1. Introduction
Neurological disorders refer to a diverse group of
conditions characterized by neurological damage and functional
impairments resulting from structural or functional abnormalities
of the central or peripheral nervous systems. Consequently, these
neurological disorders include neurodevelopmental disorders,
neurodegenerative diseases, and excitatory-inhibitory imbalances,
with complex etiologies involving genetic, traumatic, metabolic,
infectious, and degenerative factors. Recently, studies have
demonstrated that abnormal neurotransmitter release is a common
basis for the pathogenesis of various neurological conditions
(1,2). Notably, the release of
neurotransmitters is a highly coordinated process, with core
regulation fundamentally dependent on the assembly and functional
stability of the soluble N-ethylmaleimide sensitive factor
attachment protein receptor (SNARE) complex (3). STX1A is a core component of the SNARE
complex and is widely expressed in the central nervous system. It
is predominantly localized to the presynaptic membrane and is
critical for synaptic vesicle fusion and neurotransmitter
exocytosis (4). Moreover, research
indicates that STX1A regulates the speed and precision of synaptic
vesicle fusion (5) and modulates
calcium-dependent neurotransmitter release through interactions
with accessory proteins such as Munc18-1 and synaptotagmin
(6-8).
In addition to regulating neurotransmitters, STX1A is involved in
synaptic plasticity, neuronal development, and ion channel
function. Recent advances in genetics, molecular biology, and
neuroimaging have also revealed that abnormal expression,
mutations, and disorders of the regulatory mechanisms of the
STX1A gene are closely linked to various neurological
disorders, including neuropsychiatric diseases (9-12),
neurodegenerative diseases (13,14)
and ischemic stroke (IS) (15). As
such, STX1A has emerged as a prominent focus of research in
neurological disorders.
The diverse physiological functions of STX1A, along
with its dynamic changes under pathological conditions, suggest its
potential as a valuable diagnostic biomarker and therapeutic target
for neurological disorders. However, current research faces several
limitations, including insufficient understanding of
disease-specific mechanisms and limited clinical evidence for
translation. Therefore, this review systematically outlines the
structural features and physiological functions of STX1A. It
specifically focuses on recent advances in understanding its role
across various neurological diseases.
2. Structure and physiological functions of
STX1A
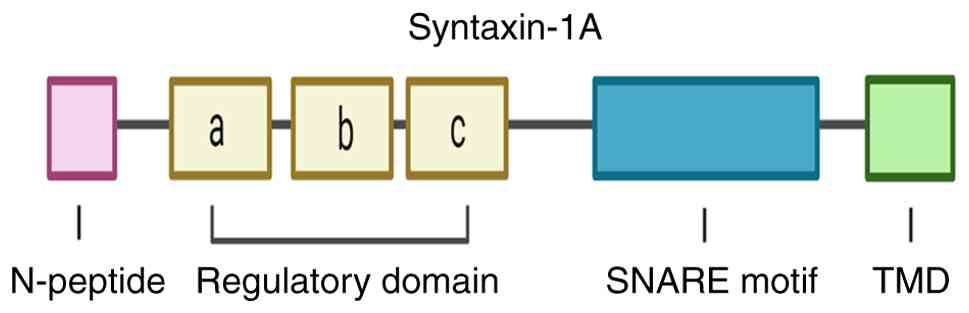
STX1A structure
STX1A is a member of the syntaxin family and is
encoded on human chromosome 7q11.23. It comprises 288 amino acids
and is one of two isoforms of Syntaxin1, the other being Syntaxin1B
(STX1B) (16). STX1A is primarily
localized on the plasma membrane. It has four major structural
components: An N-terminal peptide (N-peptide), an N-terminal
regulatory domain (Habc), a SNARE domain (also known as the H3
domain), and a C-terminal transmembrane domain (TMD) (17) (Fig.
1). The N-peptide is connected to the Habc domain by a flexible
region, and the Habc domain is linked to the H3 domain through a
linker region. Additionally, the H3 domain is connected to the TMD
by a short polybasic juxtamembrane domain. The Habc domain
(18) is a highly conserved domain
composed of three antiparallel α-helices and plays a crucial role
in synaptic transmission in mammals (18). Moreover, the Habc domain interacts
with key proteins, including synaptotagmin-1 (Syt1) (19), voltage-gated calcium channels
(VGCCs), and Munc18-1, thereby facilitating the precise recruitment
of the STX1A-Munc18-1 complex to the active zone (AZ) (20). The SNARE domain of STX1A contains a
highly conserved sequence of 60-70 residues with heptanucleotide
repeats (21). Significantly, STX1A
contributes a Qa-SNARE motif to the SNARE complex, assembling with
synaptosomal-associated protein of 25 kDa (SNAP-25; Qbc-SNARE
motifs) and synaptobrevin (VAMP; R-SNARE motif) in a 1:1:1 ratio to
form a stable four-helix bundle, a structural prerequisite for
vesicle fusion and neurotransmitter exocytosis (22). The Habc domain can also interact
with its own SNARE motif, forming a ‘closed’ conformation that
regulates STX1A activity, a process modulated by its interaction
with neuronal Sec1 (nSec1; also known as rbSec1 or Munc18-1)
(23). Furthermore, the C-terminal
TMD of STX1A primarily anchors the membrane through its hydrophobic
region.
STX1A physiological functions
STX1A is a crucial core protein involved in vesicle
fusion, primarily contributing to SNARE complex assembly (24). In mammals, vesicle trafficking is
the primary transport mechanism in eukaryotic cells, enabling
processes such as endocytosis and exocytosis through membrane
fusion. This fusion is facilitated by the formation of SNARE
complexes (25). SNARE proteins are
categorized based on their membrane localization: t-SNAREs are
found on target membranes, while v-SNAREs are located on vesicular
membranes. Moreover, VAMP belongs to v-SNARE, while SNAP-25 and
STX1A are t-SNAREs (26). The
mechanism of plasma membrane vesicle fusion in neuronal cells holds
significant physiological importance. Consequently, numerous severe
neurological diseases are associated with the improper localization
of vesicle fusion-related proteins (27).
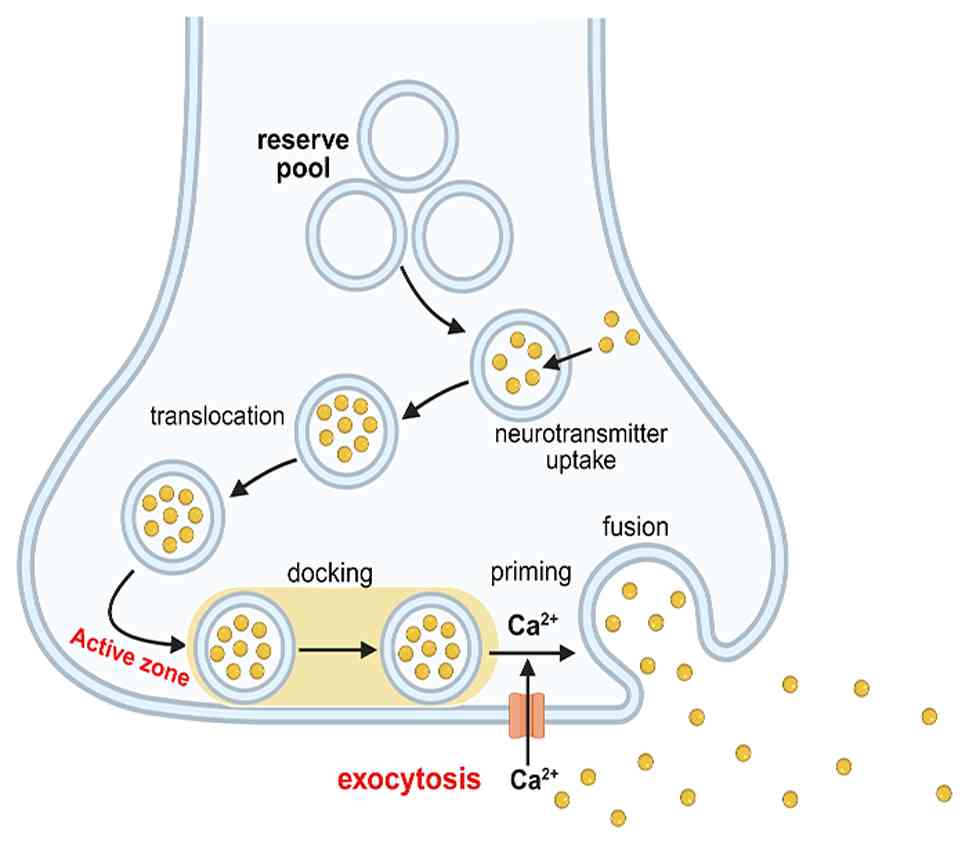
In the synapse, a small number of vesicles reside at
specific sites in the presynaptic membrane AZ. By contrast, most
vesicles are transported to areas near the cell membrane after
synthesis, forming a reserve pool. Neurotransmitter release by
neuronal exocytosis (Fig. 2) can be
divided into several steps, including vesicle mobilization,
docking, priming, fusion, and recycling (28). During rest, synaptic vesicles in the
readily releasable pool within the AZ participate in
neurotransmitter release. In addition, the AZ is enriched in
cytoskeletal and scaffold proteins, forming a dense matrix that
facilitates the anchoring, preparation, and rapid release of
synaptic vesicles. In neuronal and endocrine cells, priming is a
necessary rate-limiting step in secretion, in which vesicles are
released only after priming. Moreover, SNARE proteins mediate
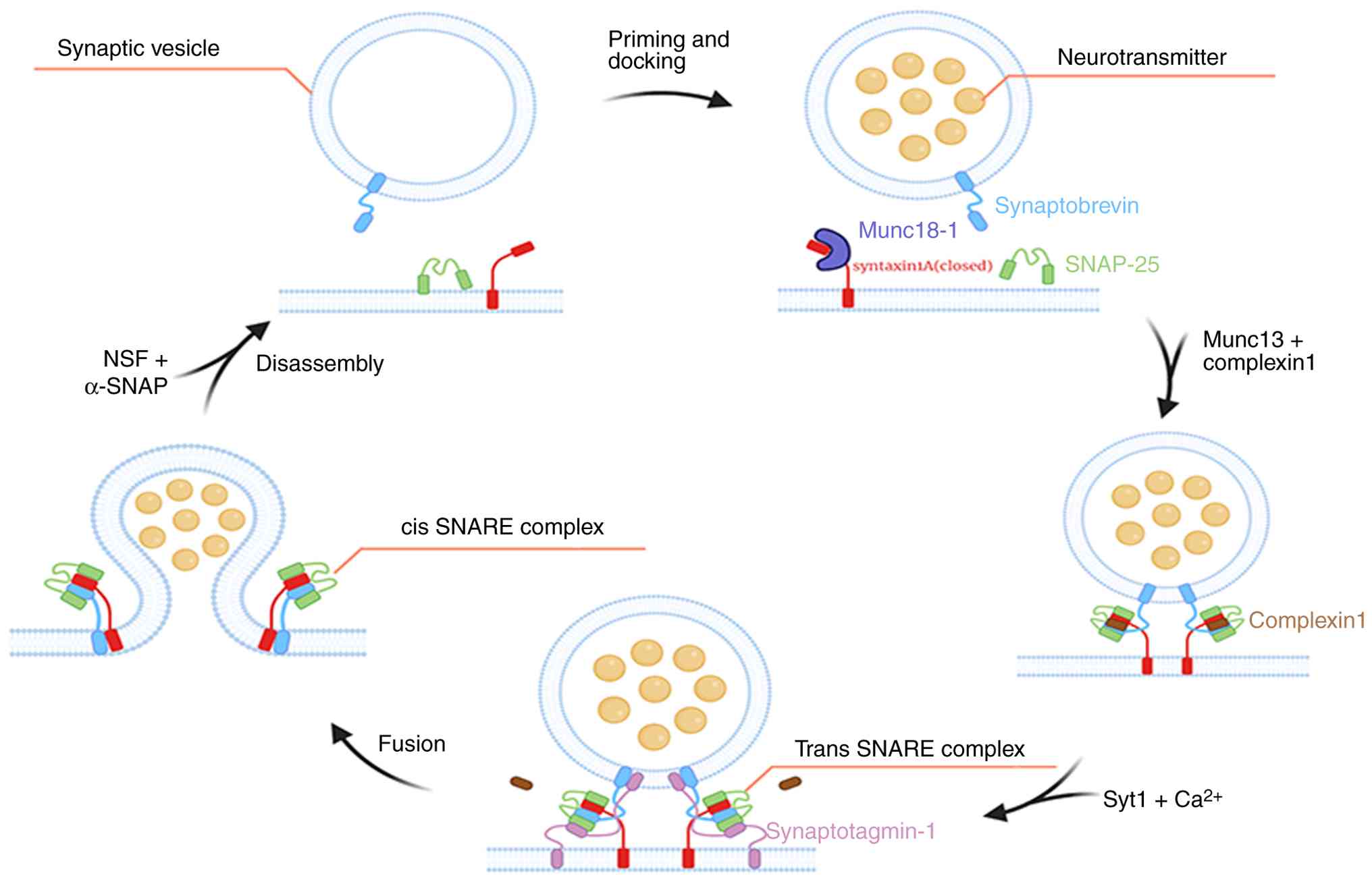
vesicle priming and membrane fusion (29). In the resting state, STX1A is in a
‘closed’ conformation and cannot participate in the assembly of the
SNARE complex. Following an action potential burst, a signal
spreads along the axon to the presynaptic membrane, depolarizing
the axon. Subsequently, this opens VGCCs, triggering
Ca²+ influx. Elevated intracellular Ca²+
facilitates the recruitment of primed vesicles from the reserve
pool to AZ through interactions with Rab proteins (30), RIM, and Munc13. These vesicles are
tethered to the plasma membrane, forming trans-SNARE complexes, a
process known as vesicle priming (31). The influx of Ca²+ binds
to synaptotagmin (a Ca²+ sensor), with the assistance of
the complexin protein, thereby interacting with STX1A to induce an
open conformation. Consequently, this promotes SNARE complex
assembly through a series of ordered and continuous reactions. In
the priming phase, STX1A and SNAP-25 form a t-SNARE complex on the
target membrane, which then assembles with VAMP2 (32,33)
(on the vesicle membrane), forming a trans-SNARE complex or
SNAREpin. This complex forms through a zipper-like mechanism from
the N-terminal to the C-terminal regions, pulling the vesicle and
plasma membranes into close apposition and providing energy for
lipid bilayer fusion (34).
