Introduction
Acute coronary syndrome (ACS) is a fatal
manifestation of coronary heart disease (CAD) comprising three main
types: Unstable angina (UA), ST-elevated myocardial infarction
(STEMI), or non-ST elevated myocardial infarction (NSTEMI)
(1,2). ACS is primarily caused by plaque
rupture, erosion, and thrombosis (3,4),
leading to a significant reduction in coronary blood flow (5). A retrospective cohort study conducted
by Kaul et al (6) found that
patients with UA, STEMI and NSTEMI had a high likelihood of
developing heart failure (16, 23.4 and 25.4% respectively),
suggesting that ACS may be a significant risk factor for the
development of heart failure. Currently, although some treatments
have been proven effective for patients with ACS in clinical
practice, recurrent events and adverse effects, such as hypotension
and hyperkalemia, remain often unavoidable (7). Therefore, identifying effective
therapies with minimal side-effects to alleviate clinical symptoms
and improve the quality of life for patients with ACS is crucial
from both medical and socioeconomic perspectives.
Notably, 12/15-lipoxygenase (ALOX15), containing 13
introns and 14 exons, is mapped at LOX gene cluster on chromosome
17 and can encode arachidonic acid 12/15-lipoxygenase (12/15-LOX)
(8). It is acknowledged that
12/15-LOX is a type of dioxygenase enzyme, which is crucial in
incorporating oxygen into polyunsaturated fatty acids to generate
biologically-active peroxide products such as
13-hydroperoxy-octadecadienoic acid and
hydroperoxy-eicosatetraenoic acid (HPETE) (8). Reports on ALOX15 in ACS are currently
extremely limited. Research on ALOX15 in cardiac disorders has
mainly focused on myocardial ischemia-reperfusion injury. Previous
studies have demonstrated that 12/15-LOX plays an essential role in
inducing the transformation of low-density lipoprotein (LDL) into
atherosclerotic lesions, indicating that ALOX15 may possess
proatherogenic properties (9-11).
Ma et al (12) found that
ALOX15-induced peroxidation of polyunsaturated fatty
acid-phospholipids increases the susceptibility to ferroptosis in
ischemia-induced myocardial damage (12). Similarly, Cai et al (13) proposed that ALOX15/HPETE-mediated
cardiomyocyte ferroptosis plays a crucial role in prolonged
myocardial ischemia-reperfusion injury (13). Additionally, ALOX15
activation-induced inflammation has been confirmed to be associated
with the development of heart failure (14). As early as 2013, Silbiger et
al (15) conducted a
transcriptional profiling analysis and indicated that ALOX15 is one
of the most significant gene expression biomarkers for the very
early stages of ACS (15). Notably,
our previous study indicated that ALOX15 can be regulated by the
long non-coding RNA ENST00000538705.1 to promote progression of ACS
(16), however the downstream
regulatory mechanisms of ALOX15 remain unclear.
Phosphoinositide 3-kinases (PI3Ks) and their
downstream target serine/threonine kinase AKT (also known as
protein kinase B) are a conserved family of signal transduction
enzymes (17). Previous research
has increasingly revealed the crucial role of the PI3K/AKT
signaling pathway in cardiac disorders. For example, the
resveratrol-mediated activation of the PI3K/AKT pathway exerts
cardioprotection by reducing mitochondrial oxidative damage during
myocardial ischemia/reperfusion injury (18). By modulating the PI3K/AKT pathway,
Qingda granule can attenuate angiotensin II-induced cardiac
hypertrophy and apoptosis (19). In
a mouse model of myocardial infarction or heart failure, activation
of the PI3K/AKT pathway has been shown to promote angiogenesis and
provide cardioprotection (20,21).
However, whether the PI3K/AKT pathway plays a regulatory role in
ACS is still in the preliminary stage of exploration. Fibroblast
growth factor receptors (FGFRs), belonging to the transmembrane
tyrosine kinase receptor family, are strongly associated with
multiple cellular and biological processes such as cell growth,
migration, angiogenesis and wound repairment (22,23).
Currently, four main types of FGFRs have been identified, including
FGFR1, FGFR2, FGFR3 and FGFR4 (24,25).
Among these subtypes, FGFR2 plays a particularly essential role in
cell differentiation, growth and migration (26). It is widely reported that
endothelial dysfunction is strongly associated with the progression
of ACS (27). A recent study
conducted by Jiao et al (28) demonstrated that FGFR2 mediates
endothelial dysfunction by regulating the AKT/Nrf2/ARE signaling
pathway. Furthermore, inhibition of FGFR2 has been proposed as
therapeutic target for cardiac fibrosis (a hallmark pathological
feature following myocardial injury) (29). Notably, FGFR2 functioning as an
activator of the PI3K/AKT pathway has been widely reported in
numerous human diseases (30,31).
Currently, there are no relevant research studies on the
interaction between ALOX15 and the FGFR2/PI3K/AKT signaling pathway
in the progression of ACS. Therefore, determining whether the
FGFR2/PI3K/AKT pathway, mediated by ALOX15, exerts regulatory
functions in the progression of ACS has sparked our great
interest.
In the present study, the effects of ALOX15 on human
primary coronary artery endothelial cells (HCAECs) were
investigated. In addition, a rat model of ACS was established. The
aim of the study was to preliminarily explore the downstream
regulatory mechanism of ALOX15 in ACS pathology.
Materials and methods
Patients
A total of 30 patients with ACS (15 women and 15
men; age range, 52-70 years; mean age, 60.5±6.4 years) were
admitted to The Third Affiliated Hospital of Shanghai University
(Wenzhou People's Hospital; Wenzhou, China). The inclusion criteria
were as follows: i) Diagnosis of UA, STEMI or NSTEMI; ii)
postoperative blood flow of the target vessel reaching thrombolysis
in myocardial infarction (TIMI) grade; iii) complete clinical
history available; iv) no surgical contraindications. The exclusion
criteria were as follows: i) Presence of congenital heart disease,
heart failure, or peripheral vascular diseases; ii) history of
prior cardiac surgery, such as coronary stent implantation or
coronary artery bypass; iii) recent history of infectious diseases;
iv) missing or incomplete case data, or lack of cooperation with
follow-up. Concurrently, 30 healthy controls (15 women and 15 men;
age range, 48-66 years; mean age, 56.7±6.4 years) were selected as
the physical examination population, with similar age and sex to
the experimental group, no symptoms of chest tightness and chest
pain, and no obvious abnormalities in biochemical tests,
electrocardiogram, chest CT, cardiac color Doppler ultrasound and
other imaging tests. Venous blood samples from patients with ACS
and healthy controls were collected, centrifuged at 3,000 x g at
4˚C for 5 min, and then stored at -80˚C for subsequent experiments.
The patients/participants provided their written informed consent
to participate in this study. The present study was conducted
according to the guidelines of the Declaration of Helsinki and
approved (approval no. 2020-351) by the Ethics Committee of The
Third Affiliated Hospital of Shanghai University (Wenzhou People's
Hospital; Wenzhou, China).
Animals, cells and reagent
A total of 18 male Sprague-Dawley rats weighing
250-280 g (8 weeks old) was obtained from Vital River Laboratory
Animal Technology Co., Ltd. HCAECs (passage 2; cat. no. CP-H087)
and rat coronary artery endothelial cells (cat. no. CP-R081) were
purchased from Procell Life Science & Technology Co., Ltd.
Approval for the use of primary cells was obtained from the Ethics
Committee of The Third Affiliated Hospital of Shanghai University
(Wenzhou People's Hospital; Wenzhou, China). 293 cells were
obtained from Thermo Fisher Scientific, Inc. Endothelial basal
medium 2 (EBM-2) and fetal bovine serum (FBS) were purchased from
Lonza Group, Ltd. The Cell Counting Kit-8 was provided by Abbkine
Scientific Co., Ltd. Small hairpin targeting ALOX15 (shRNA ALOX15),
negative control (shRNA NC), small interfering RNA (siRNA)1/2/3
ALOX15 along with their corresponding negative control (siRNA NC),
as well as pcDNA3.1-ALOX15 (OE-ALOX15), OE-FGFR2 and the pcDNA3.1
empty vector (OE-NC) were synthesized by Genomeditech (Shanghai)
Co., Ltd. The immunoprecipitation kit with protein A+G magnetic
beads (cat. no. P2179S), hematoxylin and eosin (H&E) staining
kit (cat. no. C0105S), insulin-like growth factor 1 (IGF-1; cat.
no. P5502), RIPA lysis buffer (cat. no. P0013B), ECL kit (cat. no.
P0018M) and BCA kit (cat. no. P0012) were obtained from Beyotime
Insitute of Biotechnology. Guangzhou RiboBio Co., Ltd. supplied the
riboFECT™ mRNA transfection agent (cat. no. C11055-1).
Lipofectamine 3000 (cat. no. L3000150) was obtained from Thermo
Fisher Scientific, Inc. The total cholesterol (TC) content assay
kit (cat. no. BC1980) was acquired from Beijing Solarbio Science
& Technology Co., Ltd., while the high-density
lipoprotein-cholesterol (HDL-C) assay kit (cat. no. A112-1-1) and
low-density lipoprotein-cholesterol (LDL-C) assay kit (cat. no.
