Introduction
Failure of insulin-producing β-cells is a common
feature of type 1 and type 2 diabetes mellitus. Anti-β-cell
autoimmune reactions and associated inflammation lead to β-cell
death in type 1 diabetes, whereas in type 2 diabetes the metabolic
disorders produce inflammatory mediators in insulin-sensitive
tissues leading to elevated levels of circulating inflammatory
mediators, such as interleukin-6 and tumor necrosis factor-α
(TNF-α) (1,2).
Cytokines, such as TNF-α, induce β-cell apoptosis
through the induction of signaling pathways that activate nuclear
factor-κB (NF-κB) (1–5). In rat-insulinoma (RIN) cells, which
are an insulin-producing transformed β-cell line, it was shown, by
measuring intracellular calcium [Ca2+]i and
Ca2+ buffering capacity, that exposure of RIN cells to
TNF-α caused dysregulation of [Ca2+]i
(6). It was also shown that
TNF-α-induced dysregulation of [Ca2+]i arose
from a decrease in the total content of the cytoplasmic calcium
binding protein calbindin-D28k.
The objective of the present study was to test the
hypothesis that TNF-α-induced dysregulation of
[Ca2+]i in insulin-producing β-cells causes
proteolytic degradation of IκBα and consequently leads to the
transcriptional activation of NF-κB. The NF-κB heterodimer is
localized in the cytoplasm in association with an inhibitory
subunit (IκB or inhibitory κB) (7). Following elevation of
[Ca2+]i, IκB is phosphorylated, targeting it
for ubiquitination and subsequent proteolysis. This frees NF-κB to
enter the nucleus to bind DNA and activate genes (8–10). A
key function of IκB's association with NF-κB is therefore its
prevention of DNA binding. It has been shown previously that the
sensitivity of different transcription factors to
[Ca2+]i oscillations is highly
frequency-dependent (1,11).
Since at present it is not known how the
dysregulation of [Ca2+]i caused by TNF-α
modulates the activation of NF-κB, the focus of the present study
was on the dysregulation of [Ca2+]i as a
mediator of cytokine-induced NF-κB activation via degradation of
IκBα (1,7,11–15).
To test this hypothesis, RIN cells were treated with increasing
concentrations of TNF-α, and the following were measured: i) the
degradation of IκBα using a FunctionELISA protocol, ii) the
translocation of NF-κB from the cytoplasm to the nucleus by
immunofluorescence using an antibody directed against the p65 NF-κB
subunit (16,17), and iii) NF-κB-dependent
transcription based on the DNA binding activity of NF-κB determined
using an ELISA-based kit (17).
The results are discussed in light of the dysregulation of
[Ca2+]i caused by TNF-α, which activates
NF-κB in RIN cells (6).
Materials and methods
RINr 1046-38 (abbreviated as RIN, a rat insulinoma
cell line) insulin-producing β-cells were kindly provided by Dr
Bruce Chertow (Veterans Administration Medical Center, Huntington,
WV, USA). RIN cells were grown on 25-mm diameter glass coverslips
in 6-well culture plates in a 5% CO2 incubator at 37°C.
The cell culture medium was RPMI-1640 supplemented with 10% (w/v)
fetal bovine serum, 100 U/ml penicillin and 100 μg/ml
streptomycin (6).
IκBα degradation in TNF-α-treated
β-cells
Treatment with TNF-α. TNF-α initiates a
signal transduction cascade that leads to the activation of the IκB
kinase complex (7). This complex
phosphorylates IκBα, which leads to the ubiquitination of IκBα and
its subsequent degradation by the 26S proteosome (1,7,11).
Thus, analysis of the phosphorylation state of IκBα provides a
correlation to the activity of the IκB kinase complex, as well as
the activation of NF-κB. The RIN cells were exposed to 2, 5, 10, 20
and 30 ng/ml of TNF-α at 37°C for 20 min.
Preparation of whole cell lysates. The β-cell
lysates from the control and TNF-α-treated RIN cells were prepared
by washing the cells twice with ice-cold PBS, followed by scrapping
the β-cells using a scrapper and suspending the cells in 3 ml PBS.
The cells were then centrifuged at 1,000 rpm for 10 min at 4°C. The
pellet containing β-cells was incubated in a complete cell lysis
buffer for 30 min at 4°C, followed by centrifugation at 14,000 × g
for 20 min at 4°C. The supernatant containing the cell lysate was
collected, aliquoted and stored at −80°C for further analysis. The
total protein content was determined using a BCA protein assay
protocol in a 96-well plate (Pierce; catalog no. 23225).
