1. Introduction
Pathological calcification, also known as ectopic
calcification, refers to the deposition of mineralized complexes in
soft tissues outside of bones and teeth under physiological
conditions. This condition can result from genetic factors or
chronic diseases acquired later in life (1). Research indicates that pathological
calcification shares similarities with physiological calcification
but occurs due to an imbalance between inhibitory and promoting
factors, leading to abnormal mineralization in soft tissues
(2). Key mechanisms include the
induction of bone formation, differentiation of vascular smooth
muscle cells into osteoblastic phenotypes, oxidative stress,
apoptosis, mitochondrial dysfunction, calcium-phosphorus imbalance,
and decreased levels of calcification inhibitors (3). Despite ongoing research, the molecular
mechanisms underlying pathological calcification remain poorly
understood, and effective treatments have yet to be developed.
Further investigation into these mechanisms is expected to reveal
more promising therapeutic targets.
In recent years, in-depth studies on nanobacteria
(NB) have revealed their ability to aggregate calcium and
phosphorus from the surrounding environment within the body,
forming nucleation centers. These findings highlight the
significant role of NB in the pathogenesis of pathological
calcification diseases (4).
Initially identified by Finnish scientists Kajander and Ciftçioglu
(5) as contaminants in cell
cultures, NB exhibit diameters ranging from 50 to 500 nm, with a
peak particle size of ~130 nm. Their outer layer is covered with a
dense, needle-like apatite mineralized shell, and their interior
contains cavities and inclusions. The main protein component is
fetuin-A, and they are Gram-negative. Their proliferation rate is
~10,000 times slower than that of bacteria. They can withstand
temperatures as high as 100˚C and harsh chemical conditions, while
most other bacteria cannot survive in such extreme environments,
and resistance to most antibiotics (4,6-9).
Tetracycline is one of the few drugs that can effectively reduce
the damage of NB (7). However,
debate persists regarding whether NB represent the smallest known
replicating life forms or are merely mineral-protein complexes
unrelated to bacteria. Recent findings indicate that NB lack
definitive nucleic acid sequences, leading researchers to prefer
the term ‘calcified nanoparticles’ (CNPs) (10). CNPs can accumulate calcium and
phosphorus in their surrounding environment and nucleate within the
human body, especially after being internalized by cells, where
they exert a direct toxic effect. CNPs can inflict damage upon cell
membranes, mitochondria, lysosomes, and other cellular structures
and even produce a large amount of reactive oxygen species (ROS),
ultimately leading to cell necrosis and apoptosis. This process
contributes to the formation of larger nucleation centers,
resulting in the aggregation of calcium and phosphorus into
calcified cores (11) (Fig. 1). Research has established a link
between CNPs and ectopic calcification diseases and CNPs have been
detected in various conditions involving ectopic calcification,
including kidney stones, bladder stones, dental pulp stones,
salivary gland stones, cholecystolithiasis, and testicular
microstones; calcification in hemodialysis patients,
atherosclerosis, scleroderma (systemic sclerosis), and several
malignant tumors (7). Furthermore,
fetuin-A, by binding to CNPs, enhances their solubility in
circulation, thereby inhibiting calcification (12). Consequently, some researchers
propose that increasing circulating fetuin-A levels may represent a
promising therapeutic approach for pathological calcification
diseases (13). Given these
findings, the interaction between fetuin-A and NB, as well as the
underlying mechanisms in calcification diseases, has emerged as a
focal point of current research.
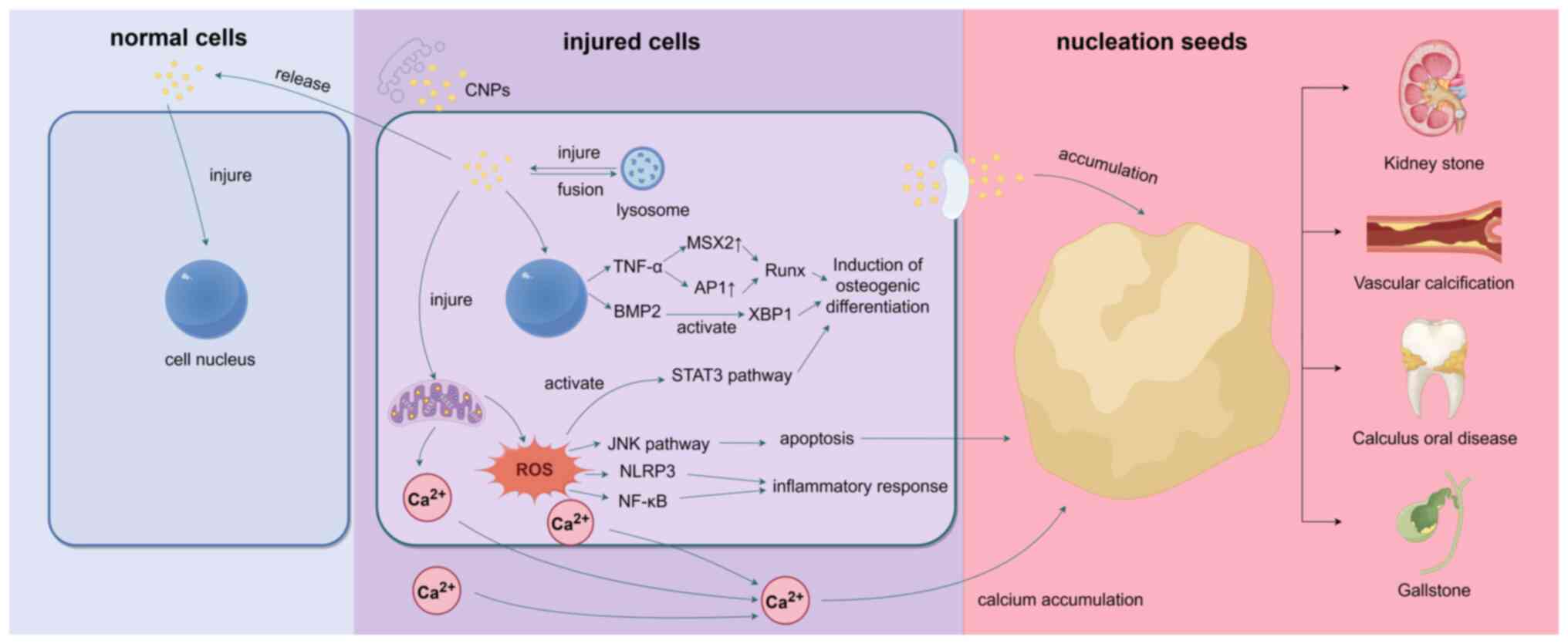 | Figure 1After CNPs enter cells through
endocytosis, they induce the expression of TNF-α and BMP2.