A113-1-1) were sourced from Nanjing Jiancheng Bioengineering
Institute. TRIzol reagent (cat. no. 10606ES60), Hifair®
Ⅱ 1st Strand cDNA Synthesis SuperMix (cat. no. 11123ES60) and
Hieff® qPCR SYBR Green Master Mix (cat. no. 11202ES08)
were procured from Shanghai Yeasen Biotechnology Co., Ltd. Primary
antibodies AKT (cat. no. 60203-2-Ig), phoshorylated (p)-AKT (cat.
no. 66444-1-Ig), GAPDH (cat. no. 60004-1-Ig) and HRP-conjugated
secondary antibodies (cat. no. SA00001-2) were obtained from
Proteintech Group, Inc. Primary antibodies p-PI3K (cat. no. 4228T)
and FGFR2 (cat. no. 23328) were purchased from Cell Signaling
Technology, Inc., and PI3K antibody (cat. no. ab191606) was sourced
from Abcam. ALOX15 antibody (cat. no. sc-133085) was obtained from
Santa Cruz Biotechnology, Inc.
Cell culture
HCAECs were cultured in EBM-2 containing 15% FBS.
The culture cells were maintained in an environment with 5%
CO2 and a temperature of 37˚C. Cells in the logarithmic
growth phase were harvested for functional experiments.
Cell transfection and treatment
Cell transfection experiments were conducted in
strict compliance with the protocols of the riboFECT™ mRNA
transfection agent. Briefly, OE-ALOX15, OE-FGFR2, siRNA1/2/3 ALOX15
and their corresponding NCs (all, 50 nM) were individually
introduced into HCAECs using the riboFECT™ mRNA transfection agent
for 48 h at 37˚C. Following a 48-h period, the cells were collected
for the subsequent experiments. The sequences of siRNA1/2/3 ALOX15
and siRNA NC were as follows: siRNA1 ALOX15 forward,
5'-GUCGAUACAUCCUAUCUUCAATT-3' and reverse,
5'-UUGAAGAUAGGAUGUAUCGACTT-3'; siRNA2 ALOX15 forward,
5'-AUGACUUCAACCGGAUUUUCUTT-3' and reverse,
5'-AGAAAAUCCGGUUGAAGUCAUTT-3'; siRNA3 ALOX15 forward:
5'-CGCUAUCAAAGACUCUCUAAATT-3' and reverse,
5'-UUUAGAGAGUCUUUGAUAGCGTT-3'; siRNA NC forward,
5'-UUCUCCGAACGUGUCACGUTT-3' and reverse:
5'-ACGUGACACGUUCGGAGAATT-3'. Additionally, to ascertain the mutual
effect between ALOX15 and the PI3K/AKT pathway through reverse
transcription-quantitative PCR (RT-qPCR), scratch tests, CCK-8
assay and western blot analysis, 4 µl of PI3K/AKT pathway specific
agonist IGF-1 was added to HCAECs for 24 h at 37˚C.
RT-qPCR
The extraction of RNA was conducted with the aid of
TRIzol reagent. The concentration of total RNA was measured using a
NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific, Inc.).
Total RNA (500 ng) was reversible transcribed into cDNA via
Hifair® II 1st Strand cDNA Synthesis SuperMix at 45˚C
for 45 min. Subsequently, PCR analysis was carried out in
accordance with the protocols of Hieff® qPCR SYBR Green
Master Mix. The reaction conditions were as follows: One cycle at
95˚C lasting 2 min, 40 cycles at 95˚C for 10 sec, 60˚C for 30 sec,
and 72˚C for 30 sec. The primers used in the present study are
listed in Table I. Relative mRNA
expression of ALOX15 and FGFR2 was normalized against GAPDH and
calculated with the 2-ΔΔCq method (32).
 | Table IReverse transcription
quantitative-PCR primer synthesis list. |
Table I
Reverse transcription
quantitative-PCR primer synthesis list.
| Gene | Sequences | |
|---|
| ALOX15 | Forward |
5'-CCGCTGCTGTTTGTGAAACT-3' |
| | Reverse |
5'-AGCGGTAACAAGGGAACCTG-3' |
| FGFR2 | Forward |
5'-TGACCAAACGTATCCCCCTG-3' |
| | Reverse |
5'-GGTGTCTGCCGTTGAAGAGA-3' |
| GAPDH | Forward |
5'-TCAAGAAGGTGGTGAAGCAGG-3' |
| | Reverse |
5'-TCAAAGGTGGAGGAGTGGGT-3' |
Scratch tests
The transfected HCAECs were seeded in 6-well plates
at a density of 3x105 cells/well. Upon reaching 100%
confluence, a scratch was created in the monolayer using a pipette
tip (10 µl), and HCAECs were then incubated in a serum-free medium
for 48 h. The width of the scratches at both 0 h and 48 h was
recorded under a light microscope (Olympus Corporation). The
migratory abilities of HCAECs were analyzed with ImageTool software
version 1.46 (University of Texas Health Science Center at San
Antonio), employing the following formula: (The width at 0 h - the
width at 48 h)/the width at 0 h x 100%.
CCK-8 assay
HCAECs were inoculated into culture plates with
96-wells (4x103 cells/well) and subsequently cultured
for 24, 36, 48, and 72 h, respectively. A total of 10 µl of CCK-8
solution was added into each well and the cells were then incubated
for 2 h at 37˚C. Cell viability was then assessed using a
microplate reader (DR-3518G, Wuxi Hiwell Diatek Instruments, Co.,
Ltd.) at an absorbance of 450 nm.
Co-immunoprecipitation (CO-IP)
assay
For per IP reaction, 1 ml of lysis buffer was added
to HCAECs (2x107 cells) and incubated on ice for 30 min.
Following centrifugation at 14,000 x g for 20 min at 4˚C, the cell
lysates from HCAECs were collected, and the resulting supernatants
were incubated with 40 µl of a 1:1 slurry of Protein G-Sepharose
beads conjugated to rabbit anti-ALOX15 (5 µg) and anti-FGFR2 (5 µg)
antibodies at 4˚C overnight. Concurrently, an anti-IgG antibody (5
µg) was used as a control. The beads were then washed 5 times with
1 ml wash buffer and centrifuged at 1,000 x g for 1 min at 4˚C
between washes. After the final wash, the supernatant was removed,
and 30 µl of elution buffer was added in the samples and boiled at
95˚C for 5 min. Subsequently, the samples were subjected to
gradient SDS-polyacrylamide gels and transferred to membranes. The
immunoprecipitates obtained were then analyzed by western
blotting.
Animal grouping and treatment
The ACS rat model was established based on
previously described protocols (33,34).
Approval for the use of animals was obtained from the Ethics
Committee of Wenzhou Medical University (Wenzhou, China). In these
previous studies, the modeling method for ACS closely resembled
that of acute myocardial infarction, as the latter is one of the
clinical manifestations of ACS. Sprague-Dawley rats were randomly
assigned to three groups: The sham group, the ACS model + shRNA NC
group, and the ACS model + shRNA ALOX15 group, with six rats in
each group. The rats were intraperitoneally injected with 50 mg/kg
of pentobarbital sodium for anesthesia. Their limbs were then
secured to a surgical table while maintaining them in a supine
position. Following thoracotomy, the heart was exposed and the
anterior descending coronary artery was ligated using 5/0 surgical
sutures. To prevent infection, penicillin (100,000 IU) was
subcutaneously injected into each rat. Meanwhile, only threading
was performed in rats in the sham group, without ligation. The
electrocardiogram results were recorded and the successfully
established ACS rat model was indicated as follows: ST segment
and/or high T wave was markedly elevated. A 3rd-generation system
was used and 9 µg lentiviral vectors were co-transfected into the
293 cells through Lipofectamine 3000 at room temperature. The rate
of lentiviral plasmid: packaging vector: envelope was 1:1:1.
Lentivirus-containing medium was collected 48 h after transfection
and used to infect rat coronary artery endothelial cells (passage
2; 5x106 cells/well) at a multiplicity of infection of
20. Subsequently, 48 h after lentiviral infection, 1 µg/ml
puromycin was used to select the infected cells. After 72 h, rats
in the ACS model + shRNA NC group received an intramyocardial
injection of shRNA NC (5'-GCTGTTCGATCGGGAAACA-3') integrated into
lentiviral vectors (50 µg), and those in the ACS model + shRNA
ALOX15 group were treated with lentivirus containing shRNA ALOX15
sequence (5'-GCTGATGCCTGATGGACAA-3') via intramyocardial injection.
After 2 weeks, the rats in the different groups were euthanized
with an overdose of pentobarbital sodium (200 mg/kg) via
intraperitoneal injection. Whole blood samples (from the eyeballs)
and heart were collected for subsequent tests. Animal experiments
were conducted in compliance with the Guidelines for the Use of
Laboratory Animals and approved (approval no. xmsq2023-1367) by the
Ethics Committee of Wenzhou Medical University (Wenzhou,
China).
Biochemical tests
The whole blood samples obtained from the eyeballs
of the rats were placed at room temperature for 2 h before being
centrifuged at 1,000 x g for 20 min at room temperature. Commercial
Kits were employed to determine the levels of TC, HDL-C and LDL-C
in serum using a fully automatic bio analysis machine.