Determination of phosphorylated IκBα using
the FunctionELISA procedure. Standard Phospho-IκBα solutions
with concentrations ranging between 0.15 and 10 ng/ml in blocking
buffer were prepared in a 96-well FunctionELISA IκBα plate from a
stock solution provided by Active Motif according to the
manufacturer's instructions (Active Motif, Carlsbad CA, USA). The
cell lysates from the control and TNF-α-treated RIN cells were
diluted to 1 mg protein/ml in blocking buffer, and 100 μl of
the lysate was pipetted into each well. The FunctionELISA IκBα
plate was sealed and incubated at 4°C for 4 h. The contents of the
FunctionELISA IκBα plate were then removed by inverting the
FunctionELISA IκBα plate, and the wells were washed four times with
250 μl of wash buffer/well. The stock IκBα-detecting
antibody was diluted to 1:250 in blocking buffer. IκBα-detecting
antibody (50 μl) was added to each well, and the plate was
incubated at room temperature for 1 h. The detecting antibody was
then removed from the FunctionELISA IκBα plate, and the plate was
washed thoroughly four times in 250 μl of wash
buffer/well.
The horseradish peroxidase (HRP)-conjugated
secondary antibody was then diluted 1:1,000 in blocking buffer, and
50 μl of this diluted secondary antibody was added to each
well. The FunctionELISA IκBα plate was incubated at room
temperature for 1 h. Following this, the FunctionELISA IκBα plate
was washed five times as described above. Freshly prepared HRP
ELISA substrate solution (50 μl) was added to each well. The
luminescence from each well of the 96-well ELISA plate was then
immediately measured using a TECAN 96-well plate reader GENious
Plus with Magellan version 6.5 software. The concentrations of
phosphorylated IκBα were calculated from the standard graph
obtained using Phospho-IκBα control solution provided by the
manufacturer Active Motif (FunctionELISA™ IκBα for the detection
and analysis of IκBα phosphorylation; catalog nos. 48005 and
48505).
Translocation of NF-κB from the
cytoplasm to the nucleus
TNF-α-induced NF-κB activation and DNA binding in
β-cells, as measured by translocation of NF-κB from the cytoplasm
to the nucleus, were assessed by immunofluorescence using an
antibody directed against the p65 NF-κB subunit as follows
(17): the RIN cells were exposed
to 10 ng/ml TNF-α for 6 h at 37°C; the control and TNF-α-treated
RIN cells on 25-mm diameter glass coverslips were then fixed by
adding Bouin; the fixed RIN cells were further treated with a
primary antibody directed against the p65 NF-κB subunit, followed
by treatment with Alexa Fluor 488-coupled fluorescent secondary
antibody; the confocal fluorescence images were scanned on a Nikon
TE2000U inverted fluorescence microscope equipped with a Nikon C1
laser scanning confocal microscope system; the ratio of
fluorescence in the nuclei to that in the cytoplasm were calculated
using Metamorph 6.1 software (Universal Imaging Corp., CA,
USA).
Transcriptional activation of
NF-κB
Transcriptional activation of NF-κB has been defined
using several methods, including the use of NF-κB-dependent
reporter assays and also a DNA binding assay (17–20).
Active Motif recently introduced the ELISA-based kit to detect and
quantify transcription factor NF-κB activation. Briefly, RIN cells
were grown on a T75 culture flask in a 5% CO2 incubator
at 37°C. The cell culture medium was RPMI-1640 supplemented with
10% (w/v) fetal bovine serum, 100 U/ml penicillin and 100
μg/ ml streptomycin. After 5 days of culture, the RIN cells
were exposed to 2, 5, 10, 20 and 30 ng/ml TNF-α for 6 h at 37°C.
The nuclear extracts from the control and TNF-α-treated RIN cells
were prepared according to the manufacturer's protocol, and the
procedure is briefly described below.
Preparation of nuclear extract. After
treatment with TNF-α, the cell culture medium was removed from each
flask, and the cells were washed thrice with ice-cold PBS buffer,
scrapped with a scrapper and suspended in PBS buffer in 1.5-ml
Eppendorf centrifuge tubes, followed by centrifugation at 500 rpm
for 5 min at 4°C. The cell pellet was suspended in 1X hypotonic
buffer as per the manufacturer's protocol. After 15 min of
incubation at 4°C, 25 μl of detergent was added to each
Eppendorf tube, followed by vortexing at high speed for 10 sec. The
suspension was centrifuged at 14,000 × g for 30 sec at 4°C. The
pellet containing the nuclear fraction was resuspended in complete
lysis buffer by pipetting up and down and was vortexed for 10 sec
followed by incubation for 30 min on ice on a rocking platform set
at 150 rpm. The contents were vortexed for 10 sec and then
centrifuged at 14,000 × g for 10 min at 4°C. The supernatant
containing the nuclear fraction was aliquoted and stored at −80°C.
The protein concentration of the nuclear extract was determined
using the BCA assay as described above.