Subsequently, primed vesicles are drawn toward the plasma membrane
and fuse with it. During this process, the trans-SNARE complex is
converted into cis-SNARE complexes, which form a fusion pore and
release neurotransmitters from synaptic vesicles into the synaptic
cleft (35). In addition, changes
in calcium ion concentration directly affect the amount of
neurotransmitter released and are necessary for vesicle fusion with
the presynaptic membrane (36).
Following membrane fusion, the resulting cis-SNARE complexes are
disassembled by the AAA+ ATPase NSF (37) and its cofactor α-soluble NSF
attachment protein (α-SNAP). This ensures that SNARE can be
recycled for the next round of fusion, thereby maintaining the
efficiency of synaptic transmission (Fig. 3).
In addition to affecting neurotransmitter release,
STX1A plays an indispensable part in neuronal development, synaptic
plasticity, and ion channel regulation. Fuschini et al
(38) demonstrated that STX1A
mediates the release of brain-derived neurotrophic factor, thereby
regulating neuronal axon growth, synapse formation, and cognitive
function. Notably, synaptic plasticity underpins higher neural
functions, including learning and memory. As such, research
indicates that STX1A plays a significant role in long-term
potentiation and low-latency inhibition (39,40).
Using the STX1A gene-mutation knock-in mouse model,
paired-pulse facilitation and enhanced short-term neuronal
plasticity (41) have been
observed. In the striatum, a previous study has also found that
STX1A expression is correlated with the acquisition of dopamine
(DA)-related reward learning (42).
Furthermore, a preliminary study discovered that STX1A interacts
with ion channels (43).
Consequently, the two conserved cysteine residues in the
transmembrane region of STX1A directly interact with VGCCs, thereby
modulating calcium influx and the excitatory coupling of
neurotransmitters (44). In
addition, STX1A binds to Kv2.1 (a voltage-gated potassium channel)
to regulate cellular excitability (45). Thus, these roles suggest that STX1A
maintains synaptic activity and functions as a key regulator in
neuronal circuit remodeling.
3. STX1A interactions with major accessory
proteins
STX1A functions within a precisely coordinated
presynaptic protein network. As such, it engages in dynamic
interactions with essential accessory proteins, including
Sec1/Munc18 (SM) proteins, complexin, and synaptotagmin. Notably,
these interactions finely regulate synaptic vesicle fusion and
sustain neurotransmitter homeostasis (Fig. 4).
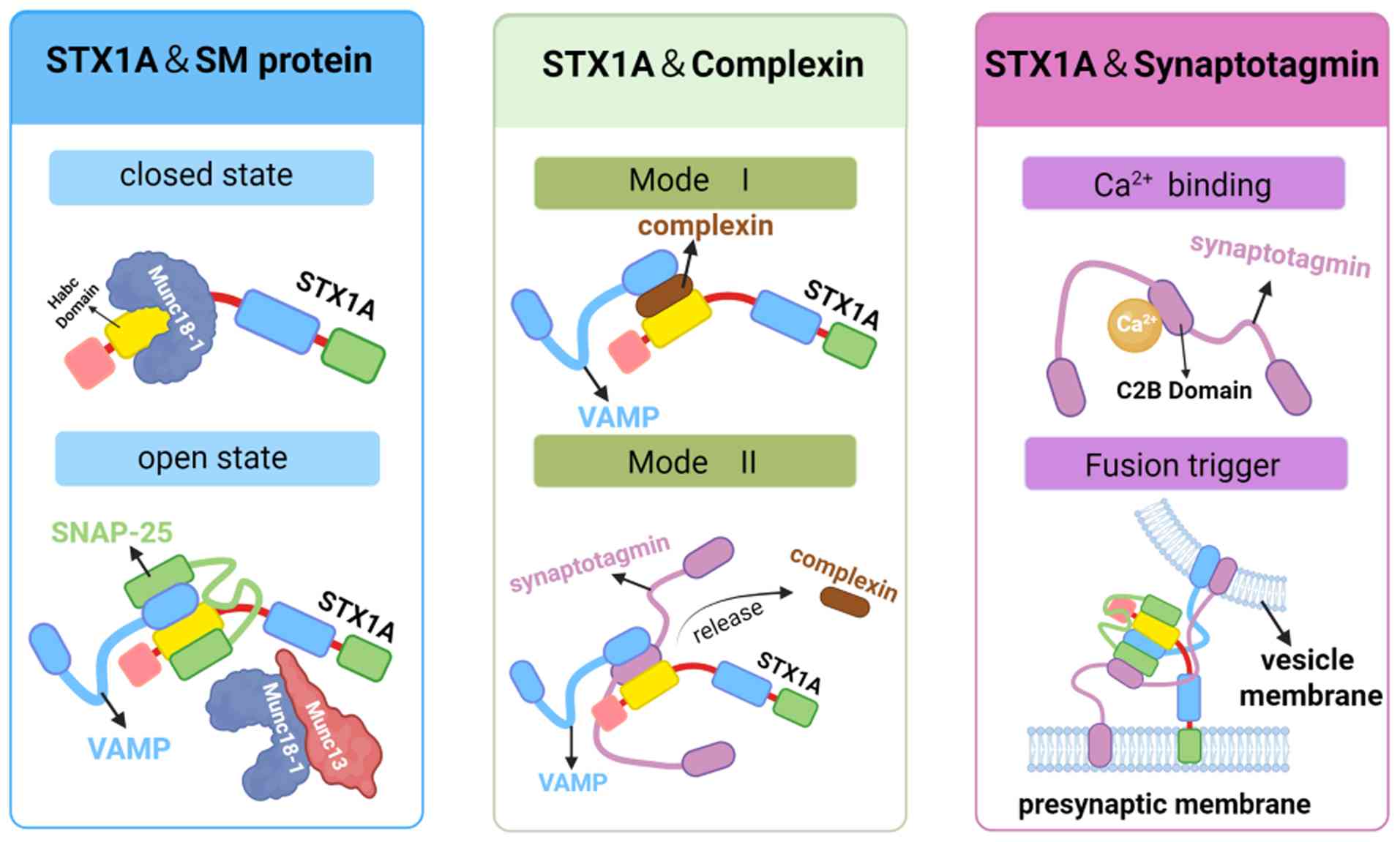
STX1A and SM protein interactions
SM family proteins (46) are essential auxiliary factors in
membrane fusion and are involved in nearly all vesicular
trafficking events. Munc18-1 (also known as STXBP1) is a key member
of the SM family and is the primary regulatory partner of STX1A. It
controls vesicle membrane fusion by modulating the conformational
states of STX1A as it transitions between open and closed
conformations. When Munc18-1 binds to the Habc domain of free
STX1A, it stabilizes the monomeric STX1A in a closed, inactive
conformation. In this state, the Habc domain interacts with the
SNARE motif, forming a closed structure that is enveloped by the
arched interface of the domains 1 and 3a of Munc18-1. As a result,
this configuration prevents premature SNARE complex formation and
inhibits vesicle fusion. Moreover, this mechanism ensures proper
STX1A folding and avoids erroneous interactions that could generate
off-pathway SNARE assemblies, thereby maintaining the precision of
neurotransmitter release. Upon activation by the MUN domain of
Munc13, Munc18-1 shifts its interaction to engage the N-terminal
region of STX1A. This conformational switch promotes the open state
of STX1A, facilitating its assembly with SNAP-25(47) and VAMP2(48) into a ternary SNARE complex (49). Finally, Munc18-1 and Munc13 are
released from the template complex. Upon doing so, the three SNARE
proteins are correctly assembled into a trans-SNARE complex, and
membrane fusion is initiated (50).
Notably, knockout experiments have shown that Munc18-1-deficient
mice exhibit a complete loss of neurotransmitter secretion, despite
normal synapse formation, underscoring the indispensable role of
Munc18-1 in exocytosis (51).
STX1A and complexin interactions
Complexin (also known as synaphin) is a small
cytosolic protein that acts as a clamp during the vesicle fusion
process. It contains an N-terminal domain, an accessory α-helix, a
central α-helix, and a C-terminal domain (52). Notably, complexin interacts with
STX1A in two distinct modes: i) Mode 1: Complexin-1 (Cpx1), the
isoform predominantly expressed in synapses, inserts its central
α-helix antiparallel into the groove between VAMP2 and STX1A,
stabilizing the vesicle in a semi-fused state and preventing
premature fusion. ii) Mode 2: Upon Ca²+ binding, Syt1
competes with complexin for binding to STX1A via its C2B domain,
relieving the inhibitory effect of complexin and initiating rapid
fusion (53).
In complexin-knockdown mice, both spontaneous and
evoked neurotransmitter release, including asynchronous and delayed
modes, were significantly reduced. This supports the critical
regulatory role of complexin in synaptic transmission (54).
STX1A and synaptotagmin
interactions
Synaptotagmin is a synaptic vesicle protein
evolutionarily conserved across species. It comprises an N-terminal
single TMD, an unstructured linker region, and two cytoplasmic
protein kinase C-like C2 domains (C2A and C2B) (53). The C2B domain has a high affinity
for Ca²+ and is primarily responsible for triggering
vesicle-plasma membrane fusion. By contrast, the C2A domain
contributes to vesicle docking and mobility. Syt1, the principal
isoform in synapses, serves as a Ca²+ sensor during
neurotransmission. Upon Ca²+ influx, Syt1 binds
phospholipids through its C2 domains, promoting membrane fusion.