H&E staining
Rat myocardial tissues were fixed in 4%
paraformaldehyde, deparaffinized and hydrated prior to paraffin
embedding. Subsequently, the tissues were sectioned into 4-µm thick
slices. The sections were stained with H&E for 3 min. Following
mounting with neutral gum, images were captured using an optical
microscope (OLYMPUS BX53; Olympus Corporation).
Western blot analysis
Total proteins were extracted from HCAECs or
myocardial tissues using RIPA lysis buffer and then measured the
concentration with a BCA Kit. All steps were conducted on ice.
Proteins (50 µg protein/lane) were then separated by 10% sodium
dodecyl sulphate-polyacrylamide gel electrophoresis and transferred
onto a polyvinylidene difluoride membrane. The membrane was blocked
using 5% nonfat milk-Tris-buffered saline with 0.05% Tween-20 for 2
h at 25˚C, on which primary antibodies (ALOX15, 1:1,000; FGFR2,
1:1,000; PI3K, 1:1,000; p-PI3K, 1:1,000; AKT, 1:5,000; p-AKT,
1:2,000; GAPDH, 1:5,0000) were incubated overnight at 4˚C.
Subsequently, the corresponding secondary antibodies (1:1,000) were
added followed by incubation for 1 h at 37˚C. GAPDH was the
normalization for proteins. Protein signals were detected using an
ECL kit under Gel-Pro analyzer (version 4.0; Media Cybernetics,
Inc.).
Statistical analysis
In vitro experiments were performed in
triplicate, and each experiment was repeated 3 times. In
vivo experiments were performed using 6 rats per group.
Unpaired Student's t-test was adopted to gauge the differences
between two groups. The variations among multiple groups were
gauged by applying one-way ANOVA, followed by Tukey's post hoc
multiple comparisons test. Data analysis was performed using SPSS
software v22.0 (IBM Corp.). The data were indicated as the mean ±
standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
High expression of ALOX15 and FGFR2 is
observed in the serum of patients with ACS
After obtaining informed consent from the patients,
blood samples were collected from individuals diagnosed with ACS.
Following centrifugation, the expression levels of ALOX15 and FGFR2
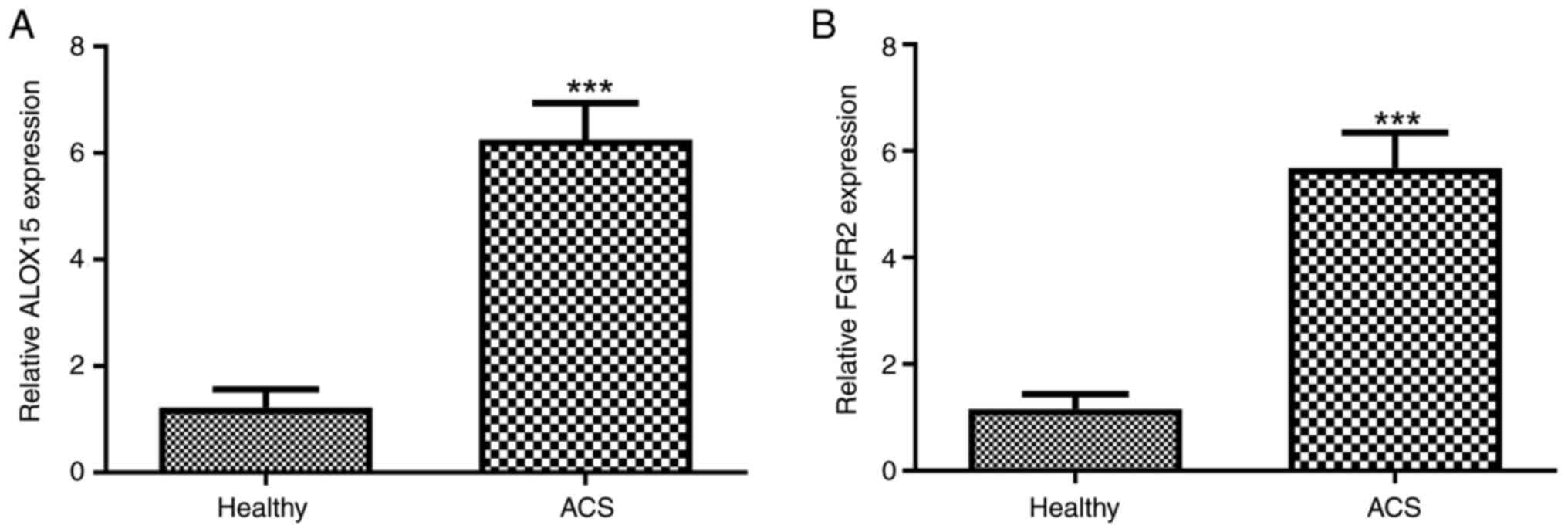
in the serum of patients with ACS were assessed. As illustrated in
Fig. 1A, a significant upregulation
of ALOX15 expression in patients with ACS compared with healthy
individuals (P<0.001) was observed. Similarly, elevated levels
of FGFR2 were also detected in patients with ACS when compared with
healthy controls (Fig. 1B;
P<0.001).
Effects of ALOX15 on ACS progression
in vitro
ALOX15 was then overexpressed or silenced to
evaluate its effects on the progression of ACS in vitro.
Following OE-ALOX15 transfection, the expression of ALOX15 in
HCAECs was significantly increased (Fig. 2A; P<0.001). Meanwhile, siRNA1
ALOX15, siRNA2 ALOX15 and siRNA3 ALOX15 were individually
transfected into HCAECs. As revealed in Fig. 2B, the expression levels of ALOX15
were significantly reduced after siRNA ALOX15 transfection
(P<0.001). Notably, siRNA3 ALOX15 was selected for the
subsequent experiments due to its relatively high transfection
efficiency. Wound healing assays were then performed and it was
revealed that the overexpressed ALOX15 significantly increased the
migratory abilities of HCAECs; conversely, silenced ALOX15
exhibited an inhibitory effect on migration in these cells
(Fig. 2C and D; P<0.01). Moreover, CCK-8 asays showed
that the proliferative ability of HCAECs were increased following
OE-ALOX15 transfection, but attenuated after siRNA ALOX15
treatment, in a time-dependent manner (Fig. 2E; P<0.01).
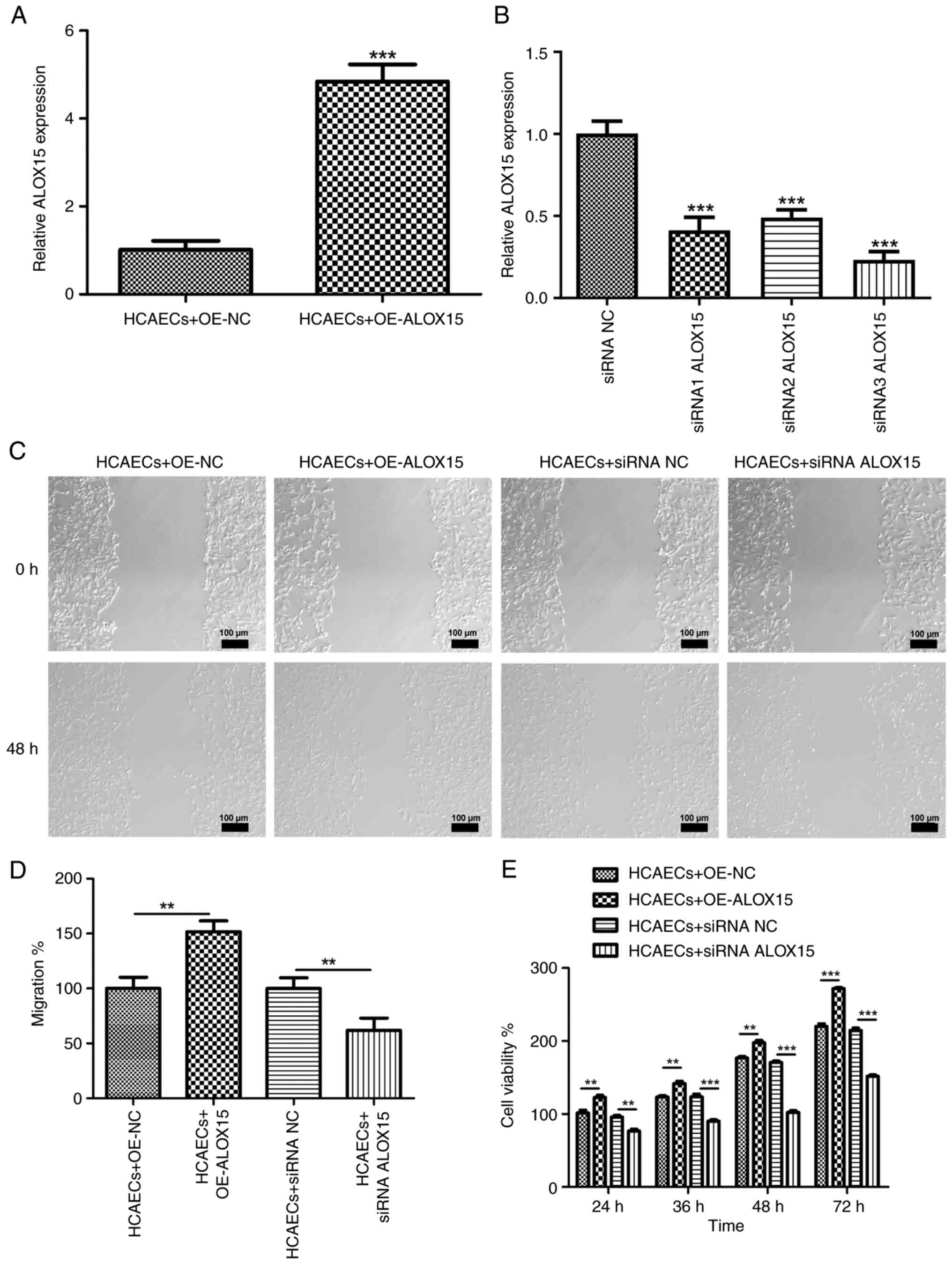 | Figure 2Effects of ALOX15 on acute coronary
syndrome progression in vitro. (A) The expression of ALOX15
in HCAECs after transfection with OE-ALOX15 or OE-NC was detected
by RT-qPCR. ***P<0.001 vs. HCAECs + OE-NC. (B) The
expression of ALOX15 in HCAECs after transfection with siRNA1
ALOX15, siRNA2 ALOX15, siRNA3 ALOX15 or siRNA NC was detected by
RT-qPCR. ***P<0.001 vs. siRNA NC. (C and D) The
migration of HCAECs transfected with OE-ALOX15/OE-NC or siRNA
ALOX15/siRNA NC was measured by scratch wound healing assays.
