Introduction
Interleukin-6 (IL-6) is a pleiotropic cytokine
integral to immune regulation, inflammatory signaling, cellular
proliferation, and survival (1,2). Upon
binding to its receptor complex, which includes gp130, IL-6
activates the Janus kinase (JAK)/signal transducer and activator of
transcription 3 (STAT3) signaling pathway, resulting in the
transcription of a wide array of target genes implicated in
inflammatory and oncogenic processes (3-7).
The activation of STAT3 is tightly regulated by phosphorylation at
two key residues, Tyrosine705 and Serine727. Specifically,
phosphorylation of STAT3 at Tyr705 is a critical event that
promotes STAT3 dimerization, nuclear translocation, and subsequent
transcriptional activation of downstream genes such as suppressor
of cytokine signaling 3 (SOCS3) and C-reactive protein (CRP), which
serve as important markers of IL-6-mediated responses. Aberrant or
sustained activation of the IL-6/STAT3 signaling axis has been
strongly associated with the pathogenesis of various chronic
inflammatory diseases, including rheumatoid arthritis (RA) and
inflammatory bowel disease (IBD), as well as malignancies such as
hepatocellular carcinoma, thus underscoring its significance as a
therapeutic target (8-10).
Given the pivotal role of IL-6/STAT3 signaling in
chronic inflammation and tumorigenesis, considerable research has
focused on identifying pharmacological inhibitors that target this
pathway. Natural products, particularly those derived from
medicinal plants, have emerged as valuable sources of bioactive
compounds capable of modulating cytokine signaling. Curcuma
longa L. (Zingiberaceae), commonly known as turmeric, has long
been used in traditional Asian medicine and is well known for its
immunoregulatory, antioxidant, antihepatotoxic,
hypocholesterolemic, and anticancer properties. These effects are
primarily attributed to curcuminoids such as curcumin,
demethoxycurcumin, and bisdemethoxycurcumin, which are abundant in
the rhizome (11-16).
While the pharmacological activities of turmeric rhizome have been
extensively investigated, the aerial parts of the plant, including
the leaves, remain largely underexplored, despite reports of their
antioxidant, antibacterial, and immuno-regulatory effects (17,18).
Recent studies have reported that C. longa
leaf extracts (CL-E) modulate inflammatory signaling and host
immunity. Phytochemical analyses of the aerial parts have
identified flavonoids, tannins, and polyphenols that underlie the
documented antioxidant and immunoregulatory activities (19-21).
Nevertheless, the molecular basis by which these constituents
affect cytokine-driven pathways remains unclear. In particular, it
is unknown whether, and how, CL-E influences the IL-6/STAT3
pathway-a central regulator of acute-phase responses, inflammation,
and tumorigenic signaling. Since STAT3 activation depends on
site-specific phosphorylation at Tyr705 and Ser727 that governs
dimerization, nuclear import, and transcriptional output, defining
the impact of CL-E on these regulatory nodes addresses this key
mechanistic gap.
In light of the ongoing need for novel natural
inhibitors of the IL-6/STAT3 axis, we utilized a STAT3 luciferase
reporter system to evaluate the effects of the ethyl acetate
fraction of C. longa leaves on STAT3 activation, including
downstream events such as STAT3 phosphorylation, nuclear
translocation, and the expression of IL-6-responsive genes such as
SOCS3 and CRP. To further elucidate the upstream regulatory
mechanisms, we investigated the involvement of the MEK-ERK pathway
employing specific pharmacological inhibitors, thereby
characterizing the signaling cascade implicated in the CL-E
mediated inhibitory effects on STAT3. Accumulating evidence
suggests that protein kinase C (PKC) can activate the Raf-MEK-ERK
cascade and that ERK-driven Ser727 phosphorylation can modulate- or
in some contexts antagonize-Tyr705-dependent STAT3 activation
(9). We therefore hypothesized that
the ethyl acetate fraction of C. longa leaves (CL-E)
attenuates IL-6/STAT3 signaling by engaging a PKC-MEK/ERK axis and
shifting the Tyr705/Ser727 phosphorylation balance.
Collectively, our findings provide novel insights
into the molecular mechanisms by which CL-E modulates IL-6/STAT3
signaling and suggest its potential as a natural therapeutic agent
for inflammation-related disorders.
Materials and methods
Reagents and chemicals
Recombinant human IL-6 was procured from R&D
Systems. Mouse anti-phospho STAT3 (Tyr705) IgG was obtained from
Calbiochem. Rabbit anti-phospho Stat3 (Ser727) IgG, anti-total
STAT3 IgG, anti-phospho ERK1/2 IgG, anti-total ERK1/2 IgG, β-actin
and secondary antibody were purchased from Cell Signaling
Technology. All other reagents, including genistein, U0126, and
bisindolylmaleimide II, were acquired from Sigma-Aldrich.
Preparation of crude extracts
Aerial parts of C. longa L. were provided by
the Rural Development Administration (RDA) of Korea. The plant
materials were extracted using methanol solvents (5 lx2 times).
Methanol extracts (1 g) were suspended in 50 ml of distilled water
and successively partitioned with ethyl acetate (100 ml x 3),
yielding the ethyl acetate-soluble fraction (227 mg, CL-E).
Cell line and cell culture
Human hepatoma Hep3B cells were obtained from the
American Type Culture Collection (ATCC; no. HB-8064) and were
maintained in DMEM supplemented with 10% fetal bovine serum, 50
U/ml penicillin, and 50 mg/ml streptomycin at 37˚C in a humidified
5% CO2 incubator. All cell culture reagents were
obtained from GibcoBRL (Life Technologies).
Establishment of stable cell line
expressing pStat3-Luc
As previously described, a stable Hep3B cell line
expressing a STAT3-responsive luciferase reporter (pStat3-Luc) was
established (22). Briefly, Hep3B
cells were co-transfected with pStat3-Luc (containing a
STAT3-binding site) and pcDNA3.1(+) carrying a hygromycin
resistance gene (Clontech Laboratories) using Lipofectamine Plus
(Invitrogen). After 48 h, cells were selected with 100 µg/ml
hygromycin, and stable clones were expanded. Luciferase expression
was confirmed by luciferase assay.
Luciferase assay
Stable pStat3-Luc-expressing Hep3B cells were seeded
in 96-well plates at a density of 2x104 cells/well.
After 24 h, the cells were serum-starved for 12 h and subsequently
treated with IL-6 (10 ng/ml) for 12 h in the presence or absence of
test compounds. Luciferase activity was quantified using a
commercial kit (Promega) according to the manufacturer's
instructions.
Cell viability
Hep3B cells were seeded at a density of
1x104 cells/well and incubated for 24 h. The medium was
then replaced with serum-free DMEM containing varying
concentrations of test samples. Following 48 h of treatment, MTT
solution (0.5 mg/ml) was added to each well and incubated for 4 h.
Subsequently, 100 µl of DMSO was added to dissolve the formazan
crystals. Absorbance was measured at 540 nm using a microplate
reader, and cell viability was calculated relative to the untreated
control.
