Introduction
Diabetes is one of the most common and serious
global health problems, as well as one of fastest growing chronic
diseases (1). For the 10th edition
of the IDF Diabetes Atlas, by 2045, the global prevalence of
diabetes among adults aged 20-79 years is projected to increase
from 9% (463 million adults) in 2019 to 12.2% (783.2 million)
(2). Among all cases of diabetes,
type 2 diabetes mellitus (T2DM) accounts for ~90% of all cases
(3). Obesity is an important risk
factor for T2D (4). The increase in
free fatty acids (FFAs) caused by obesity plays a crucial role in
the occurrence and development of T2D (4).
T2D is characterized by insufficient insulin
secretion and chronic hyperglycemia caused by pancreatic β-cell
dysfunction (5). Regulating glucose
transporter 2 (GLUT2) is a key protein for β-cell function and a
transmembrane protein expressed in pancreatic β-cells, liver,
kidneys and intestines, playing a key role in glucose sensing and
insulin secretion (6). GLUT2 has a
low affinity but high transport capacity for glucose, allowing
β-cells to rapidly take up glucose when blood levels rise. This
triggers metabolic pathways that increase intracellular ATP,
leading to insulin secretion (7).
The dysregulation of GLUT2 expression or function impairs glucose
sensing and insulin release, which has been reported to contribute
to β-cell dysfunction and the pathogenesis of T2D (8,9).
Among the various factors implicated in β-cell
dysfunction, chronic exposure to high levels of FFAs is a major
factor (10), with evidence
suggesting that elevated FFAs have deleterious effects on β-cell
function and survival, a phenomenon often referred to as
lipotoxicity (5,11). Research has shown that metabolic
stressors, such as elevated FFAs, can alter GLUT2 expression,
further exacerbating β-cell dysfunction (12). A saturated fatty acid, palmitic acid
(PA), has been shown to impair glucose-stimulated insulin release
(13-17),
and can induce lipotoxicity, leading to β-cell dysfunction and
apoptosis (often referred to as lipotoxic β-cell apoptosis)
(10,15,18,19).
PA can exert these detrimental effects on β-cells
through various mechanisms, including the induction of endoplasmic
reticulum (ER) stress, oxidative stress and inflammatory response
(20). Given their critical role in
insulin synthesis, β-cells have a highly developed ER network to
synthesize insulin. Consequently, ER stress is particularly
important in β-cell dysfunction (21). Under stress conditions, it leads to
the accumulation of misfolded proteins in the ER and the activation
of the unfolded protein response (UPR) (22). Long-term or excessive ER stress
eventually triggers β-cell apoptosis, leading to the gradual loss
of functional β-cells in patients with T2D (23).
A recent study highlighted the role of specific ER
proteins in regulating ER stress and β-cell survival (24). Endoplasmic reticulum-resident
protein 46 (ERp46), a thiol-disulfide oxidoreductase that is highly
expressed in endothelial cells, pancreatic β-cells, hepatocytes and
hypoxic tissues (25-27),
plays a crucial role in protein folding and redox regulation
(24,28). ERp46 has been identified as a key
regulator of cellular homeostasis, and its activity is particularly
important under conditions of ER stress, including maintenance of
β-cell function and involvement in insulin secretion (29). In addition, GLUT2 plays a role in
the intracellular signaling pathways involved in glucose uptake and
metabolism in ER stress and apoptosis (30), suggesting that ERp46 may also be
linked to these processes, although this has not been directly
demonstrated. Despite the potential importance of ERp46 in β-cell
function and the fact that its expression is reduced in β-cells in
diabetic mouse models and under high glucose stimulation (31), the mechanisms through which ERp46
regulates β-cells in diabetic mouse models and, particularly how it
may link ER stress to GLUT2 expression, remain unclear. The aim of
the present study was to explore the role of ERp46 in pancreatic
β-cells, particularly in the regulation of insulin secretion under
PA-induced lipotoxic stress. By elucidating the molecular mechanism
through which ERp46 affects β-cell function, this study may help
develop novel therapeutic strategies targeting ER stress to
preserve β-cell function in patients with T2D.
Materials and methods
Cell culture and PA treatment
The β-TC6 mouse insulinoma cell line was purchased
from the National Collection of Authenticated Cell Cultures. Cells
were maintained in DMEM (Sigma-Aldrich; Merck KGaA) supplemented
with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc.) at 37˚C and 5% CO2. A stock solution of PA
(Sigma-Aldrich; Merck KGaA) was prepared in 100% ethanol at 55˚C
until it was completely dissolved at a concentration of 100 mM. The
final ethanol concentration in culture did not exceed 0.1% (v/v),
and vehicle controls were included accordingly. Prior to PA
treatment, the stock solution was added to DMEM with 10% FBS at the
indicated concentrations.
AKT activator compound SC79 (SC79)
treatment
SC79 (Selleck Chemicals) was dissolved in DMSO to
prepare a 50-mM stock solution stored at -20˚C. For experiments,
the stock was diluted in DMEM to a final concentration of 5 or 10
µM, ensuring the DMSO concentration did not exceed 0.1%. β-TC6
cells at 70-80% confluency were treated with SC79 for 4 h at 37˚C,
5% CO2. DMSO-treated cells were used as controls.
Following treatment, cells were harvested for protein extraction or
insulin secretion assays.
Differential expression analysis and
functional enrichment
RNA-seq data (GSE53949) was downloaded from the GEO
database (32) and analyzed in
RStudio (version 1.4.1717; Posit PBC) using the DESeq2 package
(version 1.30.1; https://bioconductor.org/packages/release/bioc/html/DESeq2.html).
GSE53949 included five control and five PA-treated human pancreatic
islets samples; all ten samples were used for differential and
correlation analyses. Following initial quality control,
normalization was performed to adjust for library size variations.
Differential expression analysis identified genes with
|log2FC|>1 and P<0.05 as significant, and P-values were
adjusted using the Benjamini-Hochberg method to control for false
discovery rate. Volcano plots were generated with ggplot2 (version
3.3.5; http://ggplot2.tidyverse.org), where
upregulated genes were highlighted in red and downregulated genes
in green. Functional enrichment, including Gene Ontology
(http://geneontology.org) and Kyoto Encyclopedia
of genes and genomes (KEGG; https://www.genome.jp/kegg/) pathway analysis, was
conducted using the clusterProfiler package (version 4.0.5;
http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html),
and results were visualized through bar plots. To assess the
relationship between thioredoxin domain-containing protein 5
[TXNDC5 (ERp46)] and solute carrier family 2 member 2
[SLC2A2 (GLUT2)], Pearson's correlation analysis was
conducted using normalized RNA-seq expression data. A linear
regression model was applied, and the coefficient of determination
(R²) was calculated to quantify the strength of the correlation.
