Introduction
The role of mitochondria in the initiation of
apoptosis in a number of studies is well documented (1–4). A
reduction in mitochondrial transmembrane potential (ΔΨm) has been
observed before the manifestation of nuclear apoptosis in certain
cell types (2,6–11),
and nuclear apoptosis is inhibited by the stabilization of ΔΨm
(12–16). Additionally, mitochondria have been
shown to harbor apoptogenic molecules, such as SMAC/DIABLO, HTRA2,
cytochrome c, caspases and AIF (apoptosis-inducing factor),
liberating such molecules into the cytosol to participate in the
apoptotic process (13,17–22).
By contrast, there are also reports of non-ΔΨm-dependent apoptosis
(23), and studies indicating that
mitochondria may be implicated in cell death suppression (24).
Fas (CD95), a type I transmembrane protein, consists
of a cell surface receptor which transduces death signaling in a
wide variety of cells upon stimulation by the Fas ligand or
agonistic Fas antibodies (25–32).
Changes in sensitivity to apoptosis mediated by Fas have been
linked to a lack of cell surface Fas, overexpression of Bcl-2
family members, alteration in Fas intracellular signaling pathways,
existence of Fas as a soluble protein, and expression of inhibitory
factor(s) (28,33–39).
However, it has been revealed that mere expression of Fas and Bcl-2
(or Bcl-2-like molecules) is not predictive of biological
responsiveness (40).
Insensitivity of the Fas receptor to anti-Fas antibodies has been
suggested to be a consequence of mitogen-activated protein kinase
activation by the Fas receptor, which in turn interferes with
caspase activation (41). It has
also been demonstrated that Fas activates cells to die with or
without the involvement of mitochondria (42).
Proteins encoded by mitochondrial DNA (mtDNA) are
also implicated in the sensitivity to and execution of apoptosis,
and may be critical in the initiation of growth arrest and
apoptosis (43). By contrast, it
has been shown that neither the apoptosis nor the protective effect
of Bcl-2-type proteins depend on mitochondrial respiration
(44–48). The elimination of mitochondrial
oxidative metabolism has been found to inhibit not only tumor
necrosis factor (TNF)-mediated cytotoxicity, but also to reduce the
TNF-mediated gene regulatory signaling pathways (49). However, in cells depleted of mtDNA,
a diminished tumorigenic phenotype and an increased sensitivity to
cytotoxic drugs was noted (50–52).
Other studies have reported that anti-mitochondrial agents
chemosensitized glioblastoma (GBM) cells to cytotoxic agents
(52).
The present study was undertaken to investigate the
relationship between Fas and mitochondria in mediating apoptosis in
GBM cells. The cell surface expression of Fas was evaluated in GBM
cells upon the depletion of mtDNA, and in cells treated with
mitochondrial respiratory chain complex inhibitors. Sensitivity to
Fas antibodies and cis-diammine-dichloroplatinum (cisplatin) was
determined in order to evaluate whether alterations in Fas
expression lead to changes in response to the death inducers upon
mtDNA depletion. The results suggest that the expression of cell
surface Fas is not necessarily predictive of biological
responsiveness. In addition, the response of cells to cytotoxic
agents, such as cisplatin, is distinct to that of anti-Fas
antibodies, despite similar alterations at the mitochondrial
level.
Materials and methods
Cell culture
The GBM cell line DBTRG-O5MG was a gift from Dr
Carol Kruse (Sanford Burnham Institute). The U87 cell line was
purchased from ATCC (Rockville, MA, USA). The DBTRG-O5MG and U87
cell lines were cultured in RPMI-1640 supplemented with 10% FBS,
10,000 U/1 of penicillin-streptomycin, 4.5 g/1 glucose, 50
μg/ml uridine and 1 mM pyruvate. Cells were maintained at
37°C in 5% CO2. All culture mediums and supplements were
obtained from Life Technologies Inc., Gaithersburg, MD, USA.
Generation of Rho− cells
Rho− cells were generated from DBTRG-05MG
and U87 cells by depletion of their mtDNA in culture medium
containing 30 ng/ml EtBr (Sigma Chemical Company, St. Louis, MO,
USA). After at least 30 cell divisions, the Rho− status
of the cells was established by determining the loss of mtDNA by
PCR and by the auxotrophic dependence of the cells on pyruvate and
uridine. Cells were harvested and total DNA was subjected to PCR
(25 cycles) using the following set of primers: MT-B (forward)
5′-GGAACAAGCATCAAG CAC-3′ and MT-B' (reverse)
5′-GGCCATGGGTATGTTGTT-3′ (Genosys Biotechnologies Inc., The
Woodlands, TX, USA) as previously described (47). Auxotrophy on uridine and pyruvate
for survival, a characteristic feature of Rho− cells
(53), also confirmed the
Rho− status of the cells.
Anti-Fas and cisplatin-mediated
apoptosis
Cells were plated in 96-well plates at
2×104 (DBTRG-O5MG) or 4×104 (U87). After the
cells were allowed to adhere for 2–4 h, media were replaced with
fresh complete medium, and cells were incubated with various
concentrations of mouse monoclonal anti-Fas antibodies (clone
CH-11; Upstate Biotechnology, Lake Placid, NY, USA) for 24 h (U87)
or 72 h (DBTRG-O5MG). Wells containing no treatment or mouse IgM
(Sigma Chemical Company) served as controls. Cells were harvested,
washed in PBS and incubated at 4°C overnight in hypotonic
fluorochrome solution (0.1% sodium citrate, 0.1% Triton X-100 and
50 μg/ml propidium iodide). A FACScan flowcytometer
(Coulter, Miami, FL, USA) was used to quantify the percentage of
cells undergoing apoptosis as previously described (54). Nuclei that fluoresced below the G1
DNA represented apoptotic nuclei and are presented as the
percentage of apoptosis (5,000 gated events). Death induced by
cisplatin (Sigma Chemical Company) was analyzed as above by
incubating 4×104 cells with 20 μM cisplatin for
42–48 h. Wells with vehicle alone served as controls.