Detection and quantification of NF-κB
transcriptional activity using ELISA. Complete Binding Buffer
(30 μl) was added to each well of a TransAM NF-κB 96-well
ELISA plate (Active Motif). Then, 20 μl of nuclear extract
diluted in Complete Lysis Buffer to a final protein content of 5
μg was added to each well. For the blank well, only 20
μl of Complete Lysis Buffer was added. The positive control
provided as Jurkat cell nuclear extract by the manufacturer (Active
Motif) was also used. The 96-well plate was then covered with a
sealing tape and incubated for 1 h at room temperature under mild
agitation at 100 rpm on a rocking platform. Each well was then
washed three times with 200 μl of 1X wash buffer, and the
ELISA plate was inverted and tapped on an absorbent paper towel.
NF-κB primary antibody (100 μl diluted 1:1,000) was added to
each well, and the plate was incubated for 1 h at room temperature
without agitation. The wells were then washed thrice with 200
μl of wash buffer.
HRP-conjugated secondary antibody (100 μl)
was added to each well, and the plate was again incubated for 1 h
at room temperature. The plate was washed four times with 200
μl of buffer/well. The colorimetric reaction was carried out
by adding 100 μl of developing solution to each well
followed by incubation for 5 min at room temperature. The reaction
was stopped by adding 100 μl of stop solution to each well.
The absorbance of each well was then read within 5 min at 450 nm,
with a reference wavelength of 655 nm, on a Molecular Device
Spectra Max 340 PC 96-well plate reader.
Data analysis
Data are presented as the means ± standard deviation
for three individual experiments. One-way ANOVA was used, as well
as the Student's t-test, to study the statistical significance of
the data obtained from the control and TNF-α-treated RIN cells.
Results and Discussion
In β-cells, the uptake and metabolism of glucose
lead to closure of ATP-sensitive K+ channels,
depolarization of the plasma membrane and the subsequent influx of
Ca2+ through voltage-gated calcium channels. The
Ca2+ signal provided by this influx of Ca2+
is accompanied by the release of Ca2+ from the endoplasmic
reticulum (21–23). The subsequent increase in
[Ca2+]i is an important determinant for
insulin granule exocytosis. Ca2+ signals in β-cells
activate the transcription factor NF-κB involved in the regulation
of the cell cycle and apoptosis (12,24).
Our long-term goal is to characterize changes in
[Ca2+]i homeostasis as a mediator of
cytokine-induced β-cell dysfunction in diabetes (6). RINr 1046-38 (RIN), insulin-producing
β-cells that constitutively express calbindin-D28k, have
been previously used to characterize the effect of TNF-α on
[Ca2+]i homeostasis, apoptosis, replication,
insulin release and gene and protein expression (6). In its inactive form, NF-κB consists
of a three-subunit complex consisting of two (prototypical)
subunits of 50 kDa (p50) and 65 kDa (p65; RelA) and an IκB subunit
(IκBα or IκBβ) (7). Following
[Ca2+]i elevation, IκB is phosphorylated,
targeting it for ubiquitination and subsequent proteolysis without
affecting the integrity of the bound NF-κB dimer (24,25).
The liberated NF-κB dimer is capable of binding DNA and activating
genes (10). A key role of the IκB
association with NF-κB is therefore its prevention of DNA binding
(10,26).
In light of the discussion provided here, the aim
was to ascertain whether, in insulin-producing β-cells, the
dysregulation of [Ca2+]i by TNF-α leads to
the transcriptional activation of NF-κB. To test this hypothesis,
the experiments were designed and carried out as described above
and the results are described and discussed as follows.
IκBα degradation in TNF-α-treated
β-cells
Activation of the IκB kinase complex, which is
located in the cytoplasm and involved in the phosphorylation of
IκBα, occurs due to the binding of TNF-α to the TNF-α receptor
located on the plasma membrane, which in turn initiates a cascade
of signal transduction events (7).
The activated IκB kinase complex phosphorylates IκBα, which is then
followed by the ubiquitination of IκBα and its subsequent
degradation by the 26S proteosome (1,7,11).
Hence, analysis of the phosphorylation state of IκBα provides a
correlation to the activity of the IκB kinase complex, as well as
the activation of NF-κB.
The effects of TNF-α on the degradation of the IκBα
subunit were examined. RIN cells were treated with increasing
concentrations of TNF-α ranging between 2 and 30 ng/ml at 37°C for
20 min. The results are shown in Fig.
1 and indicate that, while in control RIN cell lysate there was
no Phospho-IκBα present, thereby indicating the absence of degraded
IκBα, in RIN cells exposed to 2, 5, 10, 20 and 30 ng/ ml TNF-α,
17.176±2.85, 17.292±4.35, 53.77±5.63, 30.58±4.89 and 12±3.27 ng/ml
Phospho-IκBα/mg of total cell protein was observed, respectively
(n=3, P<0.05). These observations indicate that 10 ng/ml TNF-α
caused a maximum increase in the degraded IκBα as measured by the
Phospho-IκBα value of 53.77±5.63 ng/ml Phospho-IκBα/mg of total
cell protein (n=3, P<0.05). The results also suggest that
TNF-α-induced signal transduction in β-cells results in the
activation of the IκB kinase complex.