In vitro experiments have shown that Syt1 can dock vesicles
by binding to STX1A/SNAP-25 receptor complexes, further emphasizing
its central role in Ca²+-dependent neurotransmitter
release (55).
4. Association between STX1A and
neurological disorders
There is growing evidence that the dysfunction of
STX1A is involved in the pathogenesis of various neurological
disorders. Additionally, it is closely associated with
attention-deficit/hyperactivity disorder (ADHD) (40,56),
autism spectrum disorder (ASD) (57), epilepsy (58,59),
migraine (60-62),
Alzheimer's disease (AD) (63),
Parkinson's disease (PD) and IS (Tables
I and II) (15).
 | Table IDysfunction and pathogenic mechanism
of STX1A in different neurological disorders. |
Table I
Dysfunction and pathogenic mechanism
of STX1A in different neurological disorders.
| Neurological
disorders | STX1A
expression | Pathogenic
mechanism | (Refs.) |
|---|
| Attention
deficit/hyperactivity disorder | Decreased | Dopaminergic
disorder and noradrenergic disorder | (9,39) |
| Autism spectrum
disorder | Decreased or
increased | Dopaminergic
disorder and serotonergic disorder; impaired synaptic
plasticity | (73,74,76,79) |
| Williams-Beuren
syndrome | Decreased | Unclear | (81,82) |
| Epilepsy | Decreased | Glutamatergic
disorder | (10,87,90,91) |
| Migraine | Decreased | Serotonergic
disorder and glutamatergic disorder | (59-61) |
| Alzheimer's
disease | Decreased | Synaptic
dysfunction | (62,114-117) |
| Parkinson's
disease | Decreased | Dopaminergic
disorder | (118-120) |
| Ischemic
stroke | Decreased | GABA transporter 1
disorder | (15,125) |
| Multiple
sclerosis | Decreased | Unclear | (129,130) |
| Table IISNPs of STX1A in various neurological
disorders. |
Table II
SNPs of STX1A in various neurological
disorders.
| Neurological
disorders | SNPs | (Refs.) |
|---|
| Attention
deficit/hyperactivity disorder | rs3793243,
rs875342, rs2293485 | (9,65) |
| Autism spectrum
disorder | rs4717806,
rs941298, rs4717806 | (78,79) |
| Epilepsy | rs4363087 | (87) |
| Migraine | rs941298,
rs2293489, rs6951030 | (59-61) |
| Alzheimer's
disease | rs4717806,
rs2293489, rs363050 | (113) |
| Multiple
sclerosis | rs1569061 | (129) |
STX1A and neurodevelopmental
disorders
ADHD is a common neurodevelopmental disorder that
primarily affects school-age children and adolescents. It is
characterized by inattention, hyperactivity, and impulsivity
(64). ADHD is suggested to arise
from complex interactions among genetic, neurobiological, and
environmental factors. Although its exact pathogenesis remains
unclear, genome-wide association studies (GWAS) indicate a strong
genetic component, particularly involving genes encoding components
of the SNARE complex (65). While
no single gene has been found to account for ADHD, the interaction
between polygenic susceptibility and environmental factors has been
proven to increase the risk of ADHD (66). Genetic association studies also
provide initial support for a relationship between STX1A and ADHD
susceptibility. Wang et al (9) found that STX1A variations
increase susceptibility to ADHD in Chinese Han children. Similar
findings have been reported in adult populations, linking
STX1A polymorphisms to ADHD (67). Furthermore,
neuropsychopharmacological evidence suggests that ADHD stems from
neurotransmitter system dysfunction (68), particularly imbalances in
dopaminergic and noradrenergic systems (40). Furthermore, imbalances in these
systems may lead to inattention and hyperactivity.
The most commonly prescribed treatments of ADHD are
DA agonists and norepinephrine (NE) agonists, both of which
effectively improve the symptoms of ADHD. Given the essential role
of STX1A in synaptic vesicle exocytosis, reduced expression may
result in insufficient DA release, contributing to ADHD symptoms.
Additionally, STX1A regulates DA transporter (DAT) activity, which
is responsible for DA reuptake and synaptic clearance (69). Moreover, DAT mediates the reuptake
of DA by removing it from the synaptic cleft and returning it to
presynaptic neurons. Thus, it modulates the concentration of DA in
the synaptic cleft, thereby terminating DA signal transduction and
ensuring proper nervous system function. Notably, STX1A
mutations may exacerbate ADHD by enhancing DAT reuptake (70). Similarly, NE transporter (NET)
inhibitors and α2-adrenergic receptor agonists are effective in
ADHD treatment. Methylphenidate (MPH) has been shown to improve the
symptoms of ADHD by inhibiting the reuptake of DA and NE through
its action on the DAT and NET (71). Atomoxetine (ATX), as a selective NE
reuptake inhibitor, is a non-stimulant medication commonly used in
clinical practice for the treatment of ADHD. It improves cognitive
function by modulating NE and indirectly enhancing DA signaling in
the prefrontal cortex (68). A
pharmacogenetic study assessed the therapeutic response to
immediate-release (IR)-MPH and its association with genes involved
in the SNARE complex in the treatment of ADHD. The findings
indicated that SNARE complexes mediate the response to commonly
prescribed ADHD medications (71).
Mishima et al (39) also
revealed that STX1A modulates noradrenaline transmission by
regulating dense-core vesicle secretion, further underscoring its
role in ADHD pathogenesis. Notably, the interplay between these two
mechanisms may work in concert to ultimately disrupt DA and NE
homeostasis, thereby increasing susceptibility to ADHD. Genetic
association studies have mostly focused on Chinese Han populations,
limiting generalizability (9).
However, clinical pharmacological research confirms the functional
relevance of the STX1A-associated pathway (68). This implies that modulating
presynaptic release capacity influences ADHD treatment outcomes.
Consequently, therapies targeting STX1A hold promise for improving
the clinical management of ADHD by modulating dopaminergic and
noradrenergic systems.
ASD encompasses a range of neurodevelopmental
disorders characterized by deficits in social communication,
restricted interests, and repetitive behaviors (72). Similar to ADHD, ASD is considered to
result from the interplay between genetic predisposition and
environmental influences (73).
STX1A-deficient animal models display behavioral phenotypes
analogous to human ASD, such as impaired fear memory, reduced
latent inhibition, and abnormal social behavior (74,75).
Beyond core ASD symptoms, patients with ASD may also experience
central nervous system symptoms such as epilepsy, intellectual
disability, and hyperactivity. Moreover, STX1A mutations in
ASD include splice-site mutations, missense mutations, and
frameshift variants, which lead to haploinsufficiency, impaired
synaptic plasticity, and dopaminergic and serotonergic
[5-hydroxytryptamine (5-HT)] disorders. Recently, Luppe et
al (10) reported two novel
heterozygous missense mutations in STX1A, which may perturb
SNARE complex assembly or hinder the interaction between STX1A and
STX1A-binding proteins. These two patients with epilepsy and ASD
features suggest that the dysfunction of STX1A may be the main
pathogenic mechanism for some patients with ASD. Notably, it also
suggests that ASD shares common pathogenic pathways with epilepsy
and ADHD (76). Additionally,
Cartier et al (77)
described a rare ASD-associated hypophosphorylated STX1A
mutant that reduces DAT-mediated reverse transport and is
implicated in ASD pathogenesis. Moreover, abnormalities in
serotonergic transmission are common in ASD, and STX1A directly
interacts with the serotonin transporter (5-HTT), modulating its
localization and function (78).
Genetic studies also indicate that STX1A polymorphisms,
including single-nucleotide polymorphisms (SNPs) rs4717806 and
rs941298, are associated with Asperger's syndrome (79). However, these genetic findings do
not imply that most ASD cases are driven by STX1A defect;
the alterations of STX1A in ASD show a high degree of
heterogeneity. A study in Japan reported elevated STX1A mRNA
expression in lymphocytes from individuals with high-functioning
autism (80). Furthermore,
increased STX1A expression was observed in the hippocampus
of mice prenatally exposed to bisphenol A (BPA), a model for ASD,
accompanied by impaired synaptic plasticity (57). Conversely, a study by Al-Ayadhi
et al (81) investigated the
effects of auditory integration training (AIT) in children with
ASD. It evaluated changes in plasma STX1A protein levels following
the intervention and their correlation with improvements in ASD
symptoms (81). The study observed
a significant increase in plasma STX1A levels following AIT,
accompanied by meaningful enhancements in behavioral, social, and
sensory processing scores (81).
Thus, STX1A levels may be associated with ASD symptomatology, and
plasma STX1A could potentially serve as a diagnostic biomarker for
the disorder. Moving forward, restoring or modulating STX1A
function in the brain may offer a novel therapeutic direction for
ASD.
Notably, reports on STX1A expression in ASD are
inconsistent, with both increased and decreased levels described
across studies (57,77,80,81).
Several factors may account for this discrepancy. At the genetic
level, mutations in STX1A exhibit heterogeneity in both genotype
and clinical manifestations. Loss-of-function mutations, such as
frameshift mutations or splice-site variants that lead to
haploinsufficiency) reduce the effective STX1A dosage. By contrast,
regulatory variants or compensatory responses may yield apparent
upregulation at the mRNA level. From a biological perspective,
STX1A expression is highly dependent on environmental factors. It
varies across brain regions (such as the frontal lobe and
hippocampus), cell types (including excitatory and inhibitory
neurons), and critical windows of neural development. Moreover,
most studies use peripheral tissues or homogenate samples, which
cannot capture this spatial and cell-specificity. Furthermore,
technical limitations, such as the imperfect correlation between
mRNA levels and functional protein activity, and confounding
environmental exposures (such as BPA), introduce additional
variability across studies. Consequently, the reported
inconsistencies reflect not contradiction but rather the interplay
of genetic heterogeneity, clinical heterogeneity, and
methodological limitations. Thus, future studies integrating
genotype-stratified cohorts, highly specific transcriptomics, and
functional validation are needed to elucidate the role of STX1A
across subtypes of ASDs.
Williams-Beuren syndrome (WS) is a rare genetic
neurodevelopmental disorder characterized by a hemizygous deletion
at 7q11.23. WS can affect multiple systems, particularly the
nervous, cardiovascular, endocrine, and digestive systems. It leads
to clinical manifestations of neurological abnormalities such as
intellectual disability, excessive sociability, and
attention-deficit. Notably, the STX1A gene, within the WS critical
region, has been identified as a strong candidate for WS and may
play a significant role in the neurodevelopment of the disease
(82). A previous study
demonstrated that STX1A gene transcript levels were significantly
correlated with intellectual functioning in patients with WS
(83). However, current research on
STX1A in WS remains limited. Moreover, its precise pathogenic
mechanism remains unclear.
STX1A and epilepsy
Epilepsy is a common heterogeneous neurological
disorder, whose main feature is the abnormal discharge of neurons
leading to recurrent, paroxysmal, and transient dysfunction of the
central nervous system (84). In
the past decade, growing evidence has highlighted the pivotal role
of synaptic protein-encoding genes in epilepsy pathogenesis
(85). Among these, pathogenic
variants affecting the SNARE complex and its regulatory proteins
have been incorporated into the emerging concept of ‘SNAREopathies’
(SNARE-related disease spectrum), which helps explain epilepsy and
other neurodevelopmental disorders arising from impaired
presynaptic release mechanisms (86,87).