Cell surface Fas expression
Fas expression at the cell surface was analyzed
using flow cytometry. Adherent cells were disassociated into a
single-cell suspension with 10 mM EDTA in PBS. The cells were then
pelleted, washed once in PBS and resuspended in flow cytometry
buffer (PBS containing 1% FBS). Cells (106) were
incubated with anti-Fas antibodies or an isotype (20 μg/ml)
in flow cytometry buffer for 30 min on ice. Cells were then washed
with flow cytometry buffer, incubated with FITC-conjugated
anti-mouse IgG for 30 min on ice, washed once with flow cytometry
buffer, resuspended in flow cytometry buffer and analyzed using a
FACScan flow cytometer. A minimum of 20,000 gated events per sample
was analyzed.
Treatment of cells with mitochondrial
respiratory chain complex inhibitors
DBTRG-O5MG and U87 cells were incubated with
non-lethal concentrations of rotenone (0.098 μM),
thenoyltrifluoroacetone (31.25 μM), antimycin A (12.5
μM), sodium azide (500 μM), oligomycin B (1.56
μM) and membrane potential disrupting agent valinomycin
(0.0037 μM). After 24 h, Fas expression on the cell surface
was determined as noted. A non-lethal concentration of these
inhibitors was previously determined by clonogenic survival assay
of the cells treated with various concentrations of each of the
inhibitors (data not shown). To assess the inhibition of the
mitochondrial function by mitochondrial complex inhibitors,
cytochrome c oxidase activity was measured by treating the isolated
mitochondria with one of the complex inhibitors (500 μM
sodium azide). All chemicals used were from Sigma Chemical
Company.
Isolation and purification of
mitochondria
Mitochondria were isolated as described (4). Briefly, cells were harvested, washed
and stored at −70°C until use for the isolation of mitochondria.
Cells were then resuspended in ice-cold buffer (250 mM sucrose, 20
mM HEPES-NaOH, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM
DTT, 17 μg/ml phenylmethylsulfonylfluoride, 8 μg/ml
aprotinin, 2 μg/ml leupeptin, 1 μg/ml chymostatin, 1
μg/ml pepstatin and 1 μg/ml antipain). Cells were
homogenized using a tight fitting dounce homogenizer, 20–30 strokes
on ice. After pelleting the nuclei and unlysed cells at 100 × g for
10 min at 4°C in a refrigerated microcentrifuge (Savant
Instruments, Inc., Farmingdale, NY, USA), the mitochondria were
pelleted at 10,000 × g for 25 min at 4°C in an ultracentrifuge
(Beckman Instruments Inc., Palo Alto, CA, USA). Mitochondria that
banded at a density of 1.035 (as monitored by density marker beads)
were collected.
SDS PAGE and Western blotting
Proteins of mitochondria and the whole cell
homogenate were estimated using bicinchoninic acid (Sigma Chemical
Company). Crude mitochondria, whole cell homogenate and aliquots of
purified mitochondria (50 μg) were boiled in Laemmli sample
buffer and separated using 12% SDS PAGE. Proteins were blotted onto
nitrocellulose, and the Fas protein was detected using rabbit
polyclonal anti-Fas antibody (C-20; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA) at a 1:1,000 dilution. A goat anti-rabbit
antibody conjugated with horseradish peroxidase (Santa Cruz
Biotechnology) was used at a 1:10,000 dilution to detect
immunoreactive proteins, which were visualized by enhanced
chemoluminescence (Amersham Pharmacia Biotech Inc., Piscataway, NJ,
USA).
Determination of ATP levels
ATP levels were determined using the Sigma kit 366
(Sigma Chemical Company) according to the manufacturer's
instructions, with 106 cells.
Statistics
The results were calculated as the arithmetic mean ±
SEM. The student's t-test was used to compare the groups. p<0.05
was regarded as significant.
Results
Characterization of Rho−
cells
PCR
DBTRG-05MG and U87 cells were depleted of the mtDNA
by culturing in a medium containing 30 ng/ml EtBr. The
Rho− status of the mtDNA-depleted DBTRG-05MG and U87
cells was determined by PCR using a mitochondrial-specific DNA
primer. A 2.6-kb mtDNA amplified from the DNA of Rho+
cells was not observed from the DNA of the mtDNA-depleted
DBTRG-O5MG and U87 cells by PCR; the identity of the PCR-amplified
products was confirmed by sequencing (data not shown). These
results indicate that the Rho− DBTRG-05MG and U87 cells
did not harbor mtDNA.
Auxotrophy to uridine and
pyruvate
The mtDNA-depleted cells were evaluated for
auxotrophic dependence on uridine and pyruvate. The mtDNA-depleted
cells died after 7–10 days in the culture medium lacking uridine
and pyruvate. The Rho+ cells continued to divide and
propagate during the entire period when loss of viability was noted
in the respective Rho− cells. Taken together, these
results confirm the Rho− status of EtBr-treated
DBTRG-05MG and U87 cells.
Measurement of ATP levels
In order to assess the effect of the loss of mtDNA
on ATP levels, cellular ATP was measured in the DBTRG-05MG and U87
Rho+ cells and their respective Rho− cells
(Table I). The ATP levels of
DBTRG-05MG Rho+ and Rho− cells were 14.7±0.33
and 12.7±1.33 μg/106 cells, respectively, and the
ATP levels of U87 Rho+ and Rho− cells were
16.3±1.20 and 14.3±1.45 μg/106 cells,
respectively. No significant differences were detected in the
amount of ATP in Rho− cells as compared to their
respective Rho+ cells.
 | Table I.Cellular ATP levels. |
Table I.
Cellular ATP levels.
