Introduction
Portal hypertensive gastropathy (PHG) has emerged as
a serious clinical complication associated with portal hypertension
(PHT). This condition manifests pathologically as congestive
gastropathy, primarily resulting from compromised venous drainage
in the gastric mucosal circulation (1,2). PHG
predisposes patients to acute or chronic upper gastrointestinal
bleeding, significantly compromising health outcomes and quality of
life. The pivotal pathogenic determinant of PHG is elevated
vascular resistance within the portal system, which drives PHT
(3). PHT induces venous stasis
within gastric circulation, mucosal congestion, and compromised
tissue perfusion, culminating in hypoxia-mediated injury to gastric
epithelial cells (4,5). Various pathway adaptations to hypoxic
conditions, which trigger metabolic adaptations, modulate cell
proliferation and oxidative capacity, and govern inflammatory
responses, are instrumental in detecting low oxygen levels within
cells. These hypoxia-responsive mechanisms have been pathologically
associated with diverse disease processes, including neoplastic
progression, cardiovascular pathophysiology, metabolic
dysregulation syndromes, and gastrointestinal diseases (6,7). In
addition, emerging evidence implicates that the pathogenesis of PHG
is influenced by multifactorial mechanisms, such as dysregulation
of cytokines (including endothelin-1, nitric oxide, prostaglandin
E2, and tumour necrosis factor-α), epithelial apoptosis, disruption
of the mucosal barrier, nonspecific inflammatory responses, and
mitochondrial dysfunction (4,5).
Nevertheless, the precise molecular pathogenesis remains
incompletely elucidated, necessitating further systematic
investigation.
Hypoxia induces structural and functional
adaptations in mitochondria, which exacerbates oxidative stress and
promotes anaerobic glycolysis (8).
Mitochondria, the dominant source of reactive oxygen species (ROS)
production, exhibit a close interplay between mitochondrial
oxidative stress and glycolytic dysfunction (9). ROS are frequently generated as
byproducts during the process of oxidative phosphorylation.
Effective regulation of ROS levels is crucial, as they can
potentially induce oxidative damage to DNA, proteins, and lipids
(10). Glycolysis, a highly
conserved metabolic pathway, serves as the universal initial step
in glucose metabolism. Under hypoxic conditions, glycolysis
facilitates ATP generation through glucose catabolism, yielding
lactic acid as the terminal metabolite (11). Increased ROS and excessive
glycolysis can lead to alterations in cellular function and promote
the occurrence of adverse events such as cell death. Our previous
study has demonstrated that hypoxia-mediated mitochondrial
dysfunction is a critical contributor to PHG pathogenesis (12). However, the potential causative role
of hypoxia-induced ROS overproduction and accelerated glycolytic
flux in the pathogenesis of PHG-associated mucosal injury remains
to be systematically investigated.
Therefore, to elucidate the specific molecular
mechanisms linking hypoxia-induced ROS accumulation and aberrant
glycolytic metabolism in gastric epithelial injury in PHG, gastric
mucosal biopsies from patients with PHG and healthy controls were
analysed to identify transcriptomic and ultrastructural
differences. A murine model of PHT via carbon tetrachloride
administration and an in vitro model using hypoxia-exposed
GES-1 were further established. Utilizing transmission electron
microscopy (TEM), targeted metabolomics, and molecular assays,
mitochondrial morphology, oxidative stress markers, and glycolytic
flux were assessed. Additionally, the ROS scavenger Mito-TEMPO and
the lactate dehydrogenase A (LDHA) inhibitor oxamic acid sodium
were used to validate the therapeutic potential of targeting these
pathways in preventing mucosal injury. The present study not only
has the potential to reveal the key molecular links of the
hypoxia-metabolism imbalance in PHG, but also to provide novel
experimental evidence for the development of targeted therapeutic
strategies against oxidative stress and abnormal glycolysis in
PHG.
Materials and methods
Clinical tissue specimens
Gastric mucosal biopsy specimens were collected
endoscopically from two groups: i) 10 patients diagnosed with PHG
secondary to hepatitis B virus-related liver cirrhosis, with a mean
age of 55.4±8.05 years, ranging from 43-70 years. Among these 10
patients, 5 (50.0%) were male and 5 (50.0%) were female; and ii) 10
healthy control subjects undergoing routine health examinations at
the Endoscopic Center of the Third Affiliated Hospital of Sun
Yat-sen University (Guangzhou, P.R. China) with a mean age of
53.6±7.74 years, ranging from 44-66 years. Among these 10 subjects,
5 (50.0%) were male and 5 (50.0%) were female. The study protocol
was approved by the Clinical Research Ethics Committee of the Third
Affiliated Hospital of Sun Yat-Sen University [approval no.
(2021)02-176]. Gastric biopsy specimens, collected from both
healthy controls and patients with PHG, underwent preservation
either by freezing (-80˚C) for molecular biology applications or
through formalin fixation/paraffin embedding for histopathological
evaluation. Written informed consent from the patients was
waived.
Mouse models
All animal experiments were conducted in accordance
with protocols reviewed and approved (approval no. 2022F122) by the
Institutional Animal Care and Use Committee of South China
Agricultural University (Guangzhou, P.R. China) and conducted in
accordance with relevant guidelines. Experimental groups were
randomly assigned with 92 C57BL/6 mice (male, 6-8 weeks old, 18-20
g), which were maintained in a specific pathogen-free facility
under standardized conditions (22±2˚C; 50-60% humidity; 12-h
light/dark cycle). Mice were maintained in individually ventilated
cages with sterilized wood-chip bedding and provided with
autoclaved food and water ad libitum. All experimental
procedures and assessments were performed in a blinded fashion.
Following completion of experimental protocols, all mice were
humanely euthanized via gradual-fill carbon dioxide asphyxiation,
with a volume displacement rate of 70% utilized in our experimental
procedures. Liver cirrhosis with subsequent PHT was induced in mice
via twice-weekly intraperitoneal injections of carbon tetrachloride
(20% v/v in olive oil; Sinopharm Chemical Reagent) at 5 ml/kg body
weight for 16 weeks. Control mice received equivalent volumes of
olive oil alone (5 ml/kg, intraperitoneally) on the same schedule.
To mitigate oxidative stress, mice received intraperitoneal
Mito-TEMPO (10 mg/kg solubilized in PBS; cat. no. HY-112879;
MedChemExpress) every other day. Control groups were administered
PBS alone at equivalent volumes and frequency. For LDHA
suppression, oxamic acid sodium (750 mg/kg in PBS; HY-W013032A;
MedChemExpress) was administered intraperitoneally every other day,
with PBS serving as the vehicle control.
Sample collection
Mice from both the PHT and control groups were
anesthetized with ketamine/xylazine (100 mg/kg and 10 mg/kg;
intraperitoneal injection) and were humanely euthanized following
anesthetic induction. Their abdomens were opened, and the entire
stomach was carefully dissected. The stomach was repeatedly rinsed
in ice-cold PBS solution to remove any residual contents, and then
longitudinally cut along the greater curvature to expose the
gastric mucosa. After thoroughly rinsing the gastric contents with
ice-cold PBS, gross images were obtained, and the gastric mucosal
injury was scored as follows (12):
0, normal gastric mucosa; 1, gastric mucosal erosion; 2, gastric
mucosal ulcer (<1 mm); 3, gastric mucosal ulcer (1-2 mm); 4,
gastric mucosal ulcer (3-4 mm); 5, gastric mucosal ulcer (>5
mm). Subsequently, the mucosal tissues were collected. A portion
was stored in a -80˚C freezer for subsequent protein extraction.
For paraffin section preparation, a separate portion was fixed in
10% neutral-buffered formalin.
Histopathological staining
Tissue sections (3-µm thick) were prepared for
either hematoxylin and eosin (H&E) staining or
immunohistochemical/immunofluorescence (IHC/IF) analyses. For
immunostaining, antigen-retrieved tissue sections were incubated
overnight at 4˚C with primary antibodies targeting either LDHA
(cat. no. 19987-1-AP; Proteintech Group, Inc.) or 4-HNE (cat. nos.
ab46545/ab48506; Abcam), followed by incubation with
species-matched horseradish peroxidase (HRP)-conjugated secondary
antibodies (cat. nos. A0208/A0216; Beyotime Institute of
Biotechnology) at 37˚C for 1.5 h, as previously described (13). IF sections were DAPI-counterstained
to visualize nuclei. Tissue hypoxia distribution was evaluated in
gastric mucosal sections using the Hypoxyprobe-1 Kit (cat. no.
HP3-100Kit; Hypoxyprobe, Inc.) following the manufacturer's
protocol. Briefly, 1 h before anesthetization and sampling, mice
were intraperitoneally injected with a Hypoxyprobe-1 solution at a
dose of 60 mg/kg. Subsequently, the samples underwent fixation,
embedding, and sectioning into 3 µm-thick slices. After incubation
with the IgG1 monoclonal antibody (MAb1; clone 11.23.22.R; supplied
in the Hypoxyprobe-1 Kit, cat. no. HP3-100Kit; Hypoxyprobe, Inc.)
at 4˚C overnight, the sections were incubated with HRP-labeled
anti-rat secondary antibodies (cat. no. A0192; Beyotime Institute
of Biotechnology) at room temperature for 1.5 h, followed by DAB
staining. For primary cells, they were first incubated with a
100-µM Hypoxyprobe-1 solution for 1 h, and then subjected to
permeabilization with 0.3% Triton X-100 for 20 min at room
temperature (cat. no. P0096; Beyotime Institute of Biotechnology).
After incubating with the IgG1 MAb1 at 4˚C overnight, the cells
were stained with anti-rat secondary antibodies (cat. no. A-11006;
Thermo Fisher Scientific, Inc.) at 37˚C for 1 h. Nuclei were
counterstained with DAPI for 15 min at room temperature prior to
imaging. The quantitative analysis of Hypoxyprobe-1-positive
regions was performed in multiple fields of view (>6 per
sample). The ratio of positive regions to the total area in each
image was quantified using ImageJ software (v1.54 g; National
Institutes of Health), and the area ratio was calculated and
expressed as a percentage. For TEM detection, fresh gastric tissue
samples were immediately fixed in electron microscope fixative
(cat. no. LADE035; LANDM Biotechnology Co., Ltd.; Guangzhou Ruijing
Information Technology Co., Ltd.) at 4˚C overnight (or for at least
4 h). The samples were dehydrated in a graded ethanol series
(50-100%) followed by 100% acetone, infiltrated, and embedded in
resin (polymerized at 60˚C for 48 h). Ultrathin sections (~100 nm)
were cut using a Leica UC7 ultramicrotome. Sections were stained
with uranyl acetate (cat. no. U25690; Shanghai Acmec Biochemical
Technology Co., Ltd.) for 20 min and lead citrate (cat. no.