Scatter plots with fitted regression lines were generated to
visualize the results.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from β-TC6 cells using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
First-strand complementary DNA was synthesized using the
Hifair® II 1st Strand cDNA Synthesis Kit (Shanghai
Yeasen Biotechnology Co., Ltd.), according to the manufacturer's
instructions. Subsequently, RT-qPCR was performed using the
Hieff® qPCR SYBR Green Master Mix (Shanghai Yeasen
Biotechnology Co., Ltd.) according to the standard method. The
thermocycling conditions were as follows: Initial denaturation at
95˚C for 2 min, followed by 40 cycles of denaturation at 95˚C for
10 sec and annealing/extension at 60˚C for 30 sec. A melting curve
analysis was performed to verify amplification specificity. The
relative expression (fold) was calculated using the comparative
method 2-ΔΔCq method (33). Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) was used for normalization. The primer
sequences are shown in Table I.
 | Table IPrimer sequences used for
quantitative PCR analysis of target genes in β-cells. |
Table I
Primer sequences used for
quantitative PCR analysis of target genes in β-cells.
| Primer | Sequence
(5'-3') |
|---|
| ERp46-F |
GGATGCCAAGGTCTACGTG |
| ERp46-R |
CGGAGCGAAGAACTTGATA |
| PDX1-F |
GGCGGCCGCAGGAACCAC |
| PDX1-R |
GAGGGCCCCAATACTACAAAAACC |
| GLUT2-F |
CCTGCTTGGCCTGTCTGGTGT |
| GLUT2-R |
TGTCGGTAGCTGGAATTGGTGAAG |
| GAPDH-F |
CGGGGCTCTCCAGAACATCATCC |
| GAPDH-R |
CCAGCCCCAGCGTCAAAGGTG |
Cell protein extraction
The culture dish in which β-TC6 cells were seeded
was washed twice with cold PBS to completely remove the culture
medium, and the cells were lysed using RIPA lysis buffer (Beijing
Solarbio Science & Technology Co., Ltd.) supplemented with
protease inhibitors. After gently scraping the cells with a cell
scraper, they were transferred to a 1.5 ml Eppendorf tube and
incubated on ice for 30 min for lysis. The cells were vortexed
every 5 min to enhance protein extraction. The lysate was
centrifuged at 12,000 x g for 15 min at 4˚C to precipitate cell
debris. The supernatant containing the extracted protein was
carefully transferred to a new tube for subsequent analysis.
Protein quantification
Protein concentration was determined using the
bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, Inc.). A
standard curve was prepared using bovine serum albumin (Thermo
Fisher Scientific, Inc.) as a standard. Protein samples were added
to different wells of a 96-well plate. Subsequently, 200 µl of BCA
reagent mixture (comprised of reagent A and reagent B mixed at a
ratio of 50:1) was added to each well. The plate was incubated at
37˚C for 30 min, and absorbance was measured at 562 nm using a
microplate reader. Protein concentration was calculated by
comparing the absorbance of the sample with the standard curve.
Depending on the concentration, protein samples were diluted to 1
µg/µl using RIPA and then added to protein loading buffer, boiled
at 100˚C for 5 min, aliquoted and stored in a freezer at -80˚C.
Western blotting
Protein samples (50 µg) were separated by 10%
SDS-PAGE and transferred to PVDF membranes (Wuhan Servicebio
Technology Co., Ltd.). Following blocking with 5% skim milk in TBST
for 1 h at room temperature, the membranes were incubated with
primary antibodies overnight at 4˚C: Anti-ERp46 (1:1,000; cat. no.
sc-271667; Santa Cruz Biotechnology, Inc.), anti-GLUT2 (1:1,000;
cat. no. K006592P; Beijing Solarbio Science & Technology Co.,
Ltd.), anti-p-Akt (1:1,000; cat. no. GB150002), anti-Akt (1:1,000;
cat. no. GB111114), anti-pancreatic and duodenal homeobox 1 (PDX1;
1:1,000; cat. no. GB11917), and anti-tubulin (1:2,000; cat. no.
GB11017; all from Wuhan Servicebio Technology Co., Ltd.),
anti-binding immunoglobulin protein (BiP; 1:1,000; cat. no. 3177T),
C/EBP homologous protein (CHOP; 1:1,000; cat. no. 2895T), and
anti-Na+/K+-ATPase [1:1,000; cat. no. 3010S;
all from Cell Signaling Technology, Inc.]. Following washing three
times with TBST (TBS containing 0.1% Tween-20) for 10 min each
time, the membranes were incubated with secondary antibodies:
Anti-rabbit IgG, HRP-linked antibody (cat. no. 7074) and anti-mouse
IgG, HRP-linked antibody (cat. no. 7076) (both 1:5,000; Cell
Signaling Technology, Inc.), for 1 h at room temperature. Protein
bands were visualized using enhanced chemiluminescence (Thermo
Fisher Scientific, Inc.), and band intensities were quantified
using ImageJ software (version 1.53t; National Institutes of
Health).
Insulin secretion assay
Supernatants of β-TC6 cells exposed to various
concentrations (0, 0.1, 0.25 and 0.5 mM) of PA for 24 h at 37˚C
were collected, and insulin levels were measured using a mouse
insulin ELISA kit [cat. no. 634-01481 (formerly AKRIN-011T)
FUJIFILM Wako Pure Chemical Corporation], according to the
manufacturer's instructions. Absorbance was measured at 450 nm, and
insulin concentrations were calculated using a standard curve.
Transfection
For the knockdown of ERp46, 1x106 cells
were seeded into a 6-well plate. A total of 50 pmol ERp46 small
interfering (siRNA) (sense, 5'-GUACUCGGUACGAGGUUAUTT-3' and
antisense, 5'-AUAACCUCGUACCGAGUACTT-3') or negative control siRNA
(sense, 5'-UUCUCCGAACGUGUCACGUTT-3' and antisense,
5'-ACGUGACACGUUCGGAGAATT-3') were transfected using Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions (at 3˚C for 6 h; typically 5 µl reagent
per well in Opti-MEM). The transfection medium was then replaced
with DMEM containing 12% FBS, and cells were cultured for 24 h at
37˚C. The expression of ERp46 was detected using western blotting
and RT-qPCR.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 8.0 (GraphPad Software, Inc.). Data are presented as the mean
± standard error of the mean from three to five independent
experiments, with each experiment including n=6 biological
replicates per group. Statistical comparisons between two groups
were performed using an unpaired two-tailed Student's t-test, and
multiple-group comparisons were analyzed using ANOVA followed by
Sidak's multiple-comparisons test. P<0.05 was considered to
indicate a statistically significant difference.
Results
PA activates ER stress-related
pathways and impairs β-cell function
As aforementioned, FFAs can cause lipotoxicity in
pancreatic β-cells (10,15,19),
and it was found that following PA stimulation, pancreatic β-cells
secreted less insulin, which was proportionate to the dose of PA
(Fig. 1A), consistent with previous
studies (16,17). When stimulated with 0.5 µM PA,
insulin secretion decreased over time; however, there was no
significant difference between 24 and 48 h, suggesting that the
effect reached saturation (Fig.
1A). To further investigate the cause of the reduced insulin
secretion, RNA-seq analysis of a public dataset (GSE53949) was
performed. By analyzing the RNA-seq results of PA-stimulated
β-cells compared with the control group, TXNDC5 (gene name
of ERp46) was significantly upregulated (Fig. 1B). This upregulation was supported
by enrichment analysis, which highlighted the activation of ER
stress-related pathways in β-cells following PA stimulation
(Fig. 1C). These findings were
consistent with previous reports, in which the upregulation of
TXNDC5 was associated with its role in promoting correct protein
folding and maintaining redox homeostasis under ER stress
conditions (25-27).