| Cells |
Rho+ |
Rho− |
|---|
| U87 | 16.3±1.20 | 14.3±1.45 |
| DBTRG-O5MG | 14.7±0.33 | 12.7±1.33 |
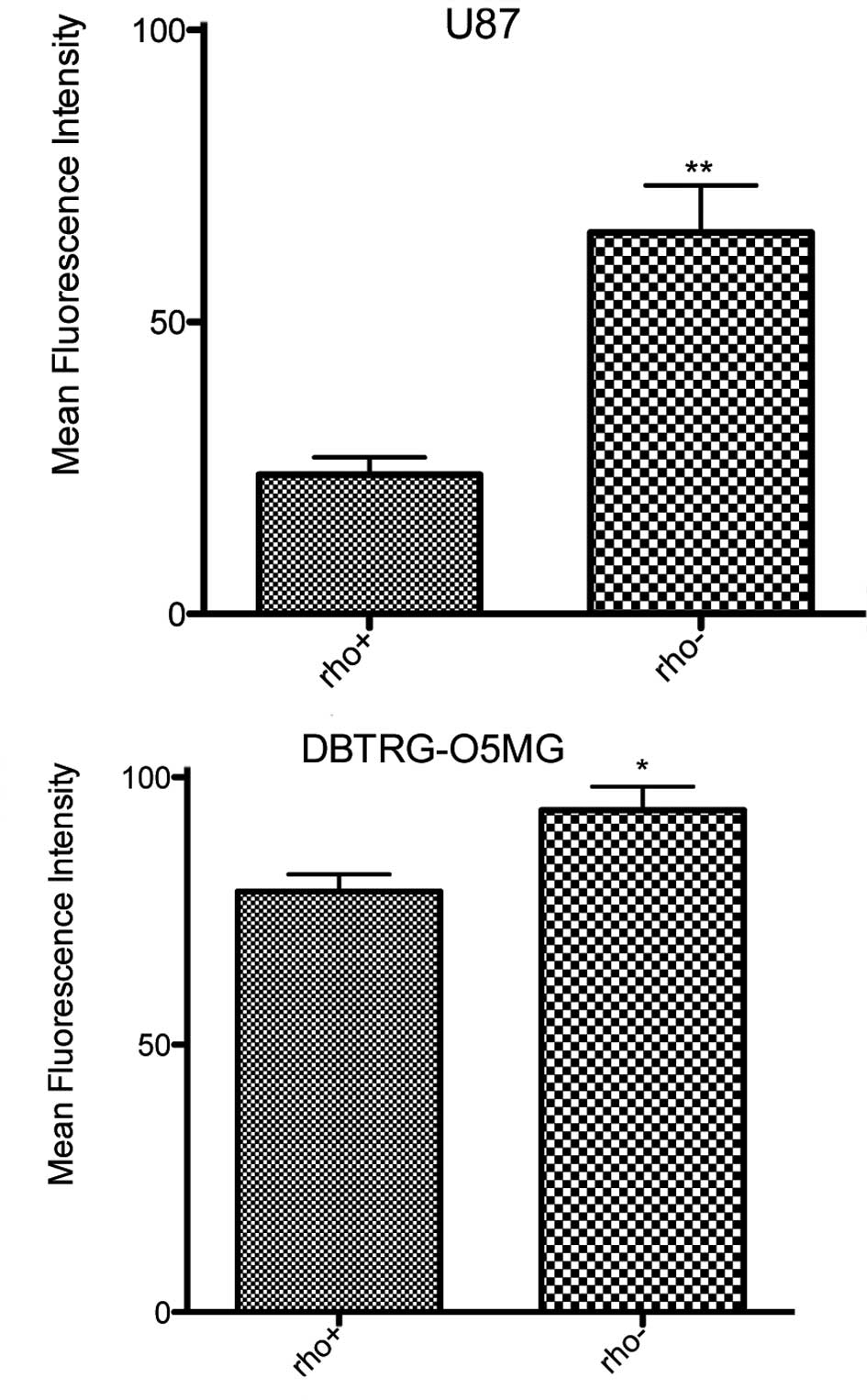
Evaluation of cell surface Fas
The cell surface expression of Fas was determined in
the Rho+ and Rho− cells in order to assess
its association with the depletion of mtDNA. Fas expression on the
cell surface was found to be enhanced in the Rho−
DBTRG-O5MG and U87 cells as compared to their respective
Rho+ cells (Fig. 1).
There was a 2.7-fold increase in the cell surface expression of Fas
in the U87 Rho− cells (p<0.0001) and a 19% increase
in surface Fas in the DBTRGO5MG Rho− cells (p<0.045)
as compared to the Rho+ cells.
DBTRG-O5MG and U87 Rho+ cells were then
treated with mitochondrial respiratory chain complex inhibitors
(Table II). Treatment with
rotenone increased the level of Fas in the U87 cells by 70%
(p<0.0001) and in the DBTRGO5MG cells by 35% (p<0.0161) as
compared to the untreated cells. Valinomycin was also observed to
increase Fas expression by over 30% in both cell lines (p<0.001,
U87 cells; p<0.007, DBTRG-05MG cells) as compared to untreated
cells. Moreover, antimycin A increased the level of Fas in the U87
cells (48%, p<0.0012), and oligomycin B increased the cell
surface Fas in the DBTRG-05MG cells (37%, p<0.001). These
results suggest that the depletion of mtDNA and the inhibition of
mitochondrial respiratory chain complex function alter the
expression of Fas at the surface of glioma cells.
| Table II.Effect of mitochondrial complex
inhibitors on the surface expression of Fas. |
Table II.
Effect of mitochondrial complex
inhibitors on the surface expression of Fas.
| Treatment | Mean % increase
over control
| Significance
(p-value vs. untreated cells)
|
|---|
| U87 | DBTRG-O5MG | U87 | DBTRG-O5MG |
|---|
| Rotenone | 70 | 35 | 0.0001 | 0.0161 |
| Antimycin A | 48 | - | 0.0012 | NS |
| Oligomycin | - | 37 | - | 0.0010 |
| Valinomycin | 43 | 32 | 0.0070 | 0.0010 |
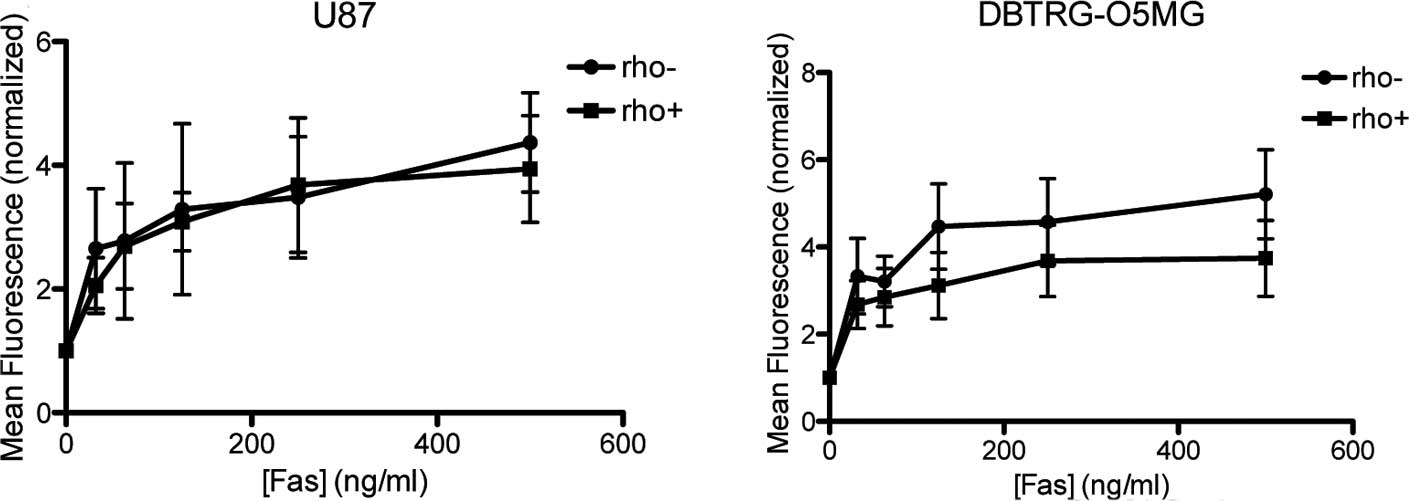
Effect of anti-Fas antibodies on
DBTRG-O5MG and U87 cells
Fas-mediated apoptosis was evaluated in the
Rho+ and mtDNA-depleted DBTRG-O5MG and U87 cells by FACS
analysis after the cells were subjected to apoptosis-inducing
anti-Fas antibodies (Fig. 2).