L885990; Macklin, Inc.) for 12 min at room temperature and imaged
using a transmission electron microscope (Tecnai G2 Spirit; FEI).
Mitochondrial morphometric analysis was performed by randomly
selecting eight mitochondria per image from each experimental
group. Mitochondrial length and width were quantified using length
parameters, while cross-sectional area was calculated via area
measurement tools.
Cell experiments
For the isolation of primary cells (14), the intact stomach was meticulously
excised and subsequently rinsed with a physiological buffer. The
pyloric region was securely ligated. Following this, the stomach
was perfused via the cardia using calcium-free Hank's Balanced Salt
Solution for a duration of 15 min to facilitate tissue preparation.
Following the initial perfusion, the stomach was subjected to
further enzymatic digestion by perfusion with 100 ml of a digestion
solution containing 0.2% pronase and 0.2% collagenase type IV (both
from Sigma-Aldrich; Merck KGaA). After enzymatic treatment, the
mucosal layer was carefully dissected and collected to generate a
single-cell suspension. The resulting suspension was then passed
through a 100-µm nylon mesh filter to remove undigested tissue
fragments and ensure a homogeneous cell population. The cell
suspension was centrifuged at 300 x g for 15 min to separate
cellular components. Following centrifugation, the resulting pellet
was carefully harvested for subsequent isolation of gastric
epithelial cells. A 30% Percoll solution (Sigma-Aldrich; Merck
KGaA) was prepared according to the manufacturer's protocol. The
cellular pellet was then carefully layered onto the Percoll density
gradient and centrifuged at 450 x g for 20 min at 4˚C to isolate
viable gastric epithelial cells. Primary cells were maintained in
RPMI-1640 medium (cat. no. 11875093; Thermo Fisher Scientific,
Inc.) supplemented with 10% heat-inactivated fetal bovine serum
(cat. no. A5256701; Thermo Fisher Scientific, Inc.) and
antibiotic-antimycotic solution (cat. no. 15240062; Thermo Fisher
Scientific, Inc.). Incubation was carried out at 37˚C in a
humidified chamber with 5% CO2 tension. For comparative
purposes, the GES-1 human gastric epithelial cell line was grown
under identical conditions, as previously established (15). For the hypoxia group, the cells were
switched to low-glucose culture medium and incubated in a tri-gas
incubator (1% oxygen, 5% CO2, 94% nitrogen) for 24 h.
The normoxia group was cultured in normal medium in a
CO2 incubator for 24 h. The working concentrations of
Mito-TEMPO and oxamic acid sodium were both set at 10 µmol/l. Using
the Cell Counting Kit-8 (CCK-8; cat. no. C0038; Beyotime Institute
of Biotechnology) assay, cellular viability across experimental
groups was systematically measured and compared after incubating
the cells with the reagent for 1.5 h at 37˚C as per the
manufacturer's instructions.
Western blotting
Total protein extracts from mucosal tissues or
isolated cells were subjected to western blot analysis following
established protocols (14,15). These protein extracts were obtained
using a total protein extraction kit (cat. no. PC201; Shanghai
EpiZyme Biomedical Technology Co., Ltd.). Protein concentration was
determined using the BCA protein assay kit (cat. no. ZJ102;
Shanghai EpiZyme Biomedical Technology Co., Ltd.). A total of 20 µg
of protein was loaded per lane and separated on a 10% SDS-PAGE gel.
Proteins were subsequently transferred to a polyvinylidene
difluoride membrane. The membrane was blocked with 5% milk in TBST
at room temperature for 1.5 h. Washing was performed three times
for 5 min each, using TBST containing 0.1% Tween-20. Membranes were
incubated overnight at 4˚C with primary antibodies against LDHA
(cat. no. 19987-1-AP; Proteintech Group, Inc.) and β-actin (cat.
no. sc-47778; Santa Cruz Biotechnology, Inc.), with β-actin serving
as the loading control. Protein bands were detected using enhanced
chemiluminescence (ECL; cat. no. E423-02; Vazyme Biotech Co., Ltd.)
after HRP-secondary antibody incubation (cat. no. RM3001/3002,
Beijing Ray Antibody Biotech) for 1 h at room temperature. Signal
acquisition was performed digitally, with band intensity
quantification conducted using ImageJ (v1.54 g; National Institutes
of Health).
ATP measurement
Gastric tissues (20 mg) were homogenized in 200 µl
of the ATP detection lysis buffer supplied with the Enhanced ATP
Assay Kit (cat. no. S0027; Beyotime Institute of Biotechnology)
using the Bead Ruptor 12 Homogenizer (Omni International, Inc.).
The homogenates were centrifuged (12,000 x g, 5 min, 4˚C), and the
supernatant was collected. Protein concentration was determined
using a BCA Protein Assay Kit (cat. no. ZJ102; Shanghai EpiZyme
Biomedical Technology Co., Ltd.). Subsequently, a 20-µl aliquot of
the supernatant was mixed with 100 µl of the ATP detection working
solution, and luminescence (RLU) was measured using a microplate
luminometer (Agilent BioTek Synergy H1 Microplate Reader; Agilent
Technologies, Inc.) with a 2 sec delay and 10 sec integration time.
ATP concentrations were calculated based on a standard curve (0,
2.5, 5, 10, 20, 40, and 50 µM) and normalized to protein
concentration (nmol/mg protein). Samples and standards were run in
triplicate.
Microarray analysis
The microarray experiment followed the same protocol
as previously described (14). The
microarray experiment was performed using the Agilent SurePrint G3
Human Gene Expression Microarray 8x60K platform (G4851B; Agilent
Technologies, Inc.). Total RNA quality and quantity were assessed
via Agilent 2100 Bioanalyzer; samples with an RNA Integrity Number
(RIN) ≥7.0, concentration ≥50 ng/µl, and 28S/18S ratio ≥1.8 were
used. Labeling was conducted using the Agilent Low Input Quick Amp
Labeling Kit (part no. 5190-2305; Agilent Technologies, Inc.)
following the manufacturer's protocol. Briefly, double-stranded
cDNA was synthesized using AffinityScript Reverse Transcriptase
(supplied in the Agilent Low Input Quick Amp Labeling Kit; part no.
5190-2305; Agilent Technologies, Inc.) and a T7 promoter primer
(supplied in the Agilent Low Input Quick Amp Labeling Kit; part no.
5190-2305; Agilent Technologies, Inc.). This was followed by in
vitro transcription (IVT) with T7 RNA Polymerase at 37˚C for 14
h to generate cRNA labeled with Cy3-CTP. Hybridization was carried
out in an Agilent hybridization oven (Model G2545A; Agilent
Technologies, Inc.) at 65˚C for 17 h using the Agilent Gene
Expression Hybridization Kit (part no. 5188-5242; Agilent
Technologies, Inc.). Subsequent data processing was conducted with
GeneSpring GX version 12.0 (Agilent Technologies, Inc.), which
executed data aggregation and normalization procedures.
Differential gene expression analysis employed the following
selection criteria: i) Absolute value of expression ratio
≥1.5-fold; ii) statistical significance threshold set at P<0.05
after Benjamini-Hochberg false discovery rate adjustment. The
expression data were log2-transformed and hierarchically clustered
via CLUSTER 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/)
Adjust Data function. Subsequently, the resulting dendrogram was
visualized with Java TreeView (v1.2.1; http://jtreeview.sourceforge.net/) for further
analysis. Gene Ontology (GO) enrichment analysis was performed
using the clusterProfiler R package (v4.14.6; https://bioconductor.org/packages//release/bioc/html/clusterProfiler.html).
GO terms with an adjusted P-value <0.05 were considered
statistically significant. The results were visualized using bubble
plots.
Energy metabolic analysis
Energy metabolism was profiled using a targeted
metabolomics platform [UPLC-MS/MS; Metabo-Profile Biotechnology
(Shanghai) Co., Ltd.] on gastric tissue. Specifically, 10 mg of
frozen sample was homogenized with ultrapure water and ceramic
grinding beads (cat. no. F6632; Beyotime Institute of
Biotechnology), and then extracted using methanol containing
internal standards. The samples were centrifuged at 18,000 x g for
15 min at 4˚C (Microfuge 20R; Beckman Coulter, Inc.). Following
centrifugation, metabolites were derivatized with
3-nitrophenylhydrazine and 1-ethyl-3-(3-dimethylaminopropyl)
carbodiimide at 30˚C for 60 min. Chromatographic separation was
performed on a BEH C18 column (2.1x100 mm, 1.7 µm; 40˚C) with a
gradient of mobile phase A (5 mM DIPEA aqueous solution) and mobile
phase B (acetonitrile/isopropanol, 7:3, v/v) at 0.3 ml/min. MS
detection was carried out in negative ESI mode (capillary voltage,
3 kV; source temperature: 150˚C; desolvation temperature, 500˚C).
Data were acquired on a Waters ACQUITY I UPLC/Xevo TQ-S system and
processed with MassLynx (v4.1; Waters Corporation). Metabolite
levels were normalized to sample weight, and missing values were
imputed as one tenth of the minimum for each metabolite. Subsequent
analyses, including principal component analysis (PCA) and
orthogonal partial least squares discriminant analysis (OPLS-DA),
were performed using iMAP [v1.0; Metabo-Profile
Biotechnology(Shanghai) Co., Ltd.]. Differentially expressed
metabolites were identified based on variable importance in
projection ≥1 (from OPLS-DA) and P<0.05 from univariate
tests.
Determination of oxidative stress
Cellular ROS production was assessed using DCFH-DA
(cat. no. S0033S; Beyotime Institute of Biotechnology). The primary
gastric epithelial cells were cultured to approximately 60-70%
confluence and subjected to the treatments. Subsequently, the cells
were incubated with DCFH-DA (diluted 1:1,000 in RPMI-1640 medium)
for 20 min at 37˚C. After probe incubation, oxidative conversion to
fluorescent DCF was measured at 488/525 nm excitation/emission.
Mitochondrial superoxide was assessed by seeding cells in confocal
dishes overnight. Cells were then stained with 5 µM MitoSOX™ Red
(cat. no. M36008; Thermo Fisher Scientific, Inc.) at 37˚C for 20
min (in the dark). Subsequently, the cells were gently washed three
times with warm PBS, followed by Hoechst 33258 (cat. no. C1017;
Beyotime Institute of Biotechnology) nuclear counterstaining at
room temperature for 20 min for localization. Fluorescence imaging
was performed using a Zeiss LSM880/800 confocal microscope (Imager
Z2; Zeiss AG). Mitochondrial DNA (mtDNA) was extracted with the
AllPrep DNA/RNA Mini Kit (cat. no. 80204; Qiagen) from the
mitochondrial fraction of cells, and oxidative damage was assessed
via 8-hydroxy-2'-deoxyguanosine (8-OHdG) quantification using the
OxiSelect™ Oxidative DNA Damage ELISA Kit (cat. no. STA-320; Cell
Biolabs, Inc.). Prior to the assay, extracted mtDNA samples were
dissolved and normalized to a uniform concentration of 1 mg/ml.