**P<0.01. Scale bar, 100 µm. (E) The viability of
HCAECs transfected with OE-ALOX15/OE-NC or siRNA ALOX15/siRNA NC
was measured by Cell Counting Kit-8 assays. **P<0.01
and ***P<0.001. ALOX15, 12/15-lipoxygenase; FGFR2,
fibroblast growth factor receptor 2; HCAECs, human primary coronary
artery endothelial cells; OE, overexpression; NC, negative control;
RT-qPCR, reverse transcription-quantitative PCR; siRNA, small
interfering RNA. |
Effects of ALOX15 on the
FGFR2/PI3K/AKT signaling pathway in HCAECs
CO-IP analysis demonstrated that ALOX15 protein
could interact with FGFR2 in HCAECs (Fig. 3A). Furthermore, following
transfection, it was demonstrated that the mRNA expression of
ALOX15 and FGFR2 was significantly upregulated by the ectopic
expression of ALOX15, while silencing of ALOX15 resulted in a
significant decrease in their expression (Fig. 3B; P<0.001). Additionally, the
effects of ALOX15 overexpression or silencing on the levels of
FGFR2/PI3K/AKT signaling pathway-related proteins were also
examined via western blotting. As illustrated in Fig. 3C, the results indicated that in
HCAECs, the protein levels of ALOX15, FGFR2, p-PI3K and p-AKT were
all markedly enhanced after OE-ALOX15 transfection, while they were
reduced after transfection with siRNA ALOX15 (P<0.001). Notably,
the protein levels of PI3K and AKT appeared unchanged following
treatment with OE-ALOX15 or siRNA ALOX15.
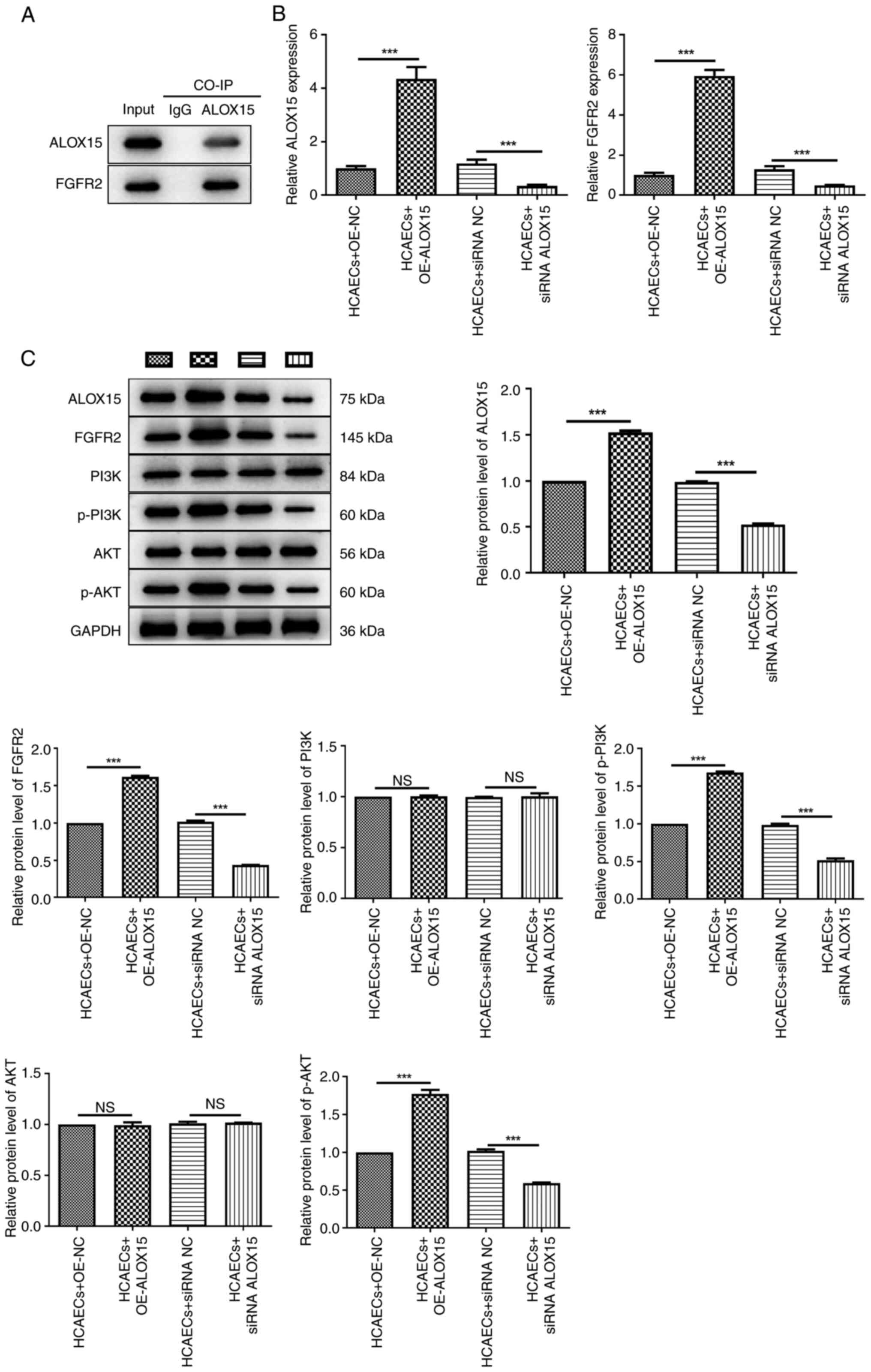 | Figure 3Effects of ALOX15 on the
FGFR2/PI3K/AKT signaling pathway in HCAECs. (A)
Co-immunoprecipitation assays of ALOX15 and FGFR2 in HCAECs. (B)
The mRNA expression of ALOX15 and FGFR2 in HCAECs after
transfection with OE-ALOX15/OE-NC or siRNA ALOX15/siRNA NC was
detected by reverse transcription-quantitative PCR.
***P<0.001. (C) The protein levels of ALOX15, FGFR2,
PI3K, p-PI3K, AKT and p-AKT in HCAECs after transfection with
OE-ALOX15/OE-NC or siRNA ALOX15/siRNA NC were determined by western
blotting. ***P<0.001. ALOX15, 12/15-lipoxygenase;
FGFR2, fibroblast growth factor receptor 2; HCAECs, human primary
coronary artery endothelial cells; OE, overexpression; NC, negative
control; siRNA, small interfering RNA; p-, phosphorylated; NS, not
significant. |
Silencing of ALOX15 mitigates ACS
progression in vitro through the FGFR2/PI3K/AKT signaling
pathway
Firstly, FGFR2 was overexpressed in HCAECs to
examine the interaction of ALOX15 with FGFR2 (P<0.001), as
depicted in Fig. 4A. Meanwhile, 4
µl of PI3K/AKT pathway specific agonist IGF-1 was added to HCAECs
to further ascertain the mutual effect between ALOX15 and the
PI3K/AKT pathway. As illustrated in Fig. 4B, it was determined that both the
overexpression of FGFR2 and the addition of IGF-1 reversed the
suppressive effects of ALOX15 knockdown on the mRNA expression of
ALOX15 and FGFR2 (P<0.05). Similarly, the inhibition of the
PI3K/AKT pathway induced by ALOX15 knockdown was also partially
counteracted following the addition of OE-FGFR2 or IGF-1 (Fig. 4C; P<0.001). Additionally, it was
further demonstrated that both the overexpression of FGFR2 and the
addition of IGF-1 significantly mitigated the inhibitory effects of
ALOX15 knockdown on the migratory and proliferative abilities of
HCAECs (Fig. 4D-F; P<0.05).
