Introduction
Hyperammonemia is a potential complication of
chronic or acute liver disease, as well as various non-hepatic
conditions. The causes of hyperammonemia in adults can generally be
divided into two main categories: Increased ammonia production
(such as via infection, high protein load, multiple myeloma, and
renal failure) and decreased ammonia elimination (such as from
liver failure, drugs, and inborn errors of metabolism) (1). However, hyperammonemia is often caused
by severe liver disease. In fact, the incidence of hyperammonemia
in hepatic encephalopathy (HE) is as high as 90% (2). Clinically, HE is a group of
neuropsychiatric signs and symptoms in patients with liver disease
or a portosystemic shunt. HE can be reversed by early treatment of
the underlying causes (3,4). Neuroimaging, particularly magnetic
resonance imaging (MRI), plays a vital role in elucidating the
neural mechanisms underlying HE (5). The brain MRI of patients with chronic
HE typically shows cerebral atrophy and bilateral symmetrical
hyperintensities of the globus pallidus on T1-weighted images
without corresponding signal intensity in T2-weighted images
(6,7). Conversely, acute hyperammonemic
encephalopathy often presents bilateral symmetrical cortical signal
abnormalities with corresponding signal intensity changes in
T2-weighted images and diffusion restriction (7-10).
The current study aims to present the clinical
characteristics and brain MRI findings of acute HE associated with
hyperammonemia in 5 adult patients.
Case report
Patients and methods
This retrospective case series study included 5
patients >18 years of age, treated between January 2016 and
December 2021 at King Fahad Specialist Hospital-Dammam (KFSH-D)
(Dammam, Saudi Arabia). The patients were all identified to have
documented hyperammonemic HE. The study was approved by the Ethics
Committee of King Fahad Specialist Hospital Dammam
(IRB-Pub-024-01). Written informed consent for publication was
obtained from all subjects. Clinical and laboratory data were
collected using electronic medical records for each patient. All 5
patients were critically ill in the intensive care unit (ICU) due
to a decline in their level of consciousness. The patients
underwent an MRI of the brain 3-10 days after the onset of
symptoms. A neuroradiologist analyzed all the images. The common
clinical features of the patients were that they presented with
decreased levels of consciousness secondary to acute hepatic
failure with hyperammonemia. The brain MRI of each patient was
performed using a 3-T MRI scanner (Magnetom Skyra; Siemens
Healthineers) with a 20-channel phased array head coil. The imaging
protocol included axial and sagittal T1-weighted images, axial and
coronal T2-weighted images, axial fluid-attenuated inversion
recovery (FLAIR), axial susceptibility weighted imaging, axial
echo-planar diffusion-weighted imaging and apparent diffusion
coefficient map. Additional axial and sagittal T1-weighted images
were recorded for 2 patients after administering the intravenous
contrast agent.
Results
A total of 5 patients (2 men and 3 women) were
included in this study (Table I).
The causes of hepatic failure were metastatic colon cancer, chronic
inflammatory hepatitis in the background of sickle cell anemia,
primary sclerosing cholangitis associated with ulcerative colitis,
autoimmune hepatitis, and post-bypass surgery steatohepatitis,
respectively. Plasma ammonia levels ranged from 165 to 512 µmol/l
(normal range, 11-32 µmol/l). Diffuse cortical swelling was
observed in the MRI brain scans of all patients, involving the
frontal, temporal, and parietal lobes, as well as the insula. The
underlying white matter, peri-rolandic area, and posterior
occipital lobe were spared. These cortical MRI abnormalities have
been shown to worsen with increased ammonia levels (Table I). In 4 of the 5 patients, the
bilateral thalami were affected, and another patient showed a few
focal ischemic lesions in the caudothalamic groove and splenium of
the corpus callosum. Of the 5 patients, 2 survived, while 3 died
due to septic shock and multiorgan failure.
 | Table IClinical summary of cases
reported. |
Table I
Clinical summary of cases
reported.