Following enzymatic digestion, a total of 50 µg of processed mtDNA
was loaded per reaction (50-µl volume). The 8-OHdG content was
determined by comparison with a predetermined standard curve (0-20
ng/ml) and is reported as ng/ml.
Statistical analysis
All results were consistently reproduced in at least
three independent experiments. Representative data from distinct
biological replicates were selected for visualization in figures
(for example microscopy images, immunoblots). Quantitative data are
reported as the mean ± standard error of the mean (SEM). All
statistical analyses were conducted using GraphPad Prism software
(version 9.5; Dotmatics). Comparisons between two groups were
analyzed using Student's two-tailed paired t-test.
Comparisons of single-factor data among more than two groups were
analyzed using one-way analysis of variance (ANOVA), while
comparisons of two-factor data among more than two groups were
analyzed using two-way ANOVA. When the F-value was <0.05,
Bonferroni's post hoc multiple comparison test was performed. A
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
Hypoxia participates in the gastric
mucosal injury of PHG in both humans and mice
To evaluate hypoxia in PHG pathogenesis, gastric
mucosal biopsies were obtained from patients who had PHG confirmed
by endoscopy, and a murine PHG model induced by PHT was developed.
Endoscopic assessment demonstrated significant mucosal
abnormalities in patients with PHG, including diffuse erythema,
vascular congestion, and oedema, which were absent in healthy
controls (Fig. 1A).
Histopathological analysis of gastric mucosal biopsies using
H&E staining demonstrated epithelial cell damage, focal
haemorrhages, and submucosal venular ectasia in the PHG group
(Fig. 1A). Hypoxyprobe-1
fluorescence signals were significantly enhanced in primary gastric
mucosal epithelial cells isolated from these tissues in the PHG
group (Fig. 1A and C). In the PHT mouse model, gross
examination showed marked mucosal congestion and even erosive
haemorrhage (Fig. 1B). Similar to
the clinical samples from the PHG group, the mice with PHT
exhibited significant epithelial cell damage, which was associated
with an elevated mucosal injury index (Fig. 1B and D). Moreover, the Hypoxyprobe-1 signal in
the PHT group was enhanced, which was positively associated with
the gastric injury index of mice with PHT (Fig. 1B and E). Collectively, these findings
demonstrated that hypoxia serves as a pathogenic factor
contributing to gastric mucosal injury in PHG.
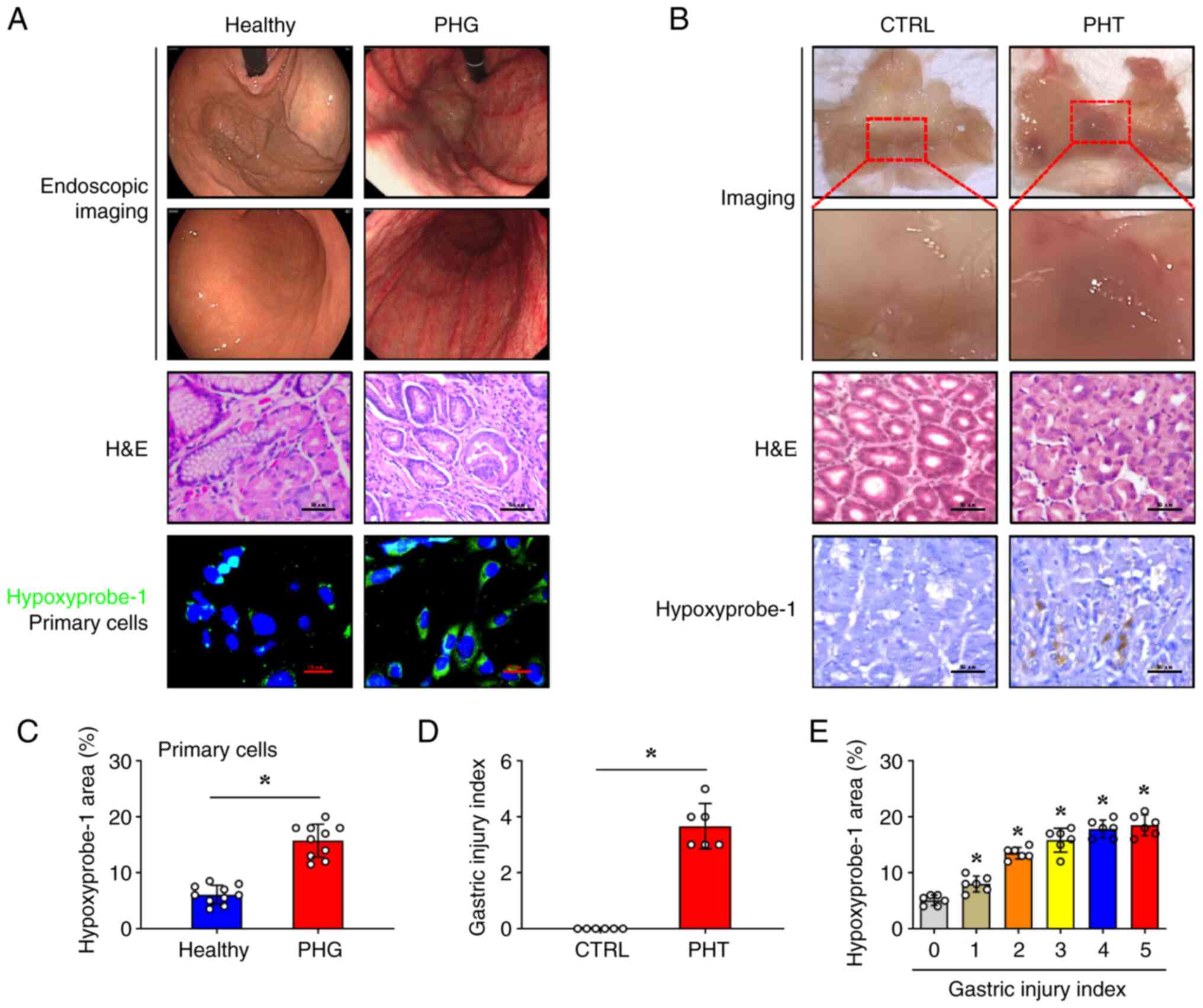 | Figure 1Hypoxia participates in gastric
mucosal injury of PHG in both humans and mice. (A) Representative
gastric endoscopic images and H&E-stained sections of gastric
mucosa from patients with PHG were compared with those from healthy
individuals (as Healthy). Scale bar, 50 µm for H&E images.
Hypoxyprobe-1 staining (green) was performed on primary gastric
mucosal epithelial cells isolated from both PHG patients and
healthy controls. Nuclei (blue) were counterstained with DAPI.
Scale bar, 10 µm. (B) Gross image, H&E staining, and
Hypoxyprobe-1 immunohistochemical staining (brown) of the gastric
mucosa in the mice with PHT compared with that in the control mice.
Scale bar, 50 µm for H&E and IHC images. (C) Quantitative
analysis of Hypoxyprobe-1 fluorescence intensity from (A), n=10 per
group; *P<0.05. (D) Mucosal injury index analysis in
the murine models is presented, n=6 per group;
*P<0.05. (E) The percentage of the
Hypoxyprobe-1-positive area corresponding to the gastric injury
index of PHT mouse models was analyzed. n=6 per group;
*P<0.05 vs. the group with a gastric injury index of
0. PHG, portal hypertensive gastropathy; H&E, hematoxylin and
eosin; DAPI, 4',6-diamidino-2-phenylindole dihydrochloride; PHT,
portal hypertension; CTRL, control. |
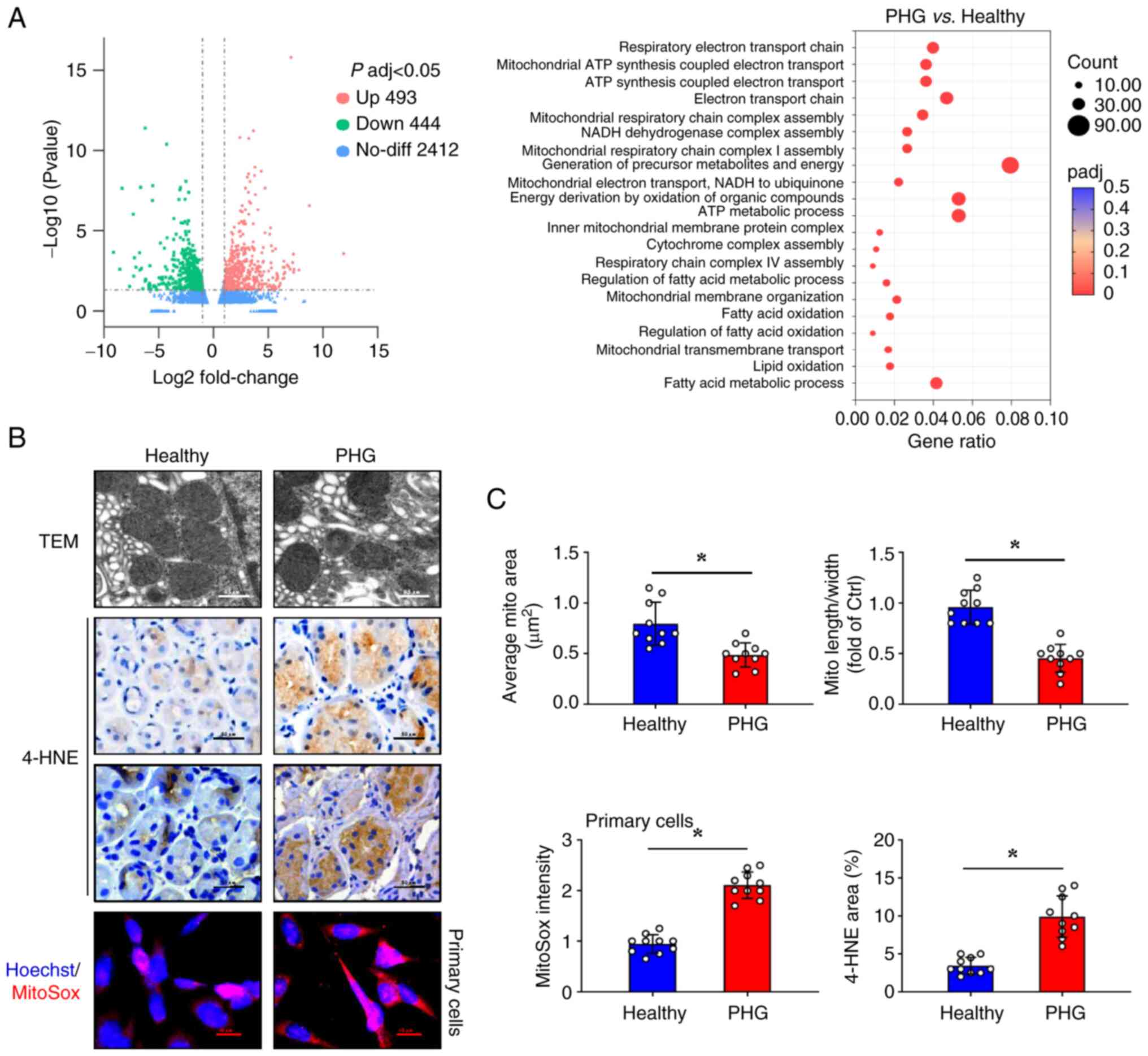
Enhanced mitochondrial oxidative
stress is identified in the gastric mucosa epithelia of PHG
To characterize the molecular alterations in PHG
pathogenesis, microarray analysis was conducted on gastric mucosal
biopsies obtained from both patients with PHG and healthy control
subjects. Comparative transcriptomic analysis revealed significant
differential gene expression profiles between PHG and normal
gastric mucosa (Fig. 2A).