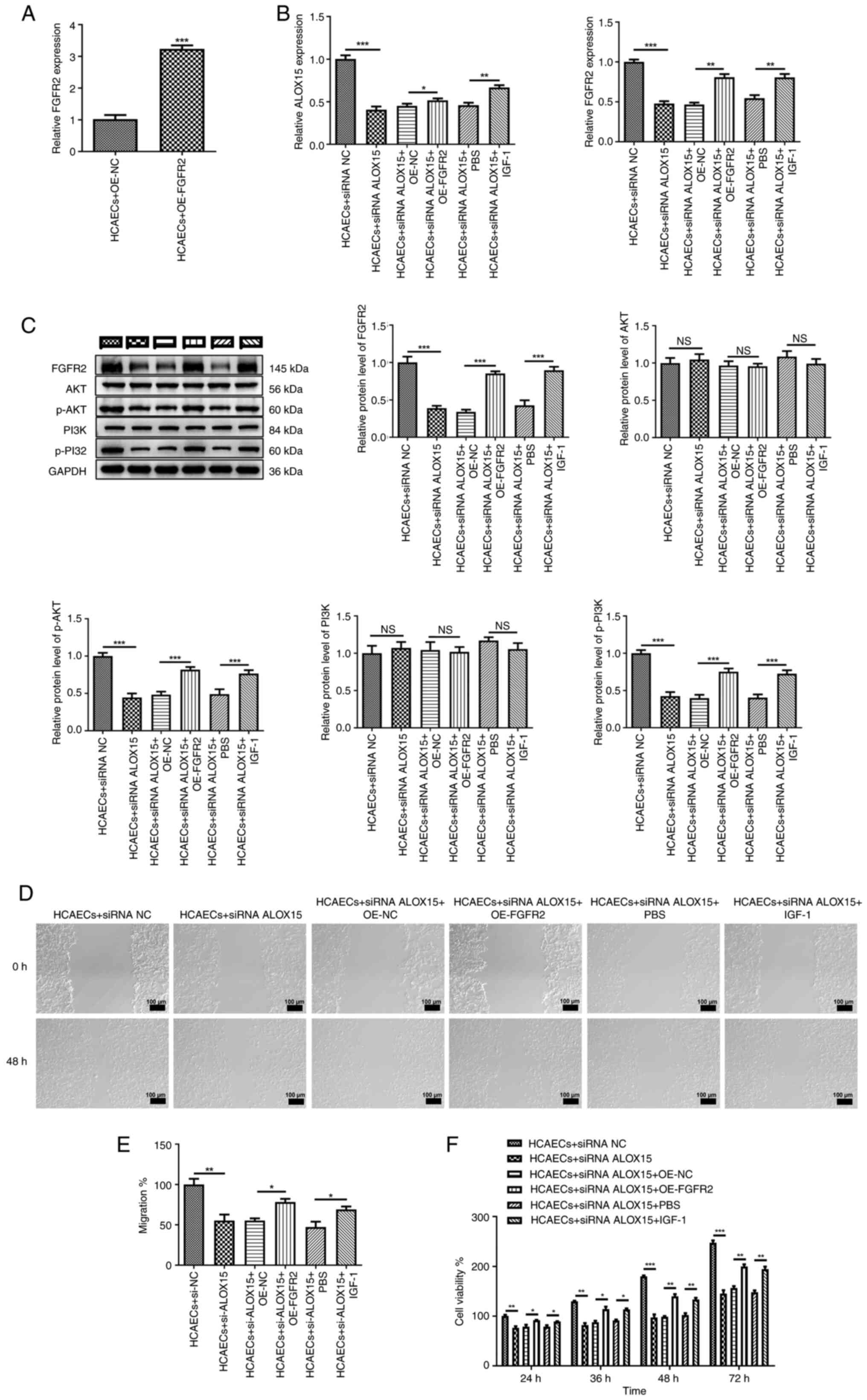 | Figure 4Silencing of ALOX15 mitigates acute
coronary syndrome progression in vitro through the
FGFR2/PI3K/AKT signaling pathway. (A) The expression of FGFR2 in
HCAECs after transfection with OE-FGFR2 or OE-NC was detected by
RT-qPCR. (B) The mRNA expression of ALOX15 and FGFR2 in HCAECs
following different treatments was detected by RT-qPCR. (C) The
protein levels of FGFR2, PI3K, p-PI3K, AKT and p-AKT in HCAECs
following various treatments were determined by western blotting.
(D, E) The migration of HCAECs following different treatments was
assessed by scratch wound healing assays. Scale bar, 100 µm. (F)
The viability of HCAECs following various treatments was measured
by Cell Counting Kit-8 assays. *P<0.05,
**P<0.01 and ***P<0.001. ALOX15,
12/15-lipoxygenase; FGFR2, fibroblast growth factor receptor 2;
HCAECs, human primary coronary artery endothelial cells; OE,
overexpression; NC, negative control; RT-qPCR, reverse
transcription-quantitative PCR; siRNA, small interfering RNA; p-,
phosphorylated; NS, not significant. |
Knockdown of ALOX15 reduces serum
lipid levels and improves myocardial injury in an ACS rat
model
An ACS rat model was subsequently established. As
shown in Fig. 5A and B, a significant elevation in the mRNA
expression levels of ALOX15 and FGFR2 in rats subjected to ACS
(P<0.001) was observed, while injection of shRNA ALOX15 reversed
the increase of ALOX15 and FGFR2 levels induced by the ACS
challenge (P<0.01). Compared with the rats in the sham group,
the serum levels of TC and LDL-C were significantly increased in
the rats with ACS, while HDL-C levels were significantly decreased
(Fig. 5C; P<0.01). Notably,
intramyocardial injection of shRNA ALOX15 not only reduced the
levels of TC and LDL-C, but also increased the levels of HDL-C in
the rats with ACS (Fig. 5C;
P<0.05). Subsequently, the heart tissues from different groups
of rats were collected for pathological examination. As illustrated
in Fig. 5D, cardiomyocytes from the
sham group exhibited a normal morphology, with a neat arrangement
and no breaks. Conversely, cardiomyocytes from the rats with ACS
became swollen and thickened, with irregular morphology and
disordered arrangement. Following intramyocardial injection of
shRNA ALOX15, a marked improvement in both the morphology and
arrangement of cardiomyocytes was noted.
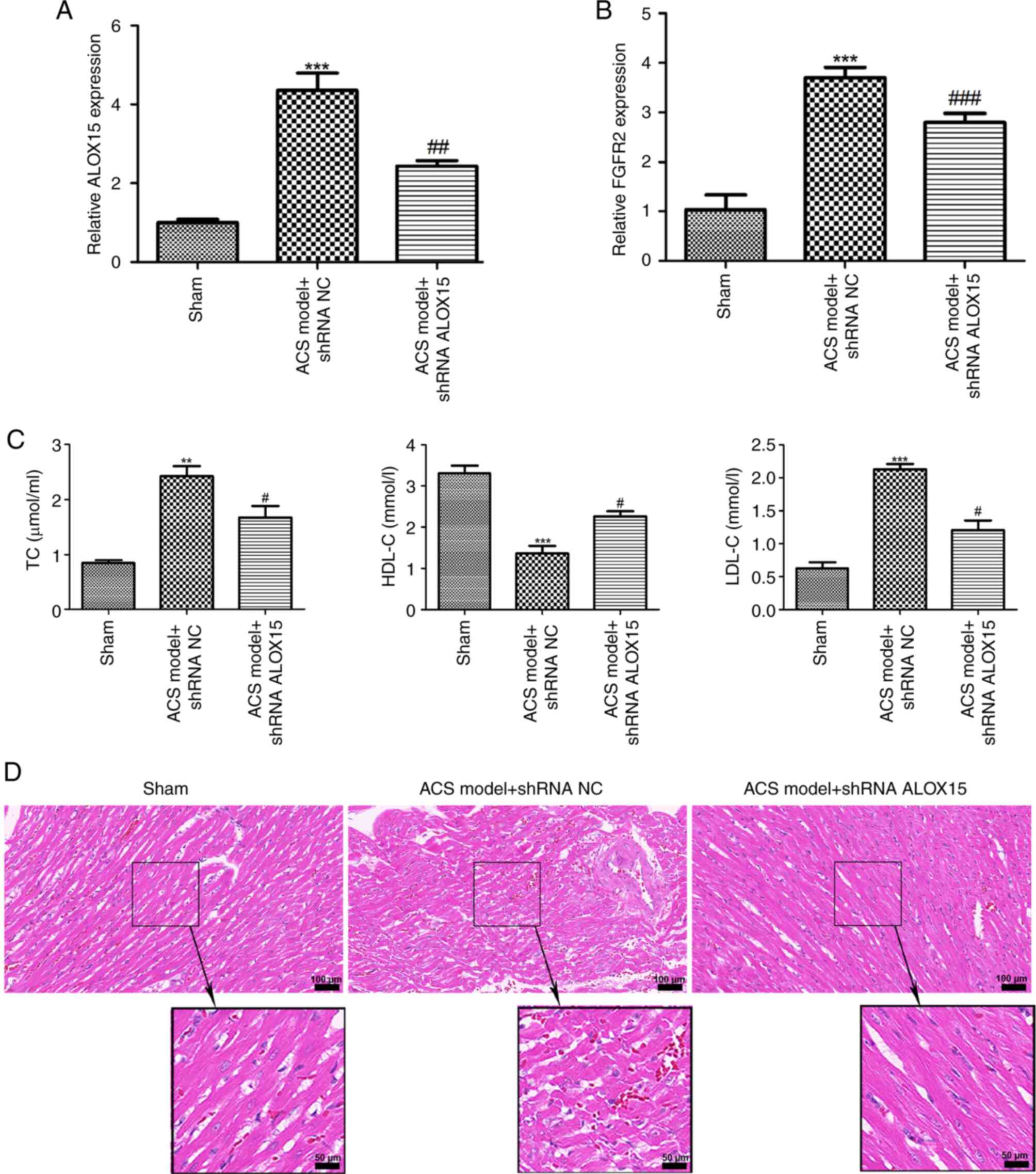 | Figure 5Knockdown of ALOX15 reduces serum
lipid levels and improves myocardial injury in an ACS rat model.
