Introduction
Acute liver failure (ALF) is a disease associated
with high mortality and characterized by massive hepatocyte
necrosis. Once liver atrophy has progressed, liver transplantation
is the only promising treatment option (1,2).
Even in such cases, it is difficult to determine the indications
for liver transplantation, since some patients recover successfully
without it. The difficulties associated with predicting the
prognosis of ALF are mainly caused by the fact that the mechanism
of the disease has not yet been fully clarified.
Recently, several authors have reported that
products derived from macrophages are increased in the serum of
patients with ALF, suggesting that overactivated macrophages in the
liver may play an important role in the progression of ALF
(3–5). However, the issue of how activated
macrophages are involved in the development of massive necrosis
remains to be clarified. On the other hand, earlier studies have
revealed deposition of fibrin in the sinusoid using rat models of
ALF, suggesting that microcirculatory disturbance may be a final
process in the development of massive liver necrosis in ALF
(6–8).
Collectively, considering these previous findings,
we subsequently hypothesized that overactivated macrophages damage
endothelial cells of the sinusoid, either directly or indirectly
via their release of cytokines, which may cause microcirculation
disturbance and lead to massive liver necrosis. Although proving
the existence of anaerobic conditions in the liver would support
our theory, it is difficult to directly measure oxygen
concentrations in the hepatic microcirculation. Therefore, we
focused on the production of lactate dehydrogenase (LDH) in
hepatocytes. LDH is an essential enzyme for anaerobic respiration,
and its production has been shown to be increased under hypoxic
conditions in various cell lines (9–12).
In the present study, we examined the amounts of LDH
production in hepatocytes using biopsy samples from patients with
ALF in comparison to samples from patients with acute hepatitis,
chronic hepatitis and liver cirrhosis.
Patients and methods
Patients
Between April 2005 and March 2007, 15 patients
suffering from ALF were admitted to our hospital. The patients
showed a prolonged prothrombin time (PT) [PT-international
normalized ratio (INR) >1.5] and encephalopathy of more than
grade 2 that occurred within 1 week of onset. Ultrasonography
guided liver biopsy was performed in 7 patients for the purpose of
the following: i) an examination for etiology, particularly to
evaluate the participation of autoimmune hepatitis (AIH); ii)
exclusion of the pre-existence of chronic liver disease; iii)
evaluation of the clinical course of the liver disease. The
etiologies of the patients were varied: 2 were hepatitis B virus
(HBV), 1 was hepatitis A virus (HAV) and 4 were undetermined. For
all patients, liver-supporting procedures, such as plasma exchange
and brain edema inhibition, were performed. Lamivudine was also
administered when HBV was detected in the serum.
To clarify the pathological characteristics of the
ALF patients, we compared their samples to liver biopsy samples
obtained from patients with other liver diseases [7 acute hepatitis
(AH) patients, 8 chronic hepatitis (CH) patients and 6 liver
cirrhosis (LC) patients] during the same period. The causes of AH
were HBV (n=3), AIH (n=2) and undetermined (n=2). These patients
maintained a PT-INR of <1.5 throughout the clinical course and
recovered within 1 month. Regarding the patients with chronic liver
diseases, the etiologies of CH were HCV (n=2), HBV (n=3) and AIH
(n=3), and those of LC were HCV (n=2), AIH (n=2), primary biliary
cirrhosis (n=1) and non-alcoholic steatohepatitis (n=1). All of the
patients with LC were classified as Child-Pugh A. The laboratory
data at the time of liver biopsy are shown in Table I.
 | Table I.Laboratory findings of the patients at
the time of liver biopsy. |
Table I.
Laboratory findings of the patients at
the time of liver biopsy.