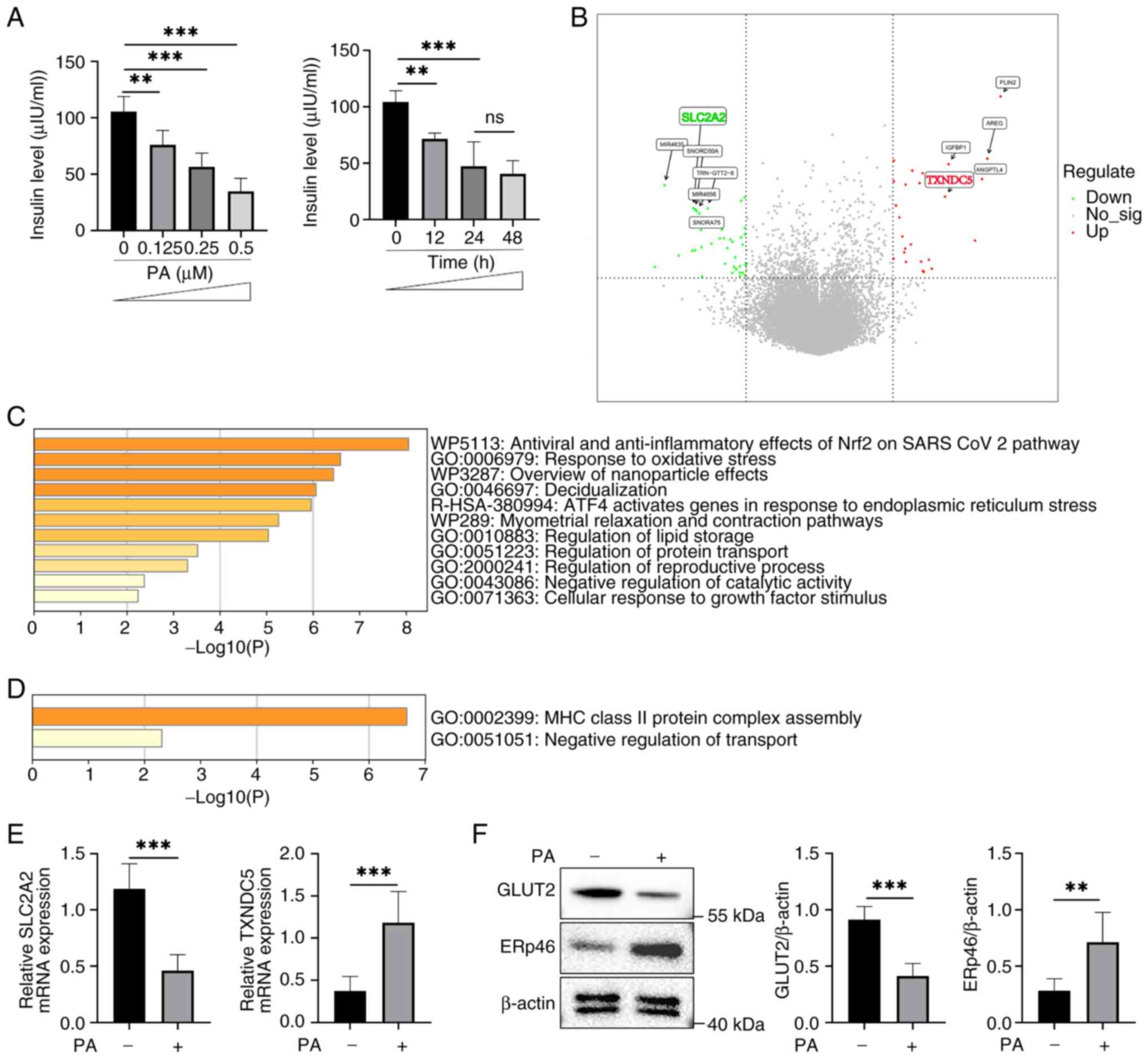 | Figure 1PA-induced ER stress reduces GLUT2
expression and impairs insulin secretion in β-cells. (A) Insulin
secretion levels under varying concentrations of PA (0, 0.125, 0.25
and 0.5 µM) and at different time points (0, 12, 24 and 48 h).
Insulin secretion decreased in a dose- and time-dependent manner.
Data are presented as the mean ± SEM (n=6). Statistical
significance was determined using one-way ANOVA. (B) Volcano plot
showing the DEGs in PA-stimulated β-cells compared with controls.
TXNDC5 (ERp46) was significantly upregulated, whereas
SLC2A2 (GLUT2) was downregulated. (C) GO and KEGG pathway
enrichment of upregulated DEGs. Key pathways included oxidative
stress response, UPR and ER stress-related pathways. (D) Enrichment
analysis of downregulated DEGs, highlighting impaired transport
regulation and immune-related processes. (E) RT-qPCR validation of
TXNDC5 and SLC2A2 expression in PA-stimulated
β-cells. (F) Western blotting showing increased ERp46 and decreased
GLUT2 protein levels following PA treatment. **P<0.01
and ***P<0.001. PA, palmitic acid; ER, endoplasmic
reticulum; GLUT2, glucose transporter 2; SEM, standard error of the
mean; DEGs, differentially expressed genes; TXNDC5,
thioredoxin domain-containing protein 5; ERp46, endoplasmic
reticulum-resident protein 46; SLC2A2, solute carrier family
2 member 2; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes
and Genomes; UPR, unfolded protein response; RT-qPCR, reverse
transcription-quantitative PCR; ns, not significant. |
By contrast, the key glucose transporter
SLC2A2 (gene name of GLUT2) in β-cells was significantly
downregulated following PA stimulation. Enrichment analysis
identified the ‘negative regulation of transport’ pathway (Fig. 1D), highlighting the importance of
GLUT2 in β-cell function. The downregulation of GLUT2 suggested a
reduced glucose-transport capacity and was compatible with the
decreased insulin secretion observed in Fig. 1A. Previous studies have demonstrated
that GLUT2 plays a critical role in glucose sensing and insulin
secretion in β-cells (8,9), although the present data did not
directly establish causality. Consistently, RT-qPCR confirmed that
SLC2A2 mRNA was decreased while TXNDC5 mRNA was
increased in PA-treated cells (Fig.
1E). Western blotting further demonstrated decreased GLUT2
protein and increased ERp46 protein levels following PA treatment
(Fig. 1F). In conclusion, PA
stimulation was associated with the activation of ER stress-related
pathways and reduced GLUT2 expression and insulin secretion in
β-cells.
ERp46 depletion exacerbates PA-induced
ER stress, reducing GLUT2 expression and insulin secretion in
β-cells
ERp46 knockdown efficiency was first verified.
Western blotting confirmed a robust reduction of ERp46 protein
following siERp46 transfection (Fig.
2A). This result validated the effectiveness of the knockdown
system for subsequent experiments. Next, GLUT2 protein was examined
across the four conditions (Control, PA, siERp46 and PA + siERp46).