Furthermore, Gene Ontology (GO) enrichment analysis demonstrated
upregulation of mitochondrial structural components (including
‘mitochondrial respiratory chain complex assembly’, ‘NADH
dehydrogenase complex assembly’, ‘inner mitochondrial membrane
protein complex’, and ‘mitochondrial membrane organization’) and
oxidative respiration (including ‘generation of precursor
metabolites and energy’, ‘energy derivation by oxidation of organic
compounds’, ‘respiratory electron transport chain’, and
‘mitochondrial ATP synthesis coupled electron transport’) in PHG
specimens (Fig. 2A). TEM analysis
identified ultrastructural mitochondrial abnormalities in the PHG
mucosa, including reduced mitochondrial size and decreased
mitochondrial cristae. The quantitative assessment indicated
decreases in the mean mitochondrial area as well as the
length-to-width ratio (Fig. 2B and
C). Immunohistochemical staining
showed that PHG tissues had elevated expression of 4-HNE, a lipid
peroxidation marker (Fig. 2B and
C). Consistently, primary gastric
epithelial cells from patients with PHG exhibited stronger MitoSox
fluorescence signals than those from healthy controls (Fig. 2B and C), indicating increased mitochondrial
superoxide production. PHT mouse models were established, and it
was verified that TEM analysis of gastric mucosa from mice with PHT
recapitulated clinical findings, demonstrating mitochondrial
shrinkage, damage, and cristae loss compared with the control mice
(Fig. 3A). In addition to the
increased MitoSox signalling and increased intracellular ROS levels
in the primary gastric epithelial cells, in mice with PHT, the
gastric mucosa also exhibited significantly increased 4-HNE
expression and elevated mitochondrial 8-OHdG (mt 8-OHdG)
concentrations (Fig. 3A and
B). Furthermore, the effects of
hypoxia were explored on the human gastric epithelial cell line
(GES-1) at the cellular level. Significant morphological changes
were observed in GES-1 cells under hypoxic conditions compared with
the controls, including reduced cell volume, enlarged central pale
areas, and increased cell death (Fig.
3C). Enhanced MitoSox signals and increased mt 8-OHdG
concentrations demonstrated that hypoxia induced mitochondrial
oxidative stress in GES-1 cells (Fig.
3C and D). In summary, these
findings indicated that mitochondrial structural alterations and
oxidative stress escalation observed in the gastric mucosa
contribute to the pathogenesis of PHG.
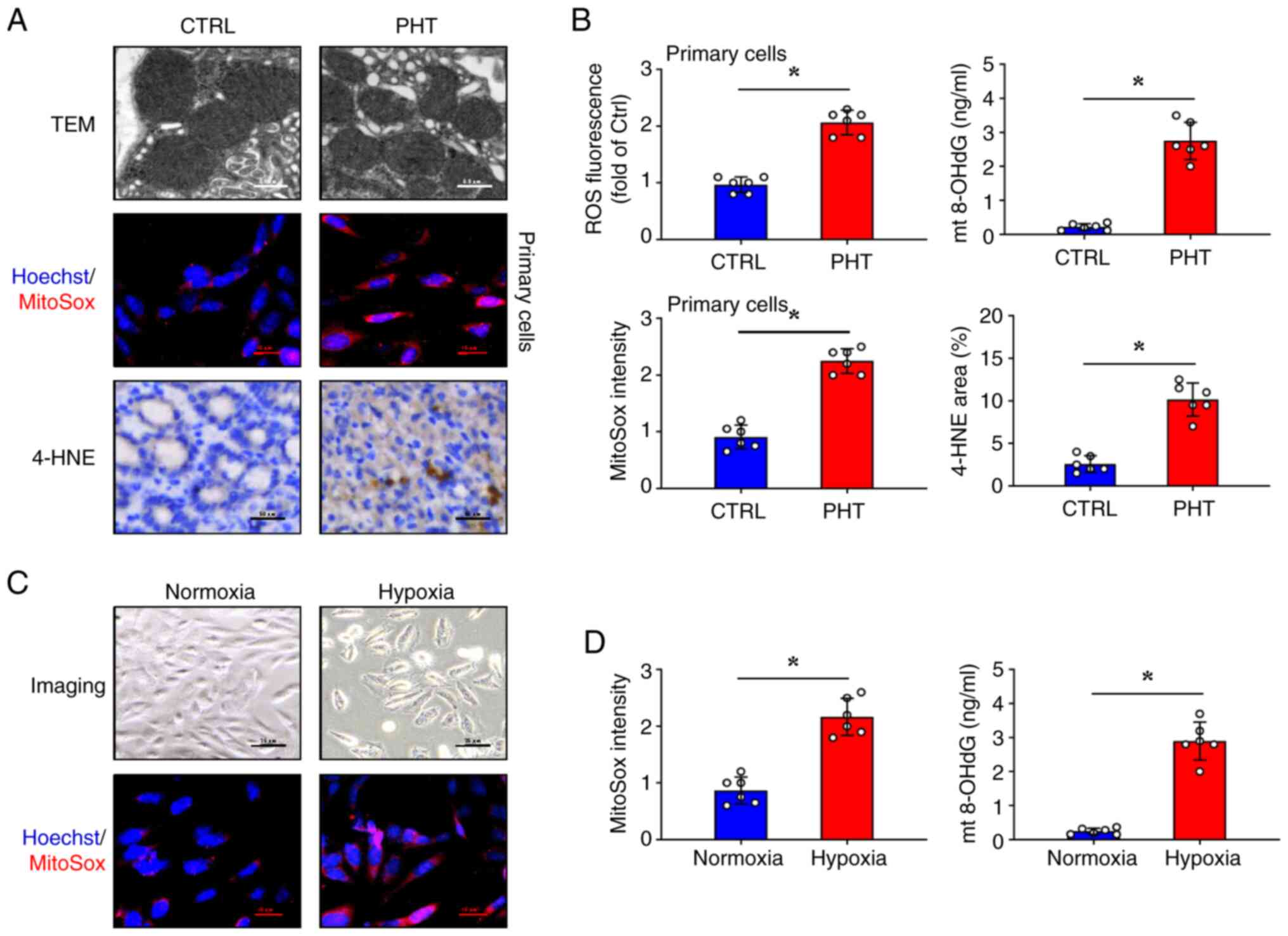 | Figure 3Mitochondrial oxidative stress is
enhanced in the in vivo and in vitro models. (A) TEM
analysis of mitochondrial morphological changes (Scale bar, 0.5
µm), MitoSox staining of primary gastric mucosal epithelial cells
(Scale bar, 10 µm), and immunohistochemical staining for 4-HNE were
performed in the gastric mucosal tissues from both control and PHT
group mice (Scale bar, 50 µm). Nuclei (blue) were counterstained
with Hoechst 33258 in MitoSox staining.(B) The quantitative
analysis of ROS fluorescence in primary gastric mucosal epithelial
cells and mt 8-OHdG concentration were determined. The MitoSox
fluorescence intensity and the quantitative analysis of 4-HNE
staining from (A) were also analyzed. n=6 per group;
*P<0.05. (C) Morphology and MitoSox fluorescence
staining of GES-1 cells under normoxic and hypoxic conditions.