While both isoforms, STX1A and STX1B, play roles in vesicle fusion,
they exhibit distinct expression patterns. In knockout mouse
models, STX1A and STX1B exhibit partial functional
compensation. However, double knockout of these isoforms results in
embryonic lethality due to the complete loss of synaptic vesicle
fusion (18). Notably, the role of
STX1B gene mutations in epileptic seizures has been
extensively studied (87). By
contrast, research on the association between STX1A
mutations and the development of epilepsy is relatively limited.
Recent human genetic studies have provided direct support for the
association between STX1A and epilepsy. In 2023, Luppe et
al (10) reported that a
missense mutation in the STX1A gene leads to
STX1A-related developmental and epileptic encephalopathy
(10). In addition to rare
variants, population-based genetic studies have identified
statistical associations between common STX1A polymorphisms and
cryptogenic epilepsy (88). For
instance, in a North Indian population, SNPs in VAMP2 and
STX1A were associated with cryptogenic epilepsy (88). However, broader population-level
validation of these associations remains limited.
Experimental and clinical evidence further suggests
that STX1A may contribute to epileptogenesis by modulating
excitatory neurotransmission and neuronal excitability. Notably,
dysregulation of glutamatergic signaling represents a key mechanism
underlying seizure generation (89), indicating that disruption of
presynaptic release machinery alters excitatory synaptic strength.
A previous study using septic rat models revealed a significant
reduction in STX1A and Munc18-1 expression in the hippocampus,
correlating with impaired glutamate release (90). Another study demonstrated that STX1A
reduces cell surface expression of the glutamate transporter
excitatory amino acid carrier 1 (EAAC1), thereby inhibiting
glutamate uptake and potentially contributing to seizure
susceptibility (91). In addition,
regulation of ion channels has been implicated in epilepsy
pathophysiology. Benign familial neonatal epilepsy (BFNE) is
commonly caused by mutations in voltage-gated potassium channels
(KCNQ2 and KCNQ3). Soldovieri et al (92) were the first to identify
dysfunctional STX1A-channel interactions in BFNE, where mutant
STX1A fails to regulate potassium channel activity.
Consequently, mutations in these channels reduce M-type
K+ currents, leading to neuronal hyperexcitability.
STX1A binding to K+ channels affects these M-currents
(93) and STX1A dysfunction
may exacerbate the effect. Another study suggested that
STX1A-related epileptic encephalopathy is not only characterized by
STX1A dysfunction but may also manifest as dysfunction of its
binding partner STXBP1 (encoding Munc-18) (94). This indicates that the epileptic
phenotype may result from the destruction of a broader presynaptic
release network rather than from the action of STX1A alone. This is
consistent with the ‘SNAREopathies’ pathological mechanism,
emphasizing co-aggregation impairment of presynaptic secretory
pathways as a common pathogenic mechanism in epilepsy and
neurodevelopmental disorders.
STX1A and migraine
Migraine is a common neurovascular disorder
characterized by recurrent headaches, often accompanied by sensory
disturbances such as photophobia and phonophobia (95,96).
Its global prevalence is higher in females than in males,
reflecting contributions from hormonal, genetic, and
neurobiological factors. From a genetic standpoint, migraine
exhibits a typical complex polygenic architecture, as supported by
additional transcriptomic and functional analyses. For instance, a
large-scale GWAS identified 123 susceptibility loci and mapped
STX1A to one of the implicated regions (97). This finding was further confirmed in
studies by Felício et al (98,99).
Furthermore, research by Quintas et al (62) indicates that the STX1A gene is
associated with migraine susceptibility and is an essential
candidate gene for migraines (100). This result has also been confirmed
in case-control studies conducted in Portugal (62) and Spain (61). These findings suggest a strong
genetic association between STX1A and migraine, but mechanistic
investigations into how STX1A variants contribute to migraine
pathophysiology remain scarce.
The leading pathophysiological hypotheses of
migraine involve cortical spreading depression (CSD), dysfunction
of serotonergic and glutamatergic neurotransmission, and neurogenic
inflammation (101-103).
Recent research has also emphasized the critical role of
neurotransmitter systems in migraine pathogenesis (1). In particular, the serotonergic system
plays a key role in pain modulation. Consequently, reduced
serotonin levels can exacerbate CSD and contribute to the onset of
migraine attacks. Clinically, triptans, selective
5-HT1B/1D receptor agonists, have proven
highly effective in treating migraine (104,105), supporting the central role of
serotonin. Therefore, STX1A mutations that reduce serotonin
release may represent a potential mechanism for migraine
development.
In parallel, glutamate and its receptors have also
been implicated in migraine in both pediatric and adult populations
(106,107). Epidemiological studies indicate a
high comorbidity between migraine and epilepsy. This is especially
true in women, individuals with temporal lobe epilepsy, or those
who experienced seizures within three months of a migraine
diagnosis (108,109). These findings suggest a shared
excitatory pathophysiology between the two disorders (110), potentially involving
STX1A-mediated regulation of glutamatergic transmission. Excessive
glutamate release can also activate N-methyl-D-aspartate receptors
and trigger CSD, a neurobiological substrate of migraine aura,
thereby promoting trigeminovascular activation and central
sensitization, which ultimately culminate in headache (107). It is hypothesized that STX1A
modulates excitatory neurotransmission, thereby participating in
the pathogenesis of both epilepsy and migraine. Given its
involvement in regulating excitatory neurotransmitter release,
STX1A could serve not only as a genetic risk factor but also as a
potential therapeutic target in migraine. Moreover, its
polymorphisms may function as disease-specific biomarkers (100).
STX1A and neurodegenerative
diseases
AD is the most common form of neurodegenerative
disorders, characterized primarily by progressive memory loss and
cognitive decline (111). Its
hallmark pathological features include extracellular accumulation
of β-amyloid (Aβ) forming neuritic plaques and intracellular
neurofibrillary tangles composed of hyperphosphorylated tau
protein. According to previous studies, synaptic dysfunction and
loss of synaptic plasticity are the fundamental pathological
processes in the early stages of a variety of neurodegenerative
diseases (112-114).
STX1A, as a necessary presynaptic t-SNARE protein, is crucial for
the secretion of synaptic vesicles. At the protein and
transcriptional levels, STX1A is commonly associated with AD.
Proteomic analysis of the prefrontal cortex in patients with AD
revealed significantly reduced STX1A levels compared with healthy
controls (115). A previous
large-scale targeted proteomic study has further revealed that the
level of STX1A in patients with AD is positively correlated with
cognitive function. By contrast, a negative correlation was found
with AD pathological burden. In cognitively impaired individuals,
the reduction in STX1A may be more pronounced than that in STX1B
(116). Collectively, these
findings provide strong evidence linking STX1A loss to AD-related
cognitive vulnerability. Complementary transcriptomic data further
support the concept that synaptic release pathways are disrupted in
AD. Significant downregulation of STX1A gene expression was
observed in the brains of an AD mouse model and Dp16 mice, which
may be a decisive factor in AD-related cognitive decline (117). In addition, emerging cerebrospinal
fluid proteomic studies in genetically AD cohorts suggest that
proteins involved in synaptic and vesicle cycling pathways exhibit
alterations early in the disease course (118). Moreover, Aβ oligomers (a
neurotoxic agent) are primary contributors to the pathological
process of AD, with the capability of damaging neurons through a
variety of mechanisms. Experimental research has also demonstrated
that Aβ oligomers directly bind to the SNARE motif of STX1A,
thereby inhibiting SNARE complex formation and blocking exocytosis
(63). This disruption of synaptic
transmission is proposed as an additional potential mechanism by
which STX1A contributes to cognitive deficits in AD. However, given
the essential and widespread role of STX1A in neurotransmission,
directly targeting STX1A carries potential safety concerns, as it
could broadly disrupt synaptic release across neurons. Thus,
therapeutic strategies may be better directed toward selectively
blocking pathological Aβ-STX1A interactions, thereby restoring
SNARE-mediated exocytosis and delaying or reversing cognitive
decline in AD while minimizing adverse effects. Current therapeutic
approaches for AD remain limited in efficacy; therefore,
elucidating the role of STX1A could offer new avenues for treatment
development.
PD is a chronic neurodegenerative disease
characterized by progressive movement disorders. The basic
pathological features of PD are the misfolding and abnormal
aggregation of α-synuclein (α-Syn) and the loss of dopaminergic
neurons in the substantia nigra. Notably, the SNARE complex is also
involved in PD pathogenesis (13).
Xiong et al (119) observed
downregulated STX1A expression in a PD rat model, suggesting that
impaired presynaptic release capacity may accompany dopaminergic
disorder. Furthermore, patients with PD exhibited elevated α-Syn
levels in serum exosomes along with higher clinical scores compared
with healthy controls (120).
Notably, Agliardi et al (121) also identified a negative
correlation between α-Syn and STX1A levels in the serum exosomes of
patients with PD. Therefore, these studies suggest an inverse
relationship between α-Syn burden and presynaptic STX1A integrity.
Collectively, STX1A may serve as a biomarker of presynaptic
vulnerability in PD diagnosis.
STX1A and IS
IS is the most prevalent type of stroke and ranks as
the third leading cause of death and disability worldwide. Previous
research has reported significant upregulation of STX1A protein
levels in the brains of IS rat models, including elevated
expression in blood samples from patients with IS (122). These findings suggest that STX1A
may serve as a potential clinical biomarker for stroke prognosis.
Additionally, elevated levels of γ-aminobutyric acid (GABA) during
the subacute phase of IS activate extra-synaptic GABA receptors,
leading to tonic inhibition that suppresses post-stroke neuronal
excitability and hinders recovery (123-125).
GABA transporter 1 (GAT-1) plays a vital role in the reuptake of
extracellular GABA, enhancing neuronal excitability and
facilitating recovery after stroke. Lin et al (15) discovered that IS induces GAT-1-STX1A
interaction, leading to GAT-1 dysfunction in the subacute phase. In
a mouse model of stroke, administration of ZLQ-3 (a small-molecule
inhibitor that disrupts the GAT-1-STX1A interaction) dissociated
the GAT-1-STX1A interaction, successfully restored GAT-1 function,
and enhanced GABA reuptake. This mechanism not only increased
cortical excitability but also strengthened GABAergic synaptic
inhibition, ultimately promoting functional recovery post-stroke
(15). Thus, STX1A is a novel
therapeutic target for enhancing post-stroke
neurorehabilitation.
In addition, Kv2.1 (a voltage-gated K+
channel) plays a key role in regulating cell excitability and is
also vital following IS. Evidence indicates that Kv2.1 can interact
with STX1A to promote K+ efflux, thereby contributing to
central neuronal apoptosis (126).
In vitro studies have shown that disrupting the Kv2.1-STX1A
interaction significantly reduces K+ efflux, exerting
neuroprotective effects (45,127).
Another study demonstrated that open-conformation STX1A appears to
inhibit Kv channel-mediated K+ currents (128), suggesting that this mechanism
could be leveraged for neuroprotection.
STX1A and multiple sclerosis (MS)
MS is a common autoimmune disease of the nervous
system. Its primary manifestation is chronic inflammation that
destroys myelin in the white matter of the brain, leading to
demyelinating disorders and progressive neurological dysfunction.
With advances in research, MS is no longer regarded as a purely
white matter demyelinating disease. Gray matter pathology and
synaptic dysfunction have also been increasingly recognized
(129). Notably, these alterations
may contribute to MS-associated cognitive impairment and
neuropsychiatric manifestations. A recent study conducted in
Turkish populations found a significant association between
STX1A gene polymorphisms and increased MS susceptibility
(130). By contrast, similar
associations were not observed in German or Egyptian populations
(131). This discrepancy suggests
that STX1A is unlikely to represent a universal, cross-population
driver of MS pathogenesis, but may instead function as a modest,
context-dependent genetic modifier.