Specifically, TNF-α enhances RUNX2 expression by regulating the
transcription factors MSX2 and AP-1, while BMP-2 activates the XBP1
signaling pathway, both contributing to osteogenic differentiation.
Additionally, CNPs stimulate ROS production, which activates STAT3
and other pathways, further promoting osteogenic differentiation.
On the other hand, CNPs can disrupt cell membrane stability,
alkalize lysosomes, and stimulate mitochondria to produce excessive
ROS, leading to damage in cellular structures such as the cell
membrane, mitochondria, and lysosomes. The generated ROS not only
activate the NLRP3 inflammasome and initiate the NF-κB signaling
pathway, causing inflammatory damage, but also trigger autophagy
and apoptosis via the ROS-JNK signaling pathway. During apoptosis,
CNPs facilitate the aggregation of Ca2+ from the
cytoplasm, mitochondria, and extracellular matrix, forming a
calcification core. Apoptotic cells release matrix vesicles
containing CNPs, which damage surrounding cells, expand the
inflammatory response, and promote continuous expansion of the
calcification core, ultimately resulting in pathological
calcification. CNPs, calcified nanoparticles; RUNX2, dwarf-related
transcription factor 2; MSX2, homeobox transcription factor muscle
segment homeobox 2; AP-1, activator protein 1; XBP1, X-box binding
protein 1; STAT3, signal transducer and activator of transcription
3; ROS, reactive oxygen species; NLRP3, NLR family pyrin domain
containing 3; BMP2, bone morphogenic protein 2. |
The nucleation of CNPs continues to gather calcium
and phosphorus from the surrounding environment, leading to the
formation of hydroxyapatite and growth of cavity-filling stones.
These stones can cause tissue damage through mechanical blockage,
resulting in colic and, in severe cases, organ failure. The high
concentration of minerals in the excretory fluids of the body
promotes the nucleation of CNPs, making the bile duct and
genitourinary tract susceptible to stone formation (14). In the urinary tract, crystallization
usually begins in the renal tubules. The gradual concentration of
glomerular filtrate and the active secretion of calcium, uric acid,
oxalate, phosphate, or drug metabolites lead to mineral
supersaturation in the renal tubules. Subsequently, stones may
develop in the larger renal pelvis, where calcified Randall plaques
on the renal papilla are formed (15). In the bile duct, bile is rich in
electrolytes, bile acids, cholesterol, phospholipids, and
conjugated bilirubin, especially in the gallbladder. The
concentration and storage of bile promote stone formation (14). In addition, CNPs can cause vascular
calcification in circulation, which is common in the elderly and
patients with uremia. This intermediate-level calcification
decreases vascular compliance and increases the risk of
cardiovascular disease (16). In
the oral cavity, CNPs contribute to calculus oral diseases by
alkalizing the environment, mediating inflammatory responses, and
promoting dental plaque mineralization (10).
The aim of the present review was to explore the
properties of CNPs and their mechanisms in pathological
calcification diseases such as kidney stones, vascular
calcification, calculus oral diseases and gallstones, offering new
insights for treating these conditions.
2. Mechanism of CNPs in calcified
diseases
Kidney stones
Kidney stones are caused by the abnormal
accumulation of crystalline substances, including calcium, oxalic
acid, uric acid, and cystine in the kidney. Among these, calcium
oxalate (CaOx) crystallization is the most prevalent, making kidney
stones a common and frequently occurring disease of the urinary
system. Approximately 10% of the global population is affected by
kidney stones, with this number continuing to rise (17). However, the mechanism of kidney
stone formation is not completely clear. Currently, numerous
theories attempt to explain the formation of renal calculi. These
theories include renal calcification spots, supersaturated
crystallization, stone matrix, crystal inhibitors, and
heterogeneous nucleation. Among them, the theory of calcified
plaques in the renal papilla (Randall's plaque) is a prominent
explanation for renal stone formation. Alexander Randall first
proposed the Randall plaque theory in 1937. Studies have shown that
renal CaOx stones originate from calcified plaques in the renal
papilla, with calcified plaques being almost universally present in
the renal papillary tips of patients with CaOx stones (18). In addition, in-depth studies of
renal papillary calcification plaques have revealed the presence of
CNPs through transmission electron microscopy observations of renal
sections and cultures of renal papillary calcification plaques
(19,20). This finding has established a
correlation between CNPs and Randall's plaque.
CNPs in the bloodstream can accumulate in the kidney
by passing through the glomerular filtration membrane, thereby
inflicting damage on renal tubular epithelial cells. In the blood,
CNPs exist as troponin granules with a diameter ranging from 30 to
100 nm. The intercellular gaps between capillary endothelial cells,
the main pore barrier of the glomerular filtration membrane,
measure between 50 and 100 nm. After intravenous injection of
99mTc-labeled CNPs into rabbits, CNPs can be detected in
the kidneys and urine (21). By
contrast, CNPs entering the urine can adhere to renal tubular
epithelial (HK-2) cells, leading to damage during urine excretion.
A study involving the co-culture of CNPs with fluorescence-labeled
HK-2 cells has observed that CNPs can enter HK-2 cells via
endocytosis mediated by membrane invagination and accumulate around
the nucleus. This accumulation results in a decrease in plasma
membrane cholesterol, compromising the stability of the cell
membrane and making cells susceptible to mechanical damage
(22). After entering HK-2 cells,
CNPs induce several adverse effects. First, CNP phagocytosis by
HK-2 cells can cause mitochondrial swelling and vacuolation around
CNPs, a decrease in mitochondrial membrane potential, and an
overload of intracellular oxygen free radicals (such as ROS). The
latter can in turn affect the stability of the mitochondrial
membrane, aggravate mitochondrial damage, and lead to persistent
overload of ROS. Internalized CNPs not only increase the activity
of NADPH oxidase but also stimulate cells to produce a large amount
of ROS (4). Second, CNPs
internalized into cells can bind to lysosomes and lead to lysosome
alkalinization, resulting in swelling of lysosomes and inhibition
of lysosomal hydrolase activity. Alkalized lysosomes can block
autophagy flux and decrease the ability of cells to digest damaged
proteins and organelles through autophagy, especially the digestion
of damaged mitochondria (22).
