantly reduces MIRI
At the end of the experiments, four rats from each
group were randomly selected, and part of their hearts was sent for
histological examination in order to identify cardiac injury. The
myocardium in the left ventricle of the rats in the Sham group
appeared normal (Fig. 4A); however,
the myocardium of rats in the CAO group exhibited interstitial
edema, myocardial cell swelling and disruption of myocardial
fibers. The myocardial injury scores of the rats in the CAO group
were significantly higher compared with those of the rats in the
Sham group (P<0.001; Fig. 4B).
However, EXC and IPC significantly reduced the extent of
histological damage, and the myocardial injury scores of the rats
in the EXC and IPC groups were significantly decreased compared
with those in the CAO group (P<0.001).
EXC and IPC interventions
significantly reduces the number of apoptotic cardiomyocytes after
myocardial I/R
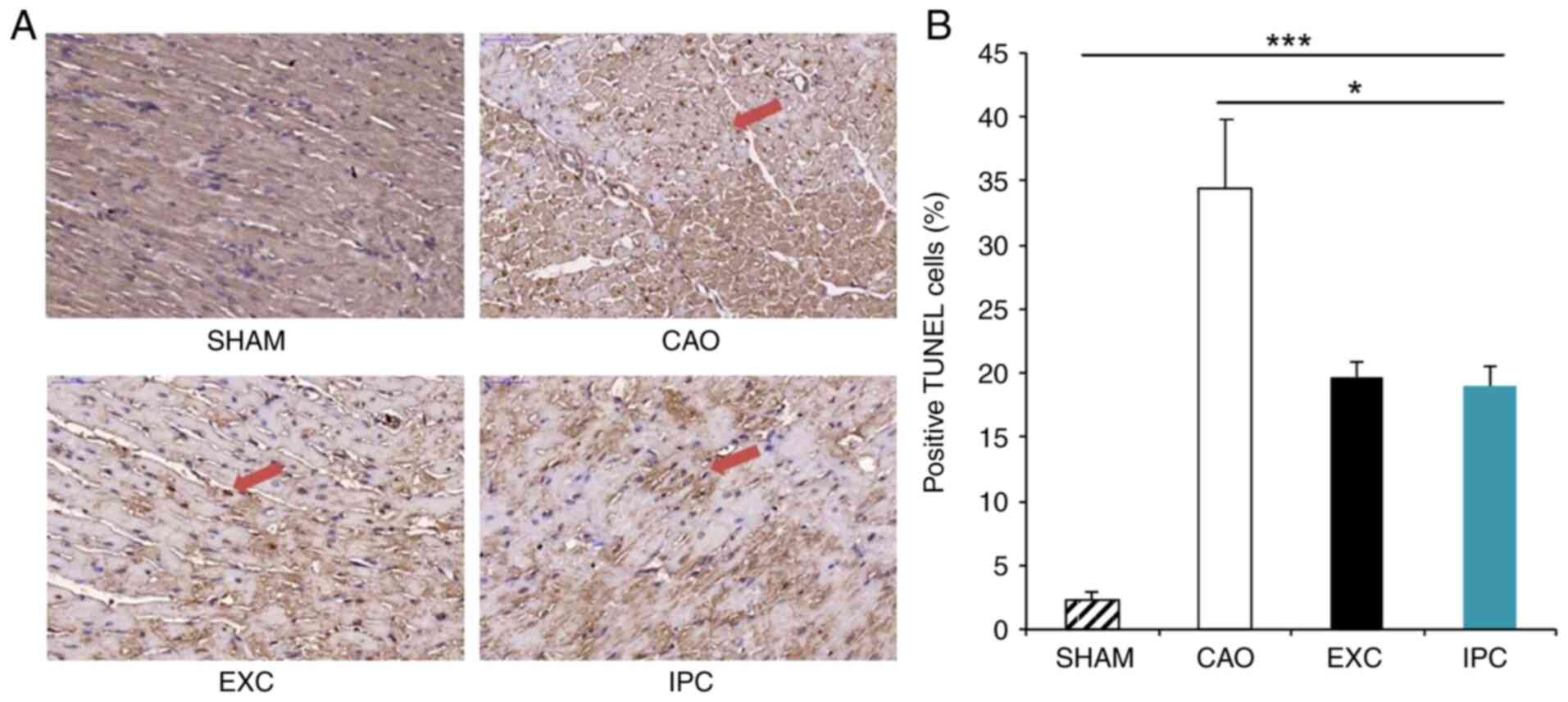
TUNEL staining is a sensitive method commonly used
for detecting apoptosis (47). At
the end of the experiment, four rats from each group were randomly
selected, and their hearts were subjected to TUNEL staining, which
was used to locate DNA fragmentation in the nuclei of apoptotic
cardiomyocytes appearing as a dark brown reaction product. Of note,
only a small portion of the hearts of the rats in the Sham group
exhibited TUNEL-stained nuclei (Fig.
5A). With regard to the rats in the CAO group, heart samples
were also found to exhibit TUNEL staining, which was induced by
myocardial I/R. In the EXC and IPC groups, dark brown staining was
observed in the heart samples in the form of scattered nuclei.
There was a significant increase in the percentage of the
TUNEL-positive nuclei against the total nuclei in the hearts of the
rats from the CAO group compared with the Sham group (34.4±5.5% vs.
2.3±0.7%, respectively; P<0.001; Fig. 5B), and the corresponding percentages
in the EXC and IPC groups were decreased significantly (19.7±1.2
and 19.0±1.5%, respectively, vs. the CAO group; P<0.05; Fig. 5B). No significant difference in the
numbers of TUNEL-positive nuclei between the EXC and IPC groups was
observed (Fig. 5B).