Sensitivity of the DBTRG-O5MG cells to anti-Fas antibodies at a
concentration ranging between 15 and 1,000 ng/ml was similar in the
Rho+ and Rho− cells (Fig. 2A). Similarly, in the U87
Rho− cells, the percentage of cells undergoing apoptosis
when treated with anti-Fas anti-bodies was not significantly
different compared to the Rho+ cells (Fig. 2B).
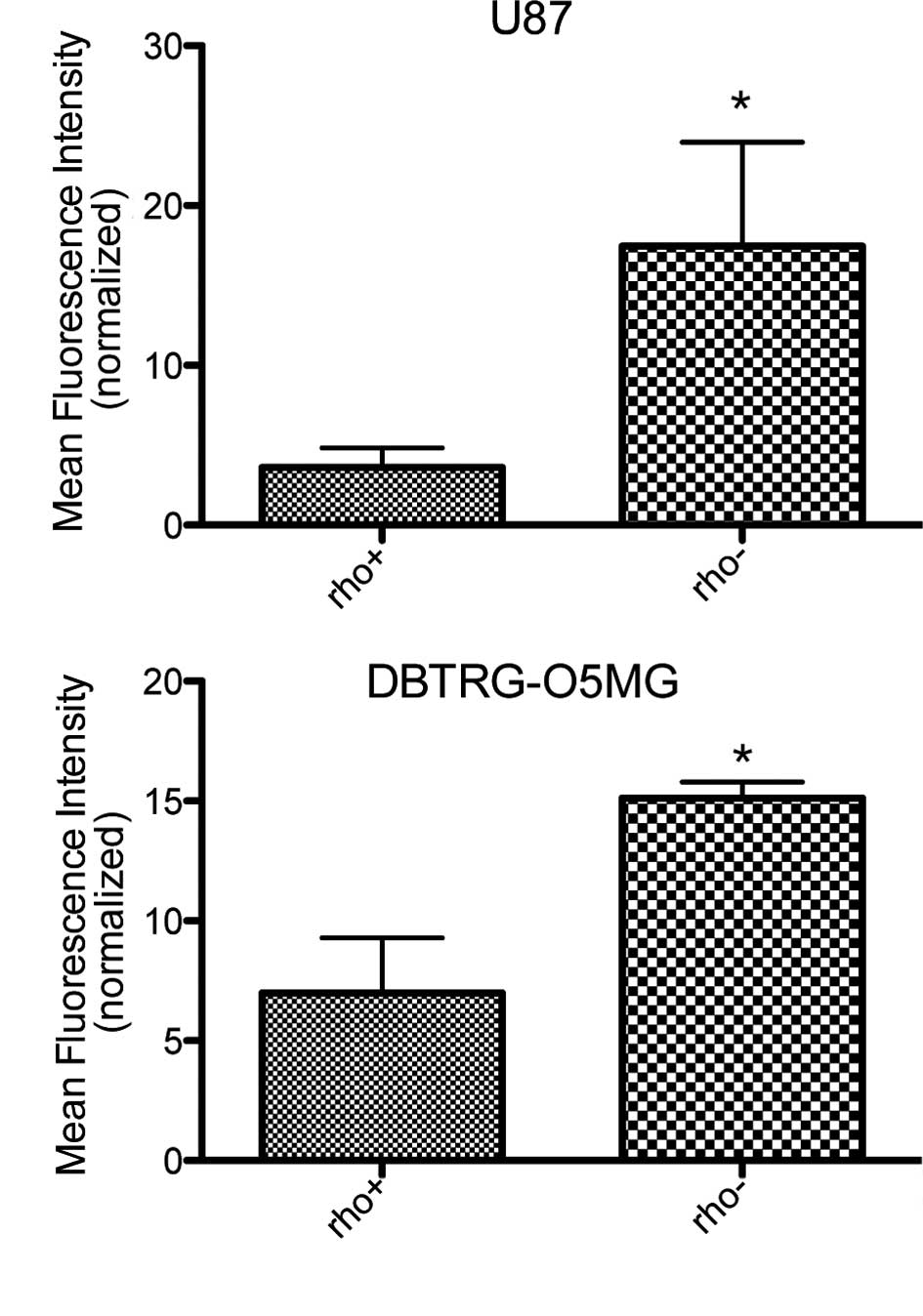
Sensitivity of DBTRG-O5MG and U87
Rho+ and their respective Rho− cells to
cisplatin
In order to assess the sensitivity of DBTRG-05MG and
U87 Rho+ and their respective Rho− cells to
cytotoxic agents, cells were treated with cisplatin (20 μM)
and subjected to FACS analysis (Fig.
3). DBTRG-O5MG and U87 Rho− cells were more
sensitive to the apoptosis-inducing effects of cisplatin; the U87
Rho− cells revealed a 4-fold and the DBTRG-O5MG cells a
2-fold increase in sensitivity to apoptosis compared to the
Rho+ cells at 20 μM. Taken together, these
results suggest that the depletion of mtDNA leads to an increased
sensitivity of Rho− cells to cisplatin.