In addition, a large amount of ROS can induce
inflammation in renal tubular epithelial cells. On the one hand, a
large amount of ROS can activate JNK through various pathways (such
as the apoptotic signal-regulating kinase 1 pathway and mixed
lineage kinase 3 pathway), leading to autophagy, apoptosis, and
even necrosis, resulting in local inflammation (23-26).
On the other hand, ROS is the key component in activating NLR
family pyrin domain containing 3 (NLRP3) inflammasomes. ROS can
activate NLRP3 inflammasomes through multiple mechanisms, promoting
the release of inflammatory cytokines such as IL-1β and
IL-18(27). Moreover, ROS can
stimulate the secretion of various cytokines by initiating the
nuclear factor-κB (NF-κB) signaling pathway, causing inflammatory
damage to renal tubular epithelial cells and renal tissue (28,29).
The inflammatory injury to renal tubular epithelial
cells may promote the formation of Randall plaques. Randall plaques
originate from the thin ring basement membrane of Henle (30). On the one hand, CNPs entering renal
tubular cells may reach the basement membrane by re-exocytosis or
colonize it by inducing autophagy, apoptosis, and necrosis. During
this process, CNPs release a large amount of calcium and phosphorus
stored within renal tubular epithelial cells, resulting in local
calcium and phosphorus supersaturation in the renal papilla.
Simultaneously, inflammatory lesions further strengthen the
deposition of hydroxyapatite, creating conditions for the formation
of calcified plaques in the renal papilla. Furthermore,
accumulating evidence indicates that extracellular vesicles (EVs)
play a critical role in the pathogenesis of pathological
calcification. Based on this, it is hypothesized that CNP-infected
cells may facilitate the formation of calcification cores by
releasing EVs enriched with CNPs into the basement membrane
(31).
Colonized CNPs in the basement membrane play a role
in biomineralization, aggregating apoptotic bodies and necrotic
cells as the core of mineralization, gradually expanding into
calcifications and promoting the formation of Randall plaques
(32). In addition, the
inflammatory injury to HK2 cells promotes calcium crystal adhesion
to the injured cells. Tamm-Horsfall Protein (THP) increases, acting
as an adhesion protein that facilitates the accumulation and growth
of calcium ions, oxalic acid, and phosphate in urine. When THP
polymerizes into macromolecular forms, it can inhibit or weaken the
inhibition of calcium crystal aggregation in urine. Moreover, THP
molecules exhibit weak binding ability to water molecules and poor
molecular rigidity. Particles such as calcium salt crystals in
urine attach easily, thus promoting stone formation (33).
ROS can upregulate the expression of hyaluronic
acid, osteopontin (OPN), and CD44 through the p38MAPK pathway,
alter the HK-2 cell adhesion to CaOx crystals and stimulate the
formation of CaOx stones. Stimulated by calcified crystals, renal
cells produce various inflammatory factors, which induce monocytes
or macrophages to migrate to sites of stone crystal deposition via
pinocytosis. Phagocytosis of stone crystals by macrophages induces
the production of inflammatory factors, including TNF-α, IL-6 and
IL-1β, which maintain and aggravate the inflammatory response,
causing severe damage to the kidney and promoting the deposition of
stone crystals (34,35).
In summary, CNPs cause severe inflammatory damage to
the kidneys by damaging HK-2 cells and inducing local inflammatory
responses. This inflammatory damage promotes the deposition of
stone crystals, forming a positive feedback loop that enhances the
formation of Randall spots.
Vascular calcification
Ectopic calcification in the cardiovascular system
is a strong predictor of cardiovascular disease morbidity and
mortality. This pathophysiological process entails the deposition
of minerals, mainly hydroxyapatite calcium apatite crystals, within
the intima and media of vascular walls and heart valve leaflets,
with calciprotein particles (CPPs) playing an important role. CPPs
are mineral-protein complexes formed by the combination of CNPs and
circulating proteins, dispersed in the blood as colloids (36). Circulating Gla-rich protein (GRP),
matrix Gla protein (MGP) and fetuin-A are the main proteins that
form CPPs. Among them, the most effective binding protein is
fetuin-A. MGP and GRP contain negatively charged γ-carboxylated
glutamic acid residues (37), which
can bind Ca2+ and calcium-containing compounds (38). By contrast, fetuin-A forms CPPs
through negatively charged β-fold binding to calcium phosphate in
the N-terminal cysteine protease inhibitor D1(39).
Under physiological conditions, initially formed
primary CPPs (CPPI) are usually harmless and contribute to the
clearance of calcium and phosphate, protecting the body from
extraosseous calcification (40).
These CPPI are cleared by macrophages, especially Kupffer cells in
the liver (41). However, under
pathological conditions, especially in patients with diabetes and
chronic kidney disease, the decrease of renal excretion of calcium
and phosphate leads to their accumulation in the body and a
relative decrease in fetuin-A. Consequently, CPPI undergo
rearrangement from a spherical structure ~75-nm in diameter to
larger secondary CPPs (CPPII), measuring ~120 nm in diameter. These
CPPII are denser, insoluble in serum and exhibit a needle-like
structure (42).
Research has demonstrated a close association
between the formation of CPPII and vascular calcification. CPPII
can induce vascular smooth muscle cell calcification and macrophage
secretion of TNF-α in vitro, but CPPI cannot. Calcification
of vascular smooth muscle cells has also been proven to be the
result of cellular uptake of CPPII, which can be detected in
calcified vascular smooth muscle cells (43). In addition, CPPs can induce
macrophages to secrete IL-1β (44).
Compared with CPPI, CPPII exhibit a more obvious pro-inflammatory
effect, which may be related to the content of crystalline
hydroxyapatite (45).