| ALF (n=7) | AH (n=7) | CH (n=8) | LC (n=6) |
|---|
| Albumin (g/dl) | 3.3±0.3 | 3.8±0.3 | 3.8±0.6 | 3.6±0.6 |
| Bilirubin
(mg/dl) | 10.2±8.2 | 7.7±7.4 | 2.0±1.9 | 1.1±0.6 |
| D/T ratio | 0.69±0.02 | 0.58±0.16 | 0.34±0.19 | 0.31±0.06 |
| AST (U/l) | 3,660.3±2,553.5 | 1,815.4±1,294.1 | 248.0±208.8 | 96.3±59.2 |
| ALT (U/l) | 3,921.9±2,249.3 | 3,079.0±2,613.0 | 310.5±238.4 | 132.2±145.0 |
| LDH (U/l) | 1,849.7±1,799.9 | 731.3±394.2 | 249.9±70.8 | 257.0±89.5 |
| PT-INR | 2.92±2.36 | 1.32±0.12 | 1.23±0.23 | 1.13±0.06 |
| Platelets
(×104/μl) | 15.2±6.5 | 23.4±12.0 | 16.0±4.7 | 12.6±3.9 |
Pathological examination
The biopsy samples were fixed with 20% formalin and
embedded in paraffin blocks. Serial sections (3 μm) were cut
from the blocks, deparaffinized and subjected to H&E staining
or immunohistochemical staining by an indirect immunoperoxidase
method using Histofine Simple Stain (Nichirei Corporation, Tokyo,
Japan). The primary antibodies used were a monoclonal antibody
against CD-68 (Abcam Inc., Cambridge, MA, USA) and a polyclonal
antibody against LDH-5 (Abcam Inc.). As normal controls for the
immunohistochemical staining, three biopsy samples from liver
transplant (LT) donors were stained using the same procedure. The
intensity of staining for LDH and the number of CD-68-positive
cells were classified into three grades: 1+, weak; 2+, moderate;
and 3+, intense/markedly increased. When the staining result did
not differ from that in the normal control samples, the sample was
graded as 1+.
Results
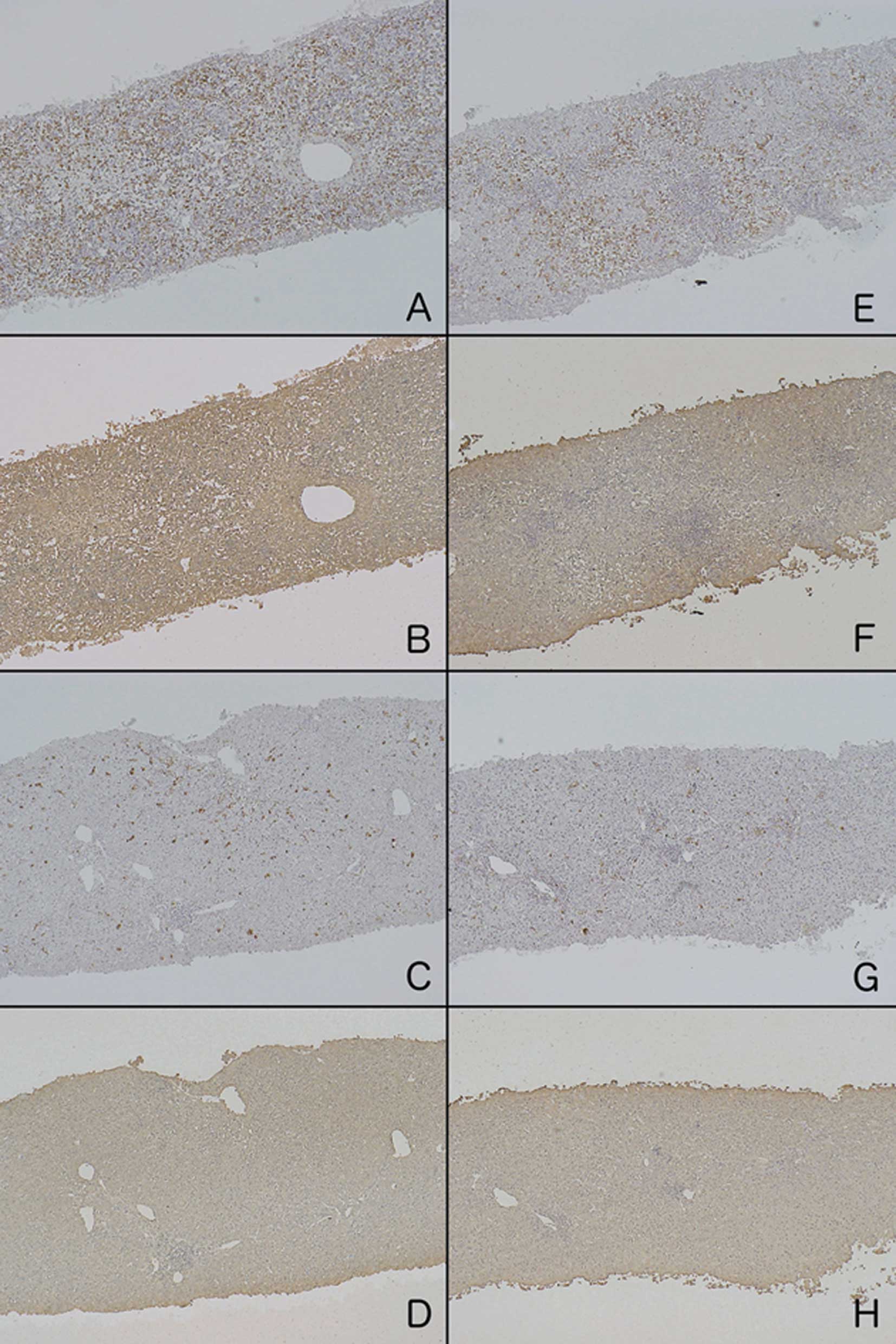
The H&E-stained sections from ALF patients on
admission revealed that >50% of the hepatocytes were necrotized.