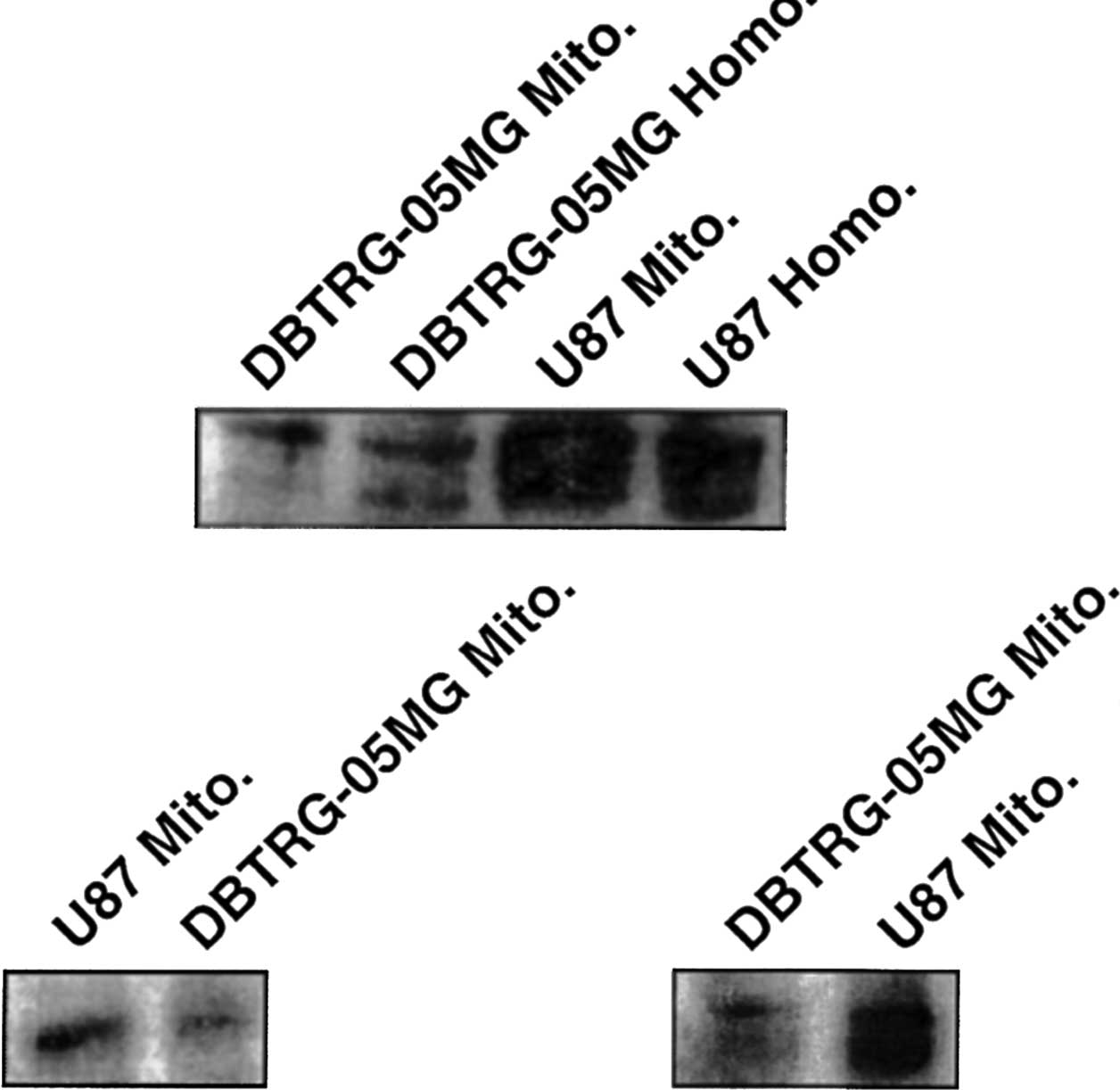
Fas protein and mitochondria
Western blot analysis of the mitochondria and whole
cell homogenate using Fas antibodies revealed a predominant 43-kDa
Fas protein band (with associated glycoforms) in both the
DBTRG-O5MG and U87 glioma cells (Fig.
4A). With percoll purification of mitochondria alone, similar
bands were noted in the immunoblots when probed with the anti-Fas
antibody (Fig. 4C); probes to
cyto-chrome c identified the mitochondrial fraction (Fig. 4B), while probes to actin did not
reveal staining (data not shown).
Discussion
Previous studies suggest an involvement of mtDNA in
sensitivity to apoptosis (43,50–52).
The present study was undertaken to assess the relationship between
mitochondria and Fas in mediating apoptosis. We found that, while
the depletion of mtDNA was associated with an increase in surface
Fas in both GBM cell lines, sensitivity to anti-Fas antibodies was
not increased in the Rho− cells as compared to the
Rho+ cells. However, the sensitivity to cisplatin was
increased in the Rho− cells as compared to the
Rho+ cells, in agreement with results from previous
studies (50,51). There may be several reasons for
this observation. The degree of sensitivity to Fas-mediated
apoptosis is reported to be dependent on several factors, including
expression of the functional Fas molecule on the cell surface
(56,57), mutations in the death domain of Fas
(33), expression of soluble Fas
(34), reduced expression of Fas
(27), intracellular signaling
cascade activation upon Fas ligation (40,58),
expression of the components of DISC and inhibition of essential
signaling pathways (28,32,35–39,41).
It has been revealed that, although Fas antigen is expressed on
numerous myeloma cell lines, only some respond to the
anti-Fas/Fas-ligand (59,60). Indeed, although the mechanism for
increased Fas expression with no concomitant increase in
sensitivity to anti-Fas antibodies is not clearly understood, it is
possible that the observed increases in Fas expression do not
result in enhanced death due to saturation of signaling pathways or
overexpression of functionally inactive Fas. It can also be
speculated that the inhibition of mitochondria via Rho−
status in these glioma cells also reduces the release of intrinsic
factors, including SMAC/DIABLO and/or Htra2; in this way, the
inhibition of XIAP is released, thus inhibiting caspase-3 and -7
either before or after Fas-induced activation via DISC/caspase-8
and -10. It would be anticipated that the linkage to the intrinsic
pathway via truncated BID to BAK and BAX might be impaired at the
level of mitochondria due to Rho− status. Nonetheless,
the fact that cisplatin-induced apoptosis still occurs – and with
higher levels of sensitivity in Rho− vs. Rho+
cells – suggests that the mechanism of at least the cytotoxic
drug-induced apoptosis is intact, and the mitochondria, acting as a
‘rheostat’ to sensitivity at least at the level of the intrinsic
pathway, continue to be maintained. The expression of components of
the Fas signaling pathway in Rho− cells is being
investigated in order to determine the mechanism of the observed
ineffective death signaling by Fas in Rho− cells.