Western blot analysis
Total protein lysates were prepared and analyzed
using Western blot as previously described (22). Briefly, Cells were lysed in Cell
Lysis Buffer (Cell Signaling Technology) supplemented with a
protease and phosphatase inhibitor cocktail (Thermo Scientific).
Protein concentrations were determined using the DC protein assay
(Bio-Rad). Equal amount of whole cell lysates (20-40 µg per lane)
were separated by SDS-PAGE on 4-12% gradient gels and transferred
to PVDF membranes (Amersham Bioscience). Membranes were blocked
with 5% (w/v) non-fat dry milk in TBS-T (20 mM Tris-HCl pH 7.5, 150
mM NaCl, 0.1% Tween-20) for 1 h at room temperature, then incubated
overnight at 4˚C with primary antibodies against p-STAT3 (Tyr705;
1:1,000; CST #9145), p-STAT3 (Ser727; 1:1,000; CST #9134), t-STAT3
(1:1,000; CST #4904), p-ERK (1:1,000; CST #4370), t-ERK (1:1,000;
CST #4695) and β-actin (1:2,000; CST #4967). After washing in
TBS-T, membranes were incubated with the appropriate horseradish
peroxidase-conjugated secondary antibody (1:2,000; CST #7074) for
30 min. Signals were developed using an enhanced chemiluminescence
(ECL) substrate (West-ZOL Plus kit; iNtRON Biotechnology) and
recorded on X-ray film (Eastman Kodak Co.). All blots were
reproduced in triplicate experiments.
Immunocytochemistry for STAT3
localization
Hep3B cells were cultured on 8-well Nunc Lab-Tek
chamber slides and treated with IL-6 (10 ng/ml) for 4 h, in the
presence or absence of CL-E. Cells were fixed with 4%
paraformaldehyde for 30 min, permeabilized with 100% cold methanol
for 5 min, and blocked with 5% BSA for 30 min. Cells were incubated
overnight at 4˚C with rabbit anti-STAT3 antibody (1:200), followed
by FITC-conjugated goat anti-rabbit IgG (1:1,000) for 1 h. Slides
were mounted using SlowFade Gold antifade reagent (Invitrogen), and
fluorescence images were acquired using a Leica DM5000B microscope
(Leica).
RNA isolation, cDNA synthesis, and
real-time RT-PCR
Total RNA was extracted from Hep3B cells using the
RNeasy MinElute Cleanup Kit (Qiagen), including on-column DNase
treatment. RNA concentration and quality were assessed using an
Agilent 2100 Bioanalyzer (Agilent Technologies). cDNA was
synthesized using the TaqMan Reverse Transcription Reagents Kit
(Applied Biosystems), and real-time PCR was performed using a
StepOnePlus Real-Time PCR System (Applied Biosystems). TaqMan
primers for human CRP (Hs04183452_g1) and SOCS3 (Hs02330328_s1)
were obtained from Applied Biosystems. 18S rRNA (Hs99999901_s1) was
used as an endogenous control. Data were analyzed using the
2-ΔΔCT method, and gene expression levels were presented
relative to those of untreated controls.
Data analysis
All experiments were performed in triplicate. Data
are presented as the mean ± standard error (SE). Statistical
analyses were conducted using GraphPad Prism 5 (GraphPad software
Inc.). One-way ANOVA followed by Tukey's post hoc test was used,
and differences were considered statistically significant at
P<0.05, P<0.01, and P<0.001.
Results
CL-E inhibits IL-6-induced STAT3
transcriptional activity without cytotoxicity
To explore natural products that inhibit
IL-6-induced activation, a luciferase reporter assay was initially
conducted using Hep3B cells stably transfected with a
STAT3-responsive luciferase construct (pSTAT3-Luc) for screening
purposes. During preliminary screening, methanol extracts of C.
longa L leaves were found to suppress IL-6-stimulated
STAT3-dependent activity (data not shown). Based on this
observation, the methanol extract was subsequently partitioned into
an ethyl acetate fraction (CL-E) and an aqueous fraction to isolate
the active component. Both fractions were evaluated for their
ability to inhibit IL-6-induced STAT3-dependent transcriptional
activity. Cells were stimulated with IL-6 (10 ng/ml) for 12 h in
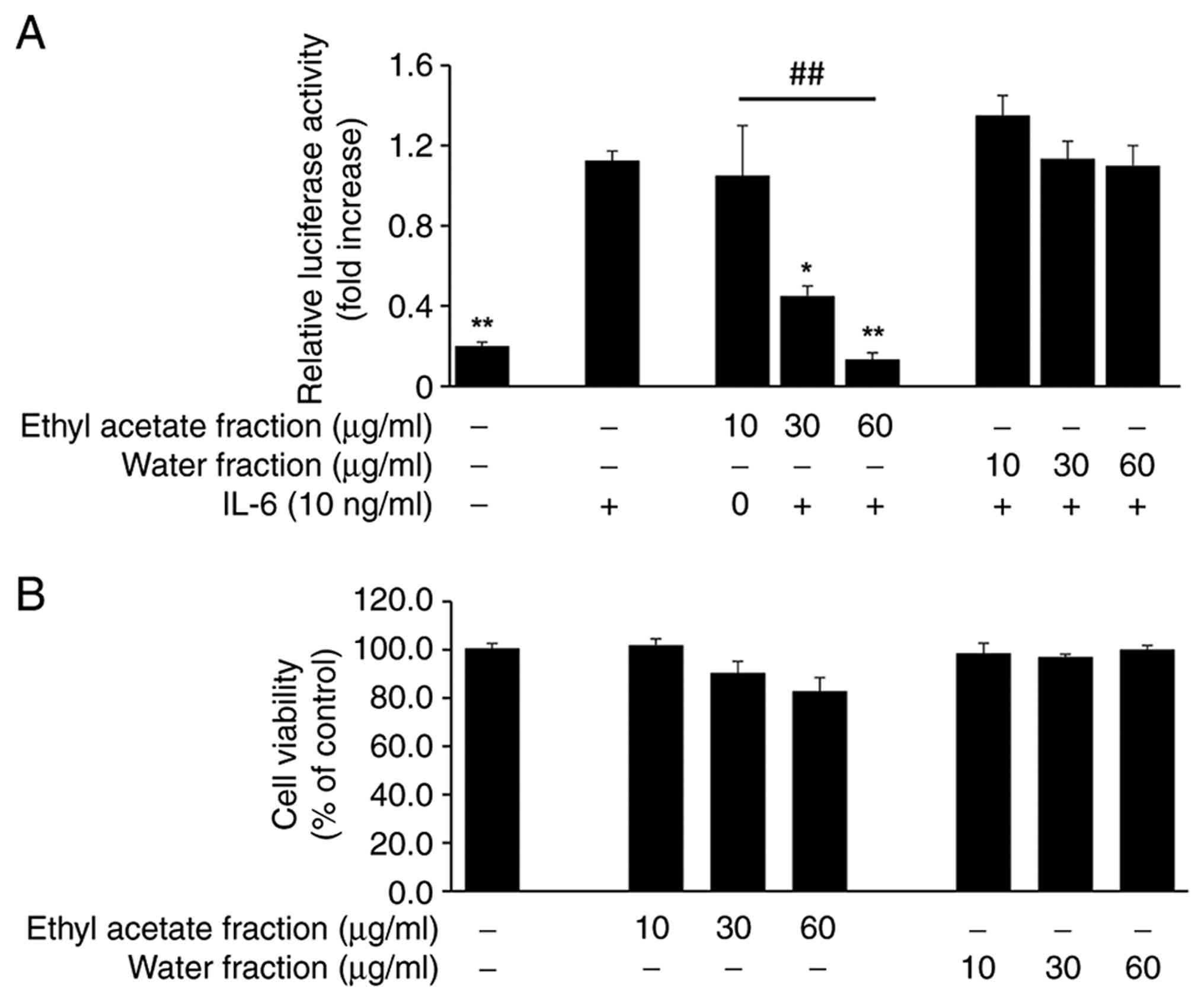
the presence or absence of each fraction. As illustrated in
Fig. 1A, CL-E significantly
inhibited IL-6-induced luciferase activity in a dose-dependent
manner (IC50: 33.9±3.7 µg/ml), with a significant
difference between the 10 and 60 µg/ml concentrations, whereas the
aqueous fraction exhibited no significant inhibitory effect.