Nuclei (blue) were counterstained with Hoechst 33258 in MitoSox
staining. Scale bars, 25 µm (upper panels) and 10 µm (lower
panels). (D) The mt 8-OHdG concentration and the quantitative
analysis of MitoSox fluorescence intensity in GES-1 cells under
normoxic and hypoxic conditions are shown. n=6 per group;
*P<0.05. TEM, transmission electron microscopy; PHT,
portal hypertension; mt 8-OhdG, mitochondrial 8-OhdG; CTRL,
control. |
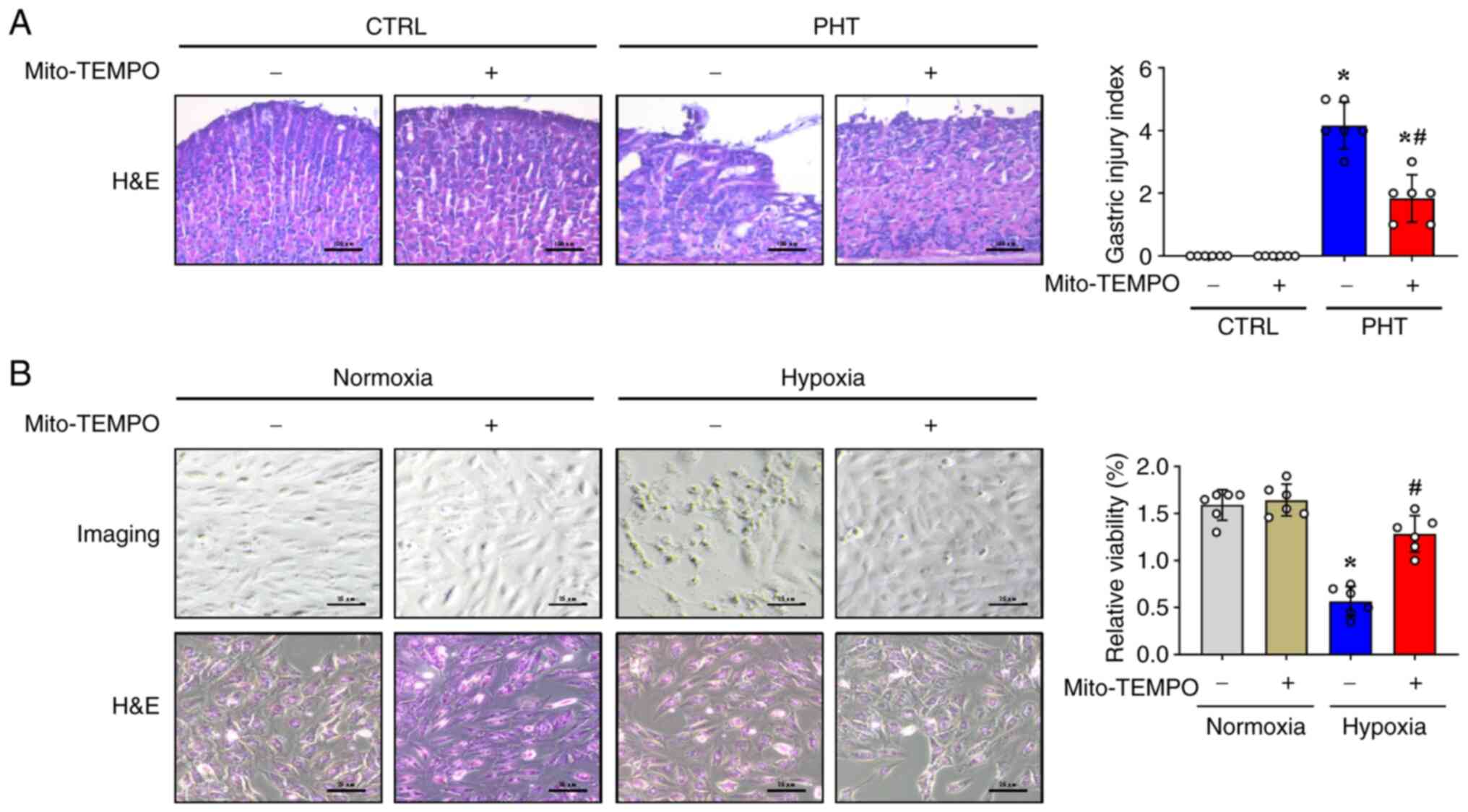
Mito-TEMPO attenuates gastric mucosal
epithelial damage in in vivo and in vitro models
The ROS scavenger Mito-TEMPO, which is an
antioxidant targeted to mitochondria, was used to investigate the
therapeutic potential of mitochondrial oxidative stress inhibition
(12). H&E staining revealed
that Mito-TEMPO relieved gastric mucosal injury, with decreased
inflammatory cell infiltration and attenuated capillary dilation in
mice with PHT (Fig. 4A). However,
no significant differences were observed in the gastric mucosa of
control mice treated with or without Mito-TEMPO (Fig. 4A). Furthermore, when used in the
in vitro cell model under normoxic and hypoxic conditions,
histological and cell viability analyses revealed that Mito-TEMPO
was able to restore the morphological alterations and increase the
relative cell viability in GES-1 cells under hypoxic conditions
(Fig. 4B), and Mito-TEMPO did not
affect the status of GES-1 cells under normoxic conditions
(Fig. 4B). These findings
demonstrated that mitochondrial-derived oxidative stress
contributes to both PHT-induced gastric mucosal damage and hypoxic
cellular injury. Notably, the mitochondrial-targeted antioxidant
Mito-TEMPO alleviated these pathological changes in murine models
and in vitro systems.
Mitochondrial oxidative stress is
associated with LDHA upregulation, which impairs ATP production and
promotes lactic acid production
Microarray analysis of clinical gastric mucosal
biopsies revealed significant differential expression of glycolytic
pathway-related genes between patients with PHG and healthy
controls. Moreover, the expression of glycolysis-related genes in
the glycolytic metabolic pathway, such as hexokinase-2
(HK2), LDHA, and pyruvate kinase isozyme type
M2 (PKM2), was increased in the gastric mucosa of
patients with PHG (Fig. 5A).
Genomic metabolic pathway analysis confirmed the enrichment of
genes that were differentially expressed in the glycolytic process,
with LDHA exhibiting the most pronounced increase in transcription
(Fig. 5B and C). Histological staining revealed that the
protein expression of LDHA was consistent with the results of
microarray analysis and was significantly upregulated in the
patients with PHG and the mice with PHT (Fig. 5D). Moreover, the protein expression
of LDHA increased in the GES-1 cell model following hypoxia
(Fig. 5D). Furthermore, western
blot analysis revealed a similar trend in LDHA expression in the
three different sections (Fig. 5E).
These findings established that mitochondrial dysfunction may drive
LDHA upregulation to facilitate glycolytic adaptation to
bioenergetic stress under hypoxia.
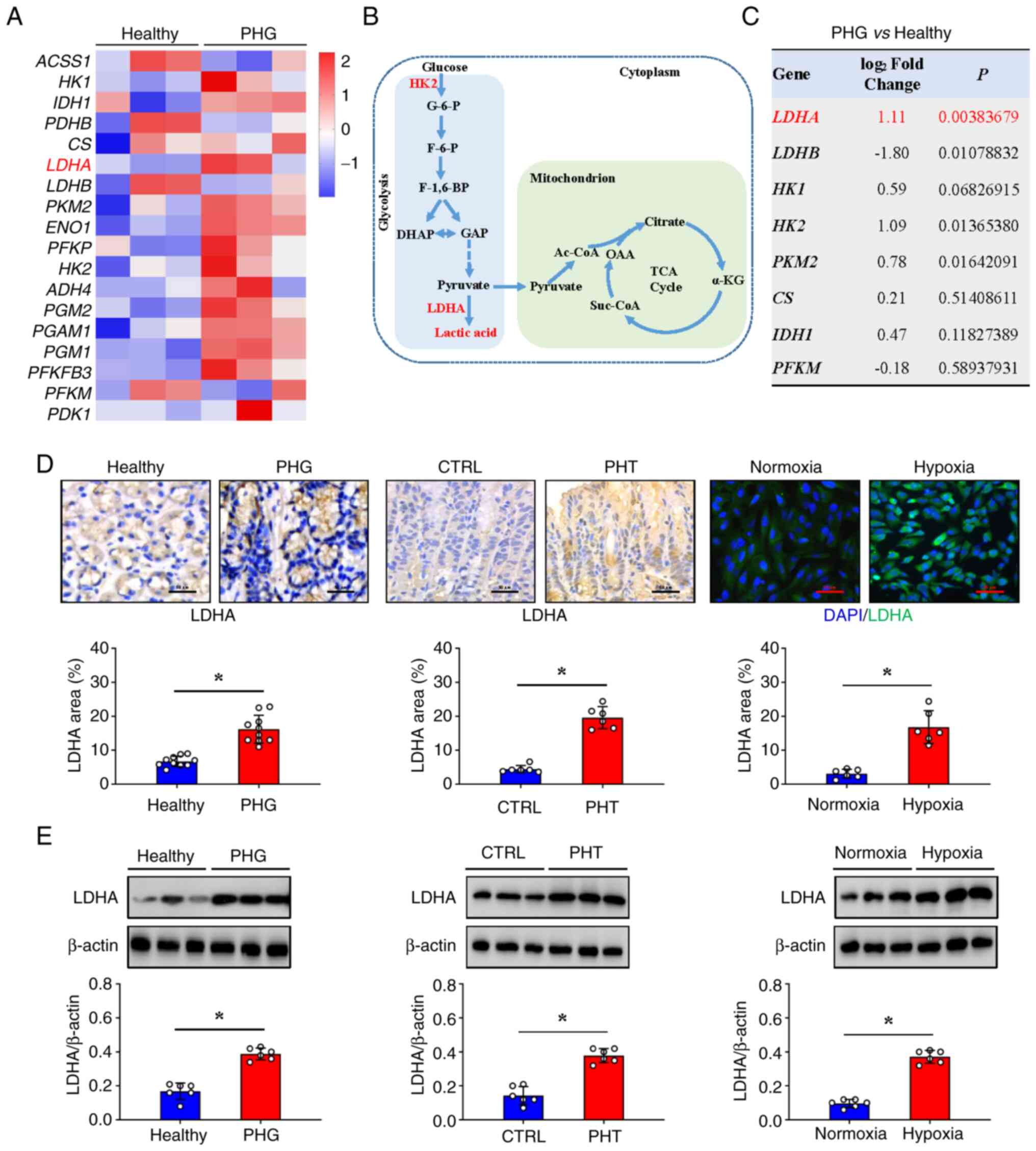 | Figure 5Mitochondrial dysfunction is
associated with the glycolytic LDHA upregulation in PHG. (A) The
heatmap analysis of differentially expressed genes between normal
gastric mucosa (healthy individuals-indicated as Healthy) and
gastric mucosa from patients with PHG (indicated as PHG) revealed
increased expression of LDHA and other genes involved in the
glycolytic metabolic pathway. (B) The glycolytic metabolic pathway
is presented. (C) The fold changes and P-values of the indicated
mRNAs in PHG tissues relative to those in normal (healthy
individuals-indicated as Healthy) tissues from the microarray
experiment are presented. (D) Immunohistochemical (brown) or
immunofluorescence (green) staining and quantitative analysis of
LDHA in the clinical tissue samples (n=10 per group), mouse models
(n=6 per group), and cellular hypoxia models (n=6 per group).
Nuclei (blue) were counterstained with DAPI in immunofluorescence
staining. Scale bars, 50 µm (IHC images) and 25 µm (IF images).
*P<0.05. (E) Western blotting detection of LDHA
protein expression levels in clinical tissue samples, mouse models,
and cellular hypoxia models. Ratios of densitometric units of
normalized LDHA/β-actin were determined from western blotting. n=6
per group. *P<0.05. PHG, portal hypertensive
gastropathy; LDHA, lactate dehydrogenase A; DAPI,
4',6-diamidino-2-phenylindole dihydrochloride; CTRL, control; PHT,
portal hypertension. |
Given the established roles of mitochondrial
oxidative stress in PHG pathogenesis and hypoxia-induced LDHA
upregulation in glycolytic reprogramming, energy metabolic analysis
of gastric mucosal tissues was performed to systematically
characterize the associated metabolic alterations. Distinct
clustering patterns were observed between the PHT and control
groups using PCA (Fig. 6A).
Metabolomic profiling revealed significant alterations in gastric
mucosal metabolism, with mice with PHT exhibiting elevated organic
acid metabolism and reduced carbohydrate metabolism compared with
the control mice (Fig. 6B). Among
these findings, the increase in the lactic acid concentration was
the most significant (Fig. 6C).