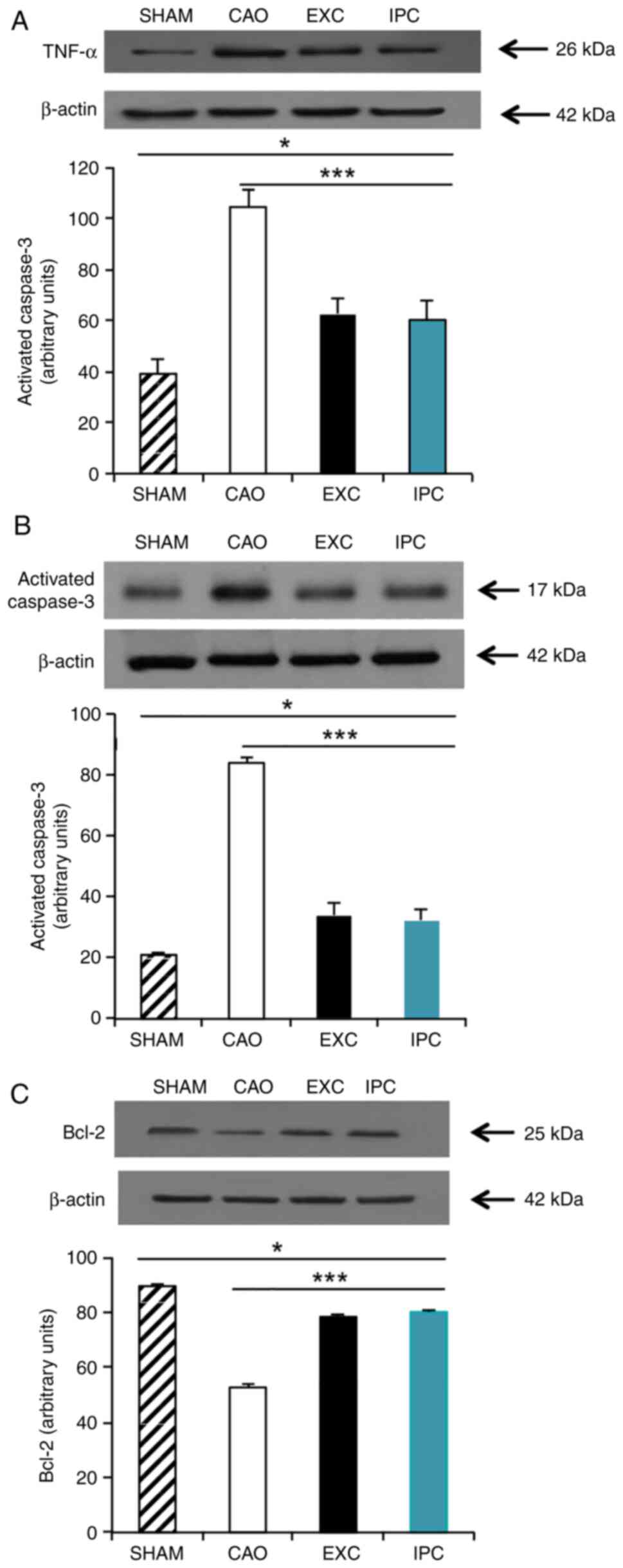
EXC and IPC interventions
significantly reduces the levels of TNF-α and activated caspase-3,
and increases the levels of Bcl-2 in cardiomyocytes
TNFs are, a family of cellular signaling proteins
that are involved in systemic inflammation, and are some of the
most critical pro-inflammatory cytokines (48). Activated caspase-3 is a universal
effector of apoptosis (49), and
Bcl-2 is an important gene family that controls the primary
apoptotic pathway (49). To measure
the levels of TNF-α, activated caspase-3 and Bcl-2, four rats from
each group were randomly selected and western blot analysis on
their heart samples was conducted at the end of the experiments.
Compared with the Sham group, myocardial I/R significantly
increased the TNF-α levels in the hearts of the rats in the CAO
group (P<0.001; lane 2, Fig.
6A). The EXC and IPC groups exhibited a significantly less
prominent increase in TNF-α levels in the hearts subjected to
myocardial I/R (P<0.001 vs. CAO group; lanes 3 and 4, Fig. 6A). There was no significant
difference in TNF-α expression between the EXC and IPC groups
(Fig. 6A). Caspase-3 is cleaved to
an active p17 subunit (17 kDa) when apoptosis is induced. According
to the results of western blot analysis, which showed a significant
increase in the activated caspase-3 band, it was inferred that
myocardial I/R could increase the levels of activated caspase-3 in
the hearts (P<0.001 vs. Sham group; lane 2, Fig. 6B); conversely, the increase in
activated caspase-3 was significantly inhibited in the EXC and IPC
groups (P<0.001 vs. CAO group; lanes 3 and 4, Fig. 6B). No significant difference in the
activated caspase-3 expression between the EXC and IPC groups was
observed (Fig. 6B). Compared with
the Sham group, CAO group hearts exhibited a significant reduction
in Bcl-2 levels (P<0.001 vs. Sham group; lane 2, Fig. 6C), while this decrease in Bcl-2
levels was significantly inhibited in the EXC and IPC groups
(P<0.001 vs. CAO group; lanes 3 and 4, Fig. 6C). No significant difference in the
Bcl-2 expression levels between the EXC and IPC groups was observed
(Fig. 6C).