Vascular calcification is the result of two main
types: Intimal and medial calcification. Intimal calcification is
related to inflammation and is the main driving factor, whereas
media calcification is mostly related to mineral disorders
(46). Intimal calcification
results from the migration of vascular smooth muscle cells during
atherosclerosis, whereas medial calcification results from the
transdifferentiation of medial vascular smooth muscle cells
(47). CPPs play an important role
in these processes.
Endothelial cells form the intima of blood vessels,
the first cell type to interact with circulating CPPs.
Internalization of CPPs by endothelial cells triggers lysosome and
cytoplasmic calcium influx, resulting in intracellular calcium
overload (48). This
internalization induces significant physiological disorders in
mitochondria and lysosomes, including oxidative stress, vacuolar
acidification, accelerated protein degradation, and increased
permeability of the mitochondrial outer membrane. In addition, the
incubation of intact endothelial cells with CPP-treated endothelial
cells in conditioned medium results in the release of
pro-inflammatory cytokines, such as upregulation of vascular cell
adhesion molecule-1 and intercellular adhesion molecule-1,
increased release of IL-6, IL-8, and monocyte chemoattractant
protein-1, ultimately triggering endothelial cell apoptosis
(49).
CPPII also affect endothelial cell function by
modulating the bioavailability and metabolism of nitric oxide (NO)
(50). Endothelial-derived NO plays
an important role in balancing and regulating vascular dilation and
contraction mediated by vascular smooth muscle cells (51). Endothelial nitric oxide synthase
(eNOS) is the principal enzyme responsible for NO production in
endothelial cells (52,53). Research has demonstrated that CPPII
can induce the dysfunction of eNOS, reducing NO production and
bioavailability, thereby impairing endothelial function. This
dysfunction affects the vasomotor regulation of vascular smooth
muscle cells and eventually leads to hypertension (50). Additionally, eNOS-derived NO is an
important antioxidant, which can scavenge a variety of formed free
radicals (54). The inhibition of
NO production aggravates intracellular oxidative stress and
accelerates cell apoptosis.
Media calcification is the main mechanism of
vascular calcification. Vascular smooth muscle cells internalizing
CPPs can increase intracellular calcium binding and induce their
differentiation into osteoblasts through various mechanisms. First,
CPPs induce vascular smooth muscle cells to express and secrete
TNF-α (43), enhancing the
expression of dwarf-related transcription factor 2 (RUNX2) through
the homeobox transcription factor muscle segment homeobox 2 (MSX2)
(55) and activator protein 1
(AP-1) (56), thus triggering
osteogenic dedifferentiation. Second, CPPs stimulate vascular
smooth muscle cells to express and secrete bone morphogenic protein
2(57), which can induce osteoblast
dedifferentiation by increasing phosphate transport (58), resulting in endoplasmic reticulum
stress and activation of osteogenic transcription factor X-box
binding protein 1 (XBP1) (59).
Third, CPPs induce oxidative stress in vascular smooth muscle cells
(43), activating downstream
signaling cascades (including Akt, p38MAPK and NF-κB), which
promotes the transcriptional activation of osteochondral
differentiation (60-63).
Alternatively, CPPs promote the secretion of IL-6 from endothelial
cells (64), which may drive the
osteochondrogenic differentiation of vascular smooth muscle cells
in a signal transducer and activator of transcription 3
(STAT3)-dependent manner (65). The
activation of osteochondral transcription eventually leads to a
decrease in the expression of contractile proteins (for example
α-smooth muscle actin, smooth muscle myosin heavy chain, smooth
muscle protein, and calmodulin) and an increase in the expression
of osteogenic markers (such as OPN, osteocalcin, alkaline
phosphatase, and collagen) (66).
The ossification of vascular smooth muscle cells aggravates the
damage of vascular homeostasis, promotes the formation of
atherosclerotic microenvironment, and exacerbates vascular
sclerosis. In addition, transformed vascular smooth muscle cells
can release EVs containing CPPs. These EVs can form the core of
vascular calcification together with apoptotic vascular smooth
muscle cells (67).
When CPPII induce endothelial cell dysfunction
through various pathways, endothelial permeability to low-density
lipoprotein is increased, causing cholesterol-rich particles to
accumulate in the subendothelial layer. Eventually, these particles
form unstable arterial plaques, and local inflammatory stimuli
further lead to plaque rupture, incorporating them into the
calcified core. In addition, CPPII induce apoptosis of vascular
smooth muscle cells and macrophages, resulting in the formation of
apoptotic bodies and necrotic cell fragments. These entities serve
as nucleation sites within ruptured plaques, initiating
microcalcification in injured sites, resulting in calcium phosphate
crystal deposition (68).
Furthermore, ossified vascular smooth muscle cells and macrophages
release EVs containing CPPs, which promote the deposition of
calcium phosphate crystals, which gradually crystallize to form
hydroxyapatite crystals. Ossified smooth muscle cells also regulate
mineralization by secreting osteogenic proteins. Finally,
hydroxyapatite crystals continue to accumulate on the calcified
core, contributing to vascular calcification.
Calculus oral disease
Calculus oral diseases include dental calculi,
dental pulp stones, and salivary gland stones. CNPs may play an
important role in these diseases due to their unique
biomineralization properties. Some scholars have proposed that CNPs
may originate from the atmosphere and can be transmitted through
the air, causing calculus oral diseases (69). Kao et al (70) observed CNPs through sampling and
imaging of salivary gland stones, whereas Zeng et al
(71) observed and cultured CNPs
from dental pulp stones. These studies have shown a large number of
CNPs in the oral cavity, which may lead to the formation of oral
calculi.
CNPs may induce calcification by damaging cells.
Through co-culture experiments of CNPs and periodontal epithelial
cells, Zhang et al (72)
found that CNPs can internalize into these cells, leading to
vacuolization of the periodontal epithelial cells. Electron
microscopy shows CNPs and calcification within vacuoles, along with
calcium deposition outside the cell membrane, which eventually
leads to apoptosis. Moreover, CNPs can use calcium and phosphorus
in periodontal crevicular fluid to form inorganic precipitates
in vivo, which are then deposited on the tooth surface as a
biomineralization centers to form dental calculus (72). In a co-culture model of human dental
pulp cells with CNPs, Yang et al (73) found that CNPs can lead to
vacuolation and mitochondrial swelling of dental pulp cells,
ultimately leading to nuclear calcification and cell necrosis.