| Characteristic | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 |
|---|
| Age, years | 37 | 27 | 36 | 18 | 51 |
| Sex | Male | Male | Female | Female | Female |
| Diagnosis | Acute liver failure
secondary to liver surgery | Decompensated liver
failure | Acute graft rejection
post-liver transplant | Acute liver failure
secondary to autoimmune hepatitis | Acute-on-chronic
steatohepatitis |
| GCS | 4/15 | 3/15 | 4/15 | 4/15 | 5/15 |
| Presence of
seizures | No | No | Yes | Yes | Yes |
| Liver enzymes,
U/la | AST, 105; ALT,
228 | AST, 263; ALT,
137 | AST, 1,856; ALT,
1,325 | AST, 1,914; ALT,
5,075 | AST, 93; ALT, 63 |
| Ammonia level,
µmol/lb | 165-188 | 193 | 512.75 | 310 | 174 |
| Total bilirubin
µmol/lc | 211 | 886 | 344.6 | 334 | 158 |
| Period between onset
of symptoms and MRI, days | 7 | 8 | 3 | 5 | 10 |
| Area of brain
involvement in MRI | Insular cortex,
frontal lobe sparing the peri-rolandic region, posterior temporal
lobe, anterior cingulate gyrus and the bilateral anterola-teral
thalami | Insular cortex,
frontal lobe sparing the peri-rolandic region, temporal lobe,
parietal lobe, and anterior cingulate gyri | Insular cortex,
frontal lobe sparing the peri-rolandic region, temporal lobe,
parietal lobe, anterior and posterior cingulate gyri, and the
bilateral thalami | Insular cortex,
frontal lobe sparing the peri-rolandic region, temporal lobe,
parietal lobe, anterior and posterior cingulate gyri, and the
bilateral postero-medial thalami | Insular cortex,
frontal lobe sparing the peri-rolandic region, posterior temporal
lobe, parietal lobe, and anterior cingulate gyrus. |
| Outcome | Recovered | Died | Died | Died | Recovered |
Case 1
A 37-year-old man with a known case of colon
adenocarcinoma with liver metastasis was admitted electively in
November 2017 under the care of the hepatobiliary team at KFSH-D
for a hepatectomy. However, the procedure was complicated by portal
vein air thrombosis and hypotension, necessitating an ICU
admission. The serum ammonia level was significantly elevated at
165 µmol/l. Additionally, liver enzymes were increased, with an
aspartate transferase (AST) level of 105 U/l (normal range, 8-38
U/l), an alanine aminotransferase (ALT) level of 228 U/l (normal
range, 17-45 U/l), a total bilirubin level of 211 µmol/l (normal
range, 4-21 µmol/l) and an international normalized ratio (INR) of
2.4 (normal range, 0.80-1.20). The patient's level of consciousness
remained low even after sedation was stopped. Initially, the
patient [Glasgow Coma Scale (GCS) score of 6/15] could open their
eyes in response to painful stimulation, but had no verbal or motor
responses, although the brainstem reflexes were intact.
Routine electroencephalography (EEG) showed
generalized slowing within the delta range and no epileptiform
discharges or electrographic seizures. The brain MRI on the seventh
day post-surgery demonstrated bilateral symmetrical cortical edema
with marked diffusion restriction involving the insula, temporal
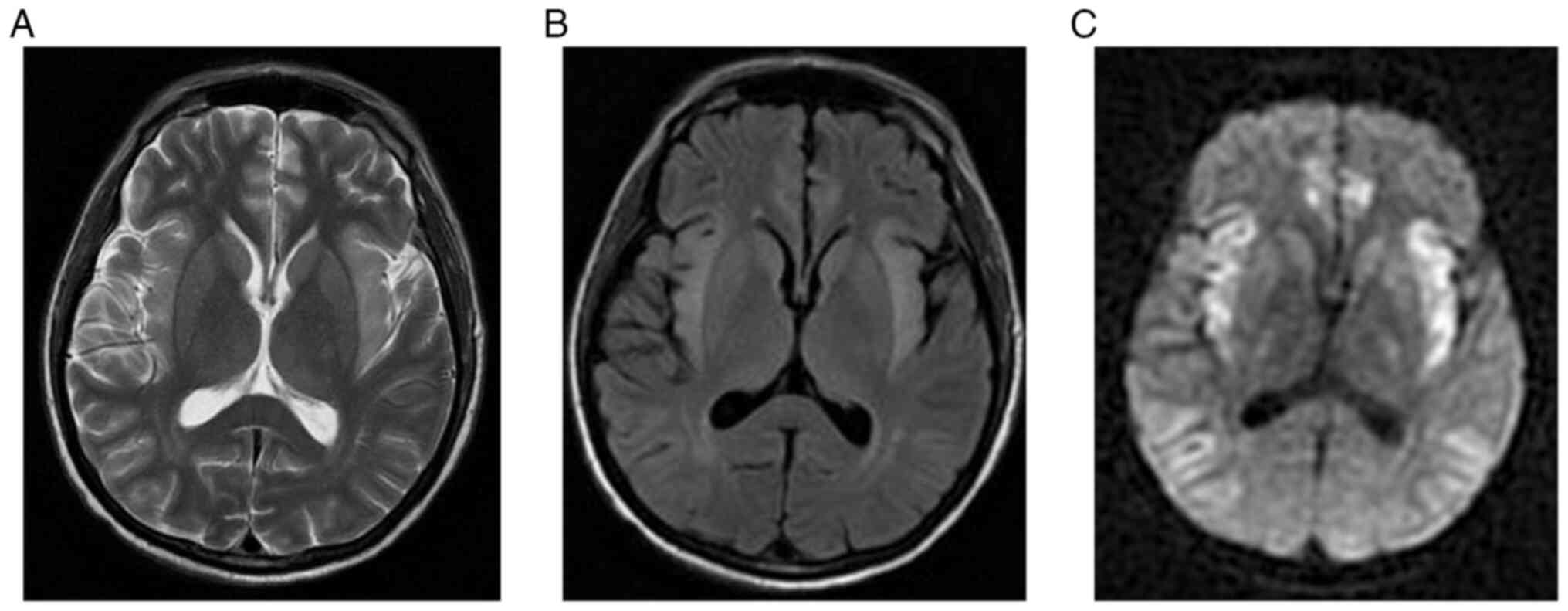
lobe, cingulate gyrus, and thalami (Fig. 1). After 10 days, the patient's
condition deteriorated even more. The liver enzymes increased (AST,
84 U/l; ALT, 139 U/l; and total bilirubin, 245 µmol/l), and the
serum ammonia level reached 188 µml/l due to acute liver failure. A
subsequent follow-up MRI revealed an increase in bilateral
symmetrical, predominantly cortical edema, with intermediate signal
intensity on T2/FLAIR, but a decrease in diffusion restriction
(Fig. 2). The patient exhibited
marked improvement gradually throughout the month following the
liver transplant. A follow-up was conducted in the clinic 2 months
after discharge, and the patient was doing well. Furthermore, the
follow-up lab results indicated a significant recovery of liver
enzymes, with AST at 23 U/l, ALT at 24 U/l, and serum ammonia at 19
µml/l. However, the patient declined further brain imaging due to
the resolution of the symptoms.