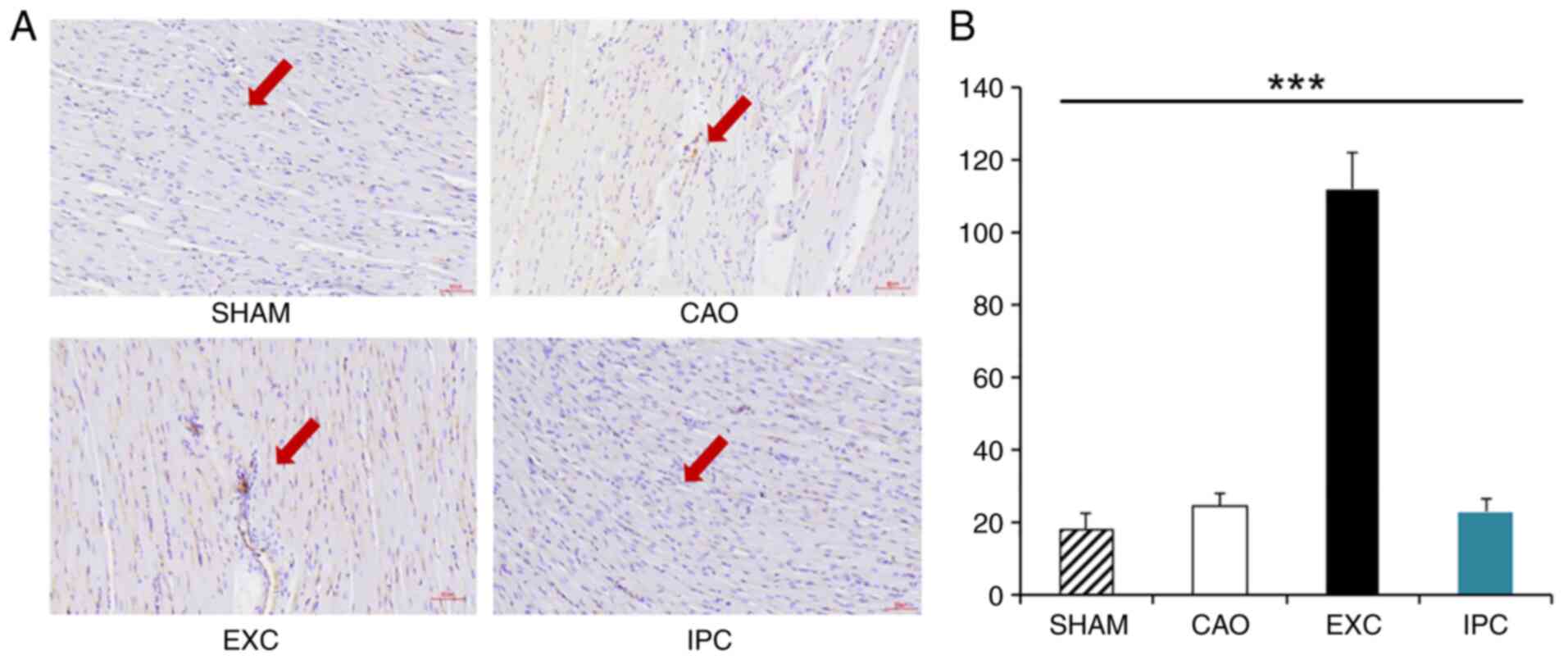
EXC significantly increases the
density of myocardial blood vessels
At the end of the experiments, four rats from each
group were randomly selected, and part of their hearts were used
for CD31 staining analysis in order to identify the density of
myocardial blood vessels. Compared with the Sham, CAO and IPC
groups, the EXC group exhibited a significant increase in the
density of myocardial blood vessels (P<0.001 vs. SHAM, CAO and
IPC groups; Fig. 7).
Discussion
In the present study, it was demonstrated that
swimming training could significantly decrease myocardial injury,
MI size, serum concentrations of MI markers (troponin I, CPK and
LDH) and cardiomyocyte apoptosis following a myocardial I/R, and
the myocardial protective effects were even comparable to those of
the commonly used method of IPC.
The intense and sustained changes of myocardial
injury that occur during the myocardial ischemic period lay the
foundation for the reperfusion injury that follows. This type of
I/R injury has been proven to be an extension of ischemic injury,
which further exacerbates irreversible myocardial injury (44,46).
Therefore, strategies for preventing reperfusion injury are the
important aspect of treatment of related heart diseases. Previous
studies have proven that IPC can significantly decrease MI size,
DNA laddering, the number of TUNEL-stained nuclei and cardiomyocyte
apoptosis to exert cardioprotective effects (50,51).
It has been demonstrated that the cardioprotective
effects of IPC can be exerted in two temporal windows. The first is
active within minutes to hours after the conditioning stress; the
second, known as ‘second window of protection’, becomes active 1-3
days later (52,53). The mechanisms underlying
training-induced cardioprotection resemble those elicited by IPC,
whereby short ischemic episodes prior to a major ischemic insult
trigger endogenous cardioprotective mechanisms. Training may induce
IPC by emulating minor local ischemic episodes, with ischemia
occurring as short periods of time during which the supply of blood
and oxygen to tissues is limited with respect to the tissue
requirements (54).
The experimental results of the present study
revealed that MI size was significantly decreased after myocardial
I/R in rats that underwent swimming training. There was no
significant difference in AAR among the three groups (groups 2-4)
and no significant hemodynamic changes were observed during the
surgical procedure. Therefore, reduced MI size could not be
attributed to differences in surgical technique or hemodynamic
changes.
Intermittent hypoxia is broadly defined as repeated
episodes of hypoxia interspersed with episodes of normoxia. The
definition of intermittent hypoxia varies widely in terms of cycle
length, number of hypoxic episodes per day and the number of days
of exposure. Nevertheless, it has been confirmed that repeated
incidents of ischemia and hypoxia may lead to physiological changes
and are considered as one of the key factors involved in
cardiovascular diseases, general inflammation and heart disease
(55,56).
Intermittent hypoxia primarily affects the cardiac
structure and function via oxidative damage caused by free radicals
(57). Myocardial I/R disrupts the
mitochondrial respiratory chain and is accompanied by ATP
depletion, resulting in the accumulation of mitochondrial ROS
(51). The effects of IPC are
mediated through activating KATP channels in the
mitochondria. Once activated, the progress of phosphorylation in
mitochondria is accelerated, which results in a reduction in ATP
consumption in mitochondria, i.e., a reduction in the metabolic
rate in cells (58). ROS levels
increase during exercise; however, mounting epidemiological data
have proven that exercise decreases the incidence of oxidative
stress-associated diseases owing to exercise-induced adaptation
(59,60). Exercise training can protect cardiac
subsarcolemmal and intermyofibrillar mitochondria, enabling them to
adapt to transient hypoxia and decreasing ROS production,
increasing their ability to tolerate high calcium levels, and
preventing oxidative damage caused by myocardial I/R to achieve
cardioprotection (61,62). In addition, mitochondria contain an
antioxidant enzyme known as Mn superoxide dismutase (MnSOD) that
can decrease intracellular oxidative stress (63). The cytoplasm also possesses a
Cu/ZnSOD that has a function similar to MnSOD (64). Research indicates that IPC can exert
protective effects through the induction of MnSOD. In addition, the
levels of MnSOD and Cu/ZnSOD were found to be increased in rat
cardiomyocytes after exercise training, which inhibited apoptosis
(65,66). Therefore, it may be hypothesized
that myocardial antioxidant effects induced by exercise can
decrease myocardial injury and cellular damage, thereby reducing MI
size.