5. Potential links between STX1A,
neuroimmune regulation, and the gut-brain axis
With continued advances in neuroscience research,
the pathophysiological mechanisms underlying neurological disorders
are no longer considered limited to simple neurotransmitter
imbalances. Instead, new evidence highlights the intricate
interplay between neuroinflammation and the gut-brain axis as
critical contributors to central nervous system homeostasis and
disease progression (132). Immune
mediators, particularly toll-like receptors (TLRs) and cytokines,
are now recognized as key modulators of neural function (133). In addition, the gut microbiota has
emerged as an important regulator of central nervous system
activity (134). Although the
present review primarily focuses on the role of STX1A in synaptic
transmission, it is increasingly important to consider its
potential links with neuroimmune signaling and gut-brain axis
dynamics.
Neuroinflammation represents an intrinsic immune
response of the central nervous system to injury, infection, or
pathological insults. It is primarily mediated by glial cells,
including microglia and astrocytes. TLRs, which belong to the
family of pattern-recognition receptors, are widely expressed on
glial cells and detect pathogen-associated molecular patterns and
damage-associated molecular patterns, thereby initiating
inflammatory signaling cascades. Activation of TLR pathways leads
to the release of pro-inflammatory cytokines, such as
interleukin-1β (IL-1β), which have been demonstrated to modulate
neuronal function and synaptic plasticity. Additionally, members of
the IL-1 cytokine family play central roles in neuroinflammatory
processes, and genetic polymorphisms within these genes can
influence the magnitude and duration of inflammatory responses,
thereby shaping susceptibility to neurological disorders (135). Moreover, dysregulated TLR
signaling has been implicated in protective and pathogenic immune
responses across diverse disease contexts, underscoring its dual
role in immune-mediated tissue homeostasis and injury (136,137). Given that STX1A is a core
component of the SNARE complex directly involved in
neurotransmitter release, inflammatory microenvironments may
influence its function by altering STX1A expression,
post-translational modification, SNARE complex assembly, or
synaptic plasticity.
In parallel, the gut microbiota interacts
bidirectionally with the central nervous system through neural,
endocrine, immune, and metabolic pathways. This is collectively
referred to as the gut-brain axis. Microbiota-derived metabolites,
microbially produced neurotransmitters, and microbiome-mediated
modulation of host immunity can exert long-range effects on brain
function and behavior (138).
Dysbiosis of the gut microbiota has been closely associated with
the onset and progression of numerous neurological disorders
(133,139). Disruption of microbial homeostasis
can compromise intestinal barrier integrity, increase gut
permeability, and permit translocation of microbial products into
the systemic circulation, thereby triggering systemic and
neuroinflammatory responses (140). Consequently, such gut-driven
inflammatory processes may profoundly affect the central nervous
system by reshaping neurotransmitter systems, inducing
neuroinflammation, and impairing synaptic integrity, ultimately
exerting indirect effects on STX1A-dependent synaptic function.
At present, direct interactions between STX1A and
immune mediators or gut microbiota-derived factors remain largely
unexplored. Nevertheless, the evidence outlined above supports the
existence of an indirect regulatory framework linking
STX1A-mediated synaptic transmission to neuroimmune and gut-brain
axis signaling. Thus, a deeper understanding of these
interconnections may open novel avenues for therapeutic strategies
targeting neuroimmune pathways or the gut-brain axis, with the
potential to improve clinical outcomes in STX1A-associated
neurological disorders.
6. Mechanistic and translational
implications of STX1A in neurological disorders
Recently, the view that synaptic structural and
functional deficits constitute a key factor in neurological
diseases has gained widespread acceptance. Disorders arising from
such synaptic dysfunctions are collectively referred to as
synaptopathies, including ADHD, ASD, epilepsy, migraine, AD, PD,
schizophrenia, as well as other disorders. Thus, these underlying
diseases may share a common pathogenic mechanism and genetic basis.
Notably, most research indicates that SNARE complexes play a
significant role in maintaining the structure and function of
synapses (58). Acting as a
‘molecular hub’ within neural networks, STX1A contributes to
synaptopathies through multiple mechanisms, including regulation of
SNARE complex assembly, coupling to accessory proteins and
transporters, and interactions with ion channels (42).
Across neurodevelopmental disorders (such as ADHD)
and neurodegenerative diseases (such as PD), STX1A may participate
in pathophysiology by mediating dopaminergic dysfunction. However,
the underlying mechanisms are fundamentally distinct. In ADHD,
STX1A dysregulation may impair synaptic vesicle exocytosis, reduce
dopamine release in prefrontal-striatal circuits, and enhance
dopamine clearance, thereby contributing to cognitive and
behavioral symptoms (67). By
contrast, STX1A-related dopaminergic impairment in PD occurs in the
context of progressive degeneration of nigrostriatal dopaminergic
neurons and aberrant α-Syn-SNARE interactions (141,142). Under these conditions, alterations
in STX1A are more likely to reflect the vulnerability and declining
function of surviving presynaptic terminals rather than serve as an
isolated driver of neurotransmitter imbalance. Thus, although both
disorders exhibit dopaminergic abnormalities, their mechanistic
bases, and consequently their translational implications, differ
substantially.
Future studies should explore therapeutic
strategies that selectively disrupt pathological STX1A-DAT coupling
to restore dopaminergic signaling in ADHD. By contrast,
PD-associated changes in STX1A may be more valuable as a biomarker
of synaptic integrity and presynaptic reserve, enabling more
accurate, cost-effective, and minimally invasive assessment of
disease progression and treatment response.
In addition, a spectrum of neurological syndromes
arising from mutations in SNARE-related genes that disrupt SNARE
complex composition and function is collectively referred to as
‘SNAREopathies’ (86).
Neurodevelopmental disorders caused by STX1A fall within this
disease spectrum and, together with other SNARE-associated genes,
contribute to disease severity, phenotypic variability, and age at
onset. Mutations in STXBP1, a master regulator required for STX1A
stabilization and SNARE complex initiation, typically result in
severe developmental and epileptic encephalopathies (143). As such, this reflects the
catastrophic consequences of early failure in SNARE assembly.
Similarly, SNAP25, a core Qbc-SNARE that directly drives membrane
fusion, is associated with profound synaptic release defects and
early-onset neurodevelopmental phenotypes when pathogenic variants
are present (144). By contrast,
STX1A occupies a more modulatory and integrative position within
the SNARE complex. Owing to its partial functional redundancy with
STX1B, STX1A dysfunction more often manifests as synaptic
vulnerability rather than complete failure of vesicle exocytosis
(17). Furthermore,
STX1A-associated disorders tend to display selective effects on
neurotransmitter release within specific neuronal populations and
often present with a relatively later onset (10). Notably, this markedly contrasts the
severe and uniform phenotypes characteristic of core SNAREopathies
(86).
Overall, elucidating the physiological and
pathophysiological roles of STX1A may serve as a critical bridge
between classical SNAREopathies and other complex synaptopathies.
Such an approach offers novel insights into the mechanistic
continuum linking rare presynaptic release disorders with polygenic
risk for common neurological diseases.
7. Conclusion and future perspectives
Over the past several decades, our understanding of
the structure and physiological functions of STX1A has
significantly advanced. As a key mediator of neuronal exocytosis,
STX1A interacts with VAMP and SNAP-25 to form the core SNARE
complex. This complex is essential for the docking and fusion of
synaptic vesicles with the presynaptic membrane, positioning STX1A
as a central component in synaptic transmission. The functional
dynamics of STX1A, including its conformational shifts between open
and closed states, its role in SNARE complex assembly, and its
interactions with various regulatory proteins, are crucial for
regulating its function.
It can be concluded that STX1A plays a crucial role
in major neurological diseases, making it a vital target for their
diagnosis and treatment. By regulating the release of
neurotransmitters, including glutamate, GABA, 5-HT, DA, and NE,
STX1A influences both the progression of these diseases and the
recovery of nervous system function. Genetic studies have strongly
indicated that mutations and dysregulation of STX1A are
linked to various neurological diseases such as ADHD, ASD,
epilepsy, migraine, AD, PD, and IS. Additionally, aberrant STX1A
expression can impair SNARE complex formation and disrupt the
release of essential neurotransmitters, resulting in cognitive,
behavioral, and neuronal signaling deficits. Since normal brain
function relies on a precise balance of neurotransmitter activity,
any disturbance in STX1A expression can lead to significant
neurological dysfunction.
To date, most research on the STX1A gene has
focused on its expression and polymorphisms. However, its potential
as a pharmacological target remains largely underexplored. While
proteomic and genomic studies have linked STX1A to various
diseases, including AD, ADHD, ASD, IS, and epilepsy, its role in
other neurological disorders has yet to be fully elucidated.
Further investigation into the mechanisms by which STX1A
contributes to neurological diseases could lead to the
identification of novel therapeutic strategies.
The targeted therapeutic strategy exemplified by
ZLQ-3 in ischemic stroke, namely, selectively disrupting
pathological STX1A protein interactions to restore synaptic balance
without globally impairing neurotransmitter release, may be
extendable to other neurological disorders. In AD, Aβ oligomers
have been shown to directly bind the SNARE motif of STX1A, thereby
inhibiting SNARE complex assembly and synaptic vesicle exocytosis.
Rather than directly targeting STX1A, peptide-based competitors
that selectively block the pathological Aβ-STX1A interaction may
represent a safer and more feasible alternative. Future studies
could focus on designing short peptides that competitively occupy
the binding interface, followed by protein modification strategies
to enhance blood-brain barrier penetration and molecular stability,
enabling targeted delivery while minimizing peripheral side
effects.
In addition, in neurological conditions driven by
aberrant overexpression of STX1A, such as certain subtypes of ASD,
gene-silencing approaches may be worth consideration. The design of
STX1A-specific siRNAs or miRNA mimics targeting the 3'-untranslated
region, delivered to defined brain regions via lipid nanoparticles
or viral vectors, could theoretically normalize excessive STX1A
expression.
Nevertheless, the development of STX1A-targeted
therapies remains a substantial challenge. This includes limited
blood-brain barrier permeability, the ubiquitous presynaptic
distribution of STX1A, and the risk of disrupting global
neurotransmitter homeostasis. Taken together, indirect modulation
of STX1A function may represent a more practical and safer
therapeutic strategy. Approaches that selectively interfere with
disease-specific protein interactions or downstream pathways appear
especially promising. Accordingly, future research should
prioritize precision-targeting approaches that restore synaptic
function while preserving the essential role of STX1A in neuronal
communication.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
YH and BB drafted the manuscript. JX, BS, PW, CX
and YY performed literature searches and contributed to manuscript
preparation and revision. XY wrote the final version of the
article. All authors have accepted responsibility for the
manuscript's content and consented to its submission. All authors
read and approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Uzay B and Kavalali ET: Genetic disorders
of neurotransmitter release machinery. Front Synaptic Neurosci.
15(1148957)2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Nimgampalle M, Chakravarthy H, Sharma S,
Shree S, Bhat AR, Pradeepkiran JA and Devanathan V:
Neurotransmitter systems in the etiology of major neurological
disorders: Emerging insights and therapeutic implications. Ageing
Res Rev. 89(101994)2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rizo J: Molecular mechanisms underlying
neurotransmitter release. Annu Rev Biophys. 51:377–408.
2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bose D, Bera M, Norman CA, Timofeeva Y,
Volynski KE and Krishnakumar SS: Minimal presynaptic protein
machinery governing diverse kinetics of calcium-evoked
neurotransmitter release. Nat Commun. 15(10741)2024.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Padmanabhan P, Bademosi AT, Kasula R,
Lauwers E, Verstreken P and Meunier FA: Need for speed:
Super-resolving the dynamic nanoclustering of syntaxin-1 at
exocytic fusion sites. Neuropharmacology.
169(107554)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Li W, Xing Y, Wang Y, Xu T, Song E and
Feng W: A non-canonical target-binding site in Munc18-1 domain 3b
for assembling the Mint1-Munc18-1-syntaxin-1 complex. Structure.