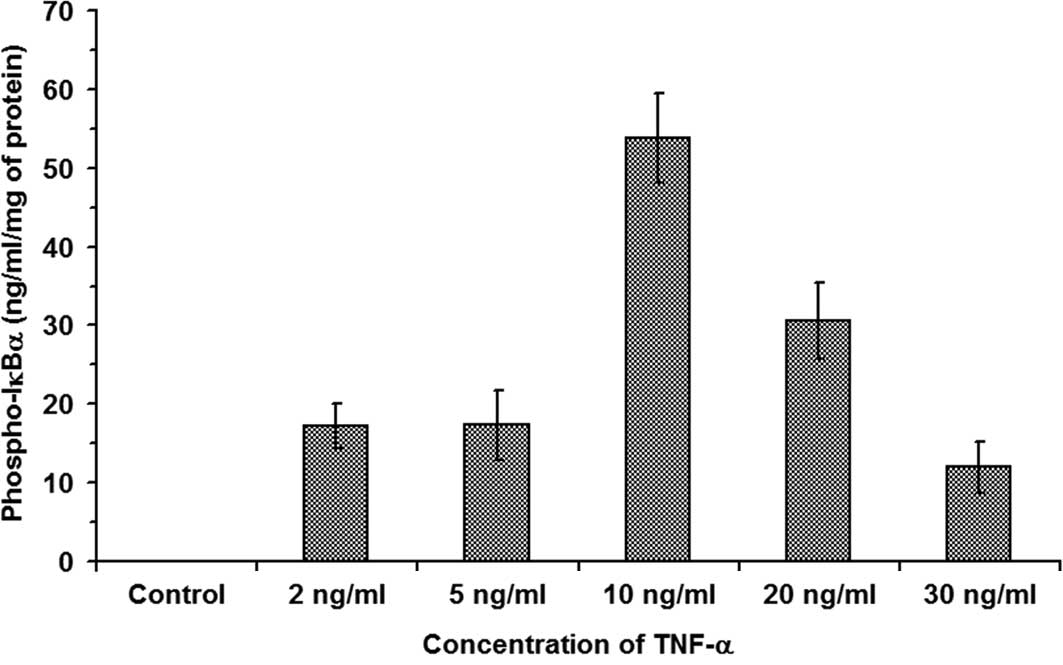 | Figure 1.TNF-α-induced degradation of IκBα in
β-cells. RIN cells were treated with increasing concentrations of
TNF-α at 37°C for 20 min. The results show that, while in control
RIN cell lysate there was no Phospho-IκBα present, thereby
indicating the absence of degraded IκBα, in RIN cells exposed to 2,
5, 10, 20 and 30 ng/ml TNF-α, 17.176±2.85, 17.292±4.35, 53.77±5.63,
30.58±4.89 and 12±3.27 ng/ml Phospho-IκBα/mg of total cell protein
was observed, respectively (n=3, P<0.05). |
Transcriptional activation of
NF-κB
As mentioned above, TNF-α-induced IκBα
phosphorylation results in its ubiquitination and subsequent
proteolysis, and therefore frees the NF-κB heterodimer bound to the
IκBα. The liberated NF-κB dimer is capable of binding DNA and
activating genes. Transcriptional activation of NF-κB has been
defined by several methods including the use of NF-κB-dependent
reporter assays and also a DNA binding assay (18–20).
Active Motif has introduced the first ELISA-based kit to detect and
quantify transcription factor NF-κB activation. This kit contains a
96-well plate, to which an oligonucleotide containing an NF-κB
consensus binding 5′-GGGACTTTCC-3′ site has been immobilized. The
activated NF-κB contained in nuclear or whole-cell extracts
specifically binds to this oligonucleotide. The primary antibodies
used to detect NF-κB recognize an epitope on p65 that is accessible
only when NF-κB is activated and bound to its target DNA. By using
an antibody that is directed against the NF-κB p65 subunit, the
NF-κB complex bound to the oligo nucleotide is detected. Addition
of a secondary antibody conjugated to HRP provides a sensitive
assay that is easily quantified by measuring the absorbance using a
96-well plate reader.