 | Figure 1Initial brain MRI for case 1. (A)
T2-weighted MRI, (B) FLAIR, (C) DWI and (D) ADC map demonstrating
asymmetric cortical high T2 and FLAIR signal intensity involving
the bilateral insular cortex, temporal lobes, cingulate gyri and
bilateral lateral thalami with corresponding diffusion restriction.
The bilateral occipital lobes are spared. At the level of vertex,
(E) T2-weighted image, (F) FLAIR image, (G) DWI and (H) ADC map
demonstrating symmetric cortical high T2 and FLAIR signal
intensities involving the frontal and parietal lobes with
corresponding restricted diffusion. The bilateral peri-Rolandic
regions are spared. FLAIR, fluid-attenuated inversion recovery;
DWI, diffusion-weighted imaging; ADC, apparent diffusion
coefficient. |
Case 2
A 27-year-old man with a known history of sickle
cell disease and chronic inflammatory hepatitis was admitted to the
ICU of the Procare Hospital (Khobar, Saudi Arabia) with low
consciousness and severe jaundice. In December 2018, the patient
was transferred to KFSH-D, where a diagnosis of decompensated liver
failure complicated by disseminated intravascular coagulation and
multiorgan failure was formed based on clinical and laboratory
findings. During the physical examination, it was observed that the
patient was in a deep coma, with a GCS score of 3 out of 15 and
intact brainstem reflexes. The patient exhibited generalized
hypotonia and did not respond to painful stimuli. The deep tendon
reflexes were decreased, and there was no bilateral reaction in the
plantar reflexes.
Laboratory tests showed leukocytosis, with a white
blood cell count of 28.3x109/l (normal range,
3.4-9.6x109/l), a low hemoglobin level of 5.8 g/dl
(normal range, 13.6-16.9 g/dl), thrombocytopenia, with a platelet
count of 58x103/l (normal range,
152-324x103/l), and a high ammonia level of 193 µmol/l.
Liver function tests indicated a total bilirubin level of 886
µmol/l, a conjugated bilirubin level of 809 µmol/l (normal range,
0.8-3.0 µmol/l), an alkaline phosphatase (ALP) level of 405 U/l
(normal range, 54-144 U/l), an ALT level of 137 U/l (normal range,
17-45 U/l) and an AST level of 263 U/l (normal range, 8-38 U/l).
Renal function was also elevated, with a blood urea nitrogen (BUN)
level of 50 mmol/l (normal range, 2.1-8.5 mmol/l) and a creatinine