31:68–77.e5. 2023.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang C, Tu J, Zhang S, Cai B, Liu Z, Hou
S, Zhong Q, Hu X, Liu W, Li G, et al: Different regions of synaptic
vesicle membrane regulate VAMP2 conformation for the SNARE
assembly. Nat Commun. 11(1531)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wang X, Gong J, Zhu L, Chen H, Jin Z, Mo
X, Wang S, Yang X and Ma C: Identification of residues critical for
the extension of Munc18-1 domain 3a. BMC Biol.
21(158)2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wang M, Gu X, Huang X, Zhang Q, Chen X and
Wu J: STX1A gene variations contribute to the susceptibility of
children attention-deficit/hyperactivity disorder: A Case-control
association study. Eur Arch Psychiatry Clin Neurosci. 269:689–699.
2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Luppe J, Sticht H, Lecoquierre F,
Goldenberg A, Gorman KM, Molloy B, Agolini E, Novelli A, Briuglia
S, Kuismin O, et al: Heterozygous and homozygous variants in STX1A
cause a neurodevelopmental disorder with or without epilepsy. Eur J
Hum Genet. 31:345–352. 2023.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Villavicencio Gonzalez E and Dhindsa RS:
Studying ultra-rare variants in STX1A uncovers a novel
neurodevelopmental disorder. Eur J Hum Genet. 31:973–974.
2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Tang BL: SNAREs and developmental
disorders. J Cell Physiol. 236:2482–2504. 2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Margiotta A: Role of SNAREs in
neurodegenerative diseases. Cells. 10(991)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Williams JB, Cao Q and Yan Z:
Transcriptomic analysis of human brains with Alzheimer's disease
reveals the altered expression of synaptic genes linked to
cognitive deficits. Brain Commun. 3(fcab123)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lin YH, Wu F, Li TY, Lin L, Gao F, Zhu LJ,
Xu XM, Chen MY, Hou YL, Zhang CJ, et al: Disrupting stroke-induced
GAT-1-syntaxin1A interaction promotes functional recovery after
stroke. Cell Rep Med. 5(101789)2024.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ke P, Gu J, Liu J, Liu Y, Tian X, Ma Y,
Meng Y and Xiao F: Syntabulin regulates neuronal
excitation/inhibition balance and epileptic seizures by
transporting syntaxin 1B. Cell Death Discov. 9(187)2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Yang X, Tu W, Gao X, Zhang Q, Guan J and
Zhang J: Functional regulation of syntaxin-1: An underlying
mechanism mediating exocytosis in neuroendocrine cells. Front
Endocrinol. 14(1096365)2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Vardar G, Salazar-Lázaro A, Brockmann M,
Weber-Boyvat M, Zobel S, Kumbol VW, Trimbuch T and Rosenmund C:
Reexamination of N-terminal domains of syntaxin-1 in vesicle fusion
from central murine synapses. Elife. 10(e69498)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ramakrishnan S, Bera M, Coleman J, Rothman
JE and Krishnakumar SS: Synergistic roles of synaptotagmin-1 and
complexin in calcium-regulated neuronal exocytosis. Elife.
9(e54506)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Astacio H, Vasin A and Bykhovskaia M:
Stochastic properties of spontaneous synaptic transmission at
individual active zones. J Neurosci. 42:1001–1019. 2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Jahn R, Cafiso DC and Tamm LK: Mechanisms
of SNARE proteins in membrane fusion. Nat Rev Mol Cell Biol.
25:101–118. 2024.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang Y and Hughson FM: Chaperoning SNARE
folding and assembly. Annu Rev Biochem. 90:581–603. 2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Stefani I, Iwaszkiewicz J and Fasshauer D:
Exploring the conformational changes of the Munc18-1/syntaxin 1a
complex. Protein Sci. 33(e4870)2023.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wu LG and Chan CY: Membrane
transformations of fusion and budding. Nat Commun.
15(21)2024.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sauvola CW and Littleton JT: SNARE
regulatory proteins in synaptic vesicle fusion and recycling. Front
Mol Neurosci. 14(733138)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yan ML, Zhang S, Zhao HM, Xia SN, Jin Z,
Xu Y, Yang L, Qu Y, Huang SY, Duan MJ, et al: MicroRNA-153 impairs
presynaptic plasticity by blocking vesicle release following
chronic brain hypoperfusion. Cell Commun Signal.
18(57)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Risselada HJ and Mayer A: SNAREs, tethers
and SM proteins: How to overcome the final barriers to membrane
fusion? Biochem J. 477:243–258. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kim N and Cousin MA: Synaptic vesicle
recycling at the developing presynapse. J Neurochem.
169(e70206)2025.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wang S and Ma C: Stability profile of the
neuronal SNARE complex reflects its potency to drive fast membrane
fusion. Biophys J. 121:3081–3102. 2022.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Prasai B, Haber GJ, Strub MP, Ahn R,
Ciemniecki JA, Sochacki KA and Taraska JW: The nanoscale molecular
morphology of docked exocytic dense-core vesicles in neuroendocrine
cells. Nat Commun. 12(3970)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Qin J, Liu Q, Liu Z, Pan YZ,
Sifuentes-Dominguez L, Stepien KP, Wang Y, Tu Y, Tan S, Wang Y, et
al: Structural and mechanistic insights into secretagogin-mediated
exocytosis. Proc Natl Acad Sci USA. 117:6559–6570. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chanaday NL and Kavalali ET:
Synaptobrevin-2 dependent regulation of single synaptic vesicle
endocytosis. Mol Biol Cell. 32:1818–1823. 2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hu Y, Zhu L and Ma C: Structural roles for
the juxtamembrane linker region and transmembrane region of
synaptobrevin 2 in membrane fusion. Front Cell Dev Biol.
8(609708)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhang Y, Ma L and Bao H: Energetics,
kinetics, and pathways of SNARE assembly in membrane fusion. Crit
Rev Biochem Mol Biol. 57:443–460. 2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Li M, Oh TJ, Fan H, Diao J and Zhang K:
Syntaxin clustering and optogenetic control for synaptic membrane
fusion. J Mol Biol. 432:4773–4782. 2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Martínez-Mármol R, Muhaisen A, Cotrufo T,
Roselló-Busquets C, Ros O, Hernaiz-Llorens M, Pérez-Branguli F,
Andrés RM, Parcerisas A, Pascual M, et al: Syntaxin-1 is necessary
for UNC5A-C/netrin-1-dependent macropinocytosis and chemorepulsion.
Front Mol Neurosci. 16(1253954)2023.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Cheppali SK, Li C, Xing W, Sun R, Yang M,
Xue Y, Lu SY, Yao J, Sun S, Chen C and Sui SF: Single-molecule two-
and three-colour FRET studies reveal a transition state in SNARE
disassembly by NSF. Nat Commun. 16(250)2025.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Fuschini G, Cotrufo T, Ros O, Muhaisen A,
Andrés R, Comella JX and Soriano E: Syntaxin-1/TI-VAMP SNAREs
interact with trk receptors and are required for
neurotrophin-dependent outgrowth. Oncotarget. 9:35922–35940.
2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Mishima T, Fujiwara T, Kofuji T and
Akagawa K: Impairment of catecholamine systems during induction of
long-term potentiation at hippocampal CA1 synapses in
HPC-1/syntaxin 1A knock-out mice. J Neurosci. 32:381–389.
2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Nakayama T, Singh AK, Fukutomi T, Uchida
N, Terao Y, Hamada H, Muraoka T, Muthusamy E, Kundu TK and Akagawa
K: Activator of KAT3 histone acetyltransferase family ameliorates a
neurodevelopmental disorder phenotype in the syntaxin 1A ablated
mouse model. Cell Rep. 43(114101)2024.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Watanabe Y, Katayama N, Takeuchi K, Togano
T, Itoh R, Sato M, Yamazaki M, Abe M, Sato T, Oda K, et al: Point
mutation in syntaxin-1A causes abnormal vesicle recycling,
behaviors, and short term plasticity. J Biol Chem. 288:34906–34919.
2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Shekar A, Mabry SJ, Cheng MH, Aguilar JI,
Patel S, Zanella D, Saleeby DP, Zhu Y, Romanazzi T, Ulery-Reynolds
P, et al: Syntaxin 1 Ser14 phosphorylation is required for
nonvesicular dopamine release. Sci Adv. 9(eadd8417)2023.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Trus M and Atlas D: Non-ionotropic
voltage-gated calcium channel signaling. Channels (Austin).
18(2341077)2024.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Vardar G, Salazar-Lázaro A, Zobel S,
Trimbuch T and Rosenmund C: Syntaxin-1A modulates vesicle fusion in
mammalian neurons via juxtamembrane domain dependent palmitoylation
of its transmembrane domain. Elife. 11(e78182)2022.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Yeh CY, Ye Z, Moutal A, Gaur S, Henton AM,
Kouvaros S, Saloman JL, Hartnett-Scott KA, Tzounopoulos T, Khanna
R, et al: Defining the Kv2.1-syntaxin molecular interaction
identifies a first-in-class small molecule neuroprotectant. Proc
Natl Acad Sci USA. 116:15696–15705. 2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Yu H and Shen J: Faithful SM proteins
chaperone SNAREs on path to successful assembly. Proc Natl Acad Sci
USA. 120(e2219769120)2023.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Papantoniou C, Laugks U, Betzin J,
Capitanio C, Ferrero JJ, Sánchez-Prieto J, Schoch S, Brose N,
Baumeister W, Cooper BH, et al: Munc13- and SNAP25-dependent
molecular bridges play a key role in synaptic vesicle priming. Sci
Adv. 9(eadf6222)2023.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Cousin MA: Synaptophysin-dependent
synaptobrevin-2 trafficking at the presynapse-mechanism and
function. J Neurochem. 159:78–89. 2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Wang S and Ma C: Neuronal SNARE complex
assembly guided by Munc18-1 and Munc13-1. FEBS Open Bio.
12:1939–1957. 2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Tomaka W, Kiessling V and Tamm LK: The
role of Munc18 in regulating the spatial arrangement of syntaxin-1A
and SNARE complex assembly. Biophys J. 123(381a)2024.
|
|
51
|
Guiberson NGL, Black LS, Haller JE,
Brukner A, Abramov D, Ahmad S, Xie YX, Sharma M and Burré J:
Disease-linked mutations in Munc18-1 deplete synaptic Doc2. Brain.
147:2185–2202. 2024.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Li YZ, Wang Y, Jiao Q, Chi J, Liang Y, Fan
B and Li GY: Complexin regulation of synaptic vesicle release:
Mechanisms in the central nervous system and specialized retinal
ribbon synapses. Cell Commun Signal. 22(581)2024.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Cui L, Li H, Xi Y, Hu Q, Liu H, Fan J,
Xiang Y, Zhang X, Shui W and Lai Y: Vesicle trafficking and vesicle
fusion: Mechanisms, biological functions, and their implications
for potential disease therapy. Mol Biomed. 3(29)2022.PubMed/NCBI View Article : Google Scholar
|
|
54
|
López-Murcia FJ, Reim K, Jahn O,
Taschenberger H and Brose N: Acute complexin knockout abates
spontaneous and evoked transmitter release. Cell Rep.
26:2521–2530.e5. 2019.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Toulmé E, Salazar Lázaro A, Trimbuch T,
Rizo J and Rosenmund C: Neurotransmitter release is triggered by a
calcium-induced rearrangement in the synaptotagmin-1/SNARE complex
primary interface. Proc Natl Acad Sci USA.
121(e2409636121)2024.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Capuzzi E, Caldiroli A, Auxilia AM,
Borgonovo R, Capellazzi M, Clerici M and Buoli M: Biological
predictors of treatment response in adult attention deficit
hyperactivity disorder (ADHD): A systematic review. J Pers Med.