In serial sections stained with anti-LDH and anti-CD-68 antibodies,
LDH was strongly and diffusely detected in the remaining
hepatocytes, and a marked increase in the number of CD-68-positive
cells was mainly observed in the sinusoid (Fig. 1). In 4 patients who survived
without LT, a second biopsy was performed during their recovery
phase at 7–10 days after the first biopsy. These second biopsies
exhibited significant decreases in LDH production in hepatocytes
and the number of CD-68-positive cells accompanied by the
regeneration of hepatocytes.
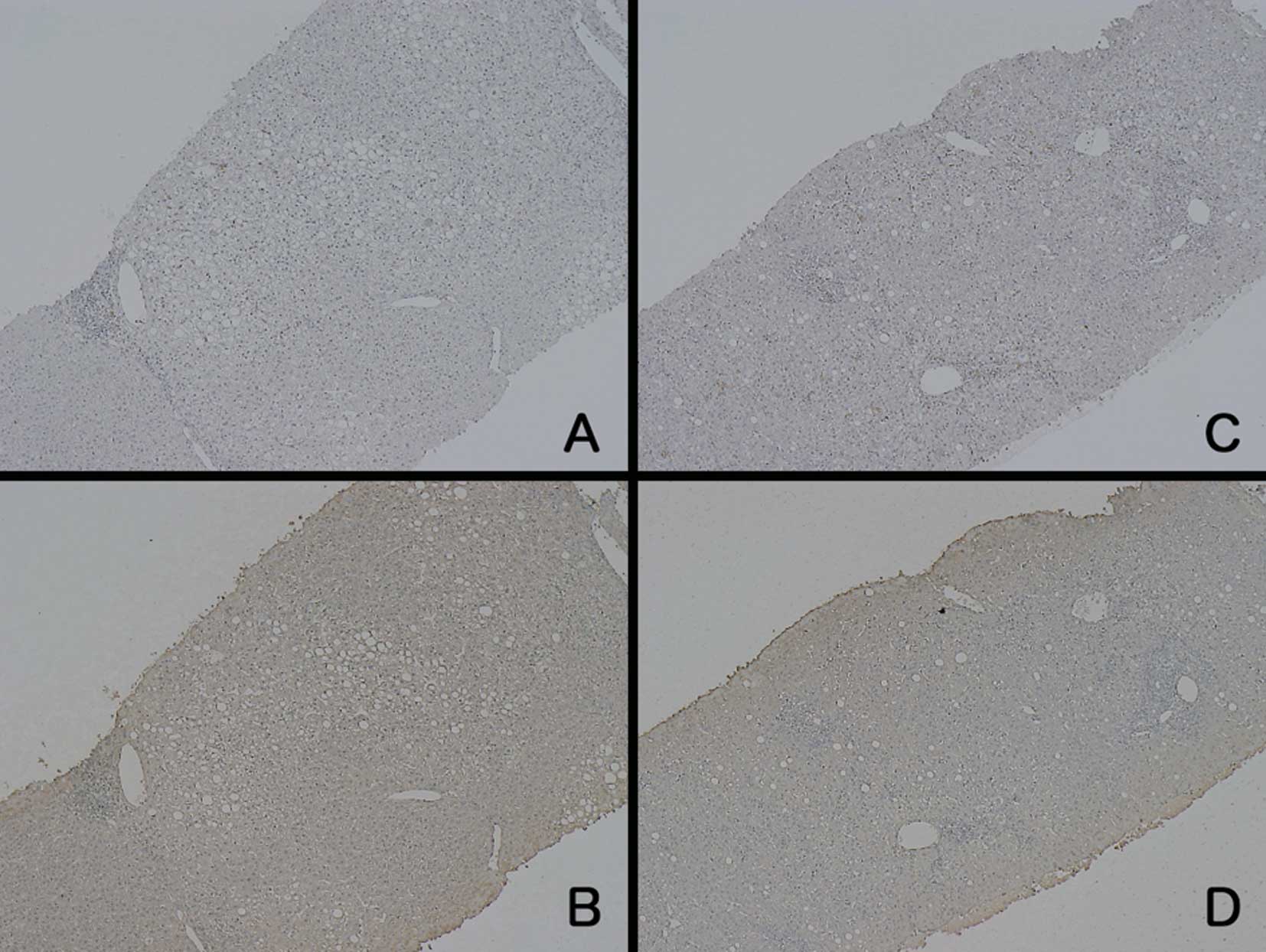
In patients with AH, the results of CD-68
immunostaining were varied (Fig.