Notably, in a functional model of Rho−
cells, Leber hereditary optic neuropathy tissues with mutations at
mtDNA positions 11778 and 3460 were found to have increased
sensitivity to the engagement of Fas compared to normal tissue
controls (61). This suggests that
mitochondrial dysfunction at the level of the tumor cell may not be
sufficient to facilitate apoptosis in such cells per se, but
may be in such mitochondrial diseases. This may be related to more
complex factors, including the BAX/BCL2 ratio (62) or the release of mitochondrial
apoptosis factors (63).
Furthermore, although the up-regulation of Fas was noted in cells
treated with cisplatin and other cytotoxic agents (64–67),
drug-induced apoptosis occurs independently of the Fas/Fas ligand
system (68–70). The increased sensitivity to
cisplatin of the Rho− DBTRG-O5MG and U87 cells appears
to be independent of the enhanced Fas expression and, in these GBM
cells, confirms previous studies in Rho− cells (50–52).
Treatment of Rho+ cells with
mitochondrial respiratory chain complex inhibitors enhanced the
levels of cell surface Fas expression. The variation of response,
both at the level of the different degrees of Fas expression noted
vs. Rho− cells and between the independent pharmacologic
inhibitors, may be a result of the dose of complex inhibitors used,
which allowed cells to survive. Moreover, differential effects of
antimycin A and oligomycin B observed on the GBM cells may be due
to the respective amounts of functional protein present and/or due
to deletions and mutations in mtDNA. Nonetheless, these
observations support the increased expression of Fas on the surface
of Rho− cells, depleted of mtDNA. Indeed, cells with
impaired mitochondrial function were found to have increased levels
of Fas mRNA (71), corroborating
the present observations.
Due to the relationship between mtDNA status and Fas
surface expression, we evaluated the localization of Fas to the
mitochondrial protein fraction as determined by whole cell
homogenates and percoll-purified mitochondria. Notably, Fas was
found to segregate with mitochondrial protein in the purified
percoll fraction. While it is only speculated on the role of Fas
therein, many proteins associated with apoptosis are found within
the mitochondria (22). The
ability of the mitochondria to regulate manifestations of the
extrinsic pathway is related to the mitochondrial distribution of
Fas; there is both supporting (42,71)
and refuting (24,34) evidence to this effect. Further
evaluation of the relationship(s) between the extrinsic and
intrinsic pathways of apoptosis, particularly in relation to Fas
protein expression, is of interest in this regard.
In conclusion, our studies show that the depletion
of mtDNA alters the expression of cell surface Fas. This is
supported by the results obtained upon treatment of the parental
cells (Rho+) with mitochondrial respiratory chain
complex inhibitors, wherein altered expression of cell surface Fas
was observed. The sensitivity of Rho− cells to
cisplatin, but not to anti-Fas antibodies, was found to be enhanced
when compared to Rho+ cells, despite similar alteration
at the mitochondrial level.
Acknowledgements
We thank C. Kruse for the DBTRG-O5MG
cell line, B. Laxman and L. Miller for technical assistance and S.
Munson for administrative assistance. This study was supported by
grants from the National Cancer Institute (CA73916) and the
American Society of Clinical Oncology.
References
|
1.
|
Henkart PA and Grinstein S: Apoptosis:
mitochondria resurrected? J Exp Med. 183:1293–1295. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Kroemer G, Zamzami N and Susin SA:
Mitochondrial control of apoptosis. Immunol Today. 18:44–51. 1997.
View Article : Google Scholar
|
|
3.
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Cavalli LR and Liang BC: Mutagenesis,
tumorigenicity and apoptosis: are the mitochondria involved? Mutat
Res. 398:19–26. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Newmeyer DD, Farschon DM and Reed JC:
Cell-free apoptosis in Xenopus egg extracts: inhibition by Bel-2
and requirement for an organelle fraction enriched in mitochondria.
Cell. 79:353–364. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Vayssiere JL, Petit PX, Risler Y and
Mignotte B: Commitment to apoptosis is associated with changes in
mitochondrial biogenesis and activity in cell lines conditionally
immortalized with simian virus 40. Proc Natl Acad Sci USA.
91:11752–11756. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Cossarizza A, Franceschi C, Monti D, et
al: Protective effect of N-acetylcysteine in tumor necrosis
factor-alpha-induced apoptosis in U937 cells: the role of
mitochondria. Exp Cell Res. 220:232–240. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Petit PX, Lecoeur H, Zorn E, et al:
Alterations in mitochondrial structure and function are early
events of dexamethasone-induced thymocyte apoptosis. J Cell Biol.
130:157–167. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Castedo M, Macho A, Zamzani N, et al:
Mitochondrial perturbations define lymphocytes undergoing apoptotic
depletion in vivo. Eur J Immunol. 25:3277–3284. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Zamzami N, Marchetti P, Castedo M, et al:
Sequential reduction of mitochondrial transmembrane potential and
generation of reactive oxygen species in early programmed cell
death. J Exp Med. 182:367–377. 1995. View Article : Google Scholar
|
|
11.
|
Zamzami N, Marchetti P, Castedo M, et al:
Mitochondrial control of nuclear apoptosis. J Exp Med.
183:1533–1544. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Shimizu S, Eguchi Y, Kamiike W, et al:
Bcl-2 blocks loss of mitochondrial membrane potential while ICE
inhibitors act at a different step during inhibition of death
induced by respiratory chain inhibitors. Oncogene. 13:21–29.