Importantly, MTT assay results demonstrated that CL-E treatment up
to 60 µg/ml did not induce cytotoxicity under the experimental
conditions (Fig. 1B), indicating
that the observed inhibition of STAT3 activity was not attributable
to reduced cell viability.
CL-E suppresses STAT3 Tyr705
phosphorylation and nuclear translocation in IL-6-stimulated
cells
To elucidate the mechanism underlying the inhibitory
effect of CL-E on STAT3 transcriptional activity, we investigated
its impact on STAT3 phosphorylation at two key regulatory residues,
Tyr705 and Ser727, which are essential for STAT3 activation and
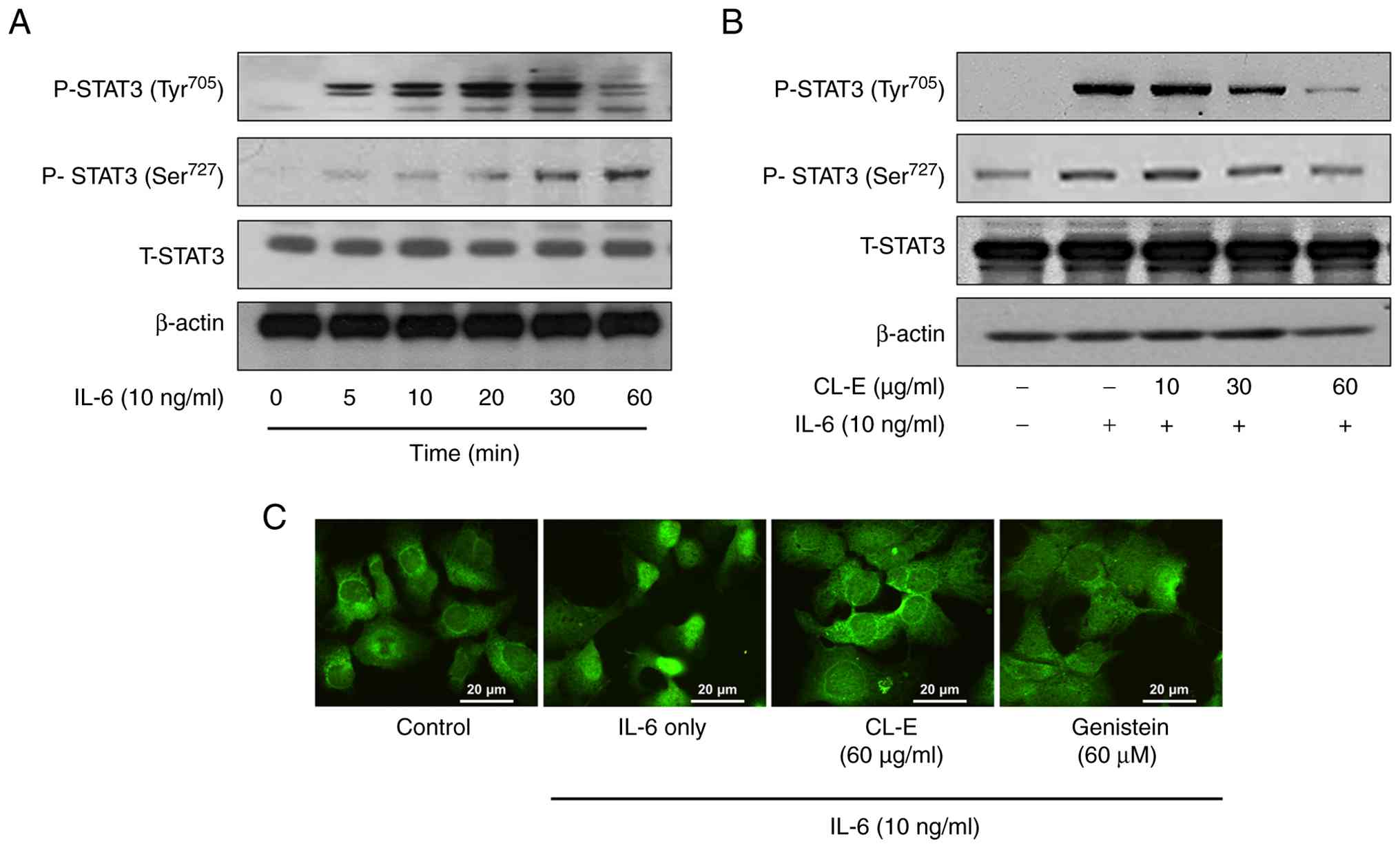
function. Initially, a time-course analysis of STAT3
phosphorylation following IL-6 stimulation was conducted. Hep3B
cells were treated with IL-6 (10 ng/ml) for varying durations (0,
5, 10, 20, 30, and 60 min), and cell lysates underwent Western blot
analysis using phospho-specific antibodies against STAT3
phosphorylated at Tyr705 and Ser727, as well as total STAT3. As
depicted in Fig. 2A, IL-6 treatment
resulted in rapid, robust phosphorylation of STAT3 at Tyr705,
peaking at approximately 20-30 min and declining thereafter. STAT3
phosphorylation at Ser727 also increased in response to IL-6 but
appeared later and with less intensity compared to that at Tyr705.
These results confirm activation of the canonical IL-6/STAT3
signaling pathway in Hep3B cells. To determine whether CL-E
treatment affects IL-6-stimulated STAT3 phosphorylation, Hep3B
cells were pretreated with CL-E (10, 30, and 60 µg/ml) for 1 h,
followed by IL-6 stimulation (10 ng/ml for 30 min).
As illustrated in Fig.