The transcriptional activation of NF-κB was measured
by Active Motif's TransAM ELISA-based kit. As described above, the
RIN cells were exposed to 2, 5, 10, 20 and 30 ng/ ml TNF-α for 6 h
at 37°C. Our measurements of the transcriptional activity of NF-κB
in the nuclear extracts of control and TNF-α-treated RIN cells as
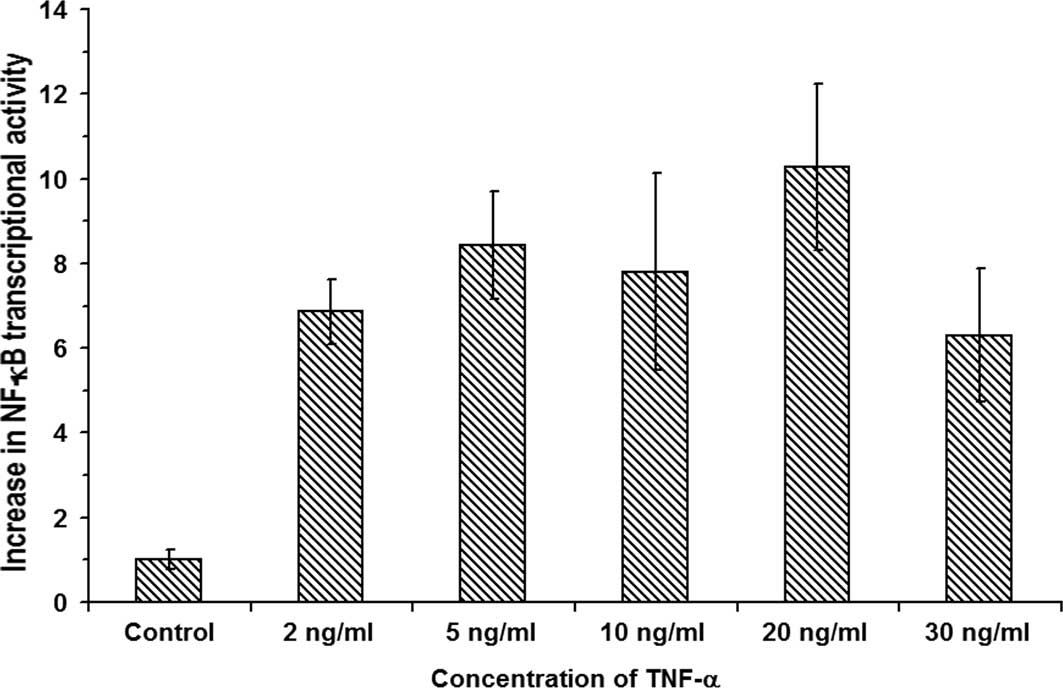
shown in Fig. 2 indicate that,
upon treatment of RIN cells with 2, 5, 10, 20 and 30 ng/ml TNF-α,
the relative increases in the NF-κB transcriptional activities were
6.86±0.76-, 8.42±1.27-, 7.8±2.32-, 10.28±1.96-and 6.3±1.57-fold,
respectively (n=3, P<0.05). The data show a maximum
10.28±1.96-fold increase in NF-κB transcription activation in RIN
cells treated with 20 ng/ml TNF-α (n=3). Therefore, the results
indicate that indeed, in insulin-producing β-cells, the
dysregulation of [Ca2+]i by TNF-α leads to
the transcriptional activation of NF-κB.
Translocation of NF-κB from the
cytoplasm to the nucleus
In its inactive form, NF-κB consists of a
three-subunit complex consisting of two (prototypical) subunits of
50 kDa (p50) and 65 kDa (p65; RelA), and an IκB subunit (IκBα or
IκBβ) (7). In the cytoplasm, the
inactive NF-κB heterodimer with two protein subunits, p50
(molecular mass 50 kDa) and p65 (molecular mass 65 kDa), is bound
to an IκB protein subunit (IκBα or IκBβ) (7). Activation of this inactive NF-κB
complex by TNF-α or elevated [Ca2+]i through
proteolysis degradation of IκBα allows the activated NF-κB to move
freely between the cytoplasm and nucleus. Upon entering the
nucleus, the activated NF-κB is capable of binding DNA and causes
the transcriptional activation of several genes.