The expression of (A) ALOX15 and (B) FGFR2 in rats with ACS after
injection of shRNA ALOX15 or shRNA NC was detected by reverse
transcription-quantitative PCR. ***P<0.001 vs. sham;
##P<0.01 and ###P<0.001 vs. ACS model +
shRNA NC. (C) The levels of TC, HDL-C and LDL-C in rats with ACS
after injection of shRNA ALOX15 or shRNA NC were determined using
biochemical tests. **P<0.01 and
***P<0.001 vs. sham; #P<0.05 vs. ACS
model + shRNA NC. (D) Hematoxylin and eosin staining for observing
the pathological condition of myocardial tissues in different
groups. Scale bars, 100 and 50 µm. ALOX15, 12/15-lipoxygenase; ACS,
acute coronary syndrome; FGFR2, fibroblast growth factor receptor
2; shRNA, short hairpin RNA; NC, negative control; TC, total
cholesterol; HDL-C, high-density lipoprotein-cholesterol; LDL-C,
low-density lipoprotein-cholesterol. |
ALOX15 silencing suppresses the
activation of the FGFR2/PI3K/AKT signaling pathway
The levels of FGFR2/PI3K/AKT pathway-related
proteins were assessed in rats with ACS, as presented in Fig. 6. Western blot analysis revealed a
pronounced upregulation of ALOX15, FGFR2, p-PI3K and p-AKT proteins
in rats with ACS compared with these levels in the sham rats
(P<0.001). Notably, intramyocardial injection of shRNA ALOX15
effectively inhibited the increase of these proteins induced by the
ACS challenge (P<0.001). Furthermore, it was determined that
both ACS challenge and shRNA ALOX15 treatment did not significantly
affect the protein levels of PI3K and AKT.
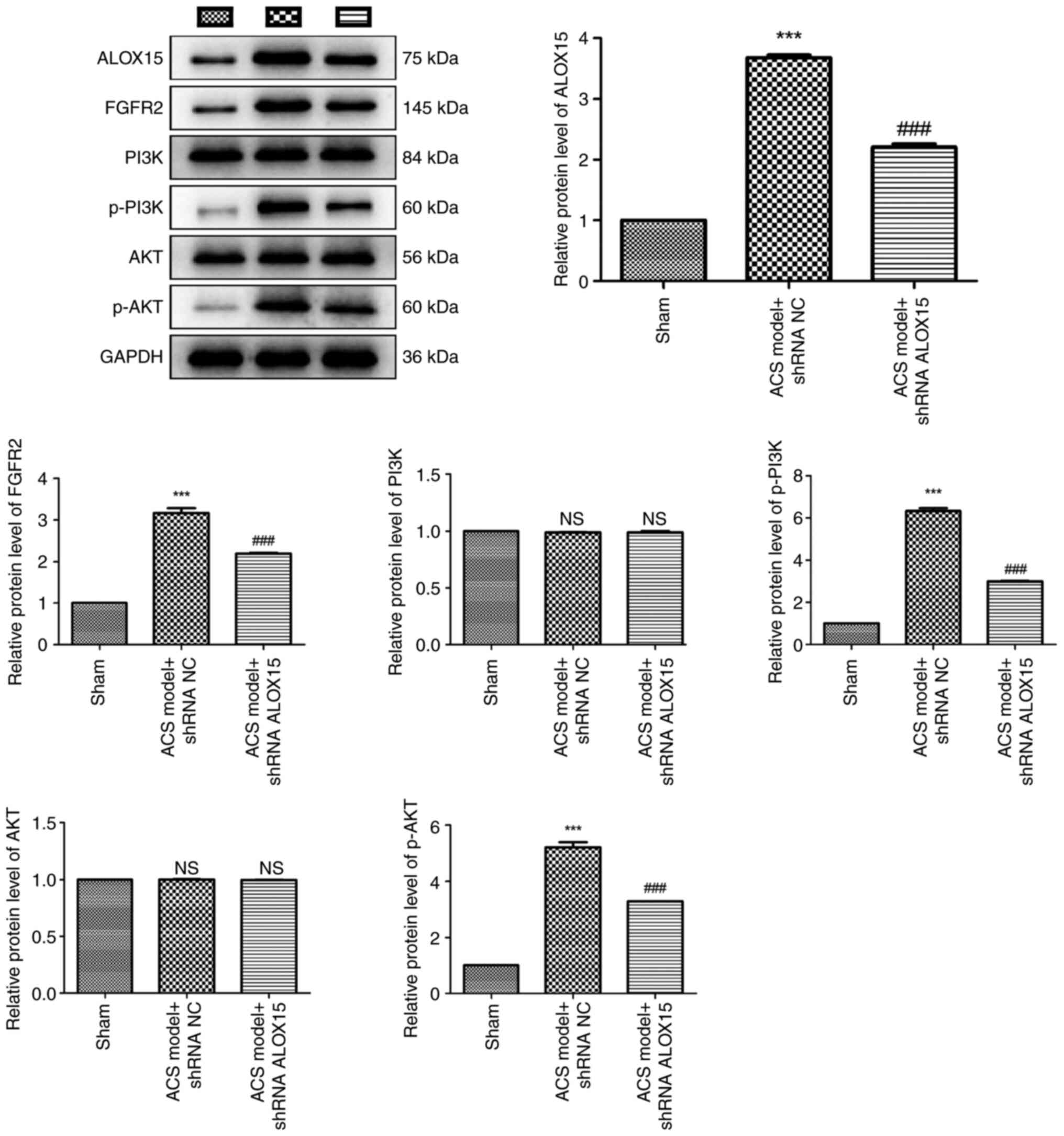 | Figure 6ALOX15 silencing suppresses the
activation of the FGFR2/PI3K/AKT signaling pathway. The protein
levels of ALOX15, FGFR2, PI3K, p-PI3K, AKT and p-AKT in rats with
ACS after injection of shRNA ALOX15 or shRNA NC were determined by
western blotting. ***P<0.001 vs. sham;
###P<0.001 vs. ACS model + shRNA NC. ALOX15,
12/15-lipoxygenase; FGFR2, fibroblast growth factor receptor 2; p-,
phosphorylated; ACS, acute coronary syndrome; shRNA, short hairpin
RNA; NC, negative control; NS, no significance. |
Discussion
Although the mortality of CAD among older patients
has recently decreased, the decrease in younger patients,
particularly in young women, has been less pronounced (35,36).
In spite of this, this condition continues to account for
approximately one-third of all deaths in individuals over the age
of 35 (37-39).
Additionally, the annual cost for each patient with ACS is
substantial and staggering, with hospitalization expenses alone
nearing $35,000 USD (40). In the
USA, the direct costs that the government spends on patients with
ACS every year amount to at least $310 billion (41). Consequently, ACS has become a heavy
burden not only for patients alone but also for the whole society.
In the present study, a novel molecular target for the potential
therapy of ACS was expounded, indicating that knockdown of ALOX15
may improve ACS by inhibiting the FGFR2/PI3K/AKT signaling
pathway.
The pathology of ACS encompasses multiple aspects,
with plaque rupture identified as one of the primary etiological
factors (3,4). The proliferation and migration of
endothelial cells following myocardial infarction are critical for
angiogenesis; however, abnormal endothelial cell behavior may
contribute to plaque instability and erosion, ultimately
precipitating the onset and progression of ACS (42-44).
In the present study, it was demonstrated that the proliferative
and migratory abilities of HCAECs were overtly weakened following
siRNA ALOX15 transfection, but enhanced by ALOX15 overexpression.
These findings indicated that the expression levels of ALOX15 may
be associated with ACS progression. Furthermore, endothelial
dysfunction is also characterized by elevated levels of TC and
LDL-C (45,46). Since it was confirmed that silencing
of ALOX15 inhibited the aberrant proliferation and migration of
HCAECs, it was further hypothesized that ALOX15 silencing may also
play a significant role in regulating blood lipid levels. As
anticipated, the findings in the present study confirmed that
injection of shRNA ALOX15 markedly reduced the levels of TC and
LDL-C, and elevated the levels of HDL-C in rats with ACS. By
determining blood lipid levels in patients with ACS, it can be
assessed whether ALOX15 is involved in ACS progression, thereby
enabling more ACS targeted clinical treatments. Evidence has
indicated that high levels of ALOX15 are correlated with
endothelial cell barrier dysfunction in rats with a high-fat diet
(47). The results of the present
study indicated that knockdown of ALOX15 can relieve endothelial
dysfunction, to some extent. In addition, silencing of ALOX15 also
alleviated the cardiac injury of rats with ACS. Therefore, it is
proposed that ALOX15 may serve as a potential therapeutic target
for ACS in clinical settings. Furthermore, the silencing of ALOX15
appears to be beneficial in mitigating the progression of ACS.