level of 256 µmol/l (normal range, 61.9-114.9 µmol/l). The brain
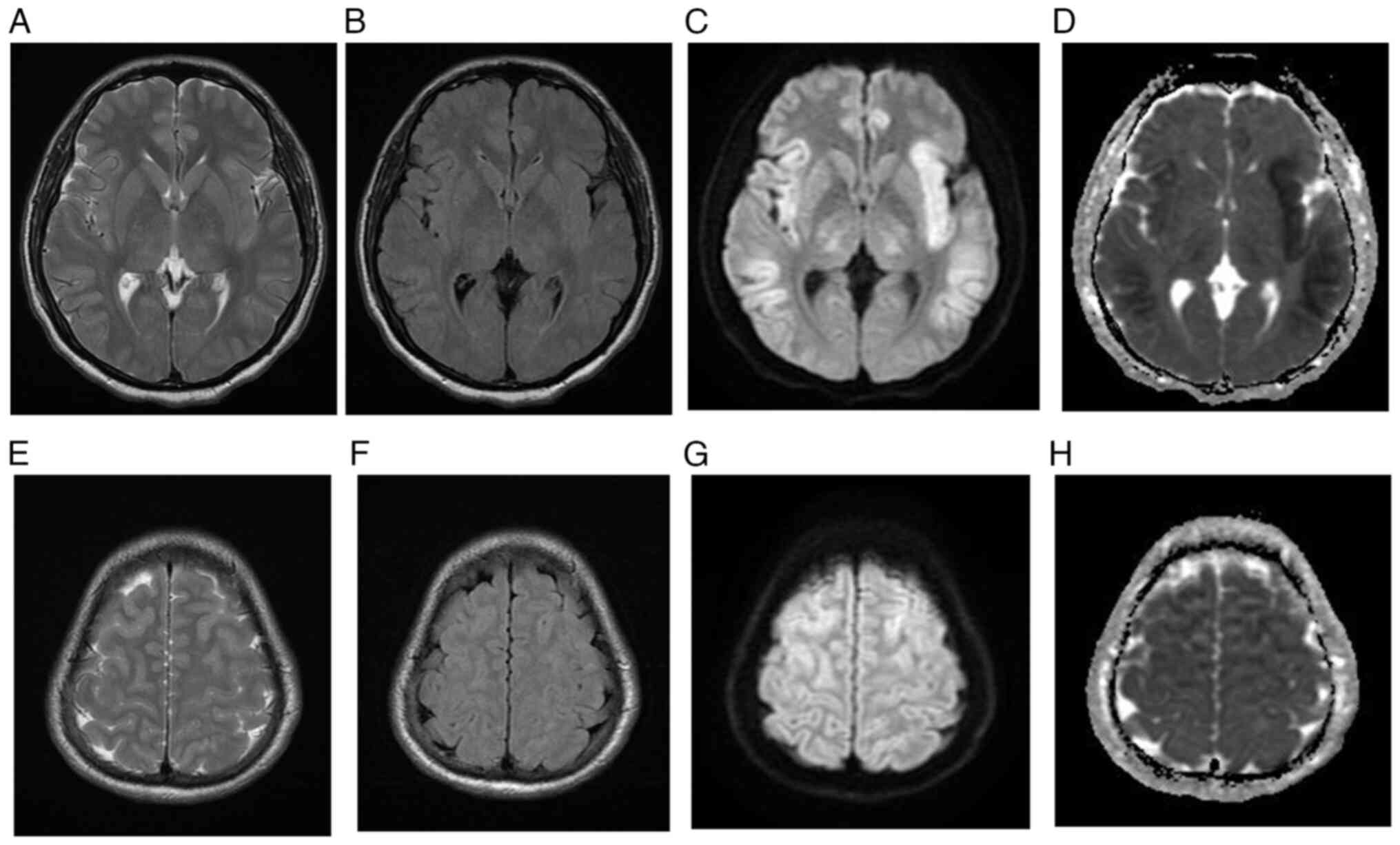
MRI, taken 8 days post-admission (Fig.
S1), showed bilateral, nearly symmetric cortical swelling, with
FLAIR/T2 high signal intensity and diffusion restriction affecting
the frontal, parietal, and temporal regions while sparing the
peri-rolandic and occipital areas. The patient's clinical condition
deteriorated steadily, and they succumbed to septic shock and
multiorgan failure 2 weeks later.
Case 3
A 36-year-old woman with ulcerative colitis (UC)
underwent a deceased donor liver transplant (split liver) smooth
surgery hepaticojejunostomy for primary sclerosing cholangitis in
Feb 2021 at KFSH-D, and recovered without any complications.
However, on the fifth day after surgery the patient's level of
consciousness began to deteriorate. During a physical examination,
while not sedated, it was noted that the patient had an elevated
temperature of 38.1˚C, generalized jaundice, and a GCS score of 4
out of 15. The patient later experienced multiple generalized
tonic-clonic convulsions, and the EEG showed generalized background
slowing with epileptiform discharges. At that time, laboratory
tests revealed high level of ammonia (512.75 µmol/l), liver enzymes
(total bilirubin, 344.6 µmol/l; conjugated bilirubin, 259 µmol/l;
ALP, 118 U/l; ALT, 1,325 U/l; and AST, 1,856 U/l) and renal
function (BUN, 10.5 mmol/l; and creatinine, 179 µmol/l). The
seizures were controlled using a midazolam infusion at 2 mg/h for 3
days. An MRI of the brain 3 days later showed diffuse symmetrical
cerebral edema with high signal intensity and diffusion
restriction, sparing the bilateral occipital lobes and
peri-rolandic region (Fig. S2).
The patient passed away on the next day.
Case 4
An 18-year-old woman with autoimmune hepatitis was
transferred from Aramco Hospital (Dhahran, Saudi Arabia) to the ICU
at KFSH-D in February 2020. The patient had acute liver failure,
decreased consciousness, and a new left-sided focal unaware
seizure. The seizure was aborted after receiving levetiracetam
loading of 3,000 mg over 10 min and a midazolam infusion of 2 mg/h
over 24 h. The initial computed tomography brain scan (CT) showed
cerebral edema, diffuse sulcal effacement, and crowding of the
foramen magnum. The patient underwent a liver transplant procedure.