Because increased glycolysis can inhibit normal cellular aerobic
oxidation and lead to reduced ATP production, the ATP levels were
assessed and it was revealed that ATP levels were decreased in
samples from patients with PHG, mice with PHT, and hypoxic GES-1
cells (Fig. 6D), whereas lactic
acid concentrations were elevated in these groups (Fig. 6E). The LDHA inhibitor oxamic acid
sodium alleviated the gastric mucosal damage in mice with PHT
(Fig. 6F). Similarly, oxamic acid
sodium reversed the morphological changes and increased the
relative cell viability of GES-1 cells under hypoxic conditions
(Fig. 6G). It is therefore
concluded that the mitochondrial oxidative stress that follows
PHT-mediated hypoxia impairs ATP production while contributing to
increases in ROS production and LDHA-mediated lactic acid, thus
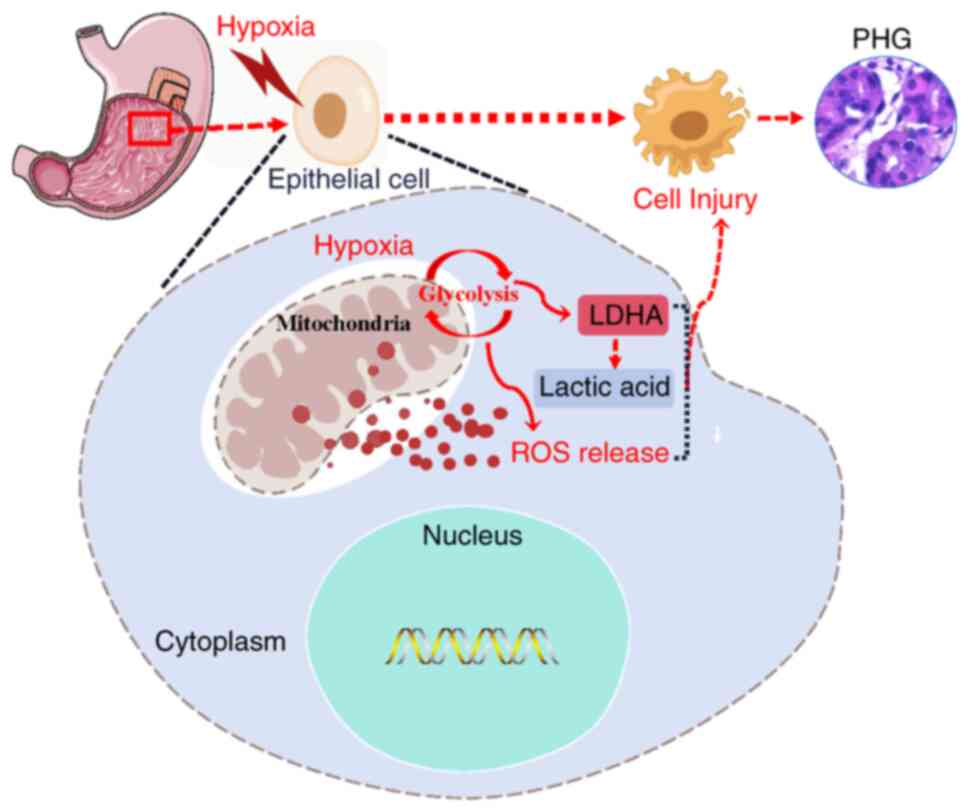
promoting mucosal epithelial injury in the PHG (Fig. 7).
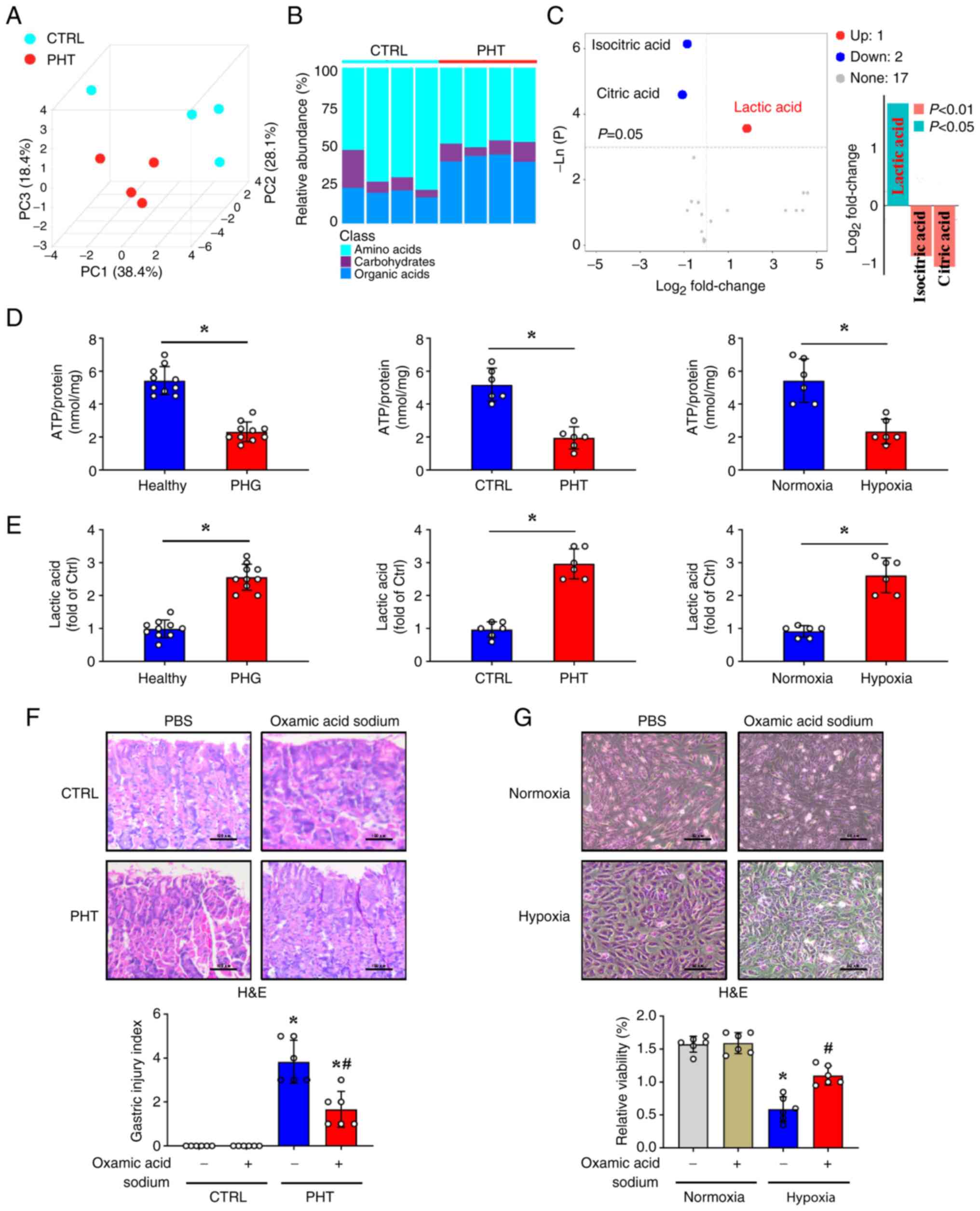 | Figure 6Mitochondrial oxidative stress in PHG
is accompanied by ATP production and promotes lactic acid
production. (A) Principal component analysis of metabolic
components in gastric mucosal tissues from the control group and
mice with PHT. n=4 per group. (B) Stacked bar plots comparing the
relative abundance of amino acid metabolism, carbohydrate
metabolism, and organic acid metabolism in the gastric mucosal
tissues between the control group and PHT mice. (C) Volcano plot
and bar chart analyses of carbohydrate metabolism biomarkers in the
indicated gastric mucosal tissues. (D) The ATP concentrations in
the clinical tissue samples (n=10 per group), murine models (n=6
per group), and cellular hypoxia models (n=6 per group) were
analyzed. *P<0.05. (E) The lactic acid concentrations
in clinical tissue samples (n=10 per group), mouse models (n=6 per
group), and cellular hypoxia models (n=6 per group) were assessed.
*P<0.05. (F) H&E staining and gastric mucosal
injury index scores in the indicated mouse models (treated with
lactate dehydrogenase A inhibitor oxamic acid sodium or not) were
determined. Scale bar, 100 µm. n=6 per group. *P<0.05
vs. the control group; #P<0.05 vs. the PHT group
without oxamic acid sodium. (G) H&E staining of the GES-1 cell
models under normoxia and hypoxia following oxamic acid sodium
administration. Cell viability from the indicated groups was also
analyzed by Cell Counting Kit-8 assay. Scale bar, 50 µm. n=6 per
group. *P<0.05 vs. the normoxia group;
#P<0.05 vs. the hypoxia group without oxamic acid
sodium. PHG, portal hypertensive gastropathy; PHT, portal
hypertension; H&E, hematoxylin and eosin; CTRL, control; PBS,
phosphate-buffered saline. |
Discussion
PHG represents a significant complication of both
cirrhotic and noncirrhotic PHT (1).
The condition is clinically defined by a portal pressure gradient
exceeding 5 mmHg, measured as the pressure differential between the
portal vein and hepatic veins (16). In cirrhotic cases, this haemodynamic
alteration primarily stems from architectural distortion of the
hepatic parenchyma and subsequent intrahepatic vascular resistance.
PHT disrupts blood flow and leads to tissue hypoxia, which
compromises mitochondrial integrity and triggers downstream
metabolic dysregulation to exacerbate the gastric mucosal
epithelial injury; the PHT-related PHG gastric mucosal lesions are
commonly defined as complications in patients with PHT (5). PHG imposes a significant burden on
health care systems and substantially reduces the quality of life
of patients. Prior studies have demonstrated that hypoxia
stabilizes hypoxia-inducible factor-1α (HIF-1α), increasing its
transcriptional activity and promoting disease progression in
gastric cancer and other related disorders (12,17,18).
Hypoxia is closely linked to gastric mucosal damage and serves as a
hallmark of gastric mucosal pathologies, spanning from inflammatory
reactions to malignancies (17,19).
Among these, PHG is the most representative benign lesion driven by
hypoxia (20).