12(1742)2022.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Henriksen AD, Andrade A, Harris EP,
Rissman EF and Wolstenholme JT: Bisphenol a exposure in utero
disrupts hypothalamic gene expression particularly genes suspected
in autism spectrum disorders and neuron and hormone signaling. Int
J Mol Sci. 21(3129)2020.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Chen F, Chen H, Chen Y, Wei W, Sun Y,
Zhang L, Cui L and Wang Y: Dysfunction of the SNARE complex in
neurological and psychiatric disorders. Pharmacol Res.
165(105469)2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Cali E, Rocca C, Salpietro V and Houlden
H: Epileptic phenotypes associated with SNAREs and related synaptic
vesicle exocytosis machinery. Front Neurol.
12(806506)2021.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Tropeano M, Wöber-Bingöl C, Karwautz A,
Wagner G, Vassos E, Campos-de-Sousa S, Graggaber A, Zesch HE,
Kienbacher C, Natriashvili S, et al: Association analysis of STX1A
gene variants in common forms of migraine. Cephalalgia. 32:203–212.
2012.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Corominas R, Ribasés M, Cuenca-León E,
Narberhaus B, Serra SA, del Toro M, Roig M, Fernández-Fernández JM,
Macaya A and Cormand B: Contribution of syntaxin 1A to the genetic
susceptibility to migraine: A case-control association study in the
Spanish population. Neurosci Lett. 455:105–109. 2009.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Quintas M, Neto JL, Sequeiros J, Sousa A,
Pereira-Monteiro J, Lemos C and Alonso I: Going deep into synaptic
vesicle machinery genes and migraine susceptibility-a case-control
association study. Headache. 60:2152–2165. 2020.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Yang Y, Kim J, Kim HY, Ryoo N, Lee S, Kim
Y, Rhim H and Shin YK: Amyloid-β oligomers may impair
SNARE-mediated exocytosis by direct binding to syntaxin 1a. Cell
Rep. 12:1244–1251. 2015.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Kessi M, Duan H, Xiong J, Chen B, He F,
Yang L, Ma Y, Bamgbade OA, Peng J and Yin F:
Attention-deficit/hyperactive disorder updates. Front Mol Neurosci.
15(925049)2022.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Bonvicini C, Faraone SV and Scassellati C:
Common and specific genes and peripheral biomarkers in children and
adults with attention-deficit/hyperactivity disorder. World J Biol
Psychiatry. 19:80–100. 2018.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Poddar A, Gaddam S, Sonnaila S, Bavaraju
VSM and Agrawal S: Unraveling attention-deficit/hyperactivity
disorder etiology: Current challenges and future directions in
treatment. Neurosci. 6(41)2025.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Sánchez-Mora C, Cormand B, Ramos-Quiroga
JA, Hervás A, Bosch R, Palomar G, Nogueira M, Gómez-Barros N,
Richarte V, Corrales M, et al: Evaluation of common variants in 16
genes involved in the regulation of neurotransmitter release in
ADHD. Eur Neuropsychopharmacol. 23:426–435. 2013.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Pliszka SR: The neuropsychopharmacology of
attention-deficit/hyperactivity disorder. Biol Psychiatry.
57:1385–1390. 2005.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Yang L, Wang X, Liu X and Chen X: Striatal
syntaxin 1A is associated with development of Tourette syndrome in
an iminodipropionitrile-induced animal model. Dis Markers.
2022(1148191)2022.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Lanzo A, Safratowich BD, Kudumala SR,
Gallotta I, Zampi G, Di Schiavi E and Carvelli L: Silencing of
syntaxin 1A in the dopaminergic neurons decreases the activity of
the dopamine transporter and prevents amphetamine-induced behaviors
in C. elegans. Front Physiol. 9(576)2018.PubMed/NCBI View Article : Google Scholar
|
|
71
|
da Silva BS, Cupertino RB, Rovaris DL,
Schuch JB, Kappel DB, Müller D, Bandeira CE, Victor MM, Karam RG,
Mota NR, et al: Exocytosis-related genes and response to
methylphenidate treatment in adults with ADHD. Mol Psychiatry.
23:1446–1452. 2018.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Özdemir Ç, Şahin N and Edgünlü T: Vesicle
trafficking with snares: A perspective for autism. Mol Biol Rep.
49:12193–12202. 2022.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Reilly J, Gallagher L, Leader G and Shen
S: Coupling of autism genes to tissue-wide expression and
dysfunction of synapse, calcium signalling and transcriptional
regulation. PLoS One. 15(e0242773)2020.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Fujiwara T, Sanada M, Kofuji T and Akagawa
K: Unusual social behavior in HPC-1/syntaxin1A knockout mice is
caused by disruption of the oxytocinergic neural system. J
Neurochem. 138:117–123. 2016.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Fujiwara T, Kofuji T and Akagawa K:
Disturbance of the reciprocal-interaction between the OXTergic and
DAergic systems in the CNS causes atypical social behavior in
syntaxin 1A knockout mice. Behav Brain Res.
413(113447)2021.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Zhuang H, Liang Z, Ma G, Qureshi A, Ran X,
Feng C, Liu X, Yan X and Shen L: Autism spectrum disorder:
Pathogenesis, biomarker, and intervention therapy. MedComm (2020).
5(e497)2024.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Cartier E, Hamilton PJ, Belovich AN,
Shekar A, Campbell NG, Saunders C, Andreassen TF, Gether U,
Veenstra-Vanderweele J, Sutcliffe JS, et al: Rare autism-associated
variants implicate syntaxin 1 (STX1 R26Q) phosphorylation and the
dopamine transporter (hDAT R51W) in dopamine neurotransmission and
behaviors. EBioMedicine. 2:135–146. 2015.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Haase J, Killian AM, Magnani F and
Williams C: Regulation of the serotonin transporter by interacting
proteins. Biochem Soc Trans. 29:722–728. 2001.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Durdiaková J, Warrier V, Banerjee-Basu S,
Baron-Cohen S and Chakrabarti B: STX1A and asperger syndrome: A
replication study. Mol Autism. 5(14)2014.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Nakamura K, Anitha A, Yamada K, Tsujii M,
Iwayama Y, Hattori E, Toyota T, Suda S, Takei N, Iwata Y, et al:
Genetic and expression analyses reveal elevated expression of
syntaxin 1A (STX1A) in high functioning autism. Int J
Neuropsychopharmacol. 11:1073–1084. 2008.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Al-Ayadhi L, Elamin NE, Halepoto DM and
Mohammed A: Efficacy of auditory integration therapy (AIT) on
plasma syntaxin1A (STX1A) levels and amelioration of behavioral,
social, and sensory symptoms in children with autism spectrum
disorder (ASD). Int J Adv Applied Sci. 10:6–11. 2023.
|
|
82
|
Zhou J, Zheng Y, Liang G, Xu X, Liu J,
Chen S, Ge T, Wen P, Zhang Y, Liu X, et al: Atypical deletion of
Williams-beuren syndrome reveals the mechanism of
neurodevelopmental disorders. BMC Med Genomics.
15(79)2022.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Gao MC, Bellugi U, Dai L, Mills DL, Sobel
EM, Lange K and Korenberg JR: Intelligence in williams syndrome is
related to STX1A, which encodes a component of the presynaptic
SNARE complex. PLoS One. 5(e10292)2010.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Milligan TA: Epilepsy: A clinical
overview. Am J Med. 134:840–847. 2021.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Melland H, Arvell EH and Gordon SL:
Disorders of synaptic vesicle fusion machinery. J Neurochem.
157:130–164. 2021.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Verhage M and Sørensen JB: SNAREopathies:
Diversity in mechanisms and symptoms. Neuron. 107:22–37.
2020.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Spoto G, Valentini G, Saia MC, Butera A,
Amore G, Salpietro V, Nicotera AG and Di Rosa G: Synaptopathies in
developmental and epileptic encephalopathies: A focus on
pre-synaptic dysfunction. Front Neurol. 13(826211)2022.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Baghel R, Grover S, Kaur H, Jajodia A,
Parween S, Sinha J, Srivastava A, Srivastava AK, Bala K, Chandna P,
et al: Synergistic association of STX1A and VAMP2 with cryptogenic
epilepsy in north Indian population. Brain Behav.
6(e00490)2016.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Gu Y, Chiu SL, Liu B, Wu PH, Delannoy M,
Lin DT, Wirtz D and Huganir RL: Differential vesicular sorting of
AMPA and GABAA receptors. Proc Natl Acad Sci USA. 113:E922–E931.
2016.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Tang F, Chen L, Gao H, Lei Y, Pan L, Xiao
D and Li X: Munc18-1 contributes to hippocampal injury in septic
rats through regulation of Syntanxin1A and synaptophysin and
glutamate levels. Neurochem Res. 48:791–803. 2023.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Yu YX, Shen L, Xia P, Tang YW, Bao L and
Pei G: Syntaxin 1A promotes the endocytic sorting of EAAC1 leading
to inhibition of glutamate transport. J Cell Sci. 119:3776–3787.
2006.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Soldovieri MV, Boutry-Kryza N, Milh M,
Doummar D, Heron B, Bourel E, Ambrosino P, Miceli F, De Maria M,
Dorison N, et al: Novel KCNQ2 and KCNQ3 mutations in a large cohort
of families with benign neonatal epilepsy: First evidence for an
altered channel regulation by syntaxin-1A. Hum Mutat. 35:356–367.
2014.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Devaux J, Dhifallah S, De Maria M,
Stuart-Lopez G, Becq H, Milh M, Molinari F and Aniksztejn L: A
possible link between KCNQ2- and STXBP1-related encephalopathies:
STXBP1 reduces the inhibitory impact of syntaxin-1A on M current.
Epilepsia. 58:2073–2084. 2017.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Taura Y, Tozawa T, Fujimoto T, Ichise E,
Chiyonobu T, Itoh K and Iehara T: Myosin Va, a Novel interaction
partner of STXBP1, is required to transport Syntaxin1A to the
plasma membrane. Neuroscience. 524:256–268. 2023.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Pleș H, Florian IA, Timis TL,
Covache-Busuioc RA, Glavan LA, Dumitrascu DI, Popa AA, Bordeianu A
and Ciurea AV: Migraine: Advances in the pathogenesis and
treatment. Neurol Int. 15:1052–1105. 2023.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Ashina M, Terwindt GM, Al-Karagholi MA, de
Boer I, Lee MJ, Hay DL, Schulte LH, Hadjikhani N, Sinclair AJ,
Ashina H, et al: Migraine: Disease characterisation, biomarkers,
and precision medicine. Lancet. 397:1496–1504. 2021.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Hautakangas H, Winsvold BS, Ruotsalainen
SE, Bjornsdottir G, Harder AVE, Kogelman LJA, Thomas LF, Noordam R,
Benner C, Gormley P, et al: Genome-wide analysis of 102,084
migraine cases identifies 123 risk loci and subtype-specific risk
alleles. Nat Genet. 54:152–160. 2022.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Felício D, Alves-Ferreira M, Santos M,
Quintas M, Lopes AM, Lemos C, Pinto N and Martins S: Integrating
functional scoring and regulatory data to predict the effect of
non-coding SNPs in a complex neurological disease. Briefings Funct
Genomics. 23:138–149. 2024.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Felício D, Dias A, Martins S, Carvalho E,
Lopes AM, Pinto N, Lemos C, Santos M and Alves-Ferreira M:
Non-coding variants in VAMP2 and SNAP25 affect gene expression:
Potential implications in migraine susceptibility. J Headache Pain.
24(78)2023.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Kowalska M, Prendecki M, Kapelusiak-Pielok
M, Grzelak T, Łagan-Jędrzejczyk U, Wiszniewska M, Kozubski W and
Dorszewska J: Analysis of genetic variants in SCN1A, SCN2A, KCNK18,
TRPA1 and STX1A as a possible marker of migraine. Curr Genomics.