Brain MRI 3 days later showed diffuse symmetrical cerebral cortical
abnormalities with high signal intensity on T2-weighted imaging and
FLAIR, affecting the splenium of the corpus callosum, the left
Globus pallidus, bilateral thalamic regions, the posterior limb of
the internal capsule, and the hippocampi. This was associated with
diffusion restriction, but the occipital lobes and peri-rolandic
region were spared (Fig. S3).
After withdrawing the midazolam infusion, the physical examination
showed a GCS score of 4 out of 15, with all brainstem reflexes
intact, generalized hypotonia and hyperreflexia, and unresponsive
plantar reflexes. Laboratory tests revealed an ammonia level of 310
µmol/l and elevated liver enzymes, with an ALT level of 5,075 U/l,
an AST level of 1,914 U/l, and a total bilirubin level of 334
µmol/l.
A follow-up EEG showed a generalized slowing in the
delta range, and no epileptiform discharges were present.
Additionally, a follow-up MRI, conducted 10 days after the initial
scan, showed interval evolution of the intraparenchymal changes
(Fig. S4). The patient passed away
due to septic shock and multiorgan failure after 39 days in the
ICU.
Case 5
A 51-year-old woman with a known case of
hypothyroidism and a history of gastric bypass surgery complicated
by steatohepatitis presented with impaired consciousness and
jaundice. The patient was admitted to KFSH-D in December 2018
through the Emergency Department with acute-on-chronic liver
failure and pulmonary edema that required intubation. The patient's
level of consciousness deteriorated within 3 days. Upon
examination, the patient was unresponsive, with a GCS of 5 out of
15, and preserved brainstem reflexes. Laboratory investigations
revealed a high serum ammonia level of 174 µmol/l, mildly elevated
liver enzymes (AST, 93 U/l; and ALT, 63 U/l), and a total bilirubin
level of 158 µmol/l. The EEG results showed generalized slowing of
the background without epileptiform discharges. A brain MRI scan
performed 10 days after the onset of symptoms showed bilateral
asymmetric cortical swelling and diffusion restriction in the
insula, cingulate gyri, and bilateral hippocampi, with
corresponding T2/FLAIR hyperintensity and T1 hyperintensity sparing
the occipital lobes and peri-rolandic region (Fig. S5). The patient experienced a
prolonged generalized convulsion for 30 min, which was controlled
with levetiracetam in a 3,000-mg intravenous infusion over 15 min,
followed by 750 mg orally twice and lamotrigine at 50 mg twice. The
patient's condition improved as the seizure subsided and the
ammonia level normalized, but residual memory impairment was still
present. A follow-up brain MRI revealed complete resolution of the
cortical and subcortical T2 and FLAIR high signal intensities,
along with marked bilateral symmetrical brain atrophy (Fig. S6). The patient was regularly
followed up by the neurology team and remained seizure-free after
adding phenytoin at 100 mg every 12 h to the treatment, but still
complains of memory impairment.
Discussion
Despite the diverse and complex presentations of the
5 patients in the present study, the clinical courses were
characterized by the interplay of acute hepatic failure
superimposed on preexisting chronic liver disease. This resulted in
a rapid rise in serum ammonia levels, leading to a marked decline
in consciousness and the development of HE. Simultaneously, brain
MRI revealed extensive cortical signal-intensity changes with
associated restricted diffusion. Regardless of the primary
etiology, the patients eventually shared similar MRI brain findings
that showed bilateral symmetrical involvement of the insular cortex
and cingulate gyri on FLAIR and T2-weighted imaging sequences, with
diffusion restriction sparing the peri-rolandic and occipital
areas. These widespread cortical lesions may represent
hyperammonemia-related brain edema (3,8) and
have been reported previously in several case series (8-12).
For example, U-King-Im et al (8) reported the cases of 4 patients with
similar findings, 3 of whom had liver damage due to acute hepatic
failure and sepsis with a background of chronic liver diseases
(8). The extensive cortical brain
MRI abnormalities seen in these patients could be indicative of
other conditions, such as hypoxic-ischemic encephalopathy, limbic
encephalitis, acute hypertension, hyponatremia, or
Creutzfeldt-Jakob disease (13).
However, none of the patients in the present study exhibited
clinical or laboratory evidence supporting these conditions.
Notably, the peri-rolandic and occipital areas are
usually relatively spared in hyerammonemic encephalopathy (10). A recent report detailed the cases of
3 patients with hyperammonemic encephalopathy stemming from
different causes, including infectious, toxic, and cirrhotic
origins, all of which demonstrated similar findings on MRI
(9). Furthermore, Takanashi et
al (14) described 3 cases of
hyperammonemia encephalopathy secondary to late-onset ornithine
transcarbamylase deficiency, each showing evidence of injury to the
cingulate and insular cortices (14). This later study also indicated that
the peri-rolandic and occipital cortices were spared. The authors
proposed the vulnerability of the cingulate gyrus and insular
cortex due to hyperammonemia-hyper glutaminergic encephalopathy
(14). This unique sparing pattern
is significant, as it can help differentiate hyperammonemic
encephalopathy from other conditions with similar MRI findings.