As shown in Fig. 2B, PA alone
decreased GLUT2, whereas siERp46 alone had no detectable effect. Of
note, the combination of siERp46 with PA further reduced GLUT2
compared with PA alone. These findings indicated that ERp46 is not
essential for basal GLUT2 expression, but contributes to the
maintenance of GLUT2 under lipotoxic stress.
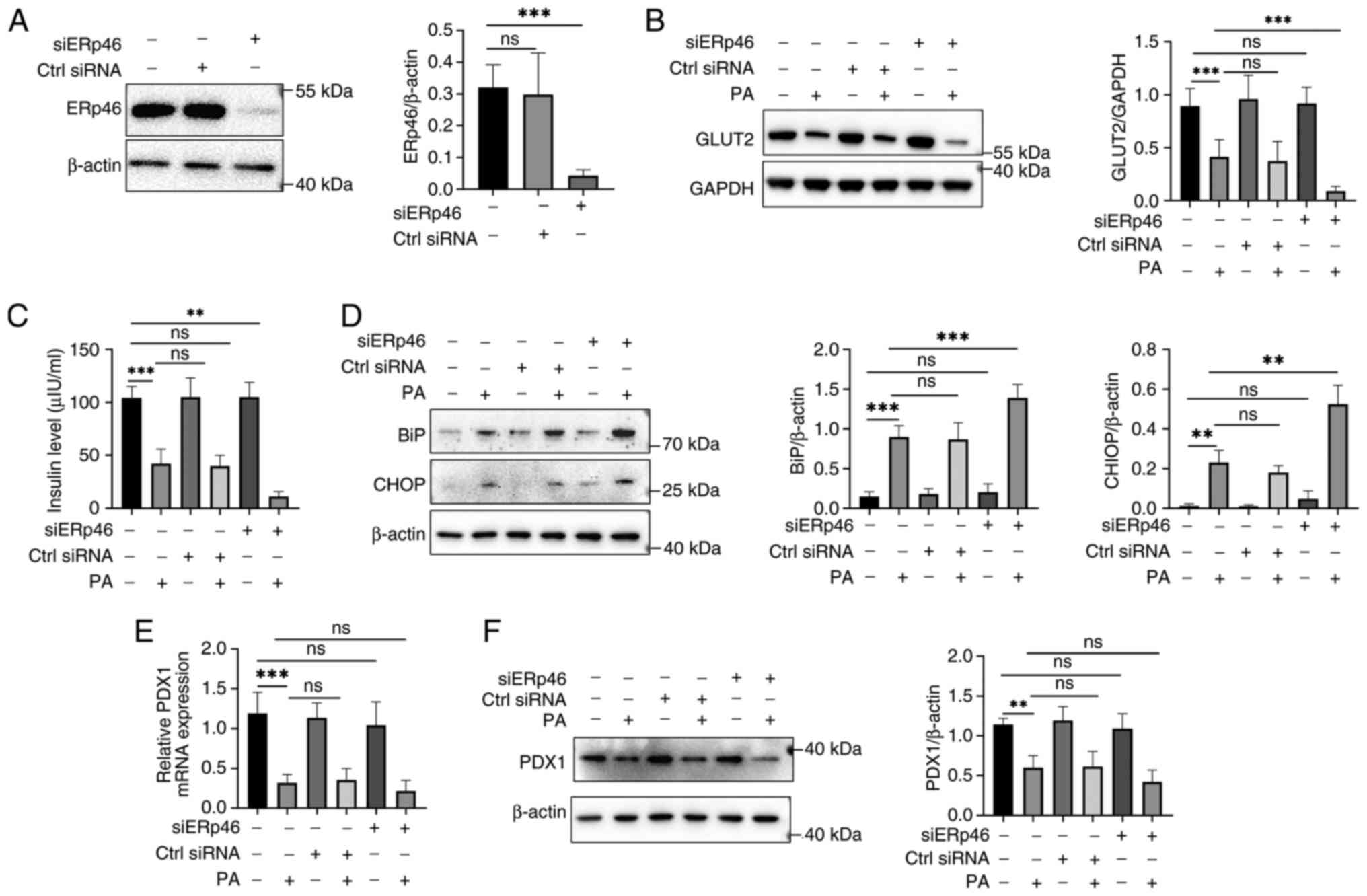 | Figure 2ERp46 regulates GLUT2 expression and
insulin secretion in β-cells under PA-induced stress. (A) Western
blotting confirming ERp46 knockdown efficiency by siRNA
transfection. (B) Western blotting showing that GLUT2 expression
was further decreased in PA-treated cells following ERp46
knockdown. (C) Insulin secretion was significantly reduced in
ERp46-depleted β-cells exposed to PA, as determined by ELISA. (D)
Western blotting of ER stress markers BiP and CHOP revealed that
ERp46 knockdown aggravated PA-induced ER stress. (E) RT-qPCR
analysis of PDX1 mRNA expression showed no significant
changes with ERp46 knockdown under PA stimulation. (F) Western
blotting confirmed that PDX1 protein levels were not significantly
altered by ERp46 knockdown. Data are presented as the mean ± SEM
(n=6). Statistical significance was determined using one-way ANOVA.
**P<0.01 and ***P<0.001. ERp46,
endoplasmic reticulum-resident protein 46; GLUT2, glucose
transporter 2; PA, palmitic acid; siRNA, small interfering RNA; ER,
endoplasmic reticulum; BiP, binding immunoglobulin protein; CHOP,
C/EBP homologous protein; RT-qPCR, reverse
transcription-quantitative PCR; PDX1, pancreatic and
duodenal homeobox 1; SEM, standard error of the mean; ns, not
significant. |
Functionally, insulin secretion followed the same
pattern (Fig. 2C): PA reduced
insulin release; siERp46 alone did not alter secretion; and siERp46
plus PA led to a further decrease relative to PA alone. This
indicates that the protective role of ERp46 becomes evident only in
the presence of PA-induced stress. Since ERp46 is an ER
oxidoreductase, ER stress markers were assessed. PA increased BiP
and CHOP, and siERp46 further increased their levels under PA,
whereas siERp46 alone showed no significant change (Fig. 2D). These results are consistent with
the interpretation that ERp46 depletion aggravates ER stress in
β-cells exposed to PA. PDX1, a β-cell identity/functional factor
(12,30), was also evaluated. PA decreased PDX1
mRNA and protein levels, while siERp46 did not produce an
additional significant reduction beyond PA in our conditions
(Fig. 2E and F). This suggested that ERp46 depletion
primarily impacted GLUT2 and insulin secretion rather than β-cell
identity markers under PA stress.
Collectively, these data indicated that ERp46
depletion exacerbates PA-induced ER stress and is associated with a
greater loss of GLUT2 and insulin secretion, whereas ERp46
knockdown alone has minimal effects at baseline. Thus, ERp46
appears to play a stress-dependent protective role in sustaining
GLUT2 expression and β-cell functionality.
AKT signaling contributes to GLUT2
maintenance under PA-induced stress; ERp46 depletion reduces AKT
phosphorylation
The relationship between TXNDC5 (ERp46) and
SLC2A2 (GLUT2) was first examined using the public RNA-seq
dataset (GSE53949). In the correlation plot, each dot represents
one RNA-seq sample from GSE53949 (PA-stimulated or control β-cell
samples). A modest positive correlation was observed (R²=0.4001,
P=0.0497; Fig. 3A), supporting an
association between ERp46 and GLUT2 under lipotoxic conditions.