Sakai et al (74) found that
the hydroxyapatite shell of CNPs can induce the expression of IL-8
in human gingival epithelial cells through the NF-κB signaling
pathway, resulting in inflammatory reactions that damage cells.
Furthermore, CNPs can increase pH during mineralization, which is
beneficial to the mineralization of dental plaque and the formation
of stones in the neutral and alkaline oral environment (75).
Therefore, when CNPs damage human gingival
epithelial cells or dental pulp cells, they induce inflammatory
responses by aggregating calcium and phosphate ions both intra- and
extracellularly and interacting with dental plaque, thereby forming
an initial calcified core. As the inflammatory response
progressively intensifies, the calcified core gradually enlarges,
ultimately leading to the development of calculus-related oral
diseases.
Gallstones
Cholecystolithiasis, a prevalent digestive system
disorder, mainly involves cholesterol stones or cholesterol-based
mixed stones and melanin stones. The cause of gallstones is
extremely complex, with various factors playing a role. Any factor
influencing the ratio of cholesterol to cholecystocholic acid
phospholipid concentration and cholestasis can contribute to
gallstone formation. Bacterial infection plays an important role in
the formation and progression of gallstones (76). CNPs can be found in the bile and
gallbladder mucosa of patients with gallstones (77). Additionally, animal models of
cholecystolithiasis have been successfully established by injecting
CNPs into the gallbladders of rabbits (78).
CNPs may interact with gallbladder bacteria to
promote stone formation. Research has shown the presence of living
bacteria in gallstones (79).
Culturing gallstone samples from patients with cholecystolithiasis
has revealed a high coinfection rate of CNPs and Escherichia
coli (E. coli) in gallstones, suggesting their
involvement in the formation of gallstones. Professor Kajander
reported that when E. coli and CNPs are mixed,
calcium-stained mineralized particles both inside and outside E.
coli are composed of CNPs (80). This finding implies that CNPs can
cause biological calcification of E. coli by directly
attaching to or invading the bacteria. In addition, the number of
calcium-stained positive particles found in the mixed culture of
CNPs and E. coli is significantly higher than that in
cultures of CNPs alone. Therefore, the existence of E. coli
provides a favorable adhesion matrix and microenvironment for CNPs,
promoting their reproduction and mineralization (78).
At the same time, CNPs can inflict damage on
gallbladder mucosal cells. Research has revealed that after the
co-culture of CNPs with human gallbladder epithelial cells, the
microvilli on the surface of gallbladder epithelial cells decrease
or even disappear, cytoplasmic endocrine granules significantly
decrease, and mitochondria swell and vacuolate (79). Over time, cell necrosis ensues,
accompanied by an increase in the mRNA levels of IL-6 and IL-1β,
indicating the onset of an inflammatory reaction (79). Liang et al (81) found that CNPs can stimulate a
decrease in the Bcl-2/Bax ratio and subsequently upregulate the
expression of cytochrome c and activated cysteine protease
(cleaved caspase-9) in gallbladder epithelial cells following
co-culture with CNPs. This finding suggests that CNPs can induce
apoptosis of gallbladder epithelial cells through the mitochondrial
pathway (81).
Therefore, during gallstone formation, the
interaction between CNPs and gallbladder bacteria may form a
calcification center. Furthermore, the injury of gallbladder
mucosal cells and the production of ROS to mediate inflammatory
reaction aggravate the damage of gallbladder mucosal cells and the
disorder of bile acid metabolism and promote the production of
cholesterol stones.
3. Conclusion
In conclusion, pathological calcification diseases
are a prevalent issue, and their diagnosis and treatment present
significant challenges. As research on CNPs continues to advance,
it is evident that CNPs plays a crucial role in various
pathological calcification diseases. In-depth investigation of CNPs
can further elucidate the mechanisms underlying pathological
calcification, which holds substantial scientific importance. The
present review provides a detailed examination of the primary
mechanisms by which CNPs contribute to four common types of
pathological calcification diseases. These mechanisms facilitate
the formation of an initial calcification core by CNPs, which
progressively expands and ultimately results in localized
calcification or stone formation. During this process, strategies
such as increasing the levels of circulating calcification
inhibitors, inhibiting cellular osteogenic differentiation, and
mitigating local inflammatory responses may offer new avenues for
the prevention and treatment of pathological calcification.
Acknowledgements
Not applicable.
Funding
Funding: The present review was supported by the Research of
Guangxi Natural Science Foundation (grant no. 2023GXNSFAA026142),
and The Outstanding Reserve Talent Fund of the Second Affiliated
Hospital of Guangxi Medical University (grant no. hbrc202101).
Availability of data and materials
Not applicable.
Authors' contributions
JW and SL conceived and designed the article. SL and
BB analysed the relevant literature. SL wrote the manuscript. JW
and SL made revisions to the manuscript. All authors read and
approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Boraldi F, Lofaro FD and Quaglino D:
Apoptosis in the extraosseous calcification process. Cells.
10(131)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kirsch T: Determinants of pathological
mineralization. Curr Opin Rheumatol. 18:174–180. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Snijders BMG, Peters MJL and Koek HL:
Ectopic calcification: What do we know and what is the way forward?
J Clin Med. 12(3687)2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wu J, Tao Z, Deng Y, Liu Q, Liu Y, Guan X
and Wang X: Calcifying nanoparticles induce cytotoxicity mediated
by ROS-JNK signaling pathways. Urolithiasis. 47:125–135.
2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kajander EO and Ciftçioglu N:
Nanobacteria: An alternative mechanism for pathogenic intra- and
extracellular calcification and stone formation. Proc Natl Acad Sci
USA. 95:8274–8279. 1998.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhu ML, Wang JJ, Pan WN, Liu SR and Zhu
YD: Significance of detection of nanobacteria in bile and
gallstones. Chin J Nosocomiol. 29:3279–3283. 2019.
|
|
7
|
Zhang Y, Zhu R, Liu D, Gong M, Hu W, Yi Q
and Zhang J: Tetracycline attenuates calcifying
nanoparticles-induced renal epithelial injury through suppression
of inflammation, oxidative stress, and apoptosis in rat models.