However, it remains unclear why certain brain areas, such as the
insular cortex or cingulate gyrus, appear especially susceptible to
the toxic effects of ammonia.
The involvement of various brain structures,
including the thalami, basal ganglia, periventricular white matter,
brainstem, and peri-rolandic and occipital cortical regions, has
been inconsistently reported in patients with hyperammonemic
encephalopathy (8,15,16).
In the present cases, in addition to the insular and cingulate
gyri, the thalami were affected in the MRI of patients 1, 3, 4, and
5, possibly due to their chronic liver diseases. However, a
premorbid brain MRI was not available to rule out this
possibility.
In the ICU, the patients experienced a range of
systemic complications, including acute renal failure, seizure, and
sepsis. These issues may have contributed to the MRI changes
observed. However, not every patient faced these complications, yet
they all displayed similar MRI cortical signal abnormalities. The
symmetrical changes in the insular and cingulate gyri suggest a
toxic effect. Additionally, the resemblance of the MRI findings in
the present cases to those reported in the previous literature
(5,8,9,10,12,14),
even in patients without hepatic failure, seizure or
hypoxic-ischemic injury, strongly supports the idea that the brain
MRI changes described are directly linked to hyperammonemia.
Ammonia is produced through protein metabolism in
the gastrointestinal tract, with the liver being the primary site
of ammonia breakdown in the urea cycle. Elevated serum ammonia
levels indicate compromised hepatic function or the presence of a
portosystemic shunt. Ammonia can cross the blood-brain barrier
through diffusion in its unionized form (NH3) and active
transport in its ionized form (NH4+)
(17). Astrocytes utilize ammonia
to generate high concentrations of glutamine (17), rapidly increasing cellular
osmolarity as glutamine accumulates, leading to astrocyte swelling
and heightened intracranial pressure (18). Furthermore, hyperammonemia disrupts
brain energy metabolism, causing an increase in the
lactate-to-pyruvate ratio (17).
Conversely, brain edema is less common in cases of chronic liver
failure, as there is sufficient time for compensatory cellular
adaptation mechanisms to respond to osmotic changes (19).
All patients in the present case series had high
serum levels of ammonia. However, in cases 1 and 4, an association
was observed between serum ammonia levels, deterioration of
consciousness, and cortical MRI changes. Higher ammonia levels
appeared to lead to deeper comas and increased cortical brain MRI
hyperintensities. This association has also been noted in previous
reports (8,20). By contrast, plasma ammonia levels
are often measured in clinical settings to assist in the diagnosis;
these results should be interpreted cautiously, as elevated ammonia
levels can arise from various conditions apart from acute liver
failure, such as the administration of drugs (e.g., sodium
valproate and chemotherapy), infections, hypothyroidism, multiple
myeloma, inborn errors of metabolism, and post-lung or -bone marrow
transplantation (11,12,20-23).
Prolonged and high serum hyperammonemia levels can
lead to significant brain injuries and devastating complications,
such as brain edema and herniation. In the long term, insular and
cingulate atrophy may occur (12,21,23-25).
Therefore, timely recognition and treatment of hyperammonemia are
crucial to avoid these complications. In the present study,
patients 2, 3, and 4 exhibited elevated serum ammonia levels and
sustained more extensive brain injuries, resulting in poor
prognoses. Conversely, patients 1 and 5, who had lower serum
ammonia levels, survived after their plasma ammonia levels
normalized, although patient 5 experienced residual memory
impairment, likely due to prolonged seizure activity and
hyperammonemia encephalopathy. The main limitations of the present
study are its retrospective design and the lack of baseline imaging
and laboratory data before the recent admission, which warrants the
need for future studies with larger cohorts and prospective designs
to perform correlational analyses between blood ammonia levels and
cerebral injury, and to strengthen the understanding of this
association in similar populations.
In conclusion, the current study presented the cases
of 5 adult patients who exhibited typical brain MRI features of
acute hyperammonemic HE. These patients demonstrated symmetrical
signal abnormalities in the insular and cingulate cortices, while
the peri-rolandic and occipital cortices were spared. Early
recognition of these diagnostic features could lead to improved
patient outcomes through proper treatment intervention.
Furthermore, an association was noted between serum ammonia levels
and brain MRI abnormalities, which warrants further investigation
in future studies.
Supplementary Material
MRI brain for case 2. (A) T2-weighted
MRI, (B) FLAIR, (C) DWI and (D) ADC map demonstrating symmetric
cortical swelling with corresponding high T2 and FLAIR signal
intensity involving bilateral insular cortex, frontal lobes,
parietal lobes, temporal lobes and cingulate gyri with
corresponding diffusion restriction. The bilateral occipital lobes
and deep grey matter nuclei are spared. At the level of the vertex,
(E) T2-wieghted image, (F) FLAIR, (G) DWI and (H) ADC map
demonstrating similar symmetric cortical swelling with high T2 and
FLAIR signal intensities involving the frontal and parietal lobes
with corresponding restricted diffusion. The bilateral
peri-Rolandic regions are spared. MRI, magnetic resonance imaging;
FLAIR, fluid-attenuated inversion recovery; DWI, diffusion-weighted
imaging; ADC, apparent diffusion coefficient.