1996.
|
|
13.
|
Susin SA, Zamzami N, Castedo M, et al:
Bcl-2 inhibits the mitochondrial release of an apoptogenic
protease. J Exp Med. 184:1331–1341. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Zamzami N, Marchetti P, Castedo M, et al:
Inhibitors of permeability transition interfere with the disruption
of the mitochondrial transmembrane potential during apoptosis. FEBS
Lett. 384:53–57. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Boise LH and Thompson CB: Bcl-x(L) can
inhibit apoptosis in cells that have undergone Fas-induced protease
activation. Proc Natl Acad Sci USA. 94:3759–3764. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Kroemer G: The proto-oncogene Bcl-2 and
its role in regulating apoptosis. Nat Med. 3:614–620. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Stuart RA and Neupert W: Apocytochrome c:
an exceptional mitochondrial precursor protein using an exceptional
import pathway. Biochimie. 72:115–121. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release of cytochrome c from mitochondria: a
primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Mancini M, Nicholson DW, Roy S, et al: The
caspase-3 precursor has a cytosolic and mitochondrial distribution:
implications for apoptotic signaling. J Cell Biol. 140:1485–1495.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Susin SA, Lorenzo HK, Zamzani N, et al:
Molecular characterization of mitochondrial apoptosis-inducing
factor. Nature. 397:441–446. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Susin SA, Lorenzo HK, Zamzani N, et al:
Mitochondrial release of caspase-2 and -9 during the apoptotic
process. J Exp Med. 189:381–394. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
D'Amelio M, Tino E and Cecconi F: The
apoptosome emerging insights and new potential targets for drug
design. Pharm Res. 25:740–751. 2008.PubMed/NCBI
|
|
23.
|
Bossy-Wetzel E, Newmeyer DD and Green DR:
Mitochondrial cytochrome c release in apoptosis occurs upstream of
DEVD-specific caspase activation and independently of mitochondrial
transmembrane depolarization. EMBO J. 17:37–49. 1998. View Article : Google Scholar
|
|
24.
|
Suzuki A, Tsutomi Y, Yamamoto N, Shibutani
T and Akahane K: Mitochondrial regulation of cell death:
mitochondria are essential for procaspase 3-p21 complex formation
to resist Fas-mediated cell death. Mol Cell Biol. 19:3842–3847.
1999.PubMed/NCBI
|
|
25.
|
Trauth BC, Klas C, Peters AM, et al:
Monoclonal antibody-mediated tumor regression by induction of
apoptosis. Science. 245:301–305. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Leithauser F, Dhein J, Mechtersheimer G,
et al: Constitutive and induced expression of APO-1, a new member
of the nerve growth factor/tumor necrosis factor receptor
superfamily, in normal and neoplastic cells. Lab Invest.
69:415–429. 1993.
|
|
27.
|
Moller P, Koretz K, Leithauser F, et al:
Expression of APO-1 (CD95), a member of the NGF/TNF receptor
superfamily, in normal and neoplastic colon epithelium. Int J
Cancer. 57:371–377. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Weller M, Malipiero U, Rensing-Ehl A, Barr
PJ and Fontana A: Fas/APO-1 gene transfer for human malignant
glioma. Cancer Res. 55:2936–2944. 1955.PubMed/NCBI
|
|
29.
|
Nagata S and Golstein P: The Fas death
factor. Science. 267:1449–1456. 1995. View Article : Google Scholar
|
|
30.
|
French LE, Hahne M, Viard I, et al: Fas
and Fas ligand in embryos and adult mice: ligand expression in
several immune-privileged tissues and coexpression in adult tissues
characterized by apoptotic cell turnover. J Cell Biol. 133:335–343.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Houghton JA, Harwood FG, Gibson AA and
Tillman DM: The fas signaling pathway is functional in colon
carcinoma cells and induces apoptosis. Clin Cancer Res.
3:2205–2209. 1997.PubMed/NCBI
|
|
32.
|
Mahmood Z and Shulka Y: Death receptors:
targets for cancer therapy. Exp Cell Res. 316:887–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Itoh N and Nagata S: A novel protein
domain required for apoptosis. Mutational analysis of human Fas
antigen. J Biol Chem. 268:10932–10937. 1993.PubMed/NCBI
|
|
34.
|
Cheng J, Zhou T, Liu C, et al: Protection
from Fas-mediated apoptosis by a soluble form of the Fas molecule.
Science. 263:1759–1762. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Sato T, Irie S, Kitada S and Reed JC:
FAP-1: a protein tyrosine phosphatase that associates with Fas.
Science. 268:411–415. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Hughes SJ, Nambu Y, Soldes OS, et al:
Fas/APO-1 (CD95) is not translocated to the cell membrane in
esophageal adenocarcinoma. Cancer Res. 57:5571–5578.
1997.PubMed/NCBI
|
|
37.
|
Irmler M, Thorne M, Hahne M, et al:
Inhibition of death receptor signals by cellular FLIP. Nature.
388:190–195. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Rokhlin OW, Bishop GA, Hostager BS, et al:
Fas-mediated apoptosis in human prostatic carcinoma cell lines.
Cancer Res. 57:1758–1768. 1997.PubMed/NCBI
|
|
39.
|
Tillman DM, Harwood FG, Gibson AA and
Houghton JA: Expression of genes that regulate Fas signalling and
Fas-mediated apoptosis in colon carcinoma cells. Cell Death Differ.
5:450–457. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Owen-Schaub LB, Radinsky R, Kruzel E,
Berry K and Yonehara S: Anti-Fas on nonhematopoietic tumors: levels
of Fas/APO-1 and bcl-2 are not predictive of biological
responsiveness. Cancer Res. 54:1580–1586. 1994.PubMed/NCBI
|
|
41.
|
Holmstrom TH, Tran SE, Johnson VL, et al:
Inhibition of mitogen-activated kinase signaling sensitizes HeLa
cells to Fas receptor-mediated apoptosis. Mol Cell Biol.
19:5991–6002. 1999.PubMed/NCBI
|
|
42.
|
Scaffidi C, Fulda S, Srinivasan A, et al:
Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17:1675–1687.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Heerdt BG, Houston MA and Augenlicht LH:
Short-chain fatty acid-initiated cell cycle arrest and apoptosis of
colonic epithelial cells is linked to mitochondrial function. Cell
Growth Differ. 8:523–532. 1997.PubMed/NCBI
|
|
44.
|
Jacobson MD, Burne JF, King MP, et al:
Bcl-2 blocks apoptosis in cells lacking mitochondrial DNA. Nature.
361:365–369. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Jacobson MD and Raff MC: Programmed cell
death and Bcl-2 protection in very low oxygen. Nature. 374:814–816.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Shimizu S, Eguchi Y, Kosaka H, et al:
Prevention of hypoxia-induced cell death by Bcl-2 and Bel-xL.