To assess the hypoxic state of the gastric mucosa in
PHG induced by PHT, gastric tissue samples were collected from
patients with an endoscopic diagnosis of PHG. Additionally, an
experimental mouse model of PHT-associated PHG was established to
investigate the underlying mechanisms involved. Using a
Hypoxyprobe-1 detection kit, it was found that the hypoxic state in
the gastric mucosa appeared in the gastric mucosal epithelia
following PHT, and a positive association between hypoxia levels
and the severity of gastric mucosal injury in mice with PHT was
also observed. The oxygen concentration directly influences
mitochondrial function, and mitochondria perform critical metabolic
and signalling functions in nearly all eukaryotic cells (21). As oxygen-sensing organelles,
mitochondria exhibit functional impairment and structural damage in
response to hypoxia, accompanied by elevated oxidative stress
(12,22). Our previous data demonstrated that
mitochondrial dysfunction is involved in mucosal epithelial injury
and the progression of benign and malignant gastric mucosal lesions
under hypoxic conditions (14). In
the present study, microarray analysis was performed on biopsied
gastric mucosal tissues. Microarray analysis revealed that compared
with healthy controls, patients with PHG exhibited increased
activation of multiple mitochondrial pathways related to structural
integrity and respiratory function. During mitochondrial ATP
synthesis, the electron transport chain generates ROS as metabolic
byproducts. Consequently, mitochondria represent the primary
endogenous source of cellular ROS, which can serve as key mediators
of oxidative stress, inflammation, and tissue damage (5,14). TEM
imaging confirmed that ultrastructural mitochondrial abnormalities,
including reduced mitochondrial size and decreased mitochondrial
cristae, were observed in the PHG mucosa. Moreover, 4-HNE
expression was significantly elevated in PHG tissues, accompanied
by increased MitoSox signalling in primary gastric epithelial cells
isolated from PHG sections. A PHT mouse model and a hypoxia-induced
GES-1 cell model were also established, and it was verified that a
significant alteration in mitochondrial structure associated with
increased mitochondrial oxidative stress and ROS levels (as assayed
by MitoSox staining, ROS detection, 4-HNE expression and mt 8-OHdG
concentration) was observed in both the gastric mucosal epithelial
cells of mice with PHT and the hypoxia-treated GES-1 cells. The
progressive accumulation of mitochondrial ROS induces oxidative
damage to cellular macromolecules, including DNA, proteins, and
lipids. These ROS-mediated modifications can trigger diverse
pathological cascades, with the magnitude and nature of their
effects being modulated by microenvironmental conditions (23). Therefore, antioxidants may have
specific therapeutic effects under various conditions to alleviate
disease progression. Mito-TEMPO, a mitochondrion-targeted ROS
scavenger, exhibits potent antioxidant activity because it
selectively accumulates within mitochondria at concentrations
several-fold higher than those in the cytosol. This compound
attenuates mitochondrial oxidative stress by suppressing superoxide
generation, thereby restoring mitophagy and counteracting excessive
mitochondrial fission (22,24,25).
In a PHT mouse model and an in vitro cell model, it was
demonstrated that Mito-TEMPO administration attenuated gastric
mucosal injury in mice with PHT and hypoxia-induced GES-1 cell
damage, suggesting that mitochondrial oxidative stress-derived ROS
resulted in gastric mucosal injury and cell damage following
hypoxia. Our aforementioned findings indicate that mitochondrial
structural derangements and oxidative stress escalation in the
gastric mucosa drive PHG pathogenesis.
Mitochondria serve as critical hubs for numerous
essential cellular processes, such as amino acid and fatty acid
metabolism, the citric acid cycle, nitrogen metabolism, and
oxidative phosphorylation, which generate the majority of cellular
ATP. Glycolysis represents a fundamental metabolic pathway mediated
by multiple glycolytic enzymes in mammalian cells. Emerging
evidence indicates that gastric mucosal disorders upregulate
glycolytic flux under hypoxic conditions to sustain cellular energy
homeostasis (26,27). Our microarray and metabolic pathway
analyses demonstrated significant activation of glycolysis in the
gastric mucosa of patients with PHG, with LDHA serving as the
critical rate-limiting regulator of this process. LDHA, a key
enzyme in glycolysis that is upregulated in various
gastrointestinal diseases, converts pyruvate to lactic acid and
produces nicotinamide adenine dinucleotide (28). Consistent with these findings, the
protein expression of LDHA was also increased in gastric mucosal
tissue samples from clinical PHG specimens, mouse PHT models and
hypoxia-induced GES-1 cells, suggesting that mitochondrial
oxidative stress drives LDHA-mediated glycolytic adaptation to
bioenergetic stress under hypoxia in PHG. Various studies have
suggested that in hypoxic tumour microenvironments, cancer cells
undergo metabolic reprogramming to preferentially utilize
glycolysis for ATP generation, enabling sustained proliferation
under oxygen-deprived conditions. This glycolytic shift not only
promotes tumour cell survival but also facilitates rapid biomass
accumulation, thereby driving aggressive tumour progression
(29). On the basis of our current
data, it is suggested that increased LDHA-mediated glycolysis in
the PHG, a representative disease of benign gastric lesions,
aggravates mucosal injury and cell damage rather than inducing cell
survival. Our previous study confirmed that HIF-1α can upregulate
the expression level of LDHA, increase glycolytic metabolism, and
increase mitochondrial oxidative stress, thereby participating in
the occurrence and development of PHG (14). Multiple studies have shown that
influenza A virus (WSN strain) impairs mitochondria, which then
generates ROS, thereby stabilizing HIF-1α and activating glycolysis
to provide energy for viral replication. Rabies virus
glycoprotein-engineered extracellular vesicles (deliver miR-137 and
downregulate HIF-1α by targeting Toll-like receptor 4 to inhibit
the NF-κB pathway, thereby improving glycolytic abnormalities and
neuroinflammation in autism model mice. Under glutamine
deprivation, extracellular cyclophilin B in glioblastoma binds to
CD147 to activate p-AKT, which upregulates HIF-1α to increase
glycolysis and inhibit mitochondrial function. In acute
pancreatitis, increased activity of xanthine oxidase (XO) generates
ROS to activate HIF-1α, which promotes lactate accumulation via
LDHA and triggers NOD-like receptor protein 3 inflammatory
responses, while the inhibition of XO can alleviate pancreatic
necrosis (30-33).
These findings confirm that mitochondrial redox stress, which is
involved in various diseases, can regulate the protein level of
HIF-1α, thereby modulating the expression of related proteins such
as LDHA, mediating changes in glycolytic metabolism, and
participating in the occurrence and development of diseases. An
in-depth exploration of the specific molecular mechanisms was not
performed in the present study, and the definite association
between the upregulation of LDHA induced by energy metabolism
alterations and mitochondrial dysfunction-mediated ROS production
under hypoxia in the PHG warrants further investigation.
Mitochondrial oxidative phosphorylation and
glycolysis constitute the two primary energy-producing pathways in
mammalian cells under physiological conditions. While glycolysis
serves to meet cellular energy demands, its upregulation also plays
a pivotal role in generating metabolic intermediates required for
macromolecular biosynthesis, particularly in highly proliferative
cells such as malignant cells (34). Although aerobic glycolysis (the
Warburg effect) can increase ATP production to sustain energy
homeostasis and supplement the tricarboxylic acid (TCA) cycle,
excessive glycolytic flux in the gastric mucosal epithelium of
patients with PHG can disrupt normal aerobic respiration. This
metabolic reprogramming ultimately results in diminished net ATP
yield despite increased glycolytic activity (14). As a glycolytic enzyme, LDHA promotes
the release of glycolysis and lactic acid, and excessive lactic
acid influences the immune microenvironment, intracellular
signalling, cellular function and homeostasis to promote cell
damage and even death (35,36). Furthermore, our metabolic profiling
of gastric mucosal epithelial cells from mice with PHT revealed
significant alterations in metabolic pathways, characterized by
increased organic acid metabolism and decreased carbohydrate
metabolism. Consistent with these findings, elevated LDHA
expression and concomitant increases in lactic acid concentrations
across multiple experimental systems, including clinical PHG
specimens, PHT mouse models, and hypoxia-exposed GES-1 cells, were
observed. These observations are particularly noteworthy given that
LDHA has emerged as a promising therapeutic target in oncology.
Several small-molecule LDHA inhibitors have been developed to
specifically block the lactate dehydrogenase activity of LDHA,
thereby suppressing glycolytic flux and inhibiting tumour
progression in various cancers (37). Notably, in the present study, it was
found that the LDHA inhibitor oxamic acid sodium not only
alleviated gastric mucosal damage in mice with PHT but also
restored morphological changes and increased relative cell
viability in GES-1 cells under hypoxic conditions. The present
study focused primarily on the role of LDHA in mitochondrial
oxidative stress in the PHG, but metabolic reprogramming affecting
mitochondrial function is complex, with other key glycolytic
enzymes, such as HK2 and PKM2, also regulating cellular redox
balance and mitochondrial stress. HK2 normally binds to the
voltage-dependent anion channel (VDAC) on the outer mitochondrial
membrane to maintain mitochondrial function, and under metabolic
disturbances, glucose-induced HK2 stabilization and subsequent
detachment from VDAC impair mitochondrial ATP disposal, cause
membrane hyperpolarization, and increase ROS levels (38). The tetrameric form of PKM2 supports
glycolytic flux and mitochondrial metabolism, whereas pathological
conditions induce its sulfenylation, disrupting tetramer formation,
reducing activity, and impairing mitochondrial biogenesis/fusion
effects, which are reversed by PKM2 activation (39). In our previous study, quantitative
protein detection of HK2 and PKM2 was conducted, and it was
revealed that the expression levels of HK2 and PKM2 were increased
in mouse models, which were involved in the occurrence and
development of PHG (14). Given
their roles in metabolic disorder-induced mitochondrial damage, HK2
and PKM2 may contribute to PHG pathogenesis, warranting further
investigation. In summary, the mechanisms underlying gastric
epithelial cell injury in the PHG warrant further investigation.
Specifically, the roles of hypoxia-induced alterations in energy
metabolism and mitochondrial dysfunction, particularly their
contribution to ROS production, remain to be fully elucidated.
The present study has several limitations. First,
the relationships between oxidative stress and various types of
programmed cell death (such as apoptosis, pyroptosis, and
ferroptosis) remain unclear. Second, the mechanism through which
LDHA affects epithelial cell death is not well defined, and future
studies need to focus on exploring more downstream mechanisms.
Third, the intervention experiments with Mito-TEMPO (for inhibiting
mitochondrial oxidative stress) and oxamic acid sodium (for
inhibiting LDHA) focus mainly on short-term effects. Although the
current experimental design included a 16-week drug administration
period, a more longer intervention study to assess their effects on
the progression of PHG has yet to be conducted. This is due to time
limitations and technical constraints in establishing a long-term
mouse PHG model in the current research, and the selectivity and
potential side effects of these agents under long-term use also
remain to be further clarified. Notably, for clinical application
of these two inhibitors, large-scale experimental verification is
still needed to investigate potential side effects and confirm
their clinical applicability.
In summary, PHT leads to congestion and hypoxia in
the gastric mucosal epithelium, and this hypoxia induces
mitochondrial oxidative stress and ROS production, which are
associated with abnormal glycolytic metabolism and increased
LDHA-mediated lactic acid production. Ultimately, these changes
result in gastric mucosal epithelial injury to promote the
progression of PHG (Fig. 7).
Acknowledgements
We thank the Institutional Animal Care and Use
Committee of South China Agricultural University (Guangzhou, P.R.
China) for providing mice breeding and experimental studies in this
experiment. We also thank the Institutional Animal Care and Use
Committee of the Third Affiliated Hospital of Sun Yat-sen
University (Guangzhou, P.R. China) for providing mouse experimental
studies and technical support in this experiment.