2B, CL-E strongly inhibited IL-6-induced STAT3 Tyr705
phosphorylation in a dose-dependent manner, with maximal
suppression at 60 µg/ml. Conversely, CL-E exerted no inhibitory
effect on IL-6-stimulated STAT3 Ser727 phosphorylation. Total STAT3
levels remained constant, indicating that the observed effects were
not due to changes in STAT3 protein expression. Give that
phosphorylation at STAT3 Tyr705 promotes STAT3 dimerization and
nuclear translocation, further examination of whether CL-E affects
STAT3 nuclear localization was conducted using immunofluorescence
microscopy. Hep3B cells cultured on chamber slides were pretreated
with CL-E (60 µg/ml) or genistein [60 µM, a known STAT3 inhibitor
(23)] for 1 h, followed by IL-6
stimulation for 4 h. Cells were fixed, permeabilized, and stained
with an anti-STAT3 antibody, followed by FITC-conjugated secondary
antibody. In cells exposed to IL-6, STAT3 was predominantly
localized within the nucleus, indicative of its activated state.
Conversely, in cells pretreated with CL-E or genistein, STAT3 was
primarily cytoplasmic (Fig. 2C),
suggesting that CL-E effectively inhibits STAT3 nuclear
translocation, likely by suppressing Tyr705 phosphorylation.
These findings collectively demonstrate that CL-E
inhibits IL-6/STAT3 signaling by suppressing STAT3 Tyr705
phosphorylation and preventing its nuclear translocation, thereby
reducing its transcriptional activity.
CL-E enhances ERK1/2 phosphorylation
and modulates STAT3 activation via the ERK signaling pathway
Previous studies have indicated that the activation
of ERK1/2 can modulate IL-6-induced STAT3 activation (24-26).
To investigate this possibility, we examined the involvement of the
ERK1/2 pathway, known to regulate STAT3 phosphorylation at Ser727.
Initially, we assessed ERK1/2 phosphorylation following IL-6
stimulation over time. Hep3B cells were treated with IL-6 (10
ng/ml) for 0, 5, 10, 30, and 60 min, and ERK1/2 phosphorylation was
analyzed via Western blotting. As depicted in Fig. 3A, ERK1/2 phosphorylation remained
low for the initial 10 min, began to increase at 30 min, and peaked
at 60 min. Subsequently, we investigated whether ERK1/2
phosphorylation was affected by CL-E treatment in the presence of
IL-6. Hep3B cells were pretreated with CL-E at concentrations of
10, 30, and 60 µg/ml for 1 h prior to IL-6 stimulation (10 ng/ml
for 30 min).
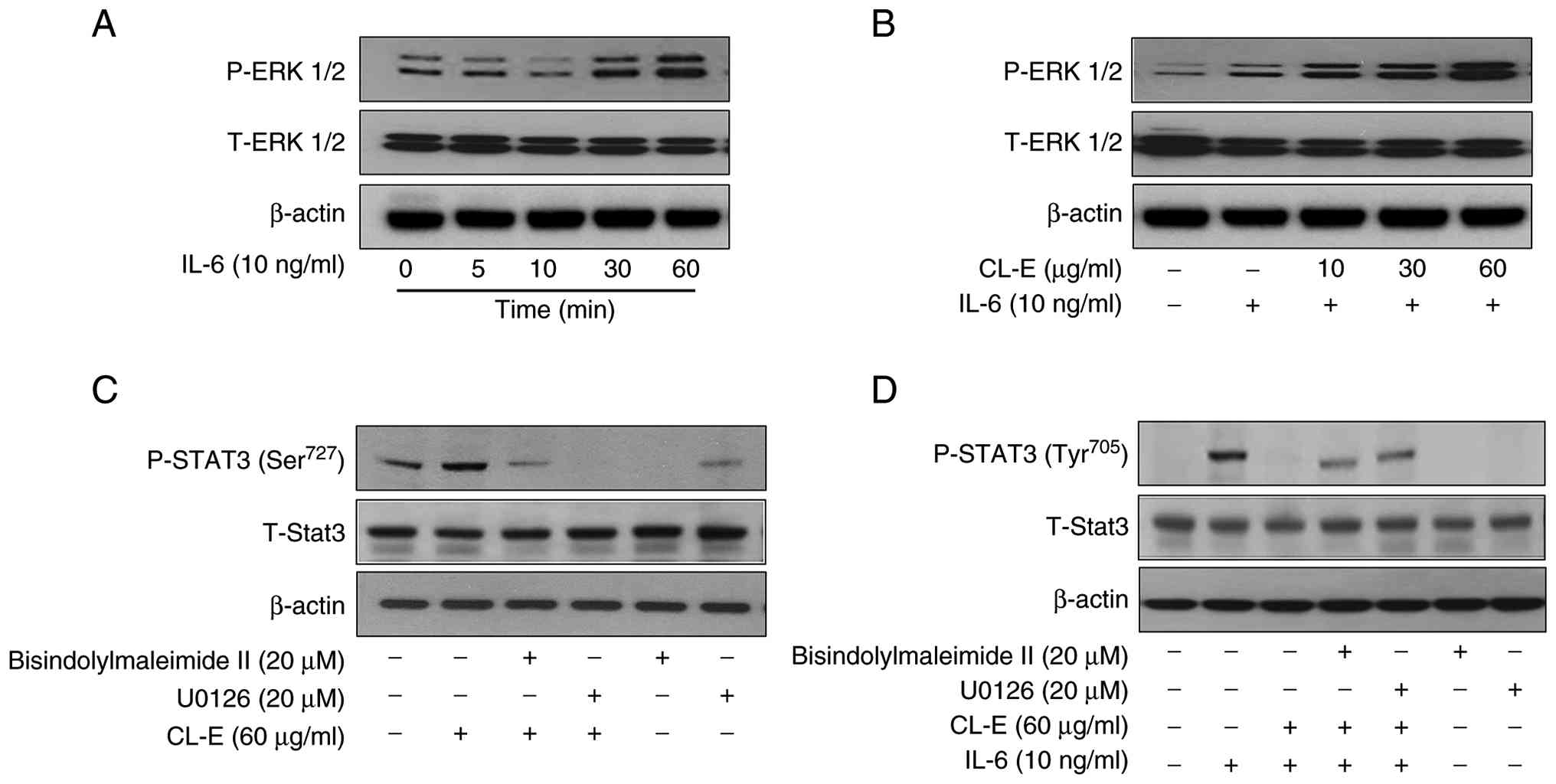 | Figure 3ERK signaling pathway contributes to
CL-E-mediated regulation of IL-6-induced STAT3 activation. (A)
Hep3B cells were stimulated with IL-6 (10 ng/ml) for the indicated
times (0, 5, 10, 30 and 60 min). Protein lysates underwent western
blotting analysis using antibodies against P-ERK1/2 and total
ERK1/2 to evaluate time-dependent ERK1/2 activation. (B) Cells were
pretreated with CL-E at 10, 30 and 60 µg/ml for 1 h, followed by
IL-6 stimulation (10 ng/ml) for 30 min. ERK1/2 phosphorylation was
analyzed by western blotting analysis. (C) To investigate the role
of ERK in STAT3 Ser727 phosphorylation, cells were treated with
CL-E (60 µg/ml), PKC inhibitor bisindolylmaleimide II (20 µM),
and/or the MEK1/2 inhibitor U0126 (20 µM) for 1 h in the absence of
IL-6. Phosphorylation of STAT3 (Ser727) and total STAT3 were
examined by western blotting analysis. (D) To determine whether ERK
activation contributes to regulation of STAT3 Tyr705
phosphorylation, cells were pretreated with CL-E, U0126 and/or
bisindolylmaleimide II (20 µM) for 1 h before IL-6 stimulation (10
ng/ml, 30 min). STAT3 Tyr705 phosphorylation was analyzed by
western blotting. CL-E, Curcuma longa L. extract; IL-6,
interleukin 6; STAT3, signal transducer and activator of
transcription 3; P-, phosphorylated. |
As shown in Fig. 3B,
CL-E significantly enhanced IL-6-induced ERK1/2 phosphorylation in
a concentration-dependent manner, with the strongest effect
observed at 60 µg/ml, suggesting that CL-E potentiates ERK
activation under IL-6 stimulation. To determine whether ERK
activation contributes to the regulation of STAT3 phosphorylation,
we assessed STAT3 Ser727 phosphorylation in the absence of IL-6.