Nature. 374:811–813. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Gamen S, Anel A, Montoya J, et al:
mtDNA-depleted U937 cells are sensitive to TNF and Fas-mediated
cytotoxicity. FEBS Lett. 376:15–18. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Marchetti P, Susin SA, Decaudin D, et al:
Apoptosis-associated derangement of mitochondrial function in cells
lacking mitochondrial DNA. Cancer Res. 56:2033–2038.
1996.PubMed/NCBI
|
|
49.
|
Schulze-Osthoff K, Beyaert R, Vandevoorde
V, Haegeman G and Fiers W: Depletion of the mitochondrial electron
transport abrogates the cytotoxic and gene-inductive effects of
TNF. EMBO J. 12:3095–3104. 1993.PubMed/NCBI
|
|
50.
|
Cavalli LR, Varella-Garcia M and Liang BC:
Diminished tumorigenic phenotype after depletion of mitochondrial
DNA. Cell Growth Differ. 8:1189–1198. 1997.PubMed/NCBI
|
|
51.
|
Liang BC and Ullyatt E: Increased
sensitivity to cis-diamminedichloroplatinum induced apoptosis with
mitochondrial DNA depletion. Cell Death Differ. 5:694–701. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Liang BC and Ullyatt E: Chemosensitization
of glioblastoma cells to bis-dichloroethyl nitrosourea with
tyrphostin AG17. Clin Cancer Res. 4:773–781. 1998.PubMed/NCBI
|
|
53.
|
King MP and Attardi G: Isolation of human
cell lines lacking mitochondrial DNA. Methods Enzymol. 264:304–313.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Nicoletti I, Migliorati G, Pagliacci MC,
Grignani F and Riccardi C: A rapid and simple method for measuring
thymocyte apoptosis by propidium iodide staining and flow
cytometry. J Immunol Methods. 139:271–279. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Vander Heiden MG, Chandel NS, Williamson
EK, Schumacker PT and Thompson CB: Bcl-xL regulates the membrane
potential and volume homeostasis of mitochondria. Cell. 91:627–637.
1997.PubMed/NCBI
|
|
56.
|
Watanabe-Fukunaga R, Brannan CI, Copeland
NG, Jenkins NA and Nagata S: Lymphoproliferation disorder in mice
explained by defects in Fas antigen that mediates apoptosis.
Nature. 356:314–317. 1992. View Article : Google Scholar
|
|
57.
|
Nagata S and Suda T: Fas and Fas ligand:
1pr and gld mutations. Immunol Today. 16:39–43. 1995. View Article : Google Scholar
|
|
58.
|
Wong GH and Goeddel DV: Fas antigen and
p55 TNF receptor signal apoptosis through distinct pathways. J
Immunol. 152:1751–1755. 1994.PubMed/NCBI
|
|
59.
|
Shima Y, Nishimoto N, Ogata A, et al:
Myeloma cells express Fas antigen/APO-1 (CD95) but only some are
sensitive to anti-Fas antibody resulting in apoptosis. Blood.
85:757–764. 1995.PubMed/NCBI
|
|
60.
|
Westendorf JJ, Lammert JM and Jelinek DF:
Expression and function of Fas (APO 1/CD95) in patient myeloma
cells and myeloma cell lines. Blood. 85:3566–3576. 1995.PubMed/NCBI
|
|
61.
|
Danielson SR, Wong A, Carelli V,
Martinuzzi A, Schapira AH and Cortopassi GA: Cells bearing
mutations causing Leber's hereditary optic neuropathy are
sensitized to Fas-Induced apoptosis. J Biol Chem. 277:5810–5815.
2002.
|
|
62.
|
Raisova M, Hossini AM, Eberle J, Riebeling
C, Wieder T, Sturm I, Daniel PT, Orfanos CE and Geilen CC: The
Bax/Bcl-2 ratio determines the susceptibility of human melanoma
cells to CD95/Fas-mediated apoptis. J Invest Dermatol. 117:333–340.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Maas C, Verbrugge I, De Vries E, Savich G,
van de Kooij LW, Tait SW and Borst J: Smac/DIABLO release from
mitochondria and XIAP inhibition are essential to limit
clonogencity of Type I tumor cells after TRAIL receptor
stimulation. Cell Death Differ. 17:1613–1623. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Friesen C, Herr I, Krammer PH and Debatin
KM: Involvement of the CD95 (APO-1/FAS) receptor/ligand system in
drug-induced apoptosis in leukemia cells. Nat Med. 2:574–577. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Matsumoto Y, Hayakawa A, Tamada Y, Mori H
and Ohashi M: Upregulated expression of Fas antigen on cultured
human keratinocytes with induction of apoptosis by cisplatin. Arch
Dermatol Res. 288:267–279. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Fulda S, Sieverts H, Friesen C, Herr I and
Debatin KM: The CD95 (APO-1/Fas) system mediates drug-induced
apoptosis in neuroblastoma cells. Cancer Res. 57:3823–3829.
1997.PubMed/NCBI
|
|
67.
|
Muller M, Strand S, Hug H, et al:
Drug-induced apoptosis in hepatoma cells is mediated by the CD95
(APO-1/Fas) receptor/ligand system and involves activation of
wild-type p53. J Clin Invest. 99:403–413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
68.
|
Eischen CM, Kottke TJ, Martins LM, et al:
Comparison of apoptosis in wild-type and Fas-resistant cells:
chemotherapy-induced apoptosis is not dependent on Fas/Fas ligand
interactions. Blood. 90:935–943. 1997.PubMed/NCBI
|
|
69.
|
Villunger A, Egle A, Kos M, et al:
Drug-induced apoptosis is associated with enhanced Fas (Apo-1/CD95)
ligand expression but occurs independently of' Fas (Apo-1/CD95)
signaling in human T-acute lymphatic leukemia cells. Cancer Res.
57:3331–3334. 1997.PubMed/NCBI
|
|
70.
|
Verbrugge I, Maas C, Heijkoop M, Verheij
and Borst J: Radiation and anticancer drugs can facilitate
mitochondrial bypass by CD95/FAS via c-FLIP downregulation. Cell
Death Differ. 17:551–561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
Asoh S, Mori T, Hayashi J and Ohta S:
Expression of the apoptosis-mediator Fas is enhanced by
dysfunctional mitochondria. J Biochem. 120:600–607. 1996.
View Article : Google Scholar : PubMed/NCBI
|