MRI brain scan for case 3. (A)
T2-weighted MRI, (B) FLAIR, (C) DWI and (D) ADC map demonstrating
symmetric cortical swelling with high T2 and FLAIR signal
intensities involving the bilateral insular cortex, frontal lobes,
temporal lobes, parietal lobes, cingulate gyri and bilateral
lateral thalami, with corresponding diffusion restriction. The
bilateral occipital lobes are spared. At the level of the vertex,
(E) T2-weighted MRI, (F) FLAIR, (G) DWI and (H) ADC map
demonstrating similar symmetric cortical swelling with high T2 and
FLAIR signal intensities affecting the frontal and parietal lobes,
with corre-sponding restricted diffusion. The bilateral
peri-rolandic regions are spared. MRI, magnetic resonance imaging;
FLAIR, fluid attenuated inversion recovery; DWI, diffusion-weighted
imaging; ADC, apparent diffusion coefficient.
MRI brain scan for case 4. (A)
T2-weighted MRI, (B) FLAIR, (C) DWI and (D) ADC map demonstrating
symmetric cortical swelling with high T2 and FLAIR signal
intensities involving the bilateral insular cortex, frontal lobes,
temporal lobes, parietal lobes and cingulate gyri. The bilateral
occipital lobes are spared. Foci of high T2 and FLAIR signal
intensities are observed in the right anterior thalamus, left
globus pallidus and splenium of the corpus callosum, with
corresponding diffusion restriction, likely representing ischemic
insults. At the level of the vertex, (E) T2-weighted MRI, (F)
FLAIR, (G) DWI and (H) ADC map demonstrating asymmetric cortical
swelling with high T2 and FLAIR signal intensities involving the
frontal and parietal lobes, with corresponding restricted
diffusion. The bilateral peri-rolandic regions are spared. MRI,
magnetic resonance imaging; FLAIR, fluid attenuated inversion
recovery; DWI, diffusion-weighted imaging; ADC, apparent diffusion
coefficient.
Follow-up MRI brain scan for case 4
after 10 days. (A) T2-weighted MRI, (B) FLAIR, (C)
diffusion-weighted imaging and (D) apparent diffusion coefficient
map demonstrating interval stability of symmetric cortical
swelling, with persistent high T2 and FLAIR signal intensities
involving the bilateral insular cortex, frontal lobes, temporal
lobes and bilateral cingulate gyri. Regression of diffusion
restriction suggests interval evolution of intraparenchymal changes
associated with acute hyperammonemic hepatic encephalopathy.
Interval regression of high T2 and FLAIR signal intensities, and
diffusion restriction in the right anterior thalamus, left globus
pallidus and splenium of the corpus callosum, indicate subacute
evolution of the ischemic insults. MRI, magnetic resonance imaging;
FLAIR, fluid attenuated inversion recovery.
MRI brain scan for case 5. (A)
T2-weighted MRI, (B) FLAIR, (C) DWI and (D) ADC map demonstrating
symmetric cortical swelling with corresponding high T2 and FLAIR
signal intensity involving the bilateral insular cortex, frontal
lobes, parietal lobes, temporal lobes and cingulate gyri, with
corresponding diffusion restriction. The bilateral occipital lobes
and deep grey matter nuclei are spared. At the level of the vertex,
(E) T2-weighted MRI, (F) FLAIR, (G) DWI and (H) ADC map
demonstrating similar asymmetric cortical swelling, with high T2
and FLAIR signal intensities involving the frontal and parietal
lobes, with corresponding restricted diffusion. The bilateral
peri-rolandic regions are spared. MRI, magnetic resonance imaging;
FLAIR, fluid attenuated inversion recovery; DWI, diffusion-weighted
imaging; ADC, apparent diffusion coefficient.
Follow-up MRI brain scan for case 5.
(A) T2-weighted MRI, (B) FLAIR and (C) diffusion-weighted imaging
demonstrating complete resolution of the cortical T2 and FLAIR high
signal intensities, with marked bilateral symmetrical brain
atrophy. MRI, magnetic resonance imaging; FLAIR, fluid attenuated
inversion recovery.
Acknowledgements
Not applicable.
Funding
Funding: The King Salman center For Disability Research funded
this work through Research Group no. KSRG-2024-307.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
AB was responsible for the design of the study and
overall project coordination. HA, SO, NA, FA and SM contributed to
data collection and assisted with the literature review and
manuscript drafting. HA and HA provided radiological and clinical
oversight and supported the validation of results. SB structured
and critically revised the manuscript and secured funding. TAH
contributed to the conceptual framework and provided overall
project supervision. AB and TAH confirm the authenticity of all the
raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
King Fahad Specialist Hospital-Dammam (IRB-Pub-024-01).
Patient consent for publication
Written informed consent for publication was
obtained from all subjects.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hussain J, Schlachterman A, Kamel A and
Gupte A: Hyperinsulinism hyperammonemia syndrome, a rare clinical
constellation. J Investig Med High Impact Case Rep.