Hep3B cells were treated with CL-E (60 µg/ml), either alone or in
combination with the MEK1/2 inhibitor U0126 or the PKC inhibitor
bisindolylmaleimide II. As shown in Fig. 3C, CL-E alone markedly increased
STAT3 Ser727 phosphorylation. However, this effect was suppressed
by co-treatment with U0126, suggesting that CL-E-induced Ser727
phosphorylation is mediated through the MEK/ERK pathway.
Interestingly, bisindolylmaleimide II also attenuated Ser727
phosphorylation, indicating a possible role for PKC upstream of ERK
in mediating the effect of CL-E.
Finally, to further ascertain whether ERK- and
PKC-dependent signaling pathways are involved in the regulation of
STAT3 Tyr705 phosphorylation, we examined the effects of these
inhibitors under IL-6 stimulation. As depicted in Fig. 3D, CL-E pretreatment suppressed
IL-6-induced STAT3 Tyr705 phosphorylation, an effect that was
reversed by U0126. Similarly, co-treatment with bisindolylmaleimide
II partially restored Tyr705 phosphorylation, further corroborating
the involvement of PKC-ERK signaling in the modulation of
IL-6/STAT3 activation by CL-E.
CL-E suppresses IL-6-induced
expression of CRP and SOCS, downstream targets of STAT3
signaling
CL-E suppresses IL-6-induced expression of CRP and
Suppressor of cytokine signaling 3 (SOCS3), downstream targets of
STAT3 signaling. CRP is primarily produced by hepatocytes in
response to IL-6 stimulation and can be transcriptionally induced
via the JAK/STAT3 pathway or a redox-sensitive NF-κB pathway
mediated by Rac1 activation (27-29).
SOCS3 acts as a classical negative feedback regulator of
IL-6-induced JAK/STAT3 signaling by binding to phosphorylated
tyrosine residues on JAK kinases (9).
Given that SOCS3 is a direct transcriptional target
of STAT3, the inhibition of STAT3 signaling results in decreased
SOCS3 expression. To assess the downstream effects of CL-E-mediated
STAT3 inhibition, we examined the expression of
inflammation-related target genes regulated by IL-6/STAT3
signaling. We focused on CRP and SOCS3, both recognized as STAT3
target genes, serving as indicators of the inflammatory response
and negative feedback regulation, respectively (30). Hep3B cells were pretreated with CL-E
(10, 30, and 60 µg/ml) for 1 h, followed by IL-6 stimulation (10
ng/ml) for 6 h. Total RNA was extracted, and mRNA levels of CRP and
SOCS3 were quantified via real-time PCR using 18S rRNA as the
internal control. As illustrated in Fig. 4, IL-6 treatment significantly
upregulated the expression of both CRP and SOCS3 mRNA levels
compared to those of untreated cells. Pretreatment with CL-E
significantly attenuated IL-6-induced expression of both genes, and
a concentration-dependent reduction was observed for SOCS-3.
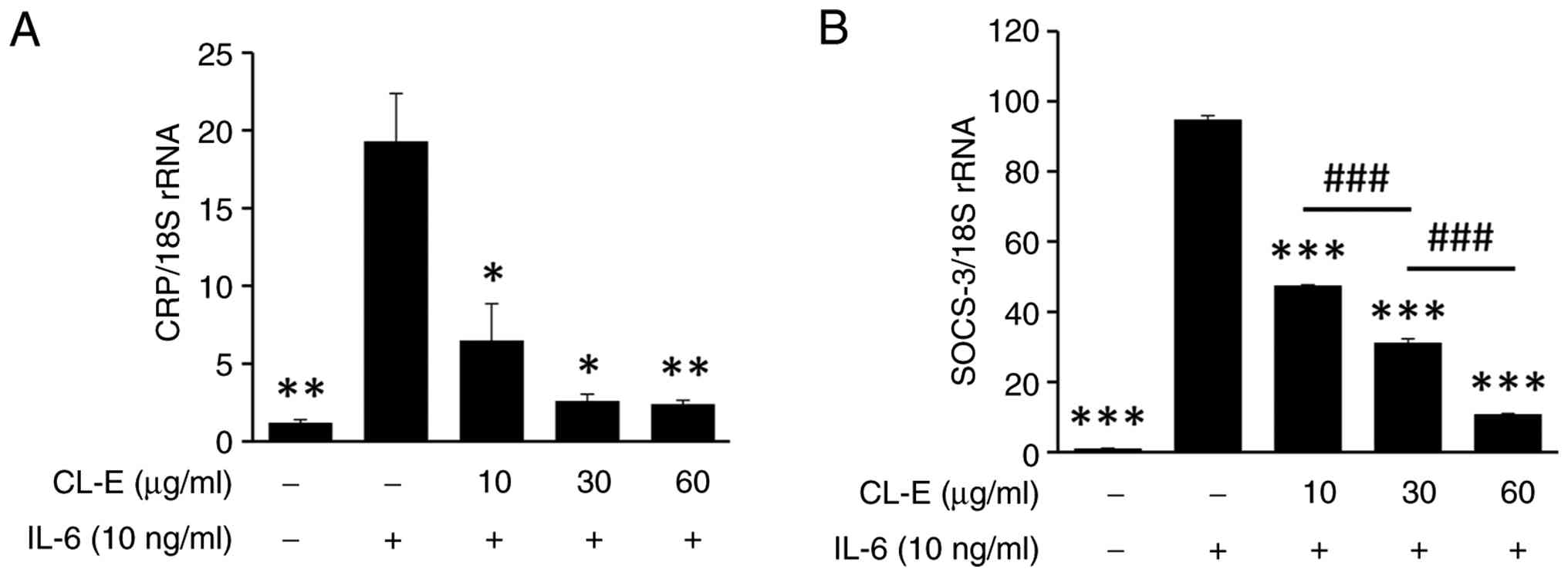 | Figure 4CL-E inhibits IL-6-induced expression
of CRP and SOCS3 mRNA in Hep3B cells. Hep3B cells were pretreated
with CL-E at concentrations of 10, 30, and 60 µg/ml for 1 h,
followed by IL-6 stimulation (10 ng/ml) for 6 h. Total RNA was
extracted, and the mRNA levels of (A) CRP and (B) SOCS3 were
measured using quantitative real-time PCR. Gene expression levels
were normalized to 18S rRNA and are presented as fold change
relative to the untreated control. Data represent the mean ± SE
(n≥3). *P<0.05, **P<0.01 and
***P<0.001 vs. the IL-6-treated group;
###P<0.001. CL-E, Curcuma longa L. extract;
IL-6, interleukin 6; CRP, C reactive protein; P-, phosphorylated;
SOCS3, suppressor of cytokine signaling 3. |
These findings indicate that CL-E effectively
suppresses IL-6/STAT3-dependent transcriptional activation of both
pro-inflammatory (CRP) and feedback inhibitor genes
(SOCS3). This transcriptional repression aligns with the
observed inhibition of STAT3 Tyr705 phosphorylation and nuclear
translocation by CL-E (Fig. 2),
further supporting its potential as an anti-inflammatory agent
targeting the IL-6/STAT3 signaling axis.