Next, AKT activation was assessed. Total AKT (t-AKT) remained
unchanged across groups, whereas PA decreased phosphorylated AKT
(p-AKT). Of note, siERp46 alone had little effect, but siERp46 in
the presence of PA further reduced p-AKT (Fig. 3B). These findings indicated that
ERp46 depletion exacerbated the PA-induced suppression of AKT
phosphorylation, while having a minimal impact under basal
conditions.
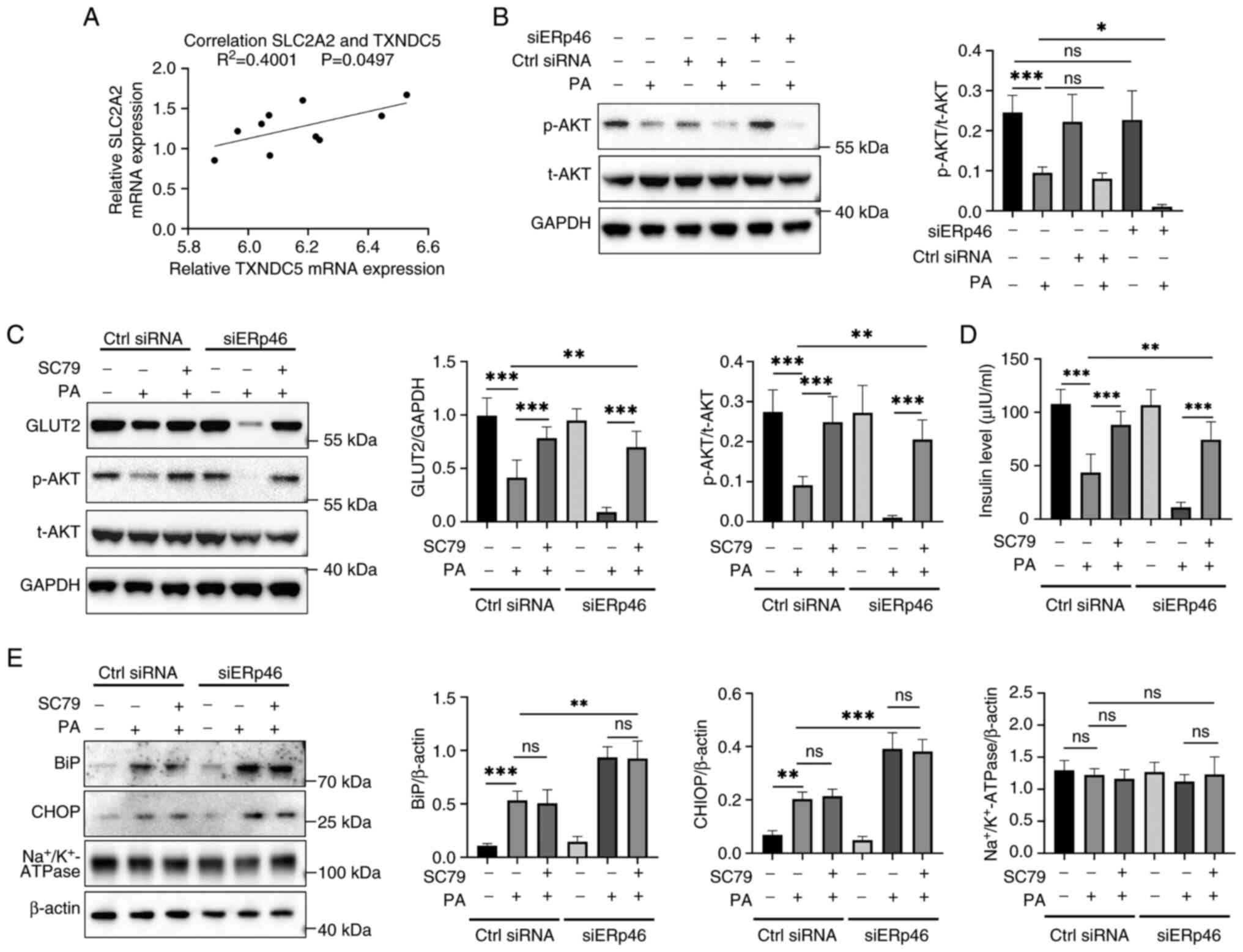 | Figure 3ERp46 regulates GLUT2 expression
through AKT activation under PA-induced stress. (A) Correlation
analysis of RNA-seq dataset (GSE53949) showing a positive
correlation between TXNDC5 (ERp46) and SLC2A2 (GLUT2)
expression in PA-stimulated and control β-cell samples. (B) Western
blotting showing that p-AKT levels were decreased by PA and further
reduced by ERp46 knockdown, while t-AKT remained unchanged. (C)
Western blotting demonstrating that treatment with the AKT
activator SC79 restored GLUT2 expression and p-AKT levels in
ERp46-depleted cells under PA stimulation. (D) ELISA assay showing
that SC79 treatment rescued insulin secretion impaired by ERp46
knockdown in PA-treated β-cells. (E) Western blotting of ER stress
markers BiP and CHOP revealed that SC79 treatment did not
significantly reverse PA-induced ER stress, indicating that AKT
activation specifically rescued GLUT2 and insulin secretion without
directly modulating ER stress. Na+/K+-ATPase
served as a membrane-protein specificity control. Data are
presented as the mean ± SEM (n=6). Statistical significance was
determined using one-way ANOVA. *P<0.05,
**P<0.01 and ***P<0.001. ERp46,
endoplasmic reticulum-resident protein 46; GLUT2, glucose
transporter 2; AKT, protein kinase B; PA, palmitic acid;
TXNDC5, thioredoxin domain-containing protein 5;
SLC2A2, solute carrier family 2 member 2; p-AKT,
phosphorylated AKT; t-AKT, total AKT; SC79, AKT activator compound
SC79; ER, endoplasmic reticulum; BiP, binding immunoglobulin
protein; CHOP, C/EBP homologous protein; SEM, standard error of the
mean; ns, not significant. |
To determine whether AKT activation is sufficient to
counteract the PA/siERp46 effects, cells were treated with the AKT
activator SC79. SC79 increased p-AKT and partially restored GLUT2
protein in PA-exposed cells under both control siRNA and siERp46
conditions (Fig. 3C). Functionally,
SC79 also improved insulin secretion that was reduced by PA and
further diminished by siERp46 (Fig.
3D). To evaluate specificity, an additional membrane protein
and ER-stress markers were probed.
Na+/K+-ATPase was selected as a
representative plasma membrane housekeeping protein whose
expression remains stable under metabolic or ER stress conditions,
serving as a control for nonspecific changes in membrane protein
abundance. Na+/K+-ATPase levels were
unchanged across treatments, and SC79 did not reduce PA-induced
increases of BiP or CHOP (Fig. 3E),
indicating that AKT activation does not broadly elevate membrane
proteins or alleviate ER stress but can still restore GLUT2 and
insulin secretion.