4(2324709616632552)2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Brusilow SW: Hyperammonemic
encephalopathy. Medicine (Baltimore). 81:240–249. 2002.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Atluri DK, Prakash R and Mullen KD:
Pathogenesis, diagnosis, and treatment of hepatic encephalopathy. J
Clin Exp Hepatol. 1:77–86. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Dharel N and Bajaj JS: Definition and
nomenclature of hepatic encephalopathy. J Clin Exp Hepatol. 5
(Suppl 1):S37–S41. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhang XD and Zhang LJ: Multimodal MR
imaging in hepatic encephalopathy: State of the art. Metab Brain
Dis. 33:661–671. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Morgan MY: Cerebral magnetic resonance
imaging in patients with chronic liver disease. Metab Brain Dis.
13:273–290. 1998.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Rosario M, McMahon K and Finelli PF:
Diffusion-weighted imaging in acute hyperammonemic encephalopathy.
Neurohospitalist. 3:125–130. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
U-King-Im JM, Yu E, Bartlett E, Soobrah R
and Kucharczyk W: Acute hyperammonemic encephalopathy in adults:
Imaging findings. AJNR Am J Neuroradiol. 32:413–418.
2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Reis E, Coolen T and Lolli V: MRI findings
in acute hyperammonemic encephalopathy: Three cases of different
etiologies: Teaching point: To recognize MRI findings in acute
hyperammonemic encephalopathy. J Belg Soc Radiol.
104(9)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Arnold SM, Els T, Spreer J and Schumacher
M: Acute hepatic encephalopathy with diffuse cortical lesions.
Neuroradiology. 43:551–554. 2001.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lora-Tamayo J, Palom X, Sarrá J, Gasch O,
Isern V, Fernández de Sevilla A and Pujol R: Multiple myeloma and
hyperammonemic encephalopathy: Review of 27 cases. Clin Lymphoma
Myeloma. 8:363–369. 2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bindu PS, Sinha S, Taly AB, Christopher R
and Kovoor JME: Cranial MRI in acute hyperammonemic encephalopathy.
Pediatr Neurol. 41:139–142. 2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Koksel Y, Benson J, Huang H, Gencturk M
and McKinney AM: Review of diffuse cortical injury on
diffusion-weighted imaging in acutely encephalopathic patients with
an acronym: ‘CRUMPLED’. Eur J Radiol Open. 5:194–201.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Takanashi J, Barkovich AJ, Cheng SF,
Weisiger K, Zlatunich CO, Mudge C, Rosenthal P, Tuchman M and
Packman S: Brain MR imaging in neonatal hyperammonemic
encephalopathy resulting from proximal urea cycle disorders. AJNR
Am J Neuroradiol. 24:1184–1187. 2003.PubMed/NCBI
|
|
15
|
McKinney AM, Lohman BD, Sarikaya B,
Uhlmann E, Spanbauer J, Singewald T and Brace JR: Acute hepatic
encephalopathy: Diffusion-weighted and fluid-attenuated inversion
recovery findings, and correlation with plasma ammonia level and
clinical outcome. AJNR Am J Neuroradiol. 31:1471–1479.
2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
McPhail MJW, Patel NR and Taylor-Robinson
SD: Brain imaging and hepatic encephalopathy. Clin Liver Dis.
16:57–72. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ott P and Vilstrup H: Cerebral effects of
ammonia in liver disease: Current hypotheses. Metab Brain Dis.
29:901–911. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hertz L and Kala G: Energy metabolism in
brain cells: Effects of elevated ammonia concentrations. Metab
Brain Dis. 22:199–218. 2007.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Rovira A, Alonso J and Córdoba J: MR
imaging findings in hepatic encephalopathy. AJNR Am J Neuroradiol.
29:1612–1621. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wang V and Saab S: Ammonia levels and the
severity of hepatic encephalopathy. Am J Med. 114:237–238.
2003.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Clay AS and Hainline BE: Hyperammonemia in
the ICU. Chest. 132:1368–1378. 2007.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lichtenstein GR, Yang YX, Nunes FA, Lewis
JD, Tuchman M, Tino G, Kaiser LR, Palevsky HI, Kotloff RM, Furth
EE, et al: Fatal hyperammonemia after orthotopic lung
transplantation. Ann Intern Med. 132:283–287. 2000.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chou HF, Yang RC, Chen CY and Jong YJ:
Valproate-induced hyperammonemic encephalopathy. Pediatr Neonatol.
49:201–204. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Nott L, Price TJ, Pittman K, Patterson K
and Fletcher J: Hyperammonemia encephalopathy: An important cause
of neurological deterioration following chemotherapy. Leuk
Lymphoma. 48:1702–1711. 2007.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Liu J, Lkhagva E, Chung HJ, Kim HJ and
Hong ST: The pharmabiotic approach to treat hyperammonemia.
Nutrients. 10(140)2018.PubMed/NCBI View Article : Google Scholar
|