Discussion
In this study, we demonstrated that CL-E effectively
inhibits IL-6-induced STAT3 activation in Hep3B cells. CL-E
suppressed STAT3-dependent transcriptional activity in a
dose-dependent manner without inducing cytotoxicity.
Mechanistically, CL-E inhibited phosphorylation of STAT3 at Tyr705,
blocked its nuclear translocation, and reduced the expression of
STAT3 downstream target genes such as CRP and SOCS3.
Notably, CL-E enhanced ERK1/2 phosphorylation, which led to
increased STAT3 Ser727 phosphorylation. Pharmacological inhibition
of the ERK and PKC pathways reversed CL-E-mediated inhibition of
STAT3 Tyr705 phosphorylation, suggesting a regulatory crosstalk
between MAPK signaling and STAT3 activation. These findings are
consistent with previous reports on natural product-derived STAT3
inhibitors. For example, manassantin A and B from Saururus
chinensis, and Kansuinine A and B from Euphorbia kansui
L. have been shown to suppress IL-6-induced STAT3
phosphorylation and nuclear translocation by targeting both STAT3
Tyr705 and ERK1/2-mediated Ser727 phosphorylation (31,32).
In agreement with previous reports, CL-E also appears to exert dual
modulatory activity, suppressing canonical STAT3 activation via
STAT3 Tyr705 phosphorylation while activating ERK signaling,
possibly as part of a feedback or compensatory mechanism. These
results suggest that CL-E may serve as a promising natural
therapeutic candidate for targeting STAT3-mediated inflammation and
cancer progression. The differential regulation of STAT3
phosphorylation at Tyr705 and Ser727 by CL-E is particularly
noteworthy, given that these two phosphorylation sites serve
distinct roles in STAT3 signaling. Tyr705 phosphorylation is
critical for STAT3 dimerization, nuclear translocation, and DNA
binding, and is typically considered a hallmark of canonical STAT3
activation in response to cytokines such as IL-6 (27,33).
In contrast, Ser727 phosphorylation, which is often mediated by
MAPK or PKC signaling, contributes to the modulation of STAT3's
transcriptional activity, interaction with co-activators, and in
certain contexts, non-canonical functions (34). To further investigate the modulation
of STAT3 signaling by CL-E, we examined previous evidence
implicating the ERK and PKC pathways in IL-6 signal transduction.
It has been documented that PKC activation by phorbol esters, such
as PMA, can influence IL-6 signaling through ERK activation
(24). While ERK signaling has been
shown to synergize with the JAK/STAT pathway in enhancing
cytokine-induced gene expression in certain contexts (35), other studies suggest that ERK
activation can also antagonize STAT3 signaling, contingent upon the
cellular environment (34,36,37).
Notably, SOCS3, a well-known negative feedback regulator of the
JAK/STAT pathway, has been reported to be upregulated by PMA via
ERK activation, and this induction was inhibited by MAPK inhibitors
(38). Based on these findings, we
hypothesized that the inhibitory effect of CL-E on STAT3 activation
may involve the activation of PKC and ERK1/2. Indeed, our results
demonstrated that CL-E significantly induced ERK1/2
phosphorylation, which was abolished by the MEK1/2 inhibitor U0126,
indicating that the effect is dependent on the MAPK/ERK
pathway.
Furthermore, CL-E enhanced STAT3 Ser727
phosphorylation even in the absence of IL-6, and this effect was
diminished by co-treatment with the PKC inhibitor
bisindolylmaleimide II, suggesting that PKC activity contributes to
this mechanism. Given previous reports that ERK-mediated STAT3
Ser727 phosphorylation can interfere with STAT3 Tyr705
phosphorylation (34,36,37),
our findings support a model in which CL-E-mediated ERK activation
leads to a shift in STAT3 phosphorylation balance, thereby
attenuating its activation. Collectively, these results highlight
ERK and PKC signaling as key modulators involved in the suppression
of IL-6/STAT3 signaling by CL-E. The ability of CL-E to suppress
IL-6-induced expression of CRP and SOCS3 further supports its
anti-inflammatory potential. CRP is a classical acute-phase protein
synthesized in hepatocytes under IL-6/STAT3 regulation, and
elevated CRP levels are widely used as biomarkers for systemic
inflammation, cardiovascular risk, and cancer prognosis (39,40).
In our study, CL-E treatment markedly reduced CRP mRNA expression
in IL-6-stimulated Hep3B cells, suggesting that CL-E may attenuate
systemic inflammatory responses at the transcriptional level.
SOCS3, another STAT3 target gene, acts as a negative
feedback regulator by binding to JAKs and preventing further
cytokine signaling. Although SOCS3 is classically viewed as an
inhibitor of STAT3 signaling, paradoxically, its overexpression in
chronic inflammatory states can desensitize anti-inflammatory
signals and contribute to persistent disease persistence (41,42).
Our finding that CL-E downregulates SOCS3 expression suggests that
it may restore homeostatic feedback mechanisms by reprogramming the
inflammatory environment. These findings are consistent with
previous studies on flavonoids and diarylheptanoids derived from
natural products, which have demonstrated suppression of
IL-6-stimulated inflammatory markers and STAT3 target genes
(30,43). Collectively, our results highlight
the therapeutic potential of CL-E as a botanical modulator of
inflammatory signaling, particularly in diseases characterized by
hyperactivation of the IL-6/STAT3 axis, such as rheumatoid
arthritis, inflammatory bowel disease, metabolic syndrome, and
hepatocellular carcinoma. The rhizome of C. longa has been
extensively studied and commercialized for its curcuminoid content
and associated pharmacological activities, including
anti-inflammatory, antioxidant, and anticancer effects (8,44,45).
In contrast, the aerial parts, particularly the
leaves, have been relatively understudied in scientific research.