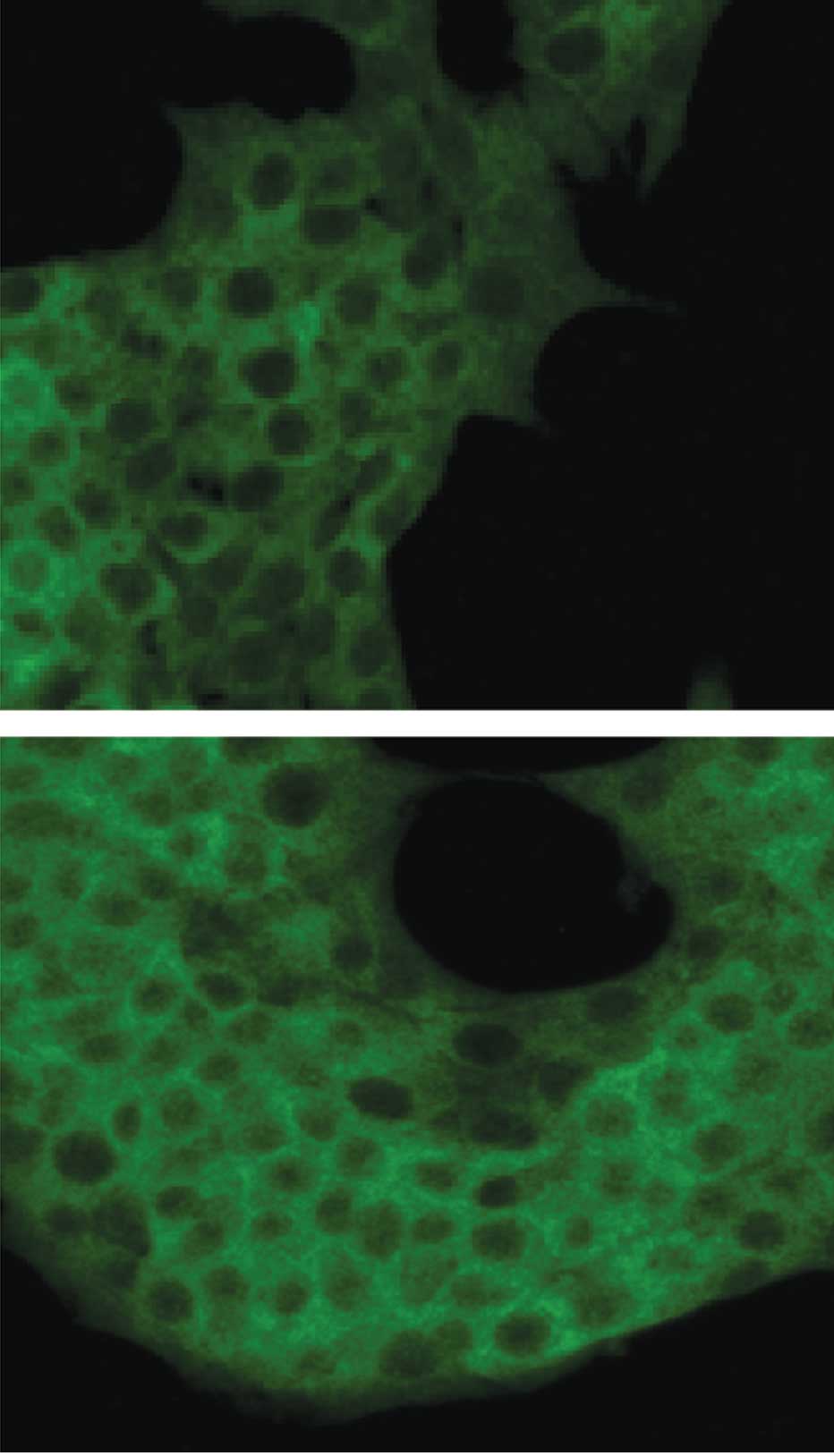
In insulin-producing β-cells, nuclear translocation
of the activated NF-κB from the cytoplasm was measured by
immunofluorescence confocal microscopy in conjunction with an
antibody directed against the p65 NF-κB subunit. The confocal
fluorescence images presented in Fig.
3 show the cytoplasmic and nuclear distribution of NF-κB
fluorescence in the control (Fig.
3A) and TNF-α-treated (Fig.
3B) insulin-producing β-cells.
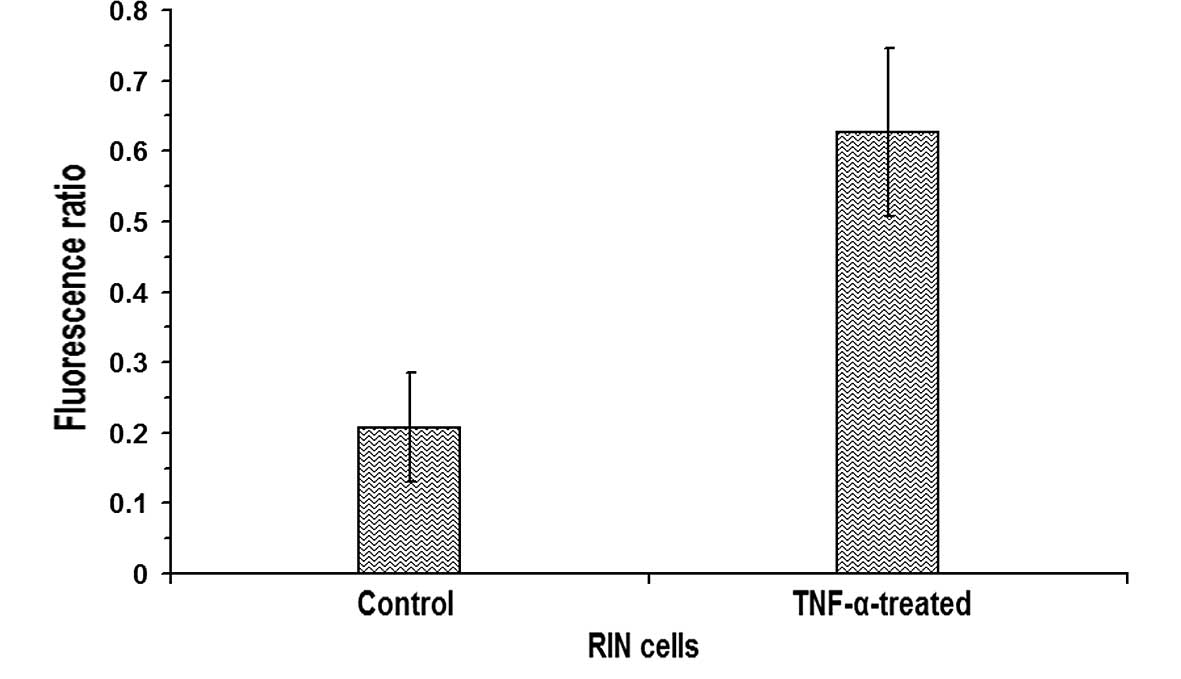
Subsequently, the ratio of fluorescence intensities
in nuclear regions to cytoplasmic regions was calculated by drawing
regions of interests in confocal images of control and
TNF-α-treated β-cells using Metamorph 6.1 software (Universal
Imaging Corp.). The ratio of fluorescence in the nuclei to that in
the cytoplasm of the untreated RIN cells was 0.2078±0.0778 (n=11),
whereas in RIN cells treated with 10 ng/ml TNF-α, the ratio was
0.6267±0.1186 (n=11), indicating a statistically significant
increase (P<0.05) in the nuclear translocation of NF-κB in the
TNF-α-treated RIN cells (Fig. 4).
The results shown in Figs. 3 and
4 indicate that indeed, in
insulin-producing β-cells, the dysregulation of
[Ca2+]i by TNF-α leading to the
transcriptional activation of NF-κB occurs due to the increased
nuclear translocation of NF-κB.
To elucidate the structural and functional
parameters that define the relationship between calcium and the
activation of NF-κB, Lewis (11)
and Dolmetsch et al (1)
conducted experiments using a ‘calcium clamp’, and showed that the
sensitivity of different transcription factors, including NF-κB to
[Ca2+]i oscillations, is highly
frequency-dependent. [Ca2+]i oscillations of
different frequencies lead to the expression of different sets of
genes, presumably as a consequence of their effects on the
underlying transcription factors. By differentially controlling the
activation of distinct sets of transcription factors and the
expression of different genes, the [Ca2+]i
oscillation frequency may direct cells along specific developmental
pathways. The ability to decode [Ca2+]i
oscillation frequencies results from the kinetics of transcription