When cardiomyocytes are exposed to ischemia or
hypoxia, the mitochondrial matrix shrinks, leading to an
enlargement of the intermembrane space and resulting in a
disorientation of cardiomyocytes and disruption of the myocardial
structure, and a release of endocardial substances (67). In addition, ischemia in the
myocardium can cause the membrane potential of mitochondria to
dissipate, leading to an increase in the permeability and an
opening in mitochondrial permeability transition pore (mPTP),
resulting in a release of apoptosis-inducing factors from
mitochondria (67). Therefore, the
evaluation of cardiomyocyte injury could be performed by
determining the plasma levels of specific and sensitive biomarkers,
such as troponin I, CPK and LDH (68-70).
It has been found that IPC, by activating protein kinase C, can
open KATP channels, reduce the release of myocardial
enzymes by reducing the consumption of ATP, and create an inflow of
potassium ions and other ions, which may help the myocardium reach
a balance in osmotic pressure and potential equilibrium, so as to
mitigate myocardial injury (71).
Theoretically, the cardioprotective effects of exercise may be
attributed to the fact that exercise training induces a
mitochondrial phenotype, including increases in the protein levels
of primary antioxidant enzymes in both subsarcolemmal and
interfibrillar mitochondria, attenuation of ROS-induced cytochrome
c release, and reduced maximal rates of mPTP opening
(72,73). Furthermore, the ratio of blood
vessels/muscle fibers increases in healthy hearts after exercise
intervention. In addition, exercise training in advance can also
increase angiogenesis after MI (74,75).
Similar results were obtained in the present study, which showed
that swimming intervention increased angiogenesis in the heart. The
results of the present study showed that cardiomyocytes released
troponin I, LDH and CPK into the blood after I/R. Therefore, these
three enzymes were found to be significantly elevated in the CAO
group. On the other hand, there were significantly lower levels of
the aforementioned three enzymes in the EXC and IPC groups compared
with those in the CAO group. Therefore, it can be deduced that
exercise training decreased I/R-induced damage by exerting
cardioprotective effects through protecting myocardial
mitochondria, increasing myocardial vascularization and maintaining
cardiomyocyte integrity, thereby decreasing the release of the
aforementioned enzymes. In addition, examination of HE-stained
myocardial sections revealed that the myocardium exhibited
deformation and expansion, and the muscle fibers were arranged in a
disorderly manner after myocardial I/R, and these changes were
significantly reduced following treatment intervention with EXC and
IPC.
The free radicals produced during myocardial I/R can
activate NF-κB. Subsequently, TNF-α, which induces the
transcription of NF-κB, can directly damage vascular endothelial
cells and cardiomyocytes (76).
Functionally, TNF-α could trigger cellular responses via
interacting with its transmembrane receptors, such as TNF receptor
1 (TNFR1) (77). Upon activation by
TNF-α, TNFR1 can activate the death induction signal mediated by
TNF receptor-associated death domain and Fas-associated death
domain in order to activate caspase-3, -6 and -8, and reduce the
production of the anti-apoptotic protein Bcl-2, eventually causing
apoptosis (78,79).
NF-κB is a family of inducible transcription factors
that serve a variety of evolutionarily conserved roles in the
immune system. Cytokines belonging to the TNF family induce rapid
transcription of genes regulating inflammation, cell survival,
proliferation and differentiation, primarily through activation of
the NF-κB pathway (80). IPC can
impose mild oxidative stress on cells, which can activate NF-κB and
induce Bcl-2, which in-turn reduce the extent of cardiomyocyte
damage and apoptosis during the subsequent I/R process (81). Exercise can trigger short-term
inflammation, alongside an increase in white blood cells, oxidative
stress and the levels of C-reactive protein (CRP). Regular exercise
reduces CRP, IL-6 and TNF-α levels, and also increases the levels
of anti-inflammatory factors, such as IL-4 and IL-10(82). The results of the present study
demonstrated that the concentration of TNF-α in the myocardial
cells of both EXC and IPC group rats decreased significantly,
indicating that exercise reduced the concentration of TNF-α and
reduced the inflammatory and apoptotic responses to achieve
cardioprotective effects.
Apoptosis and necrosis are two types of cell death
induced by myocardial I/R. Apoptosis-related genes, such as those
of the Bcl-2 or caspase families, are known components of the
signaling pathway controlling apoptosis. The caspase family is a
protease system that directly causes apoptosis. Activation of both
the extrinsic and the intrinsic apoptotic pathway results in
cleavage of caspase-3, which activates the execution phase of
apoptosis (82). Bcl-2 enhances
proton release from the mitochondria and exerts a regulatory effect
on the opening of the mPTP, and therefore has an anti-apoptotic
effect (83). Caspase-3 is
activated by intracytoplasmic release of mitochondrial proteins,
such as cytochrome c and apoptosis-inducing factor, which is
restrained by Bcl-2 and Bcl-xL upstream of caspase. Furthermore,
the expression of antiapoptotic genes, such as Bcl-2 or Bcl-xL, is
activated by the suppression of the transcription factor NF-κB,
which restrains apoptosis (81). It
was previously demonstrated that IPC could attenuate caspase-3
activation and increase Bcl-2 performance (84,85).
These findings suggest that prevention of caspase activation may
contribute to myocardial protection.
The heat shock response is a common cellular
reaction to external stimuli, such as ischemia, hypoxia, acidosis,
oxidative stress, protein degradation, increased intracellular
calcium and energy depletion; therefore, IPC can also activate the
release of heat shock proteins (HSPs) (86,87).
Exercise also increases the expression of cardiac HSPs, as a
variety of stressors associated with exercise, including heat
stress and hypoxia, reduced intracellular pH, ROS and reactive
nitrogen species production, depletion of glucose and glycogen
stores, increase in cytosolic calcium levels and cardiomyocyte
stretching, can all contribute to HSP elevation in cardiac muscle
(88). The protective effects of
HSPs on cardiomyocytes include maintenance of myocardial calcium
ion concentration and a decrease in the release of pro-inflammatory
factors. HSPs can also regulate ATP synthesis in the mitochondria
and affect signal transduction through the MAPK/JNK pathway to
inhibit apoptosis and increase the anti-apoptotic capacity in
cardiomyocytes (89-91).