Nonetheless, recent investigations have begun to reveal that C.
longa leaves contain bioactive compounds such as polyphenols,
flavonoids, and tannins, which exhibit antioxidant and
antimicrobial properties (19-21).
The present study further corroborates the potential of CL-E as an
effective modulator of IL-6/STAT3 signaling. Notably, CL-E was able
to modulate the STAT3 signaling pathway without inducing
cytotoxicity, underscoring its potential as a safe, mechanism-based
botanical regulator of inflammatory responses. Furthermore, our
data suggest that CL-E operates through ERK- and PKC-dependent
pathways, offering new insights into how its phytochemicals may
regulate the IL-6 signaling cascade. Future research should focus
on isolating and structurally characterizing the active
constituents of CL-E, and on evaluating their in vivo
efficacy in various IL-6–driven animal models of rheumatoid
arthritis and inflammatory bowel disease, such as collagen-induced
arthritis (RA) and DSS-induced colitis (IBD). Additionally,
comparative studies with turmeric rhizome extracts, regarding
bioactivity and chemical composition, may elucidate whether CL-E
offers distinct advantages in terms of potency, selectivity, or
safety. From a resource utilization perspective, the use of C.
longa aerial parts, which are often discarded during
cultivation, could contribute to more sustainable and comprehensive
utilization of this medicinal plant.
Acknowledgements
The authors would like to acknowledge the late Dr
Jong Sun Chang, formerly with the Eco-friendly Biomaterial Research
Center, Korea Research Institute of Bioscience and Biotechnology,
Jeongeup, Republic of Korea, for her valuable contributions and
unwavering support to this work. This study is respectfully
dedicated to her memory.
Funding
Funding: This research was supported by the Regional Innovation
System & Education (RISE) program through the Daejeon RISE
Center, funded by the Ministry of Education and the Daejeon
Metropolitan City, Republic of Korea (grant no. 2025-RISE-06-013);
the Korea Research Institute of Bioscience and Biotechnology
Research Initiative Program (grant no. KGM1052612) and
Global-Learning & Academic Research Institution for Master's
and PhD students, the Postdocs Program of the National Research
Foundation of Korea grant funded by the Ministry of Education
(grant no. RS-2023-00301914); and an NRF grant funded by the Korean
Government (grant no. RS-2024-00338822).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SWL and SL conceived and designed the study. HJJ,
EJP and YK performed the experiments and analyzed the data. EP and
WK contributed to the design of the experiments, and to the
analysis and interpretation of the data, and, together with SWL and
SL, supervised the overall research. HJJ and YK wrote the original
manuscript. EP and WK, together with SWL and SL, revised the
manuscript critically for important intellectual content and
confirmed the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Strassmann G, Masui Y, Chizzonite R and
Fong M: Mechanisms of experimental cancer cachexia. Local
involvement of IL-1 in colon-26 tumor. J Immunol. 150:2341–2345.
1993.PubMed/NCBI
|
|
2
|
Kishimoto T: Interleukin-6: From basic
science to medicine-40 years in immunology. Annu Rev Immunol.
23:1–21. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lütticken C, Wegenka UM, Yuan J, Buschmann
J, Schindler C, Ziemiecki A, Harpur AG, Wilks AF, Yasukawa K, Taga
T, et al: Association of transcription factor APRF and protein
kinase Jak1 with the interleukin-6 signal transducer gp130.
Science. 263:89–92. 1994.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ihle JN: STATs: Signal transducers and
activators of transcription. Cell. 84:331–334. 1996.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sebti SM and Der CJ: Opinion: Searching
for the elusive targets of farnesyltransferase inhibitors. Nat Rev
Cancer. 3:945–951. 2003.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Darnell JE Jr: STATs and gene regulation.
Science. 277:1630–1635. 1997.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Aaronson DS and Horvath CM: A road map for
those who don't know JAK-STAT. Science. 296:1653–1655.
2002.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Aggarwal BB, Kunnumakkara AB, Harikumar
KB, Gupta SR, Tharakan ST, Koca C, Dey S and Sung B: Signal
transducer and activator of transcription-3, inflammation, and
cancer: How intimate is the relationship? Ann N Y Acad Sci.
1171:59–76. 2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Heinrich PC, Behrmann I, Haan S, Hermanns
HM, Müller-Newen G and Schaper F: Principles of interleukin
(IL)-6-type cytokine signalling and its regulation. Biochem J.
374:1–20. 2003.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zhang D, Sun M, Samols D and Kushner I:
STAT3 participates in transcriptional activation of the C-reactive
protein gene by interleukin-6. J Biol Chem. 271:9503–9509.
1996.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Nishiyama T, Mae T, Kishida H, Tsukagawa
M, Mimaki Y, Kuroda M, Sashida Y, Takahashi K, Kawada T, Nakagawa K
and Kitahara M: Curcuminoids and sesquiterpenoids in turmeric
(Curcuma longa L.) suppress an increase in blood glucose
level in type 2 diabetic KK-Ay mice. J Agric Food Chem. 53:959–963.
2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Asai A, Nakagawa K and Miyazawa T:
Antioxidative effects of turmeric, rosemary and capsicum extracts
on membrane phospholipid peroxidation and liver lipid metabolism in
mice. Biosci Biotechnol Biochem. 63:2118–2122. 1999.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Samaha HS, Kelloff GJ, Steele V, Rao CV
and Reddy BS: Modulation of apoptosis by sulindac, curcumin,
phenylethyl-3-methylcaffeate, and 6-phenylhexyl isothiocyanate:
Apoptotic index as a biomarker in colon cancer chemoprevention and
promotion. Cancer Res. 57:1301–1305. 1997.PubMed/NCBI
|
|
14
|
Jayaprakasha GK, Jena BS, Negi PS and
Sakariah KK: Evaluation of antioxidant activities and
antimutagenicity of turmeric oil: A byproduct from curcumin
production. Z Naturforsch C J Biosci. 57:828–835. 2002.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hong CH, Noh MS, Lee WY and Lee SK:
Inhibitory effects of natural sesquiterpenoids isolated from the
rhizomes of Curcuma zedoaria on prostaglandin E2 and nitric oxide
production. Planta Med. 68:545–547. 2002.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pothitirat W and Gritsanapan W: Variation
of bioactive components in Curcuma longa in Thailand. Curr
Sci. 91:1397–1400. 2006.
|
|
17
|
Bhardwaj KS, Bhardwaj RS, Ranjeet D and
Ganesh N: Curcuma longa leaves exhibits a potential
antioxidant, antibacterial and immunomodulating properties. Int J
Phytomed. 3:270–278. 2011.
|
|
18
|
Abida Y, Tabassum F, Zaman S, Chhabi SB
and Islam N: Biological screening of Curcuma longa L. for
insecticidal and repellent potentials against Tribolium castaneum
(Herbst) adults. Univ J Zool Rajshahi Univ. 28:69–71. 2009.
|
|
19
|
Kim DW, Lee SM, Woo HS, Park JY, Ko BS,
Heo JD, Ryu YB and Lee WS: Chemical constituents and
anti-inflammatory activity of the aerial parts of Curcuma
longa. J Funct Foods. 26:485–493. 2016.
|
|
20
|
Ahn D, Lee EB, Ahn MS, Lim HW, Xing MM,
Tao C, Yang JH and Kim DK: Antioxidant constituents of the aerial
parts of Curcuma longa Kor. J Pharmacogn. 43:274–278.