It is well-known that FGFR2 is strongly associated
with endothelial dysfunction, including aberrant cell proliferation
and migration (26,28). Given the significant effects of
ALOX15 on the viability and migration of HCAECs, it was
hypothesized that ALOX15 may interact with FGFR2 in ACS progression
in vitro. In the present study, CO-IP confirmed that ALOX15
protein interacts with FGFR2 in HCAECs. Furthermore, it was
demonstrated that ALOX15 overexpression increases both the mRNA
expression and protein levels of FGFR2, while ALOX15 silencing
leads to their reduction. These results indicated that ALOX15 may
synergistically interact with FGFR2 to affect the development of
ACS. Notably, FGFR2 has been widely reported as an activator of the
PI3K/AKT pathway in various human diseases (30,31).
Additionally, the PI3K/AKT pathway has been demonstrated to be
involved in regulating the growth and function of cardiomyocytes,
thrombogenesis and vascular homeostasis (48). Previous studies have reported that
PI3K/AKT is a classical signaling pathway involved in numerous
cardiovascular disorders such as myocardial ischemia/reperfusion
injury, cardiac hypertrophy, myocardial infarction and heart
failure (18-21).
It was therefore further hypothesized that PI3K/AKT can be
modulated by ALOX15 in ACS pathobiology. As anticipated,
overexpression of ALOX15 markedly activated the PI3K/AKT pathway.
Conversely, silencing of ALOX15 inhibited the PI3K/AKT pathway both
in HCAECs and rats with ACS. Notably, both the overexpression of
FGFR2 and the addition with IGF-1 significantly mitigated the
inhibitory effects of ALOX15 knockdown on the migratory and
proliferative abilities of HCAECs. It was therefore concluded that
silencing of ALOX15 may suppress the progression of ACS by
inhibiting the FGFR2/PI3K/AKT pathway.
Some limitations of the present study should be
mentioned. First, in addition to the FGFR2/PI3K/AKT pathway, other
potential pathways or mechanisms regulated by ALOX15 in the
progression of ACS may exist and should be further investigated in
depth. Second, with the exception of the rat model that was
established in the present study, similar models have also been
extensively used in other animal species, including rabbits
(49), mice (50), and pigs (51). In future studies, the findings of
the present study will be validated in other animal models. Third,
this study was conducted in a specific population in Wenzhou, which
may limit the generalizability of the results. In future studies,
the research will be expanded to include diverse populations across
China. Fourth, elevated levels of ALOX15 may also be influenced by
unrelated inflammatory processes. Future studies will better
control for such potential confounders.
Additionally, several potential challenges or
limitations may arise when translating the current findings from
preclinical models to clinical settings. For example, a)
physiological differences: i) Targeted drugs can be effectively
metabolized in animals, but may be metabolized slowly or produce
different metabolites in humans, resulting in differences in
efficacy and toxicity; ii) animal models are difficult to fully
simulate the complexity of human diseases. b) Drug response
differences: i) Targeted drugs that have shown promising efficacy
in preclinical models may have reduced efficacy in clinical
settings due to various factors; ii) animals often differ in their
tolerance and response to drug toxicity compared with humans. Some
drugs that may not show significant toxicity in animal experiments,
may cause serious adverse reactions in humans. c) External
environmental differences: i) Preclinical animals are typically
housed in controlled environments with consistent temperature,
humidity, light cycles, and diets, however, human lifestyles and
environments are highly variable, which can affect treatment
outcomes; ii) preclinical models cannot simulate the psychological
and social factors of human patients. The psychological state of
human patients, such as stress, anxiety, depression, and other
emotions, can have an impact on the development and treatment
response of the disease.
In summary, the present study provides preliminary
insights into the underlying mechanism of ALOX15 in the
pathogenesis of ACS. The findings, for the first time, to the best
of the authors' knowledge indicate that silencing of ALOX15 may
mitigate ACS progression by inhibiting the FGFR2/PI3K/AKT pathway.
This research offers a foundational basis and perspectives on
potential clinical therapeutic strategies for ACS. However, further
investigations to validate these findings in clinical settings are
urgently warranted.
Acknowledgements
Not available.
Funding
Funding: The present study was supported by Zhejiang Provincial
Hygiene and Health Plan in 2021 (grant no. 2021RC126).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LY made substantial contributions to the conception
and design of the study. HC, NZ, SH, FG, SH and FY made substantial
contributions to the acquisition, analysis and interpretation of
the data. HC drafted the manuscript. NZ, SH, FG, SH and FY revised
the manuscript critically for important intellectual content. All
authors confirm the authenticity of all the raw data, as well as
read and approved the final version of the manuscript, and agree to
be accountable for all aspects of the work in ensuring that
questions related to the accuracy or integrity of any part of the
work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The patients/participants provided their written
informed consent to participate in the present study. The study was
conducted according to the guidelines of the Declaration of
Helsinki and approved (approval no. 2020-351) by the Ethics
Committee of The Third Affiliated Hospital of Shanghai University
(Wenzhou People's Hospital; Wenzhou, China). Animal experiments
were conducted in compliance with the Guidelines for the Use of
Laboratory Animals and approved (approval no. xmsq2023-1367) by the
Ethics Committee of Wenzhou Medical University (Wenzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dong CH, Wang ZM and Chen SY: Neutrophil
to lymphocyte ratio predict mortality and major adverse cardiac
events in acute coronary syndrome: A systematic review and
meta-analysis. Clin Biochem. 52:131–136. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bob-Manuel T, Ifedili I, Reed G, Ibebuogu
UN and Khouzam RN: Non-ST elevation acute coronary syndromes: A
comprehensive review. Curr Probl Cardiol. 42:266–305.
2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Collet JP, Thiele H, Barbato E, Barthélémy
O, Bauersachs J, Bhatt DL, Dendale P, Dorobantu M, Edvardsen T,
Folliguet T, et al: 2020 ESC Guidelines for the management of acute
coronary syndromes in patients presenting without persistent
ST-segment elevation. Eur Heart J. 42:1289–1367. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Libby P and Pasterkamp G: Requiem for the
‘vulnerable plaque’. Eur Heart J. 36:2984–2987. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Makki N, Brennan TM and Girotra S: Acute
coronary syndrome. J Intensive Care Med. 30:186–200.
2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kaul P, Ezekowitz JA, Armstrong PW, Leung
BK, Savu A, Welsh RC, Quan H, Knudtson ML and McAlister FA:
Incidence of heart failure and mortality after acute coronary
syndromes. Am Heart J. 165:379–385 e2. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
O'Gara PT, Kushner FG, Ascheim DD, Casey
DE Jr, Chung MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM,
Franklin BA, et al: 2013 ACCF/AHA guideline for the management of
ST-elevation myocardial infarction: Executive summary: A report of
the American college of cardiology foundation/American heart
association task force on practice guidelines: Developed in
collaboration with the American college of emergency physicians and
society for cardiovascular angiography and interventions. Catheter
Cardiovasc Interv. 82:E1–E27. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Oliw EH: Diversity of the manganese
lipoxygenase gene family-A mini-review. Fungal Genet Biol.
163(103746)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hiltunen T, Luoma J, Nikkari T and
Yla-Herttuala S: Induction of 15-lipoxygenase mRNA and protein in
early atherosclerotic lesions. Circulation. 92:3297–3303.
1995.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kuhn H, Heydeck D, Hugou I and Gniwotta C:
In vivo action of 15-lipoxygenase in early stages of human
atherogenesis. J Clin Invest. 99:888–893. 1997.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhang K, Wang YY, Liu QJ, Wang H, Liu FF,
Ma ZY, Gong YQ and Li L: Two single nucleotide polymorphisms in
ALOX15 are associated with risk of coronary artery disease in a
Chinese Han population. Heart Vessels. 25:368–373. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ma XH, Liu JH, Liu CY, Sun WY, Duan WJ,
Wang G, Kurihara H, He RR, Li YF, Chen Y and Shang H:
ALOX15-launched PUFA-phospholipids peroxidation increases the
susceptibility of ferroptosis in ischemia-induced myocardial
damage. Signal Transduct Target Ther. 7(288)2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Cai W, Liu L, Shi X, Liu Y, Wang J, Fang
X, Chen Z, Ai D, Zhu Y and Zhang X: Alox15/15-HpETE aggravates
myocardial ischemia-reperfusion injury by promoting cardiomyocyte
ferroptosis. Circulation. 147:1444–1460. 2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kayama Y, Minamino T, Toko H, Sakamoto M,
Shimizu I, Takahashi H, Okada S, Tateno K, Moriya J, Yokoyama M, et
al: Cardiac 12/15 lipoxygenase-induced inflammation is involved in
heart failure. J Exp Med. 206:1565–1574. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Silbiger VN, Luchessi AD, Hirata RD,
Lima-Neto LG, Cavichioli D, Carracedo A, Brión M, Dopazo J,
García-García F, dos Santos ES, et al: Novel genes detected by
transcriptional profiling from whole-blood cells in patients with
early onset of acute coronary syndrome. Clin Chim Acta.