Certain researchers even consider HSPs, in addition to Bcl-2, as a
type of anti-apoptosis protein (90,92,93).
The present study found that, in addition to a smaller infarct size
in the EXC and the IPC groups, the extent of apoptosis and the
expression of caspase-3 in cardiomyocytes decreased considerably,
while the expression of Bcl-2 in cardiomyocytes in both groups
increased substantially. By inference, this may result from an
increase in HSP levels after swimming training and preconditioning
of the rats, which strengthens the effect of cardioprotection
induced by exercise, so as to improve the endurance of
cardiomyocytes to ischemia and hypoxia, resulting in less severe
MIRI. Future studies may combine measuring HSPs levels with other
related signaling elements such that the cardioprotective mechanism
of exercise can be further elucidated.
In this study, histomorphology, blood biochemistry,
TUNEL staining and cardiomyocyte TNF-α, caspase-3, and Bcl-2
expression were used to measure the severity of MIRI. The results
proved that swimming for 3 h daily for 4 continuous weeks before MI
decreased MI size, myocardial injury and cardiomyocyte apoptosis
after myocardial I/R to achieve cardioprotective effects that were
comparable to IPC. In the future, swimming training can be included
in health promotion to decrease mortality rates due to
cardiovascular diseases and improve the health of people.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by Ministry of Science and
Technology (MOST; grant no. 109-2410-H-845-038) and partly
supported by the University of Taipei (Taipei, Taiwan).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
KWT designed the experiments, and wrote and revised
the manuscript. CCL performed the primary experimental work and was
involved in drafting the manuscript. CCL and CYT contributed to
data acquisition and analysis. SKF and WCT made contributions to
the conception and design of the study, manuscript revision, and
confirmed the authenticity of all the raw data. All authors have
read and approved the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal
Experiment Committee of University of Taipei (Taipei, Taiwan; IACUC
approval no. UT110001). The animals used in the study were managed
in a humane manner in accordance with the Guide for the Care and
Use of Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Balakumar P, Maung UK and Jagadeesh G:
Prevalence and prevention of cardiovascular disease and diabetes
mellitus. Pharmacol Res. 113:600–609. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Nowbar AN, Gitto M, Howard JP, Francis DP
and Al-Lamee R: Mortality from ischemic heart disease. Circ
Cardiovasc Qual Outcomes. 12(e005375)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Patterson SW and Starling EH: On the
mechanical factors which determine the output of the ventricles. J
Physiol. 48:357–379. 1914.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Buja LM: Myocardial ischemia and
reperfusion injury. Cardiovasc Pathol. 14:170–175. 2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Singhal AK, Symons JD, Boudina S, Jaishy B
and Shiu YT: Role of endothelial cells in myocardial
ischemia-reperfusion injury. Vasc Dis Prev. 7:1–14. 2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sheehan FH, Doerr R, Schmidt WG, Bolson
EL, Uebis R, von Essen R, Effert S and Dodge HT: Early recovery of
left ventricular function after thrombolytic therapy for acute
myocardial infarction: An important determinant of survival. J Am
Coll Cardiol. 12:289–300. 1988.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Reeve JL, Duffy AM, O'Brien T and Samali
A: Don't lose heart-therapeutic value of apoptosis prevention in
the treatment of cardiovascular disease. J Cell Mol Med. 9:609–622.
2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bolli R and Marbán E: Molecular and
cellular mechanisms of myocardial stunning. Physiol Rev.
79:609–634. 1999.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Basso C and Thiene G: The pathophysiology
of myocardial reperfusion: A pathologist's perspective. Heart.
92:1559–1562. 2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ruiz-Meana M, Abellán A, Miró-Casas E and
Garcia-Dorado D: Opening of mitochondrial permeability transition
pore induces hypercontracture in Ca2+ overloaded cardiac
myocytes. Basic Res Cardiol. 102:542–552. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Argaud L, Loufouat J, Gateau-Roesch O,
Gomez L, Robert D and Ovize M: Persistent inhibition of
mitochondrial permeability transition by preconditioning during the
first hours of reperfusion. Shock. 30:552–556. 2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ishida T, Yarimizu K, Gute DC and Korthuis
RJ: Mechanisms of ischemic preconditioning. Shock. 8:86–94.
1997.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gross ER and Gross GJ: Ischemic
preconditioning and myocardial infarction: An update and
perspective. Drug Discov Today Dis Mech. 4:165–174. 2007.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Murry CE, Jennings RB and Reimer KA:
Preconditioning with ischemia: A delay of lethal cell injury in
ischemic myocardium. Circulation. 74:1124–1136. 1986.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Vegh A, Szekeres L and Parratt JR:
Transient ischaemia induced by rapid cardiac pacing results in
myocardial preconditioning. Cardiovasc Res. 25:1051–1053.
1991.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Cumming DV, Heads RJ, Brand NJ, Yellon DM
and Latchman DS: The ability of heat stress and metabolic
preconditioning to protect primary rat cardiac myocytes. Basic Res
Cardiol. 91:79–85. 1996.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Huang CH, Kim SJ, Ghaleh B, Kudej RK, Shen
YT, Bishop SP and Vatner SF: An adenosine agonist and
preconditioning shift the distribution of myocardial blood flow in
conscious pigs. Am J Physiol. 276:H368–H375. 1999.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Schott RJ, Rohmann S, Braun ER and Schaper
W: Ischemic preconditioning reduces infarct size in swine
myocardium. Circ Res. 66:1133–1142. 1990.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Liu GS, Thornton J, Van Winkle DM, Stanley
AW, Olsson RA and Downey JM: Protection against infarction afforded
by preconditioning is mediated by A1 adenosine receptors in rabbit
heart. Circulation. 84:350–356. 1991.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li Y and Kloner RA: The cardioprotective
effects of ischemic ‘preconditioning’ are not mediated by adenosine
receptors in rat hearts. Circulation. 87:1642–1648. 1993.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Sumeray MS and Yellon DM: Ischaemic
preconditioning reduces infarct size following global ischaemia in
the murine myocardium. Basic Res Cardiol. 93:384–390.