In conclusion, ERp46 depletion was demonstrated to
enhance the PA-induced loss of p-AKT, GLUT2 and insulin secretion,
while pharmacological AKT activation rescued GLUT2 and insulin
without reducing ER-stress markers. These findings supported a
model in which AKT acts downstream or in parallel to ER-stress
pathways, with ERp46 helping to preserve AKT activity and β-cell
function specifically under lipotoxic stress.
Discussion
The present study revealed the critical role of
ERp46 in regulating GLUT2 expression and insulin secretion in
pancreatic β-cells under PA-induced lipotoxic stress. These
findings demonstrated that ERp46 exerted a protective effect on
β-cell function, primarily by alleviating PA-induced ER stress,
which in turn helps sustain AKT phosphorylation and maintain GLUT2
expression. These observations suggested that AKT may act
downstream or in parallel to ER stress, rather than being directly
regulated by ERp46 under basal conditions. However, the possibility
that ERp46 may influence AKT signaling more directly through
effects on the folding or stability of upstream signaling
components cannot be excluded and should be further investigated.
The findings of the present study provide novel insights into the
molecular mechanisms underlying β-cell dysfunction in T2DM.
PA-induced ER stress is a well-known contributor to
β-cell dysfunction, leading to impaired insulin secretion and
increased cell death through mechanisms such as protein misfolding
and oxidative stress (23,30). Consistent with these observations,
the present study showed that PA stimulation significantly
decreased GLUT2 expression and insulin secretion in β-cells.
Previous studies have shown that ER stress disrupts glucose sensing
by reducing GLUT2 expression and impairing insulin granule
exocytosis (22,23). Furthermore, prolonged ER stress was
shown to contribute to β-cell apoptosis, further limiting insulin
secretion capacity (10,18). The chronic activation of the UPR has
been demonstrated to further promote β-cell apoptosis and
functional decline under lipotoxicity (10,34).
Of note, ERp46 expression was upregulated in response to PA,
suggesting a compensatory mechanism to counteract ER stress.
However, the knockdown of ERp46 further reduced GLUT2 expression
and exacerbated the decline in insulin secretion, highlighting its
protective role in maintaining β-cell functionality. As a member of
the protein disulfide isomerase (PDI) family, ERp46 is essential
for protein folding and redox homeostasis in the ER (6,12).
This is consistent with its reported ability to mitigate cellular
stress by ensuring proteostasis under adverse conditions.
Concordantly, multiple PDI paralogs (such as PDI family A member 1
(PDIA1)/pyruvate dehydrogenase E1 component subunit beta and
ERp57/PDIA3 modulate β-cell stress adaptation and insulin
biosynthesis, underscoring a conserved ER proteostasis axis in the
endocrine pancreas (24,35). Previous studies have further
emphasized the role of ERp46 in alleviating ER stress and apoptosis
in β-cells under diabetic conditions (22,31),
suggesting its broader function in metabolic regulation. A recent
comprehensive review of TXNDC5/ERp46 across diseases also
highlighted its metabolic relevance and therapeutic tractability
(29).
In addition to its role in maintaining ER
homeostasis, ERp46 may influence GLUT2 expression, at least partly
through the modulation of the AKT signaling pathway. AKT activation
has been widely reported as a crucial regulator of glucose uptake
and insulin secretion in β-cells (30,36).
In the present study, PA stimulation significantly reduced p-AKT,
and this effect was further exacerbated by ERp46 knockdown,
suggesting that ERp46 supports AKT activation under lipotoxic
conditions, which in turn may help preserve GLUT2 expression.
Although total AKT levels remained unchanged across experimental
groups, PA stimulation significantly reduced p-AKT levels, and this
reduction was further exacerbated by ERp46 knockdown. This
suggested that ERp46 primarily sustains AKT phosphorylation
indirectly by alleviating ER stress, as siERp46 had no effect under
baseline conditions but markedly suppressed AKT activity in the
presence of PA. Thus, AKT is more likely to act downstream or in
parallel to ER stress rather than being directly controlled by
ERp46. Of note, treatment with SC79, a direct AKT activator,
successfully rescued GLUT2 expression and restored insulin
secretion in ERp46-depleted cells. This is consistent with broader
evidence that phosphoinositide 3-kinase (PI3K)/AKT signaling is a
central survival and metabolic pathway in β-cells and a candidate
lever to counter lipotoxic dysfunction (37,38).
These findings provided direct evidence that ERp46 supports GLUT2
expression and β-cell functionality by modulating AKT
activation.
UPR signaling has been reported to intersect with
AKT activity in other cellular contexts. For example, PERK-eIF2α
signaling can suppress insulin biosynthesis, whereas adaptive
inositol-requiring enzyme 1-X-box binding protein 1 and activating
transcription factor 6 branches promote β-cell survival and
proteostasis (39,40). Additional studies have suggested
that maladaptive ER stress can impair AKT phosphorylation, while
protective UPR responses may sustain it (41,42).
Although these findings indicated potential crosstalk between UPR
and AKT, whether this mechanism operates in β-cells under lipotoxic
stress remains uncertain. The present data that ERp46 depletion
reduces p-AKT under PA stress raise the possibility that ERp46 may
act by stabilizing ER proteostasis, thereby preventing maladaptive
UPR activation and indirectly supporting AKT activity.
Nevertheless, the alternative hypothesis that ERp46 may also
regulate AKT through more direct mechanisms, cannot be
excluded.
The positive correlation between ERp46 and GLUT2
highlights the importance of maintaining ER homeostasis in
preserving β-cell function under lipotoxic stress. Furthermore, the
ability of AKT activation to mitigate the negative effects of ERp46
depletion suggests that targeting the ERp46-AKT-GLUT2 axis could
offer a therapeutic strategy for improving β-cell survival and
insulin secretion in T2DM. In parallel, interventions that lower ER
stress, such as chemical chaperones or UPR modulators, have
demonstrated the protection of β-cell mass and function in
preclinical models and early translational studies (43-46),
supporting an ‘ER-proteostasis-first’ approach that aligned with
the present model. These findings supported the rationale that
enhancing ERp46 activity may represent a novel strategy to
stabilize β-cell proteostasis and insulin secretory function.
Beyond diabetes, ER stress is a convergent mechanism
in multiple chronic diseases, including cardiovascular disease,
neurodegeneration and cancer (40,47-50).
Placing these results in this broader context underscores the
translational significance of modulating ER proteostasis; targeting
ERp46 or allied PDI nodes could complement metabolic therapies and
potentially benefit co-morbid conditions characterized by secretory
stress. Of note, the spleen has recently been recognized as an
active immunometabolic hub that communicates with the gut and liver
to shape lipid and glucose metabolism, as well as systemic
inflammation, forming distinct spleen-organ axes (51). In addition, in a
high-fat/streptozotocin rat model with splenectomy, adipose
tissue-derived stem cells protected against T2D through the
induction of spleen-derived IL-10; this benefit was blunted by
splenectomy, highlighting a spleen-IL-10 anti-inflammatory circuit
with a metabolic impact (52). An
independent study further showed that spleen-derived or exogenous
IL-10 can dampen obesity-associated inflammation and insulin
resistance in liver and adipose tissues (53). Of note, IL-10 has also been
demonstrated to alleviate ER stress and apoptosis in non-islet
tissues, such as cardiomyocytes under doxorubicin challenge
(54) and skeletal muscle of aged
mice (55). These findings
suggested that splenic IL-10-driven anti-inflammatory circuits
could, in principle, buffer β-cell ER stress and indirectly
preserve AKT phosphorylation and GLUT2 expression, consistent with
our ERp46-proteostasis model.