factor regulation.
In light of the observations presented above as well
as those of previous studies, we conclude that the subcellular
localization and kinetics of the Ca2+ signal affect how
increases in [Ca2+]i induced by different
physiological stimuli are translated into a specific cellular
response. Additionally, it is likely that the spatiotemporal nature
of β-cell Ca2+ signaling plays an important role in
Ca2+-dependent NF-κB signaling (6,24).
Acknowledgements
This study was supported by an
NIH/NIGMS grant to J.P. (award no. SC3GM084751). The content is
solely the responsibility of the author and does not necessarily
represent the official views of the National Institute of General
Medical Sciences or the National Institute of Health.
References
|
1.
|
Dolmetsch RE, Xu K and Lewis RS: Calcium
oscillations increase the efficiency and specificity of gene
expression. Nature. 392:933–936. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Donath MY and Halban PA: Decreased β-cell
mass in diabetes: significance, mechanisms and therapeutic
implications. Diabetologia. 47:581–589. 2004.
|
|
3.
|
Donath MY, Storling J, Maedler K and
Mandrup-Poulsen T: Inflammatory mediators and islet β-cell failure:
a link between type 1 and type 2 diabetes. J Mol Med. 81:455–470.
2003.
|
|
4.
|
Flodstrom M, Welsh N and Eizirik DL:
Cytokines activate the nuclear factor κB (NF-κB) and induce nitric
oxide production in human pancreatic islets. FEBS Lett. 385:4–6.
1996.
|
|
5.
|
Sekine N, Ishikawa T, Okazaki T, Hayashi
M, Wollheim CB and Fujita T: Synergistic activation of NF-κB and
inducible isoform of nitric oxide synthase induction by
interferon-gamma and tumor necrosis factor-alpha in INS-1 cells. J
Cell Physiol. 184:46–57. 2000.
|
|
6.
|
Parkash J, Chaudhry MA and Rhoten WB:
Tumor necrosis factor-α-induced changes in insulin-producing
β-cells. Anat Rec. 286A:982–993. 2005.
|
|
7.
|
Hoffman A and Baltimore D: Circuitry of
nuclear factor κB signaling. Immunol Rev. 210:171–186. 2006.
|
|
8.
|
Dolmetsch RE, Lewis RS, Goodnow CC and
Healy JI: Differential activation of transcription factors induced
by Ca2+ response amplitude and duration. Nature.