1998.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Wilmore JH: Aerobic exercise and
endurance: Improving fitness for health benefits. Phys Sportsmed.
31:45–51. 2003.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Swain DP and Franklin BA: Comparison of
cardioprotective benefits of vigorous versus moderate intensity
aerobic exercise. Am J Cardiol. 97:141–147. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Khot UN, Khot MB, Bajzer CT, Sapp SK,
Ohman EM, Brener SJ, Ellis SG, Lincoff AM and Topol EJ: Prevalence
of conventional risk factors in patients with coronary heart
disease. JAMA. 290:898–904. 2003.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lawson WE, Hui JC, Zheng ZS, Burgen L,
Jiang L, Lillis O, Oster Z, Soroff H and Cohn P: Improved exercise
tolerance following enhanced external counterpulsation: cardiac or
peripheral effect? Cardiology. 87:271–275. 1996.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Demirel HA, Powers SK, Zergeroglu MA,
Shanely RA, Hamilton K, Coombes J and Naito H: Short-term exercise
improves myocardial tolerance to in vivo ischemia-reperfusion in
the rat. J Appl Physiol (1985). 91:2205–2212. 2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Perrault H and Turcotte RA:
Exercise-induced cardiac hypertrophy. Fact or fallacy? Sports Med.
17:288–308. 1994.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jin H, Yang R, Li W, Lu H, Ryan AM,
Ogasawara AK, Van Peborgh J and Paoni NF: Effects of exercise
training on cardiac function, gene expression, and apoptosis in
rats. Am J Physiol Heart Circ Physiol. 279:H2994–H3002.
2000.PubMed/NCBI View Article : Google Scholar
|
|
29
|
McElroy CL, Gissen SA and Fishbein MC:
Exercise-induced reduction in myocardial infarct size after
coronary artery occlusion in the rat. Circulation. 57:958–962.
1978.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Bowles DK, Farrar RP and Starnes JW:
Exercise training improves cardiac function after ischemia in the
isolated, working rat heart. Am J Physiol. 263:H804–H809.
1992.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gul M, Demircan B, Taysi S, Oztasan N,
Gumustekin K, Siktar E, Polat MF, Akar S, Akcay F and Dane S:
Effects of endurance training and acute exhaustive exercise on
antioxidant defense mechanisms in rat heart. Comp Biochem Physiol A
Mol Integr Physiol. 143:239–245. 2006.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Freimann S, Scheinowitz M, Yekutieli D,
Feinberg MS, Eldar M and Kessler-Icekson G: Prior exercise training
improves the outcome of acute myocardial infarction in the rat:
Heart structure, function, and gene expression. J Am Coll Cardiol.
45:931–938. 2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ozturk N, Olgar Y, Er H, Kucuk M and
Ozdemir S: Swimming exercise reverses aging-related contractile
abnormalities of female heart by improving structural alterations.
Cardiol J. 24:85–93. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Nagaraja HS and Jeganathan PS: Forced
swimming stress-induced changes in the physiological and
biochemical parameters in albino rats. Indian J Physiol Pharmacol.
43:53–59. 1999.PubMed/NCBI
|
|
35
|
Popgeorgiev N, Sa JD, Jabbour L, Banjara
S, Nguyen TTM, Akhavan-E-Sabet A, Gadet R, Ralchev N, Manon S,
Hinds MG, et al: Ancient and conserved functional interplay between
Bcl-2 family proteins in the mitochondrial pathway of apoptosis.
Sci Adv. 6(eabc4149)2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Chou PL, Chen KH, Chang TC and Chien CT:
Repetitively hypoxic preconditioning attenuates
ischemia/reperfusion-induced liver dysfunction through upregulation
of hypoxia-induced factor-1 alpha-dependent mitochondrial Bcl-xl in
rat. Chin J Physiol. 63:68–76. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Murphy MP and Hartley RC: Mitochondria as
a therapeutic target for common pathologies. Nat Rev Drug Discov.
17:865–886. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Jiang T, Ma X, Chen H, Jia H and Xiong Y:
Diazepam ameliorated myocardial ischemia-reperfusion injury via
inhibition of C-C chemokine receptor type 2/Tumor necrosis
factor-alpha/Interleukins and Bcl-2-associated X protein/Caspase-3
pathways in experimental rats. J Vet Med Sci. 83:1965–1976.
2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Verboven M, Cuypers A, Deluyker D,
Lambrichts I, Eijnde BO, Hansen D and Bito V: High intensity
training improves cardiac function in healthy rats. Sci Rep.
9(5612)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Clark JD, Gebhart GF, Gonder JC, Keeling
ME and Kohn DF: The 1996 guide for the care and use of laboratory
animals. ILAR Journal. 38:41–48. 1997.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Gazdag P, Oravecz K, Acsai K,
Demeter-Haludka V, Ördög B, Szlovák J, Kohajda Z, Polyák A, Barta
BA, Oláh A, et al: Increased Ca2+ content of the
sarcoplasmic reticulum provides arrhythmogenic trigger source in
swimming-induced rat athlete's heart model. Sci Rep.
10(19596)2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Lee HW, Han TH, Yi KJ, Choi MC, Lee SY and
Ryu PD: Time course of diurnal rhythm disturbances in autonomic
function of rats with myocardial infarction. Auton Neurosci.
179:28–36. 2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Zhao C, Yin M, Li F, Ling W, Luo C and Qin
S: Mechanisms of Paeoniflorin against myocardial ischemia
reperfusion injury based on network pharmacology. Mater Exp.
11:1505–1515. 2021.
|
|
44
|
El-Shitany NA, Tolba OA, El-Shanshory MR
and El-Hawary EE: Protective effect of carvedilol on
adriamycin-induced left ventricular dysfunction in children with
acute lymphoblastic leukemia. J Card Fail. 18:607–613.
2012.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Sabatasso S, Mangin P, Fracasso T, Moretti
M, Docquier M and Djonov V: Early markers for myocardial ischemia
and sudden cardiac death. Int J Legal Med. 130:1265–1280.