Despite these insights, the precise mechanisms
through which ERp46 regulates AKT activation remain to be fully
elucidated. It is possible that ERp46 interacts directly with
components of the PI3K/AKT pathway, but it is also plausible that
the effects are mediated indirectly through ER stress and UPR
signaling. Further studies are warranted to dissect these molecular
interactions and to explore the in vivo role of ERp46,
particularly in diabetic animal models. In addition, complementary
approaches such as conditional β-cell-specific ERp46-knockout
models or the pharmacological modulation of ER stress may provide
more definitive evidence. In addition, the involvement of other ER
stress-related pathways in regulating GLUT2 expression and insulin
secretion deserves further investigation. Collectively, these
findings not only enhanced our understanding of β-cell dysfunction
under lipotoxic stress but also underscored the potential of ERp46
as a promising therapeutic target for diabetes and possibly other
ER stress-related diseases.
In conclusion, the present study demonstrated that
ERp46 is a key regulator of GLUT2 expression and insulin secretion
in β-cells under lipotoxic stress. Rather than directly activating
AKT, ERp46 primarily alleviated PA-induced ER stress, which in turn
supported AKT phosphorylation and helped preserve GLUT2 expression.
These findings provided a mechanistic basis for the development of
therapeutic strategies targeting ER stress modulators in
combination with ERp46- and AKT-related pathways to combat β-cell
dysfunction in T2DM.
Acknowledgements
The authors would like to thank Zhongshan Hospital
of Xiamen University (Xiamen, China) for providing laboratory
facilities and technical support.
Funding
Funding: The present study was supported by the Guidance Project
of Xiamen Science and Technology Bureau (grant no.
3502Z20189038).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
DC acquired the funding and contributed to the
conceptualization, data curation, formal analysis, investigation,
methodology, and drafting and revision of the manuscript. CH
contributed to the conceptualization, investigation, methodology,
project administration, supervision and manuscript revision. XC
contributed to the investigation, data curation and formal
analysis. YT and KW contributed to data curation and formal
analysis. DC and CH confirm the authenticity of all the raw data.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Heald AH, Stedman M, Davies M, Livingston
M, Alshames R, Lunt M, Rayman G and Gadsby R: Estimating life years
lost to diabetes: Outcomes from analysis of National diabetes audit
and office of National statistics data. Cardiovasc Endocrinol
Metab. 9:183–185. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Sun H, Saeedi P, Karuranga S, Pinkepank M,
Ogurtsova K, Duncan BB, Stein C, Basit A, Chan JCN, Mbanya JC, et
al: IDF diabetes atlas: Global, regional and country-level diabetes
prevalence estimates for 2021 and projections for 2045. Diabetes
Res Clin Pract. 183(109119)2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Saeedi P, Petersohn I, Salpea P, Malanda
B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA,
Ogurtsova K, et al: Global and regional diabetes prevalence
estimates for 2019 and projections for 2030 and 2045: Results from
the International diabetes federation diabetes atlas, 9th edition.
Diabetes Res Clin Pract. 157(107843)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Piché ME, Tchernof A and Després JP:
Obesity phenotypes, diabetes, and cardiovascular diseases. Circ
Res. 126:1477–1500. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Eizirik DL, Pasquali L and Cnop M:
Pancreatic β-cells in type 1 and 2 diabetes mellitus: Different
pathways to failure. Nat Rev Endocrinol. 16:349–362.
2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Thorens B: GLUT2, glucose sensing and
glucose homeostasis. Diabetologia. 58:221–232. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mueckler M and Thorens B: The SLC2 (GLUT)
family of membrane transporters. Mol Aspects Med. 34:121–138.
2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Thorens B, Guillam MT, Beermann F,
Burcelin R and Jaquet M: Transgenic reexpression of GLUT1 or GLUT2
in pancreatic beta cells rescues GLUT2-null mice from early death
and restores normal glucose-stimulated insulin secretion. J Biol
Chem. 275:23751–23758. 2000.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Guillam MT, Dupraz P and Thorens B:
Glucose uptake, utilization, and signaling in GLUT2-null islets.
Diabetes. 49:1485–1491. 2000.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Lytrivi M, Castell AL, Poitout V and Cnop
M: Recent insights into mechanisms of β-cell lipo- and
glucolipotoxicity in type 2 diabetes. J Mol Biol. 432:1514–1534.
2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Prentki M, Peyot ML, Masiello P and
Madiraju SRM: Nutrient-induced metabolic stress, adaptation,
detoxification, and toxicity in the pancreatic β-cell. Diabetes.
69:279–290. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ashcroft FM and Rorsman P: Diabetes
mellitus and the β cell: The last ten years. Cell. 148:1160–1171.
2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Xie T, So WY, Li XY and Leung PS:
Fibroblast growth factor 21 protects against lipotoxicity-induced
pancreatic β-cell dysfunction via regulation of AMPK signaling and
lipid metabolism. Clin Sci (Lond. 133:2029–2044. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Biden TJ, Robinson D, Cordery D, Hughes WE
and Busch AK: Chronic effects of fatty acids on pancreatic
beta-cell function: New insights from functional genomics.
Diabetes. 53 (Suppl 1):S159–S165. 2004.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Cnop M: Fatty acids and glucolipotoxicity
in the pathogenesis of type 2 diabetes. Biochem Soc Trans.
36:348–352. 2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Barlow J, Jensen VH, Jastroch M and
Affourtit C: Palmitate-induced impairment of glucose-stimulated
insulin secretion precedes mitochondrial dysfunction in mouse
pancreatic islets. Biochem J. 473:487–496. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Manukyan L, Ubhayasekera SJ, Bergquist J,
Sargsyan E and Bergsten P: Palmitate-induced impairments of β-cell
function are linked with generation of specific ceramide species
via acylation of sphingosine. Endocrinology. 156:802–812.
2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Cnop M, Foufelle F and Velloso LA:
Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med.
18:59–68. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
El-Assaad W, Buteau J, Peyot ML, Nolan C,
Roduit R, Hardy S, Joly E, Dbaibo G, Rosenberg L and Prentki M:
Saturated fatty acids synergize with elevated glucose to cause
pancreatic beta-cell death. Endocrinology. 144:4154–4163.
2003.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wang XY, Zhu BR, Jia Q, Li YM, Wang T and
Wang HY: Cinnamtannin D1 protects pancreatic β-cells from
glucolipotoxicity-induced apoptosis by enhancement of autophagy in
vitro and in vivo. J Agric Food Chem. 68:12617–12630.
2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Omar-Hmeadi M and Idevall-Hagren O:
Insulin granule biogenesis and exocytosis. Cell Mol Life Sci.