Funding
Funding: The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 82170569
and 82470589), and the Science and Technology Planning Projects of
Guangzhou City (grant no. 2025A03J3193).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author. Datasets from this study
are available in the public database Science Data Bank (ScienceDB)
at https://www.scidb.cn/detail?dataSetId=59ef752d1c60447a978aa3a62ad83c44.
Authors' contributions
JL performed the mouse experiments and analyzed the
data of the patients. KX contributed essential reagents and
conducted the cell experiments. XO planned and conducted primary
cell isolation and signalling pathway study. ST designed the whole
project, supervised the research and wrote the paper. JL and KX
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The study protocol was approved [approval no.
(2021)02-176] by the Clinical Research Ethics Committee of the
Third Affiliated Hospital of Sun Yat-Sen University (Guangzhou,
P.R. China). Written informed consent from the patients was waived.
All animal experimental studies were approved (approval no.
2022F122) by the Institutional Animal Care and Use Committee of
South China Agricultural University (Guangzhou, P.R. China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cubillas R and Rockey DC: Portal
hypertensive gastropathy: A review. Liver Int. 30:1094–1102.
2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
McCormack TT, Sims J, Eyre-Brook I,
Kennedy H, Goepel J, Johnson AG and Triger DR: Gastric lesions in
portal hypertension: Inflammatory gastritis or congestive
gastropathy? Gut. 26:1226–1232. 1985.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Guixé-Muntet S, Quesada-Vázquez S and
Gracia-Sancho J: Pathophysiology and therapeutic options for
cirrhotic portal hypertension. Lancet Gastroenterol Hepatol.
9:646–663. 2024.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Urrunaga NH and Rockey DC: Portal
hypertensive gastropathy and colopathy. Clin Liver Dis. 18:389–406.
2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Tan SW: Molecular mechanism of portal
hypertensive gastropathy: An update. Clin Res Hepatol
Gastroenterol. 48(102423)2024.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Luo Z, Tian M, Yang G, Tan QR, Chen YB, Li
G, Zhang QW, Li YK, Wan P and Wu JG: Hypoxia signaling in human
health and diseases: Implications and prospects for therapeutics.
Signal Transduct Target Ther. 7(218)2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yuan XY, Ruan W, Bobrow B, Carmeliet P and
Eltzschig HK: Targeting hypoxia-inducible factors: Therapeutic
opportunities and challenges. Nat Rev Drug Discov. 23:175–200.
2024.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lee P, Chandel NS and Simon MC: Cellular
adaptation to hypoxia through hypoxia inducible factors and beyond.
Nat Rev Mol Cell Biol. 21:268–283. 2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Brooks GA: Lactate as a fulcrum of
metabolism. Redox Biol. 35(101454)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sies H: Oxidative stress: A concept in
redox biology and medicine. Redox Biol. 4:180–183. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kierans SJ and Taylor CT: Glycolysis: A
multifaceted metabolic pathway and signaling hub. J Biol Chem.
300(107906)2024.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Xiao YL, Zhang YW, Xie KD, Huang XL, Liu
XZ, Luo JJ and Tan SW: Mitochondrial dysfunction by FADDosome
promotes gastric mucosal injury in portal hypertensive gastropathy.
Int J Biol Sci. 20:2658–2685. 2024.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tan S, Chen X, Xu M, Huang X, Liu H, Jiang
J, Lu Y, Peng X and Wu B: PGE2/EP4 receptor attenuated mucosal
injury via β-arrestin1/Src/EGFR-mediated proliferation in portal
hypertensive gastropathy. Br J Pharmacol. 174:848–866.
2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Xiao YL, Liu XZ, Xie KD, Luo JJ, Zhang YW,
Huang XL, Luo JN and Tan SW: Mitochondrial dysfunction induced by
HIF-1α under hypoxia contributes to the development of gastric
mucosal lesions. Clin Transl Med. 14(e1653)2024.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tan S, Li L, Chen T, Chen X, Tao L, Lin X,
Tao J, Huang X, Jiang J, Liu H and Wu B: β-Arrestin-1 protects
against endoplasmic reticulum stress/p53-upregulated modulator of
apoptosis-mediated apoptosis via repressing p-p65/inducible nitric
oxide synthase in portal hypertensive gastropathy. Free Radic Biol
Med. 87:69–83. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Gana JC, Cifuentes LI, Gattini D,
Villarroel Del Pino LA, Peña A and Torres-Robles R: Band ligation
versus beta-blockers for primary prophylaxis of oesophageal
variceal bleeding in children with chronic liver disease or portal
vein thrombosis. Cochrane Database Syst Rev.
9(CD010546)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Noto JM, Piazuelo MB, Romero-Gallo J,
Delgado AG, Suarez G, Akritidou K, Girod Hoffman M, Roa JC, Taylor
CT and Peek RM Jr: Targeting hypoxia-inducible factor-1 alpha
suppresses Helicobacter pylori-induced gastric injury via
attenuation of both cag-mediated microbial virulence and
proinflammatory host responses. Gut Microbes.
15(2263936)2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lin ZH, Song JL, Gao YK, Huang SH, Dou RZ,
Zhong PY, Huang GQ, Han L, Zheng JS, Zhang XY, et al:
Hypoxia-induced HIF-1α/lncRNA-PMAN inhibits ferroptosis by
promoting the cytoplasmic translocation of ELAVL1 in peritoneal
dissemination from gastric cancer. Redox Biol.
52(102312)2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
He C, Wang LB, Zhang JT and Xu H:
Hypoxia-inducible microRNA-224 promotes the cell growth, migration
and invasion by directly targeting RASSF8 in gastric cancer. Mol
Cancer. 16(35)2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang YF, Lu HW, Ji H and Li YM: p53
upregulated by HIF-1α promotes gastric mucosal epithelial cells
apoptosis in portal hypertensive gastropathy. Dig Liver Dis.
55:81–92. 2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Winter JM, Yadav T and Rutter J: Stressed
to death: Mitochondrial stress responses connect respiration and
apoptosis in cancer. Mol Cell. 82:3321–3332. 2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Fuhrmann DC and Brüne B: Mitochondrial
composition and function under the control of hypoxia. Redox Biol.
12:208–215. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang BY, Pan CY, Feng C, Yan CQ, Yu YJ,
Chen ZL, Guo CJ and Wang XX: Role of mitochondrial reactive oxygen
species in homeostasis regulation. Redox Rep. 27:45–52.
2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yang SG, Bae JW, Park HJ and Koo DB:
Mito-TEMPO protects preimplantation porcine embryos against
mitochondrial fission-driven apoptosis through DRP1/PINK1-mediated
mitophagy. Life Sci. 315(121333)2023.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Shetty S, Kumar R and Bharati S:
Mito-TEMPO, a mitochondria-targeted antioxidant, prevents
N-nitrosodiethylamine-induced hepatocarcinogenesis in mice. Free
Radic Biol Med. 136:76–86. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Liu X, Wang X, Zhang J, Lam EK, Shin VY,
Cheng AS, Yu J, Chan FK, Sung JJ and Jin HC: Warburg effect
revisited: An epigenetic link between glycolysis and gastric
carcinogenesis. Oncogene. 29:442–450. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Cheng CT, Kuo CY, Ouyang C, Li CF, Chung
Y, Chan DC, Kung HJ and Ann DK: Metabolic Stress-induced
phosphorylation of KAP1 Ser473 blocks mitochondrial fusion in
breast cancer cells. Cancer Res. 76:5006–5018. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yi Y, Wu MY, Chen KT, Chen AH, Li LQ,
Xiong Q, Wang XR, Lei WB, Xiong GX and Fang SB: LDHA-mediated
glycolysis in stria vascularis endothelial cells regulates
macrophages function through CX3CL1-CX3CR1 pathway in noise-induced
oxidative stress. Cell Death Dis. 16(65)2025.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kooshan Z, Cárdenas-Piedra L, Clements J
and Batra J: Glycolysis, the sweet appetite of the tumor
microenvironment. Cancer Lett. 600(217156)2024.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang YJ, Chang LF, Xin X, Qiao YX, Qiao
WN, Ping JH, Xia J and Su J: Influenza A virus-induced glycolysis
facilitates virus replication by activating ROS/HIF-1α pathway.
Free Radic Biol Med. 250:910–924. 2024.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Qin Q, Li MY, Fan LL, Zeng X, Zheng DY,
Wang H, Jiang YT, Ma XR, Xing L, Wu LJ, et al: RVG engineered
extracellular vesicles-transmitted miR-137 improves autism by
modulating glucose metabolism and neuroinflammation. Mol
Psychiatry. 30:4072–4084. 2025.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yin H, Liu Y, Dong Q, Wang HY, Yan YJ,
Wang XQ, Wan XQ, Yuan GQ and Pan YW: The mechanism of extracellular
Cyp B promotes glioblastoma adaptation to glutamine deprivation
microenvironment. Cancer Lett. 597(216862)2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Rong J, Han CX, Huang Y, Wang YQ, Qiu Q,
Wang M, Wang SS, Wang R, Yang JQ, Li X, et al: Inhibition of
xanthine oxidase alleviated pancreatic necrosis via HIF-1
α-regulated LDHA and NLRP3 signaling pathway in acute pancreatitis.
Acta Pharm Sin B. 14:3591–3604. 2024.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Chelakkot C, Chelakkot VS, Shin Y and Song
K: Modulating glycolysis to improve cancer therapy. Int J Mol Sci.
24(2606)2023.PubMed/NCBI View Article : Google Scholar
|
|
35
|
An S, Yao Y, Hu HB, Wu JJ, Li JX, Li LL,
Wu J, Sun MM, Deng ZY, Zhang YY, et al: PDHA1
hyperacetylation-mediated lactate overproduction promotes
sepsis-induced acute kidney injury via Fis1 lactylation. Cell Death
Dis. 14(457)2023.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Rabinowitz JD and Enerbäck S: Lactate: The
ugly duckling of energy metabolism. Nat Metab. 2:566–571.
2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Liu J, Zhang C, Zhang TL, Chang CY, Wang
JM, Bazile L, Zhang LJ, Haffty BG, Hu WW and Feng ZH: Metabolic
enzyme LDHA activates Rac1 GTPase as a noncanonical mechanism to
promote cancer. Nat Metab. 4:1830–1846. 2022.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Rabbani N, Xue M and Thornalley PJ:
Hexokinase-2-linked glycolytic overload and unscheduled
Glycolysis-driver of insulin resistance and development of vascular
complications of diabetes. Int J Mol Sci. 23(2165)2022.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Qi W, Keenan HA, Li Q, Ishikado A, Kannt
A, Sadowski T, Yorek MA, Wu I, Lockhart S, Coppey LJ, et al:
Pyruvate kinase M2 activation may protect against the progression
of diabetic glomerular pathology and mitochondrial dysfunction. Nat
Med. 23:753–762. 2017.PubMed/NCBI View Article : Google Scholar
|