2012.
|
|
21
|
Jiang CL, Tsai SF and Lee SS: Flavonoids
from Curcuma longa leaves and their NMR assignments. Nat
Prod Commun. 10:63–66. 2015.PubMed/NCBI
|
|
22
|
Lee SJ, Jang HJ, Kim Y, Oh HM, Lee S, Jung
K, Kim YH, Lee WS, Lee SW and Rho MC: Inhibitory effects of
IL-6-induced STAT3 activation of bio-active compounds derived from
Salvia plebeia R.Br. Process Biochem. 51:2222–2229.
2016.
|
|
23
|
Li HC and Zhang GY: Inhibitory effect of
genistein on activation of STAT3 induced by brain
ischemia/reperfusion in rat hippocampus. Acta Pharmacol Sin.
24:1131–1136. 2003.PubMed/NCBI
|
|
24
|
Sengupta TK, Talbot ES, Scherle PA and
Ivashkiv LB: Rapid inhibition of interleukin-6 signaling and Stat3
activation mediated by mitogen-activated protein kinases. Proc Natl
Acad Sci USA. 95:11107–11112. 1998.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Alessi DR, Cuenda A, Cohen P, Dudley DT
and Saltiel AR: PD 098059 is a specific inhibitor of the activation
of mitogen-activated protein kinase kinase in vitro and in vivo. J
Biol Chem. 270:27489–27494. 1995.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Dudley DT, Pang L, Decker SJ, Bridges AJ
and Saltiel AR: A synthetic inhibitor of the mitogen-activated
protein kinase cascade. Proc Natl Acad Sci USA. 92:7686–7689.
1995.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Heinrich PC, Behrmann I, Müller-Newen G,
Schaper F and Graeve L: Interleukin-6-type cytokine signalling
through the gp130/Jak/STAT pathway. Biochem J. 334:297–314.
1998.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ochrietor JD, Harrison KA, Zahedi K and
Mortensen RF: Role of STAT3 and C/EBP in cytokine-dependent
expression of the mouse serum amyloid P-component (SAP) and
C-reactive protein (CRP) genes. Cytokine. 12:888–899.
2000.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhang D, Jiang SL, Rzewnicki D, Samols D
and Kushner I: The effect of interleukin-1 on C-reactive protein
expression in Hep3B cells is exerted at the transcriptional level.
Biochem J. 310:143–148. 1995.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lim HJ, Jang HJ, Bak SG, Lee S, Lee SW,
Lee KM, Lee SJ and Rho MC: In vitro inhibitory effects of cirsiliol
on IL-6-induced STAT3 activation through anti-inflammatory
activity. Bioorg Med Chem Lett. 29:1586–1592. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chang JS, Lee SW, Kim MS, Yun BR, Park MH,
Lee SG, Park SJ, Lee WS and Rho MC: Manassantin A and B from
Saururus chinensis inhibit interleukin-6-induced signal
transducer and activator of transcription 3 activation in Hep3B
cells. J Pharmacol Sci. 115:84–88. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chang JS, Lee SW, Park MH, Kim MS, Hudson
BI, Park SJ, Lee WS and Rho MC: Kansuinine A and Kansuinine B from
Euphorbia kansui L. inhibit IL-6-induced Stat3 activation.
Planta Med. 76:1544–1549. 2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hodge DR, Hurt EM and Farrar WL: The role
of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer.
41:2502–2512. 2005.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Chung J, Uchida E, Grammer TC and Blenis
J: STAT3 serine phosphorylation by ERK-dependent and -independent
pathways negatively modulates its tyrosine phosphorylation. Mol
Cell Biol. 17:6508–6516. 1997.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Stancato LF, Sakatsume M, David M, Dent P,
Dong F, Petricoin EF, Krolewski JJ, Silvennoinen O, Saharinen P,
Pierce J, et al: Beta interferon and oncostatin M activate Raf-1
and mitogen-activated protein kinase through a JAK1-dependent
pathway. Mol Cell Biol. 17:3833–3840. 1997.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Beadling C, Ng J, Babbage JW and Cantrell
DA: Interleukin-2 activation of STAT5 requires the convergent
action of tyrosine kinases and a serine/threonine kinase pathway
distinct from the Raf1/ERK2 MAP kinase pathway. EMBO J.
15:1902–1913. 1996.PubMed/NCBI
|
|
37
|
Lee HK, Jung J, Lee SH, Seo SY, Suh DJ and
Park HT: Extracellular signal-regulated kinase activation is
required for serine 727 phosphorylation of STAT3 in Schwann cells
in vitro and in vivo. Korean J Physiol Pharmacol. 13:161–168.
2009.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Terstegen L, Gatsios P, Bode JG, Schaper
F, Heinrich PC and Graeve L: The inhibition of
interleukin-6-dependent STAT activation by mitogen-activated
protein kinases depends on tyrosine 759 in the cytoplasmic tail of
glycoprotein 130. J Biol Chem. 275:18810–18817. 2000.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Pepys MB and Hirschfield GM: C-reactive
protein: A critical update. J Clin Invest. 111:1805–1812.
2003.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Ridker PM: From C-reactive protein to
interleukin-6 to interleukin-1: Moving upstream to identify novel
targets for atheroprotection. Circ Res. 118:145–156.
2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Yasukawa H, Ohishi M, Mori H, Murakami M,
Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, et al:
IL-6 induces an anti-inflammatory response in the absence of SOCS3
in macrophages. Nat Immunol. 4:551–556. 2003.PubMed/NCBI View
Article : Google Scholar
|
|
42
|
El Kasmi KC, Holst J, Coffre M, Mielke L,
de Pauw A, Lhocine N, Smith AM, Rutschman R, Kaushal D, Shen Y, et
al: General nature of the STAT3-activated anti-inflammatory
response. J Immunol. 177:7880–7888. 2006.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Jang HJ, Park EJ, Lee SJ, Lim HJ, Jo JH,
Lee SW and Rho MC: Diarylheptanoids from Curcuma phaeocaulis
suppress IL-6-induced STAT3 activation. Planta Med. 85:94–102.
2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Rajasingh J, Raikwar HP, Muthian G,
Johnson C and Bright JJ: Curcumin induces growth-arrest and
apoptosis in association with the inhibition of constitutively
active JAK-STAT pathway in T cell leukemia. Biochem Biophys Res
Commun. 340:359–368. 2006.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Saydmohammed M, Joseph D and Syed V:
Curcumin suppresses constitutive activation of STAT-3 by
up-regulating protein inhibitor of activated STAT-3 (PIAS-3) in
ovarian and endometrial cancer cells. J Cell Biochem. 110:447–456.
2010.PubMed/NCBI View Article : Google Scholar
|