2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Thornberry NA and Lazebnik Y: Caspases:
Enemies within. Science. 281:1312–1316. 1998.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Guski H, Meerson FZ and Wassilew G:
Comparative study of ultrastructure and function of the rat heart
hypertrophied by exercise or hypoxia. Exp Pathol. 20:108–120.
1981.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Frank A, Bonney M, Bonney S, Weitzel L,
Koeppen M and Eckle T: Myocardial ischemia reperfusion injury: From
basic science to clinical bedside. Semin Cardiothorac Vasc Anesth.
16:123–132. 2012.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Pluijmert NJ, Bart CI, Bax WH, Quax PH and
Atsma DE: Effects on cardiac function, remodeling and inflammation
following myocardial ischemia-reperfusion injury or unreperfused
myocardial infarction in hypercholesterolemic APOE*
3-Leiden mice. Sci Rep. 10(16601)2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Hong XY, Hong X, Gu WW, Lin J and Yin WT:
Cardioprotection and improvement in endothelial-dependent
vasodilation during late-phase of whole body hypoxic
preconditioning in spontaneously hypertensive rats via VEGF and
endothelin-1. Eur J Pharmacol. 842:79–88. 2019.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Bolli R: The late phase of
preconditioning. Circ Res. 87:972–983. 2000.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Marini M, Lapalombella R, Margonato V,
Ronchi R, Samaja M, Scapin C, Gorza L, Maraldi T, Carinci P,
Ventura C and Veicsteinas A: Mild exercise training,
cardioprotection and stress genes profile. Eur J Appl Physiol.
99:503–510. 2007.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Labarca G, Gower J, Lamperti L, Dreyse J
and Jorquera J: Chronic intermittent hypoxia in obstructive sleep
apnea: A narrative review from pathophysiological pathways to a
precision clinical approach. Sleep Breath. 24:751–760.
2020.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Sanderson JE, Fang F, Lu M, Ma CY and Wei
YX: Obstructive sleep apnoea, intermittent hypoxia and heart
failure with a preserved ejection fraction. Heart. 107:190–194.
2021.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Nanduri J and Nanduri RP: Cellular
mechanisms associated with intermittent hypoxia. Essays Biochem.
43:91–104. 2007.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Chen L, Shi D and Guo M: The roles of
PKC-delta and PKC-epsilon in myocardial ischemia/reperfusion
injury. Pharmacol Res. 170(105716)2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Radak Z, Chung HY and Goto S: Systemic
adaptation to oxidative challenge induced by regular exercise. Free
Radic Biol Med. 44:153–159. 2008.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Powers SK, Deminice R, Ozdemir M,
Yoshihara T, Bomkamp MP and Hyatt H: Exercise-induced oxidative
stress: Friend or foe? J Sport Health Sci. 9:415–425.
2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Soukhova-O'Hare GK, Ortines RV, Gu Y,
Nozdrachev AD, Prabhu SD and Gozal D: Postnatal intermittent
hypoxia and developmental programming of hypertension in
spontaneously hypertensive rats: The role of reactive oxygen
species and L-Ca2+ channels. Hypertension. 52:156–162.
2008.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Boulghobra D, Coste F, Geny B and Reboul
C: Exercise training protects the heart against
ischemia-reperfusion injury: A central role for mitochondria? Free
Radic Biol Med. 152:395–410. 2020.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Lee MG, Park KS, Kim DU, Choi SM and Kim
HJ: Effects of high-intensity exercise training on body
composition, abdominal fat loss, and cardiorespiratory fitness in
middle-aged Korean females. Appl Physiol Nutr Metab. 37:1019–1027.
2012.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Pattwell DM, McArdle A, Morgan JE,
Patridge TA and Jackson MJ: Release of reactive oxygen and nitrogen
species from contracting skeletal muscle cells. Free Radic Biol
Med. 37:1064–1072. 2004.PubMed/NCBI View Article : Google Scholar
|
|
64
|
McArdle A, van der Meulen J, Close GL,
Pattwell D, Van Remmen H, Huang TT, Richardson AG, Epstein CJ,
Faulkner JA and Jackson MJ: Role of mitochondrial superoxide
dismutase in contraction-induced generation of reactive oxygen
species in skeletal muscle extracellular space. Am J Physiol Cell
Physiol. 286:C1152–C1158. 2004.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Rinaldi B, Corbi G, Boccuti S, Filippelli
W, Rengo G, Leosco D, Rossi F, Filippelli A and Ferrara N: Exercise
training affects age-induced changes in SOD and heat shock protein
expression in rat heart. Exp Gerontol. 41:764–770. 2006.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Dasgupta A, Wu D, Tian L, Xiong PY,
Dunham-Snary KJ, Chen KH, Alizadeh E, Motamed M, Potus F, Hindmarch
CCT and Archer SL: Mitochondria in the pulmonary vasculature in
health and disease: Oxygen-sensing, metabolism, and dynamics. Compr
Physiol. 10:713–765. 2020.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Siu PM, Bryner RW, Martyn JK and Always
SE: Apoptotic adaptations from exercise training in skeletal and
cardiac muscles. FASEB J. 18:1150–1152. 2004.PubMed/NCBI View Article : Google Scholar
|
|
68
|
O'Brien PJ, Smith DE, Knechtel TJ, Marchak
MA, Pruimboom-Brees I, Brees DJ, Spratt DP, Archer FJ, Butler P,
Potter AN, et al: Cardiac troponin I is a sensitive, specific
biomarker of cardiac injury in laboratory animals. Lab Anim.
40:153–171. 2006.PubMed/NCBI
|
|
69
|
Evran B, Karpuzoğlu H, Develi S, Kalaz EB,
Soluk-Tekkeşin M, Olgaç V, Doğru-Abbasoğlu S and Uysal M: Effects
of carnosine on prooxidant-antioxidant status in heart tissue,
plasma and erythrocytes of rats with isoproterenol-induced
myocardial infarction. Pharmacol Rep. 66:81–86. 2014.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Hausenloy DJ, Schulz R, Girao H, Kwak BR,
De Stefani D, Rizzuto R, Bernardi P and Di Lisa F: Mitochondrial
ion channels as targets for cardioprotection. J Cell Mol Med.