21:224–236. 2020.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Kitamura E and Imai N: Molecular and
cellular neurobiology of spreading depolarization/depression and
migraine: A narrative review. Int J Mol Sci.
25(11163)2024.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Vongseenin S, Ha-Ji-A-Sa N, Thanprasertsuk
S and Bongsebandhu-Phubhakdi S: Deciphering migraine pain
mechanisms through electrophysiological insights of trigeminal
ganglion neurons. Sci Rep. 13(14449)2023.PubMed/NCBI View Article : Google Scholar
|
|
103
|
O'Hare L, Tarasi L, Asher JM, Hibbard PB
and Romei V: Excitation-inhibition imbalance in migraine: From
neurotransmitters to brain oscillations. Int J Mol Sci.
24(10093)2023.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Juhasz G, Gecse K and Baksa D: Towards
precision medicine in migraine: Recent therapeutic advances and
potential biomarkers to understand heterogeneity and treatment
response. Pharmacol Ther. 250(108523)2023.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Ashina M, Buse DC, Ashina H, Pozo-Rosich
P, Peres MFP, Lee MJ, Terwindt GM, Halker Singh R, Tassorelli C, Do
TP, et al: Migraine: Integrated approaches to clinical management
and emerging treatments. Lancet. 397:1505–1518. 2021.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Bell T, Stokoe M, Khaira A, Webb M, Noel
M, Amoozegar F and Harris AD: GABA and glutamate in pediatric
migraine. Pain. 162:300–308. 2021.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Karsan N, Luiza Bastos A and Goadsby PJ:
Glutamate as a therapeutic substrate in migraine. Int J Mol Sci.
26(3023)2025.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Shi W, Sun H, Peng W, Chen Z, Wang Q, Lin
W, Ding M, Sun H, Wang X, Wang T, et al: A cross-sectional,
multicenter survey of the prevalence and influencing factors for
migraine in epilepsy. Epilepsia Open. 9:1406–1415. 2024.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Wang L, Cai XT, Zu MD, Zhang J and Wang Y:
Decreased resting-state functional connectivity of periaqueductal
gray in temporal lobe epilepsy comorbid with migraine. Front
Neurol. 12(636202)2021.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Engstrand H, Revdal E, Argren MB, Hagen K,
Zwart JA, Brodtkorb E and Winsvold BS: Relationship between
migraine and epilepsy in a large population-based cohort: The HUNT
study. Eur J Neurol. 31(e16496)2024.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Graff-Radford J, Yong KXX, Apostolova LG,
Bouwman FH, Carrillo M, Dickerson BC, Rabinovici GD, Schott JM,
Jones DT and Murray ME: New insights into atypical alzheimer's
disease in the era of biomarkers. Lancet, Neurol. 20:222–234.
2021.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Das S, Goossens J, Jacobs D, Dewit N,
Pijnenburg YAL, In ‘t Veld SGJG, Teunissen CE and Vanmechelen E:
Synaptic biomarkers in the cerebrospinal fluid associate
differentially with classical neuronal biomarkers in patients with
Alzheimer's disease and frontotemporal dementia. Alzheimers Res
Ther. 15(62)2023.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Sze CI, Bi H, Kleinschmidt-DeMasters BK,
Filley CM and Martin LJ: Selective regional loss of exocytotic
presynaptic vesicle proteins in Alzheimer's disease brains. J
Neurol Sci. 175:81–90. 2000.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Costa AS, Guerini FR, Arosio B, Galimberti
D, Zanzottera M, Bianchi A, Nemni R and Clerici M: SNARE complex
polymorphisms associate with alterations of visual selective
attention in Alzheimer's disease. J Alzheimers Dis. 69:179–188.
2019.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Hasan B, Khan A, Lenz C, Asif AR and Ahmed
N: Characterization of functional protein complexes from
Alzheimer's disease and healthy brain by mass spectrometry-based
proteome analysis. Sci Rep. 11(13891)2021.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Ramos-Miguel A, Jones AA, Petyuk VA,
Barakauskas VE, Barr AM, Leurgans SE, De Jager PL, Casaletto KB,
Schneider JA, Bennett DA and Honer WG: Proteomic identification of
select protein variants of the SNARE interactome associated with
cognitive reserve in a large community sample. Acta Neuropathol.
141:755–770. 2021.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Chen XQ, Zuo X, Becker A, Head E and
Mobley WC: Reduced synaptic proteins and SNARE complexes in Down
syndrome with Alzheimer's disease and the Dp16 mouse down syndrome
model: Impact of APP gene dose. Alzheimers Dementc. 19:2095–2116.
2023.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Shen Y, Ali M, Timsina J, Wang C, Do A,
Western D, Liu M, Gorijala P, Budde J, Liu H, et al: Systematic
proteomics in autosomal dominant Alzheimer's disease reveals
decades-early changes of CSF proteins in neuronal death, and immune
pathways. medRxiv: Jan 13, 2024 doi:
10.1101/2024.01.12.24301242.
|
|
119
|
Xiong Y, Zhang Y, Iqbal J, Ke M, Wang Y,
Li Y, Qing H and Deng Y: Differential expression of synaptic
proteins in unilateral 6-OHDA lesioned rat model-a comparative
proteomics approach. Proteomics. 14:1808–1819. 2014.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Niu M, Li Y, Li G, Zhou L, Luo N, Yao M,
Kang W and Liu J: A longitudinal study on α-synuclein in plasma
neuronal exosomes as a biomarker for Parkinson's disease
development and progression. Eur J Neurol. 27:967–974.
2020.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Agliardi C, Meloni M, Guerini FR,
Zanzottera M, Bolognesi E, Baglio F and Clerici M: Oligomeric α-syn
and SNARE complex proteins in peripheral extracellular vesicles of
neural origin are biomarkers for Parkinson's disease. Neurobiol
Dis. 148(105185)2021.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Cappelletti P, Filareti M, Masuelli L, Bei
R, Hassanzadeh K, Corbo M and Feligioni M: Syntaxin-1a and SNAP-25
expression level is increased in the blood samples of ischemic
stroke patients. Sci Rep. 12(14483)2022.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Lin YH, Yang D, Ni HY, Xu XM, Wu F, Lin L,
Chen J, Sun YY, Huang ZQ, Li SY, et al: Ketone bodies promote
stroke recovery via GAT-1-dependent cortical network remodeling.
Cell Rep. 42(112294)2023.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Joy MT and Carmichael ST: Encouraging an
excitable brain state: Mechanisms of brain repair in stroke. Nat
Rev Neurosci. 22:38–53. 2021.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Perovic M, Pavlovic D, Palmer Z, Udo MSB,
Citadin CT, Rodgers KM, Wu CY, Zhang Q, Lin HW and Tesic V:
Modulation of GABAergic system as a therapeutic option in stroke.
Exp Neurol. 384(115050)2025.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Schulien AJ, Yeh CY, Orange BN, Pav OJ,
Hopkins MP, Moutal A, Khanna R, Sun D, Justice JA and Aizenman E:
Targeted disruption of Kv2.1-VAPA association provides
neuroprotection against ischemic stroke in mice by declustering
Kv2.1 channels. Sci Adv. 6(eaaz8110)2020.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Yeh CY, Schulien AJ, Molyneaux BJ and
Aizenman E: Lessons from recent advances in ischemic stroke
management and targeting Kv2.1 for neuroprotection. Int J Mol Sci.
21(6107)2020.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Chow LWC and Leung YM: The versatile kv
channels in the nervous system: Actions beyond action potentials.
Cell Mol Life Sci. 77:2473–2482. 2020.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Schwarz K and Schmitz F: Synapse
dysfunctions in multiple sclerosis. Int J Mol Sci.
24(1639)2023.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Yalın OÖ, Gökdoğan Edgünlü T, Karakaş
Çelik S, Emre U, Güneş T, Erdal Y and Eroğlu Ünal A: Novel SNARE
complex polymorphisms associated with multiple sclerosis: Signs of
synaptopathy in multiple sclerosis. Balk Med J. 36:174–178.
2019.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Oraby MI, Soliman RH, Abdel Kader NA,
Abdul Galil EM and Masoud MM: Syntaxin 1A gene polymorphism in
multiple sclerosis: A case-control study. Egypt J Neurol Psychiatry
Neurosurg. 60(47)2024.
|
|
132
|
Behzadi P, Dodero VI and Golubnitschaja O:
Systemic inflammation as the health-related communication tool
between the human host and gut microbiota in the framework of
predictive, preventive, and personalized medicine. In: All Around
Suboptimal Health: Advanced Approaches by Predictive, Preventive
and Personalised Medicine for Healthy Populations. Wang W (ed).
Springer Nature Switzerland, Cham, pp203-241, 2024.
|
|
133
|
Dodero VI, Morré SA and Behzadi P:
Editorial: Gut microbiota and immunity in health and disease:
Dysbiosis and Eubiosis's effects on the human body. Front Immunol.
15(1536258)2024.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Loh JS, Mak WQ, Tan LKS, Ng CX, Chan HH,
Yeow SH, Foo JB, Ong YS, How CW and Khaw KY: Microbiota-gut-brain
axis and its therapeutic applications in neurodegenerative
diseases. Signal Transduction Targeted Ther. 9(37)2024.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Behzadi P, Sameer AS, Nissar S, Banday MZ,
Gajdács M, García-Perdomo HA, Akhtar K, Pinheiro M, Magnusson P,
Sarshar M and Ambrosi C: The interleukin-1 (IL-1) superfamily
cytokines and their single nucleotide polymorphisms (SNPs). J
Immunol Res. 2022(2054431)2022.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Behzadi P, Chandran D, Chakraborty C,
Bhattacharya M, Saikumar G, Dhama K, Chakraborty A, Mukherjee S and
Sarshar M: The dual role of toll-like receptors in COVID-19:
Balancing protective immunity and immunopathogenesis. Int J Biol
Macromol. 284(137836)2025.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Mukherjee S, Patra R, Behzadi P, Masotti
A, Paolini A and Sarshar M: Toll-like receptor-guided therapeutic
intervention of human cancers: Molecular and immunological
perspectives. Front Immunol. 14(1244345)2023.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Petakh P, Duve K, Oksenych V, Behzadi P
and Kamyshnyi O: Molecular mechanisms and therapeutic possibilities
of short-chain fatty acids in posttraumatic stress disorder
patients: A mini-review. Front Neurosci. 18(1394953)2024.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Petakh P, Behzadi P, Oksenych V and
Kamyshnyi O: Current treatment options for leptospirosis: A
mini-review. Front Microbiol. 15(1403765)2024.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Behzadi P, Kim CH, Pawlak EA and Algammal
A: Editorial: The innate and adaptive immune system in human
urinary system. Front Immunol. 14(1294869)2023.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Kaur U and Lee JC: Unroofing site-specific
α-synuclein-lipid interactions at the plasma membrane. Proc Natl
Acad Sci USA. 117:18977–18983. 2020.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Meloni M, Agliardi C, Guerini FR, Saibene
FL, Milner AV, Zanzottera M, Bolognesi E, Puligheddu M, Figorilli
M, Navarro J and Clerici M: Oligomeric alpha-synuclein and STX-1A
from neural-derived extracellular vesicles (NDEVs) as possible
biomarkers of REM sleep behavior disorder in Parkinson's disease: A
preliminary cohort study. Int J Mol Sci. 24(8839)2023.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Freibauer A, Wohlleben M and Boelman C:
STXBP1-related disorders: Clinical presentation, molecular
function, treatment, and future directions. Genes.
14(2179)2023.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Klöckner C, Sticht H, Zacher P, Popp B,
Babcock HE, Bakker DP, Barwick K, Bonfert MV, Bönnemann CG,
Brilstra EH, et al: De novo variants in SNAP25 cause an early-onset
developmental and epileptic encephalopathy. Genet Med. 23:653–660.
2021.PubMed/NCBI View Article : Google Scholar
|