78:1957–1970. 2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Back SH, Kang SW, Han J and Chung HT:
Endoplasmic reticulum stress in the β-cell pathogenesis of type 2
diabetes. Exp Diabetes Res. 2012(618396)2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Eizirik DL, Cardozo AK and Cnop M: The
role for endoplasmic reticulum stress in diabetes mellitus. Endocr
Rev. 29:42–61. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Lee JH and Lee J: Endoplasmic reticulum
(ER) stress and its role in pancreatic β-cell dysfunction and
senescence in type 2 diabetes. Int J Mol Sci.
23(4843)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hung CT, Tsai YW, Wu YS, Yeh CF and Yang
KC: The novel role of ER protein TXNDC5 in the pathogenesis of
organ fibrosis: mechanistic insights and therapeutic implications.
J Biomed Sci. 29(63)2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang X, Li H and Chang X: The role and
mechanism of TXNDC5 in diseases. Eur J Med Res.
27(145)2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Okumura M, Kadokura H and Inaba K:
Structures and functions of protein disulfide isomerase family
members involved in proteostasis in the endoplasmic reticulum. Free
Radic Biol Med. 83:314–322. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Duivenvoorden WCM, Hopmans SN, Austin RC
and Pinthus JH: Endoplasmic reticulum protein ERp46 in prostate
adenocarcinoma. Oncol Lett. 13:3624–3630. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Bidooki SH, Navarro MA, Fernandes SCM and
Osada J: Thioredoxin domain containing 5 (TXNDC5): Friend or Foe?
Curr Issues Mol Biol. 46:3134–3163. 2024.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Rutter GA, Pullen TJ, Hodson DJ and
Martinez-Sanchez A: Pancreatic β-cell identity, glucose sensing and
the control of insulin secretion. Biochem J. 466:203–218.
2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lampropoulou E, Lymperopoulou A and
Charonis A: Reduced expression of ERp46 under diabetic conditions
in β-cells and the effect of liraglutide. Metabolism. 65:7–15.
2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Cnop M, Abdulkarim B, Bottu G, Cunha DA,
Cunha DA, Igoillo-Esteve M, Masini M, Turatsinze JV, Griebel T,
Villate O, Santin I, et al: RNA sequencing identifies dysregulation
of the human pancreatic islet transcriptome by the saturated fatty
acid palmitate. Diabetes. 63:1978–1993. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Chen CW, Guan BJ, Alzahrani MR, Gao Z, Gao
L, Bracey S, Wu J, Mbow CA, Jobava R, Haataja L, et al: Adaptation
to chronic ER stress enforces pancreatic β-cell plasticity. Nat
Commun. 13(4621)2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Jiang H, Thapa P, Hao Y, Ding N,
Alshahrani A and Wei Q: Protein disulfide isomerases function as
the missing link between diabetes and cancer. Antioxid Redox
Signal. 37 (16-18):1191–1205. 2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Taniguchi CM, Emanuelli B and Kahn CR:
Critical nodes in signalling pathways: Insights into insulin
action. Nat Rev Mol Cell Biol. 7:85–96. 2006.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Camaya I, Donnelly S and O'Brien B:
Targeting the PI3K/Akt signaling pathway in pancreatic β-cells to
enhance their survival and function: An emerging therapeutic
strategy for type 1 diabetes. J Diabetes. 14:247–260.
2022.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Dalle S and Abderrahmani A: Receptors and
signaling pathways controlling beta-cell function and survival as
targets for anti-diabetic therapeutic strategies. Cells.
13(1244)2024.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Back SH and Kaufman RJ: Endoplasmic
reticulum stress and type 2 diabetes. Annu Rev Biochem. 81:767–793.
2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Hetz C and Papa FR: The unfolded protein
response and cell fate control. Mol Cell. 69:169–181.
2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Sano R and Reed JC: ER stress-induced cell
death mechanisms. Biochim Biophys Acta. 1833:3460–3470.
2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Hetz C, Zhang K and Kaufman RJ:
Mechanisms, regulation and functions of the unfolded protein
response. Nat Rev Mol Cell Biol. 21:421–438. 2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Ozcan U, Yilmaz E, Ozcan L, Furuhashi M,
Vaillancourt E, Smith RO, Görgün CZ and Hotamisligil GS: Chemical
chaperones reduce ER stress and restore glucose homeostasis in a
mouse model of type 2 diabetes. Science. 313:1137–1140.
2006.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Gao Y, Ryu H, Lee H, Kim YJ, Lee JH and
Lee J: ER stress and unfolded protein response (UPR) signaling
modulate GLP-1 receptor signaling in the pancreatic islets. Mol
Cells. 47(100004)2024.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Xing D, Zhou Q, Wang Y and Xu J: Effects
of tauroursodeoxycholic acid and 4-phenylbutyric acid on selenium
distribution in mice model with type 1 diabetes. Biol Trace Elem
Res. 201:1205–1213. 2023.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Yong J, Johnson JD, Arvan P, Han J and
Kaufman RJ: Therapeutic opportunities for pancreatic β-cell ER
stress in diabetes mellitus. Nat Rev Endocrinol. 17:455–467.
2021.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Celik C, Lee SYT, Yap WS and Thibault G:
Endoplasmic reticulum stress and lipids in health and diseases.
Prog Lipid Res. 89(101198)2023.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Chen X and Cubillos-Ruiz JR: Endoplasmic
reticulum stress signals in the tumour and its microenvironment.
Nat Rev Cancer. 21:71–88. 2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Ren J, Bi Y, Sowers JR, Hetz C and Zhang
Y: Endoplasmic reticulum stress and unfolded protein response in
cardiovascular diseases. Nat Rev Cardiol. 18:499–521.
2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Tarantino G and Citro V: Crosstalk between
the spleen and other organs/systems: Downstream signaling events.
Immuno. 4:479–501. 2024.
|
|
52
|
Zhang J, Deng Z, Jin L, Yang C, Liu J,
Song H, Han W and Si Y: Spleen-derived anti-inflammatory cytokine
IL-10 stimulated by adipose tissue-derived stem cells protects
against type 2 diabetes. Stem Cells Dev. 26:1749–1758.
2017.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Gotoh K, Inoue M, Masaki T, Chiba S,
Shimasaki T, Ando H, Fujiwara K, Katsuragi I, Kakuma T, Seike M, et
al: A novel anti-inflammatory role for spleen-derived
interleukin-10 in obesity-induced inflammation in white adipose
tissue and liver. Diabetes. 61:1994–2003. 2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Malik A, Bagchi AK, Jassal DS and Singal
PK: Interleukin-10 mitigates doxorubicin-induced endoplasmic
reticulum stress as well as cardiomyopathy. Biomedicines.
10(890)2022.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Marafon BB, Pinto AP, de Sousa Neto IV, da
Luz CM, Pauli JR, Cintra DE, Ropelle ER, Simabuco FM, Pereira de
Moura L, de Freitas EC, et al: The role of interleukin-10 in
mitigating endoplasmic reticulum stress in aged mice through
exercise. Am J Physiol Endocrinol Metab. 327:E384–E395.
2024.PubMed/NCBI View Article : Google Scholar
|