Transl Androl Urol. 8:619–630. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wu JH, Deng YL, Liu Q, Yu JC, Liu YL, He
ZQ and Guan XF: Induction of apoptosis and autophagy by calcifying
nanoparticles in human bladder cancer cells. Tumour Biol.
39(1010428317707688)2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Erdemir F, Karabulut A, Ozveren B and
Kocagoz T: How much do we know about nanobacteria? Ecotoxicol
Environ Saf. 288(117415)2024.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wang S, Yang L, Bai G, Gu Y, Fan Q, Guan
X, Yuan J and Liu J: A preliminary study on calcifying
nanoparticles in dental plaque: Isolation, characterization, and
potential mineralization mechanism. Clin Exp Dent Res.
10(e885)2024.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Liu Y, Sun Y, Kang J, He Z, Liu Q, Wu J,
Li D, Wang X, Tao Z, Guan X, et al: Role of ROS-induced NLRP3
inflammasome activation in the formation of calcium oxalate
nephrolithiasis. Front Immunol. 13(818625)2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Jahnen-Dechent W, Heiss A, Schäfer C and
Ketteler M: Fetuin-A regulation of calcified matrix metabolism.
Circ Res. 108:1494–1509. 2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Jahnen-Dechent W, Schäfer C, Ketteler M
and McKee MD: Mineral chaperones: A role for fetuin-A and
osteopontin in the inhibition and regression of pathologic
calcification. J Mol Med (Berl). 86:379–389. 2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mulay SR and Anders HJ: Crystallopathies.
N Engl J Med. 374:2465–2476. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Leaf DE: Calcium kidney stones. N Engl J
Med. 363:2470–2471. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hutcheson JD, Goettsch C, Rogers MA and
Aikawa E: Revisiting cardiovascular calcification: A multifaceted
disease requiring a multidisciplinary approach. Semin Cell Dev
Biol. 46:68–77. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Mager R and Neisius A: Current concepts on
the pathogenesis of urinary stones. Urologe A. 58:1272–1280.
2019.PubMed/NCBI View Article : Google Scholar : (In German).
|
|
18
|
Randall A: The origin and growth of renal
calculi. Ann Surg. 105:1009–1027. 1937.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wong TY, Wu CY, Martel J, Lin CW, Hsu FY,
Ojcius DM, Lin PY and Young JD: Detection and characterization of
mineralo-organic nanoparticles in human kidneys. Sci Rep.
5(15272)2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ciftçioğlu N, Vejdani K, Lee O, Mathew G,
Aho KM, Kajander EO, McKay DS, Jones JA and Stoller ML: Association
between Randall's plaque and calcifying nanoparticles. Int J
Nanomedicine. 3:105–115. 2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Akerman KK, Kuikka JT, Ciftcioglu N,
Parkkinen J, Bergstroem KA, Kuronen I and Kajander EO:
Radiolabeling and in vivo distribution of nanobacteria in rabbits.
Proc SPIE Int Soc Opt Eng. 3111:436–442. 1997.
|
|
22
|
Kunishige R, Mizoguchi M, Tsubouchi A,
Hanaoka K, Miura Y, Kurosu H, Urano Y, Kuro-O M and Murata M:
Calciprotein particle-induced cytotoxicity via lysosomal
dysfunction and altered cholesterol distribution in renal
epithelial HK-2 cells. Sci Rep. 10(20125)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ray PD, Huang BW and Tsuji Y: Reactive
oxygen species (ROS) homeostasis and redox regulation in cellular
signaling. Cell Signal. 24:981–990. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chen K, Vita JA, Berk BC and Keaney JF Jr:
c-Jun N-terminal kinase activation by hydrogen peroxide in
endothelial cells involves SRC-dependent epidermal growth factor
receptor transactivation. J Biol Chem. 276:16045–16050.
2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Thévenin AF, Zony CL, Bahnson BJ and
Colman RF: GST pi modulates JNK activity through a direct
interaction with JNK substrate, ATF2. Protein Sci. 20:834–848.
2011.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Van den B MCW, Van Gogh IJA, Smits AMM,
van Triest M, Dansen TB, Visscher M, Polderman PE, Vliem MJ,
Rehmann H and Burgering BMT: The small GTPase RALA controls c-Jun
N-terminal kinase-mediated FOXO activation by regulation of a JIP1
scaffold complex. J Biol Chem. 288:21729–21741. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Song H, Liu B, Huai W, Yu Z, Wang W, Zhao
J, Han L, Jiang G, Zhang L, Gao C and Zhao W: The E3 ubiquitin
ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting
proteasomal degradation of NLRP3. Nat Commun.
7(13727)2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Khan SR and Thamilselvan S:
Nephrolithiasis: A consequence of renal epithelial cell exposure to
oxalate and calcium oxalate crystals. Mol Urol. 4:305–312.
2000.PubMed/NCBI
|
|
29
|
Huang HS, Ma MC and Chen J: Chronic
L-arginine administration increases oxidative and nitrosative
stress in rat hyperoxaluric kidneys and excessive crystal
deposition. Am J Physiol Renal Physiol. 295:F388–F396.
2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Verkoelen CF, van der Boom BG, Houtsmuller
AB, Schröder FH and Romijn JC: Increased calcium oxalate
monohydrate crystal binding to injured renal tubular epithelial
cells in culture. Am J Physiol. 274:F958–F965. 1998.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Golub EE: Biomineralization and matrix
vesicles in biology and pathology. Semin Immunopathol. 33:409–417.
2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Vicencio JM, Galluzzi L, Tajeddine N,
Ortiz C, Criollo A, Tasdemir E, Morselli E, Ben Younes A, Maiuri
MC, Lavandero S and Kroemer G: Senescence, apoptosis or autophagy?
When a damaged cell must decide its path-a mini-review.
Gerontology. 54:92–99. 2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Liu X, Su H, Chen J, Zhu Y, Luo S, Ji M,
Chen K and Tang Y: Effects of Tamm-Horsfall protein on kidney stone
formation. J Med Postgrad. 922–925. 2017.
|
|
34
|
Umekawa T, Chegini N and Khan SR:
Increased expression of monocyte chemoattractant protein-1 (MCP-1)
by renal epithelial cells in culture on exposure to calcium
oxalate, phosphate and uric acid crystals. Nephrol Dial Transplant.