421:184–190. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Chen H, Huang S, Guan F, Han S, Ye F, Li X
and You L: Targeting circulating lncRNA ENST00000538705.1 relieves
acute coronary syndrome via modulating ALOX15. Dis Markers.
2022(8208471)2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Feng J, Yang F, Wu H, Xing C, Xue H, Zhang
L, Zhang C, Hu G and Cao H: Selenium protects against
cadmium-induced cardiac injury by attenuating programmed cell death
via PI3K/AKT/PTEN signaling. Environ Toxicol. 37:1185–1197.
2022.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yu D, Xiong J, Gao Y, Li J, Zhu D, Shen X,
Sun L and Wang X: Resveratrol activates PI3K/AKT to reduce
myocardial cell apoptosis and mitochondrial oxidative damage caused
by myocardial ischemia/reperfusion injury. Acta Histochem.
123(151739)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cheng Y, Shen A, Wu X, Shen Z, Chen X, Li
J, Liu L, Lin X, Wu M, Chen Y, et al: Qingda granule attenuates
angiotensin II-induced cardiac hypertrophy and apoptosis and
modulates the PI3K/AKT pathway. Biomed Pharmacother.
133(111022)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Han X, Zhang G, Chen G, Wu Y, Xu T, Xu H,
Liu B and Zhou Y: Buyang Huanwu Decoction promotes angiogenesis in
myocardial infarction through suppression of PTEN and activation of
the PI3K/Akt signalling pathway. J Ethnopharmacol.
287(114929)2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chang H, Li C, Wang Q, Lu L, Zhang Q,
Zhang Y, Zhang N, Wang Y and Wang W: QSKL protects against
myocardial apoptosis on heart failure via PI3K/Akt-p53 signaling
pathway. Sci Rep. 7(16986)2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ruan R, Li L, Li X, Huang C, Zhang Z,
Zhong H, Zeng S, Shi Q, Xia Y, Zeng Q, et al: Unleashing the
potential of combining FGFR inhibitor and immune checkpoint
blockade for FGF/FGFR signaling in tumor microenvironment. Mol
Cancer. 22(60)2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Takahashi M, Umehara Y, Yue H,
Trujillo-Paez JV, Peng G, Nguyen HLT, Ikutama R, Okumura K, Ogawa
H, Ikeda S and Niyonsaba F: The antimicrobial peptide human
β-defensin-3 accelerates wound healing by promoting angiogenesis,
cell migration, and proliferation through the FGFR/JAK2/STAT3
signaling pathway. Front Immunol. 12(712781)2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Weaver A and Bossaer JB: Fibroblast growth
factor receptor (FGFR) inhibitors: A review of a novel therapeutic
class. J Oncol Pharm Pract. 27:702–710. 2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Farooq M, Khan AW, Kim MS and Choi S: The
role of fibroblast growth factor (FGF) signaling in tissue repair
and regeneration. Cells. 10(3242)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mieczkowski K, Popeda M, Lesniak D, Sadej
R and Kitowska K: FGFR2 Controls growth, adhesion and migration of
nontumorigenic human mammary epithelial cells by regulation of
integrin β 1 degradation. J Mammary Gland Biol Neoplasia.
28(9)2023.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yoshii T, Matsuzawa Y, Kato S, Sato R,
Hanajima Y, Kikuchi S, Nakahashi H, Konishi M, Akiyama E,
Minamimoto Y, et al: Endothelial dysfunction predicts bleeding and
cardiovascular death in acute coronary syndrome. Int J Cardiol.
376:11–17. 2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jiao K, Su P and Li Y: FGFR2 modulates the
Akt/Nrf2/ARE signaling pathway to improve angiotensin II-induced
hypertension-related endothelial dysfunction. Clin Exp Hypertens.
45(2208777)2023.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Huang C, Wang R, Lu J, He Y, Wu Y, Ma W,
Xu J, Wu Z, Feng Z and Wu M: MicroRNA-338-3p as a therapeutic
target in cardiac fibrosis through FGFR2 suppression. J Clin Lab
Anal. 36(e24584)2022.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Riccetti MR, Green J, Taylor TJ and Perl
AT: Prenatal FGFR2 signaling via PI3K/AKT specifies the
PDGFRA+ myofibroblast. Am J Respir Cell Mol Biol.
70:63–77. 2024.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Yang J, Xin C, Yin G and Li J:
Taraxasterol suppresses the proliferation and tumor growth of
androgen-independent prostate cancer cells through the
FGFR2-PI3K/AKT signaling pathway. Sci Rep. 13(13072)2023.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Wu S, Sun H and Sun B: MicroRNA-145 is
involved in endothelial cell dysfunction and acts as a promising
biomarker of acute coronary syndrome. Eur J Med Res.
25(2)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li J, Gong L, Zhang R, Li S, Yu H, Liu Y,
Xue Y, Huang D, Xu N, Wang Y, et al: Fibroblast growth factor 21
inhibited inflammation and fibrosis after myocardial infarction via
EGR1. Eur J Pharmacol. 910(174470)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Roth GA, Huffman MD, Moran AE, Feigin V,
Mensah GA, Naghavi M and Murray CJ: Global and regional patterns in
cardiovascular mortality from 1990 to 2013. Circulation.
132:1667–1678. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Gupta A, Wang Y, Spertus JA, Geda M,
Lorenze N, Nkonde-Price C, D'Onofrio G, Lichtman JH and Krumholz
HM: Trends in acute myocardial infarction in young patients and
differences by sex and race, 2001 to 2010. J Am Coll Cardiol.
64:337–345. 2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Rosamond W, Flegal K, Furie K, Go A,
Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, et
al: Heart disease and stroke statistics-2008 update: A report from
the American heart association statistics committee and stroke
statistics subcommittee. Circulation. 117:e25–e146. 2008.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Nichols M, Townsend N, Scarborough P and
Rayner M: Cardiovascular disease in Europe 2014: Epidemiological
update. Eur Heart J. 35(2929)2014.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ferreira-Gonzalez I: The epidemiology of
coronary heart disease. Rev Esp Cardiol (Engl Ed). 67:139–144.
2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Menzin J, Wygant G, Hauch O, Jackel J and
Friedman M: One-year costs of ischemic heart disease among patients
with acute coronary syndromes: Findings from a multi-employer
claims database. Curr Med Res Opin. 24:461–468. 2008.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al:
Executive summary: Heart disease and stroke statistics-2013 update:
A report from the American heart association. Circulation.
127:143–152. 2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bonetti PO, Lerman LO and Lerman A:
Endothelial dysfunction: A marker of atherosclerotic risk.
Arterioscler Thromb Vasc Biol. 23:168–175. 2003.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Kinlay S and Ganz P: Role of endothelial
dysfunction in coronary artery disease and implications for
therapy. Am J Cardiol. 80:11I–16I. 1997.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhu F, Wang Q, Guo C, Wang X, Cao X, Shi
Y, Gao F, Ma C and Zhang L: IL-17 induces apoptosis of vascular
endothelial cells: A potential mechanism for human acute coronary
syndrome. Clin Immunol. 141:152–160. 2011.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Stein RA: Endothelial dysfunction,
erectile dysfunction, and coronary heart disease: The
pathophysiologic and clinical linkage. Rev Urol. 5 (Suppl
7):S21–S27. 2003.PubMed/NCBI
|
|
46
|
Ling L, Zhao SP, Gao M, Zhou QC, Li YL and
Xia B: Vitamin C preserves endothelial function in patients with
coronary heart disease after a high-fat meal. Clin Cardiol.
25:219–224. 2002.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Singh NK and Rao GN: Emerging role of
12/15-Lipoxygenase (ALOX15) in human pathologies. Prog Lipid Res.
73:28–45. 2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Eisenreich A and Rauch U: PI3K inhibitors
in cardiovascular disease. Cardiovasc Ther. 29:29–36.
2011.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Ozkaynak B, Sahin I, Ozenc E, Subaşı C,
Oran DS, Totoz T, Tetikkurt ÜS, Mert B, Polat A, Okuyan E and
Karaöz E: Mesenchymal stem cells derived from epicardial adipose
tissue reverse cardiac remodeling in a rabbit model of myocardial
infarction. Eur Rev Med Pharmacol Sci. 25:4372–4384.
2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Chen L, Li S, Zhu J, You A, Huang X, Yi X
and Xue M: Mangiferin prevents myocardial infarction-induced
apoptosis and heart failure in mice by activating the Sirt1/FoxO3a
pathway. J Cell Mol Med. 25:2944–2955. 2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Nagy RN, Makkos A, Baranyai T, Giricz Z,
Szabó M, Kravcsenko-Kiss B, Bereczki Z, Ágg B, Puskás LG, Faragó N,
et al: Cardioprotective microRNAs (protectomiRs) in a pig model of
acute myocardial infarction and cardioprotection by ischaemic
conditioning: MiR-450a. Br J Pharmacol. 182:396–416.
2025.PubMed/NCBI View Article : Google Scholar
|