386:855–858. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Hoffmann A, Levchenko A, Scott ML and
Baltimore D: The IκB-NF-κB signaling module: temporal control and
selective gene activation. Science. 298:1241–1245. 2002.
|
|
10.
|
Lee SH and Hannink M: Characterization of
the nuclear import and export functions of Ikappa B(epsilon). J
Biol Chem. 277:23358–23366. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Lewis RS: Calcium oscillations in T-cells:
mechanisms and consequences for gene expression. Biochem Soc Trans.
31:925–929. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Parnaud G, Hammar E, Ribaux P, Donath MY,
Berney T and Halaban PA: Signaling pathways implicated in the
stimulation of beta-cell proliferation by extracellular matrix. Mol
Endocrinol. 23:1264–1271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Parkash J, Chaudhry MA, Amer AS,
Christakos S and Rhoten WB: Intracellular calcium ion response to
glucose in β-cells of calbindin-D28k nullmutant mice and
in βHC13 cells over-expressing calbindin-D28k.
Endocrine. 18:221–229. 2002.
|
|
14.
|
Parkash J, Chaudhry MA and Rhoten WB:
Ca2+ sensing receptor activation by CaCl2
increases [Ca2+]i resulting in enhanced
spatial interaction with calbindin-D28k protein. Int J
Mol Med. 13:3–11. 2004.
|
|
15.
|
Parkash J: Inflammatory cytokine signaling
in insulin producing β-cells enhances the colocalization
correlation coefficient between L-type voltage-dependent calcium
channel and calcium-sensing receptor. Int J Mol Med. 22:155–163.
2008.
|
|
16.
|
Cnop M, Welsh N, Jones JC, Jorns A, Lenzen
S and Eizirik DL: Mechanisms of pancreatic β-cell death in type 1
and type 2 diabetes. Diabetes. 54:S97–S107. 2005.
|
|
17.
|
Hammar EB, Irminger JC, Rickenbach K,
Parnaud G, Ribaux P, Bosco D, Rouiller DG and Halban PA: Activation
of NF-κB by extracellular matrix is involved in spreading and
glucosestimulated insulin secretion of pancreatic beta cells. J
Biol Chem. 280:30630–30637. 2005.
|
|
18.
|
Bergmann M, Hart L, Lindsay M, Barnes PJ
and Newton R: IκBα degradation and nuclear factor-κB DNA binding
are insufficient for interleukin-1β and tumor necrosis
factor-α-induced κB-dependent transcription. Requirement for an
additional pathway. J Biol Chem. 273:6607–6610. 1998.
|
|
19.
|
Hu Q, Deshpande S, Irani K and Ziegelstein
RC: Ca2+ oscillation frequency regulates
agonist-stimulated NF-κB transcriptional activity. J Biol Chem.
274:33995–33998. 1999.
|
|
20.
|
Trushin SA, Pennington KN,
Algeciras-Schimnich A and Paya CV: Protein kinase C and calcineurin
synergize to activate IκB kinase and NF-κB in T lymphocytes. J Biol
Chem. 274:22923–22931. 1999.
|
|
21.
|
Ammala C, Larsson O, Berggren PO, Bokvist
K, Juntti-Berggren L, Kindmark H and Rorsman P: Inositol
trisphosphate-dependent periodic activation of a
Ca2+-activated K+-conductance in
glucose-stimulated pancreatic β-cells. Nature. 353:849–852. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Berggren PO and Larsson O: Ca2+
and pancreatic β-cell function. Biochem Soc Trans. 22:12–18.
1994.
|
|
23.
|
Lemmens R, Larsson O, Berggren PO and
Islam MS: Ca2+-induced Ca2+ release from the
endoplasmic reticulum amplifies the Ca2+ signal mediated
by activation of voltage-gated L-type Ca2+ channels in
pancreatic β-cells. J Biol Chem. 276:9971–9977. 2001.
|
|
24.
|
Bernal-Mizrachi E, Wen W, Shornick M and
Permutt AM: Activation of nuclear factor-κB by depolarization and
Ca2+-influx in MIN6 insulinoma cells. Diabetes. 51(Suppl
3): 484–488. 2002.
|
|
25.
|
Claudio E, Brown K, Park S, Wang H and
Siebenlist U: BAFF-induced NEMO-independent processing of NF-kappa
B2 in maturing B cells. Nat Immun. 3:958–965. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Scott ML, Fujita T, Liou HC, Nolan GP and
Baltimore D: The p65 subunit of NF-kappa B regulates I kappa B by
two distinct mechanisms. Genes Dev. 7:1266–1276. 1993. View Article : Google Scholar : PubMed/NCBI
|