18:664–669. 2003.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Deng Y, Sun B and Li C: UP-3.135: COM
crystals stimulate the expression and activity of NADPH oxidase in
macrophage. Urology. 74 (Suppl)(S337)2009.
|
|
36
|
Mukai H, Miura Y, Kotani K, Kotoda A,
Kurosu H, Yamada T, Kuro-O M and Iwazu Y: The effects for
inflammatory responses by CPP with different colloidal properties
in hemodialysis patients. Sci Rep. 12(21856)2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Viegas CSB, Rafael MS, Enriquez JL,
Teixeira A, Vitorino R, Luís IM, Costa RM, Santos S, Cavaco S,
Neves J, et al: Gla-rich protein acts as a calcification inhibitor
in the human cardiovascular system. Arterioscler Thromb Vasc Biol.
35:399–408. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Tesfamariam B: Involvement of vitamin
K-dependent proteins in vascular calcification. J Cardiovasc
Pharmacol Ther. 24:323–333. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Koeppert S, Ghallab A, Peglow S, Winkler
CF, Graeber S, Büscher A, Hengstler JG and Jahnen-Dechent W: Live
imaging of calciprotein particle clearance and receptor mediated
uptake: Role of calciprotein monomers. Front Cell Dev Biol.
9(633925)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kutikhin AG, Feenstra L, Kostyunin AE,
Yuzhalin AE, Hillebrands JL and Krenning G: Calciprotein particles:
Balancing mineral homeostasis and vascular pathology. Arterioscler
Thromb Vasc Biol. 41:1607–1624. 2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Herrmann M, Schäfer C, Heiss A, Gräber S,
Kinkeldey A, Büscher A, Schmitt MM, Bornemann J, Nimmerjahn F,
Herrmann M, et al: Clearance of fetuin-A-containing calciprotein
particles is mediated by scavenger receptor-A. Circ Res.
111:575–584. 2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Heiss A, DuChesne A, Denecke B, Grötzinger
J, Yamamoto K, Renné T and Jahnen-Dechent W: Structural basis of
calcification inhibition by alpha 2-HS glycoprotein/fetuin-A.
Formation of colloidal calciprotein particles. J Biol Chem.
278:13333–13341. 2003.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Aghagolzadeh P, Bachtler M, Bijarnia R,
Jackson C, Smith ER, Odermatt A, Radpour R and Pasch A:
Calcification of vascular smooth muscle cells is induced by
secondary calciprotein particles and enhanced by tumor necrosis
factor-α. Atherosclerosis. 251:404–414. 2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Smith ER, Hanssen E, McMahon LP and Holt
SG: Fetuin-A-containing calciprotein particles reduce mineral
stress in the macrophage. PLoS One. 8(e60904)2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Silaghi CN, Ilyés T, Van Ballegooijen AJ
and Crăciun AM: Calciprotein particles and serum calcification
propensity: Hallmarks of vascular calcifications in patients with
chronic kidney disease. J Clin Med. 9(1287)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Blaser MC and Aikawa E: Roles and
regulation of extracellular vesicles in cardiovascular mineral
metabolism. Front Cardiovasc Med. 5(187)2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Disthabanchong S and Srisuwarn P:
Mechanisms of vascular calcification in kidney disease. Adv Chronic
Kidney Dis. 26:417–426. 2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Shishkova DK, Velikanova EA, Bogdanov LA,
Sinitsky MY, Kostyunin AE, Tsepokina AV, Gruzdeva OV, Mironov AV,
Mukhamadiyarov RA, Glushkova TV, et al: Calciprotein particles link
disturbed mineral homeostasis with cardiovascular disease by
causing endothelial dysfunction and vascular inflammation. Int J
Mol Sci. 22(12458)2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Shishkova D, Lobov A, Zainullina B,
Matveeva V, Markova V, Sinitskaya A, Velikanova E, Sinitsky M,
Kanonykina A, Dyleva Y and Kutikhin A: Calciprotein particles cause
physiologically significant pro-inflammatory response in
endothelial cells and systemic circulation. Int J Mol Sci.
23(14941)2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Feenstra L, Kutikhin AG, Shishkova DK,
Buikema H, Zeper LW, Bourgonje AR, Krenning G and Hillebrands JL:
Calciprotein particles induce endothelial dysfunction by impairing
endothelial nitric oxide metabolism. Arterioscler Thromb Vasc Biol.
43:443–455. 2023.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Durand MJ and Gutterman DD: Diversity in
mechanisms of endothelium-dependent vasodilation in health and
disease. Microcirculation. 20:239–247. 2013.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Zhao Y, Vanhoutte PM and Leung SWS:
Vascular nitric oxide: Beyond eNOS. J Pharmacol Sci. 129:83–94.
2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Förstermann U and Sessa WC: Nitric oxide
synthases: Regulation and function. Eur Heart J. 33:829–837,
837a-837d. 2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Oe Y, Mitsui S, Sato E, Shibata N, Kisu K,
Sekimoto A, Miyazaki M, Sato H, Ito S and Takahashi N: Lack of
endothelial nitric oxide synthase accelerates ectopic calcification
in uremic mice fed an adenine and high phosphorus diet. Am J
Pathol. 191:283–293. 2021.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Lee HL, Woo KM, Ryoo HM and Baek JH: Tumor
necrosis factor-alpha increases alkaline phosphatase expression in
vascular smooth muscle cells via MSX2 induction. Biochem Biophys
Res Commun. 391:1087–1092. 2010.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Zickler D, Luecht C, Willy K, Chen L,
Witowski J, Girndt M, Fiedler R, Storr M, Kamhieh-Milz J, Schoon J,
et al: Tumour necrosis factor-alpha in uraemic serum promotes
osteoblastic transition and calcification of vascular smooth muscle
cells via extracellular signal-regulated kinases and activator
protein 1/c-FOS-mediated induction of interleukin 6 expression.
Nephrol Dial Transplant. 33:574–585. 2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Sage AP, Lu J, Tintut Y and Demer LL:
Hyperphosphatemia-induced nanocrystals upregulate the expression of
bone morphogenetic protein-2 and osteopontin genes in mouse smooth
muscle cells in vitro. Kidney Int. 79:414–422. 2011.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Li X, Yang HY and Giachelli CM: BMP-2
promotes phosphate uptake, phenotypic modulation, and calcification
of human vascular smooth muscle cells. Atherosclerosis.