24:7102–7114. 2020.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Marcil M, Bourduas K, Ascah A and Burelle
Y: Exercise training induces respiratory substrate-specific
decrease in Ca2+-induced permeability transition pore
opening in heart mitochondria. Am J Physiol Heart Circ Physiol.
290:H1549–H1557. 2006.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Kavazis AN, McClung JM, Hood DA and Powers
SK: Exercise induces a cardiac mitochondrial phenotype that resists
apoptotic stimuli. Am J Physiol Heart Circ Physiol. 294:H928–H935.
2008.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Mnafgui K, Hajji R, Derbali F, Khlif I,
Kraiem F, Ellefi H, Elfeki A, Allouche N and Gharsallah N:
Protective effect of hydroxytyrosol Against cardiac remodeling
after isoproterenol-induced myocardial infarction in rat.
Cardiovasc Toxicol. 16:147–155. 2016.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Leite CF, Lopes CS, Alves AC, Fuzaro CS,
Silva MV, Oliveira LF, Garcia LP, Farnesi TS, Cuba MB, Rocha LB, et
al: Endogenous resident c-kit cardiac stem cells increase in mice
with an exercise-induced, physiologically hypertrophied heart. Stem
Cell Res. 15:151–164. 2015.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Pan C, Yuan Q and Xu F: Progress in
cardiorespiratory ischemia-reperfusion injury. In: Sudden Death. pp
79-92, 2021.
|
|
76
|
Rath PC and Aggarwal BB: TNF-induced
signaling in apoptosis. J Clin Immunol. 19:350–364. 1999.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Cook AD, Lee MC, Saleh R, Khiew HW,
Christensen AD, Achuthan A, Fleetwood AJ, Lacey DC, Smith JE,
Förster I and Hamilton JA: TNF and granulocyte macrophage-colony
stimulating factor interdependence mediates inflammation via CCL17.
JCI Insight. 3(e99249)2018.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Sheikh MS and Huang Y: Death receptor
activation complexes: It takes two to activate TNF receptor 1. Cell
Cycle. 2:550–552. 2003.PubMed/NCBI
|
|
79
|
Balzano T, Arenas YM, Dadsetan S, Forteza
J, Gil-Perotin S, Cubas-Nuñez L, Casanova B, Gracià F,
Varela-Andrés N, Montoliu C, et al: Sustained hyperammonemia
induces TNF-α IN Purkinje neurons by activating the TNFR1-NF-κB
pathway. J Neuroinflammation. 17(70)2020.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Takeshita M, Tani T, Harada S, Hayashi H,
Itoh H, Tajima H, Ohnishi I, Takamura H, Fushida S and Kayahara M:
Role of transcription factors in small intestinal
ischemia-reperfusion injury and tolerance induced by ischemic
preconditioning. Transplant Proc. 42:3406–3413. 2010.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Huldani Pattelongi I, Massi MN, Idris I,
Bukhari A, Widodo ADW, Uinarni H, Carmelita AB, Trisia A, Gunma S,
Prayudhistya BKA and Achmad H: Cortisol, IL-6, TNF Alfa, Leukocytes
and DAMP on Exercise. Sys Rev Pharm. 11:474–485. 2020.
|
|
82
|
Wertz IE: TNFR1-activated NF-κB signal
transduction: Regulation by the ubiquitin/proteasome system. Curr
Opin Chem Biol. 23:71–77. 2014.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Valen G: The basic biology of apoptosis
and its implications for cardiac function and viability. Ann Thorac
Surg. 75:S656–S660. 2003.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Ji HB, Zhai QW, Liu XY and Zheng ZC:
Transcription regulation of bcl-2 gene. Sheng Wu Hua Xue Yu Sheng
Wu Wu Li Xue Bao (Shanghai). 32:95–99. 2000.PubMed/NCBI
|
|
85
|
Yaoita H, Ogawa K, Maehara K and Maruyama
Y: Attenuation of ischemia/reperfusion injury in rats by a caspase
inhibitor. Circulation. 97:276–281. 1998.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Shimamoto A, Matsuo E, Kaneda S, Ito A,
Kawaguchi K and Takao M: Heat shock protein 70 performs as
pharmacological preconditioning to protect against lung ischemia
reperfusion injury through toll-like receptor 4 signaling. J Heart
Lung Transplant. 40(S69)2021.
|
|
87
|
Mitra S, Dasgupta R and Bagchi A: Heat
shock proteins and their associated oxidative stress-induced heart
disease. Modulation of Oxidative Stress in Heart Disease, 215-235,
2019.
|
|
88
|
Shamsi MM, Hassan ZM and Gharakhanlou R:
Exercise-induced chaperokine activity of hsp70: Possible role in
chronic diseases. In: Chaperokine Activity of Heat Shock Proteins.
Springer, Cham, pp193-209, 2019.
|
|
89
|
Milne KJ, Thorp DB, Krause M and Noble EG:
Core temperature is a greater influence Than endogenous
17β-estradiol on the exercise-induced accumulation of myocardial
heat shock protein mRNA. Can J Physiol Pharmacol. 89:855–860.
2011.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Liu X, Zhang C, Zhang C, Li J, Guo W, Yan
D, Yang C, Zhao J, Xia T, Wang Y, et al: Heat shock protein 70
inhibits cardiomyocyte necroptosis through repressing autophagy in
myocardial ischemia/reperfusion injury. In Vitro Cell Dev Biol
Anim. 52:690–698. 2016.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Rani N, Bharti S, Manchanda M, Nag TC, Ray
R, Chauhan SS, Kumari S and Arya DS: Regulation of heat shock
Proteins 27 and 70, p-Akt/p-eNOS and MAPKs by naringin dampens
myocardial injury and dysfunction in vivo after
ischemia/reperfusion. PLoS One. 8(e82577)2013.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Wu J, Chen S, Liu Y, Liu Z, Wang D and
Cheng Y: Therapeutic perspectives of heat shock proteins and their
protein-protein interactions in myocardial infarction. Pharmacol
Res. 160(105162)2020.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Hsu SF, Hsu CC, Cheng BC and Lin CH:
Cathepsin B is involved in the heat shock induced cardiomyocytes
apoptosis as well as the anti-apoptosis effect of HSP-70.
Apoptosis. 19:1571–1580. 2014.PubMed/NCBI View Article : Google Scholar
|