199:271–277. 2008.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Liberman M, Johnson RC, Handy DE, Loscalzo
J and Leopold JA: Bone morphogenetic protein-2 activates NADPH
oxidase to increase endoplasmic reticulum stress and human coronary
artery smooth muscle cell calcification. Biochem Biophys Res
Commun. 413:436–441. 2011.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Byon CH, Javed A, Dai Q, Kappes JC,
Clemens TL, Darley-Usmar VM, McDonald JM and Chen Y: Oxidative
stress induces vascular calcification through modulation of the
osteogenic transcription factor Runx2 by AKT signaling. J Biol
Chem. 283:15319–15327. 2008.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Liu H, Li X, Qin F and Huang K: Selenium
suppresses oxidative-stress-enhanced vascular smooth muscle cell
calcification by inhibiting the activation of the PI3K/AKT and ERK
signaling pathways and endoplasmic reticulum stress. J Biol Inorg
Chem. 19:375–388. 2014.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Blanc A, Pandey NR and Srivastava AK:
Distinct roles of Ca2+, calmodulin, and protein kinase C in
H2O2-induced activation of ERK1/2, p38 MAPK, and protein kinase B
signaling in vascular smooth muscle cells. Antioxid Redox Signal.
6:353–366. 2004.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Zhao MM, Xu MJ, Cai Y, Zhao G, Guan Y,
Kong W, Tang C and Wang X: Mitochondrial reactive oxygen species
promote p65 nuclear translocation mediating high-phosphate-induced
vascular calcification in vitro and in vivo. Kidney Int.
79:1071–1079. 2011.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Shishkova D, Velikanova E, Sinitsky M,
Tsepokina A, Gruzdeva O, Bogdanov L and Kutikhin A: Calcium
phosphate bions cause intimal hyperplasia in intact aortas of
normolipidemic rats through endothelial injury. Int J Mol Sci.
20(5728)2019.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Kurozumi A, Nakano K, Yamagata K, Okada Y,
Nakayamada S and Tanaka Y: IL-6 and sIL-6R induces STAT3-dependent
differentiation of human VSMCs into osteoblast-like cells through
JMJD2B-mediated histone demethylation of RUNX2. Bone. 124:53–61.
2019.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Tintut Y, Parhami F, Boström K, Jackson SM
and Demer LL: cAMP stimulates osteoblast-like differentiation of
calcifying vascular cells. Potential signaling pathway for vascular
calcification. J Biol Chem. 273:7547–7553. 1998.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Turner ME, Bartoli-Leonard F and Aikawa E:
Small particles with large impact: Insights into the unresolved
roles of innate immunity in extracellular vesicle-mediated
cardiovascular calcification. Immunol Rev. 312:20–37.
2022.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Ewence AE, Bootman M, Roderick HL, Skepper
JN, McCarthy G, Epple M, Neumann M, Shanahan CM and Proudfoot D:
Calcium phosphate crystals induce cell death in human vascular
smooth muscle cells: A potential mechanism in atherosclerotic
plaque destabilization. Circ Res. 103:e28–e34. 2008.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Wang S and Zhang Z: Biological
characteristics of nanobacteria and the relationship between
nanobacteria and oral stone diseases. J Oral Sci Res. 35:827–829.
2019.
|
|
70
|
Kao WK, Chole RA and Ogden MA: Evidence of
a microbial etiology for sialoliths. Laryngoscope. 130:69–74.
2020.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Zeng J, Yang F, Zhang W, Gong Q, Du Y and
Ling J: Association between dental pulp stones and calcifying
nanoparticles. Int J Nanomedicine. 6:109–118. 2011.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Zhang SM, Tian F, Jiang XQ, Li J, Xu C,
Guo XK and Zhang FQ: Evidence for calcifying nanoparticles in
gingival crevicular fluid and dental calculus in periodontitis. J
Periodontol. 80:1462–1470. 2009.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Yang F, Zeng J, Zhang W, Sun X and Ling J:
Evaluation of the interaction between calcifying nanoparticles and
human dental pulp cells: A preliminary investigation. Int J
Nanomedicine. 6:13–18. 2010.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Sakai Y, Nemoto E, Kanaya S, Shimonishi M
and Shimauchi H: Calcium phosphate particles induce interleukin-8
expression in a human gingival epithelial cell line via the nuclear
factor-κB signaling pathway. J Periodontol. 85:1464–1473.
2014.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Demir T: Is there any relation of
nanobacteria with periodontal diseases? Med Hypotheses. 70:36–39.
2008.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Sun H, Warren J, Yip J, Ji Y, Hao S, Han W
and Ding Y: Factors influencing gallstone formation: A review of
the literature. Biomolecules. 12(550)2022.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Wen Y, Li Y, Yang Z, Wang XJ, Wei H, Liu
W, Tan AL, Miao XY, Wang QW, Huang SF, et al: Nanobacteria in
serum, bile and gallbladder mucosa of cholecystolithiasis patients.
Zhonghua Wai Ke Za Zhi. 41:267–270. 2003.PubMed/NCBI(In Chinese).
|
|
78
|
Wang L, Shen W, Wen J, An X, Cao L and
Wang B: An animal model of black pigment gallstones caused by
nanobacteria. Dig Dis Sci. 51:1126–1132. 2006.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Wen Y, Li YG, Yang ZL, Wang XJ, Wei H, Liu
W, Miao XY, Wang QW, Huang SF, Yang J, et al: Detection of
nanobacteria in serum, bile and gallbladder mucosa of patients with
cholecystolithiasis. Chin Med J (Engl). 118:421–424.
2005.PubMed/NCBI
|
|
80
|
Kajander EO: Nanobacteria-propagating
calcifying nanoparticles. Lett Appl Microbiol. 42:549–552.
2006.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Liang L, Jin Z, Xu B, Guo Y, Peng M and
Wen Z: Mechanism of nanobacteria promoting apoptosis of human
gallbladder epithelial cells. Chin J Exp Surg. 36:1410–1413.
2019.
|