2). Two patients exhibited a marked increase in CD-68-positive
cells, similar to the case for the ALF patients, whereas the other
patients showed less intense staining. In contrast to the CD-68
staining, the intensity of LDH staining was generally weak. None of
the CD-68-stained samples from the patients with AH were comparable
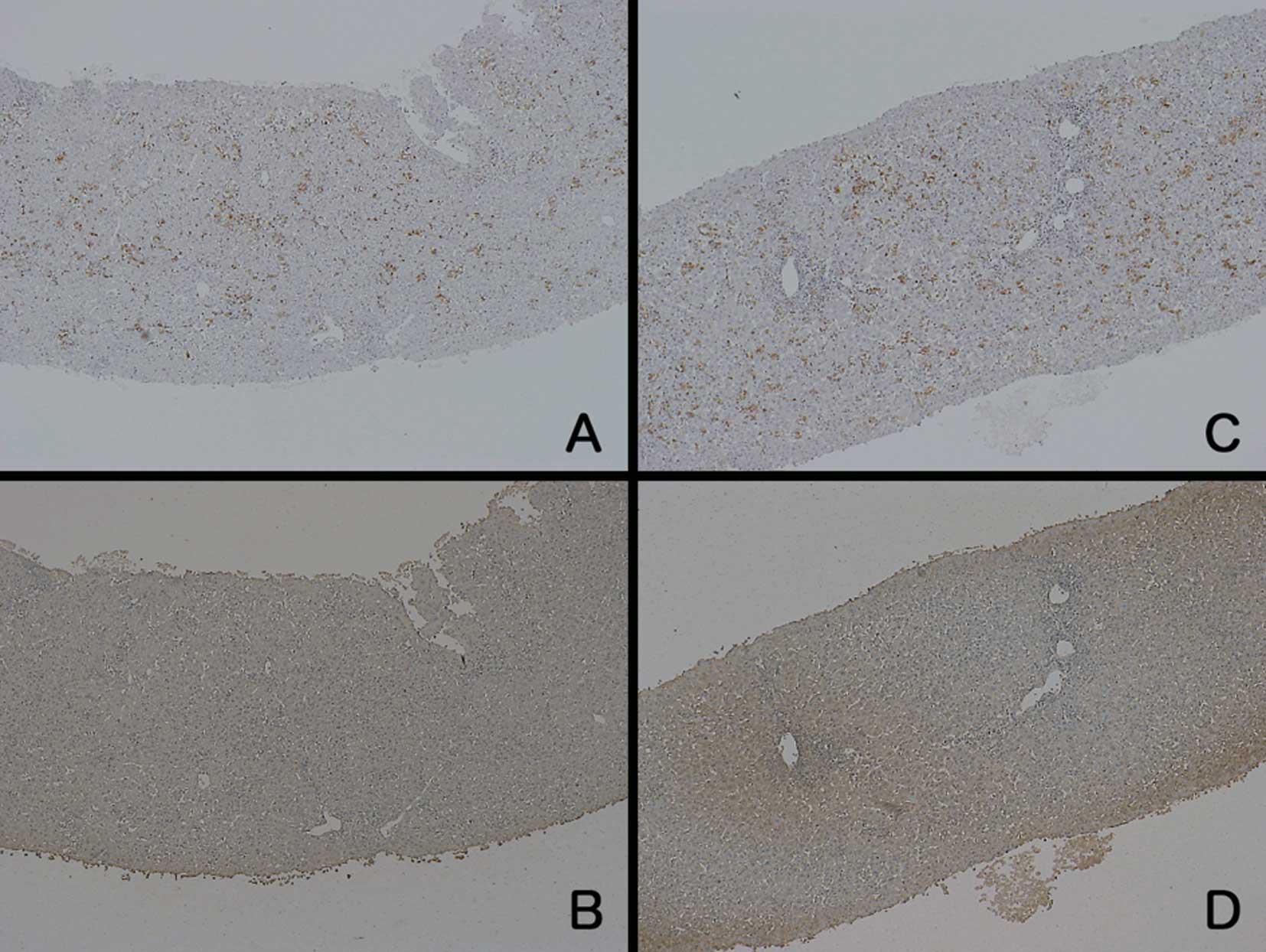
to those from the ALF patients. Regarding the patients with CH, the
staining intensities of both LDH and CD-68 were similar to those in
the normal control samples (Fig.
3).
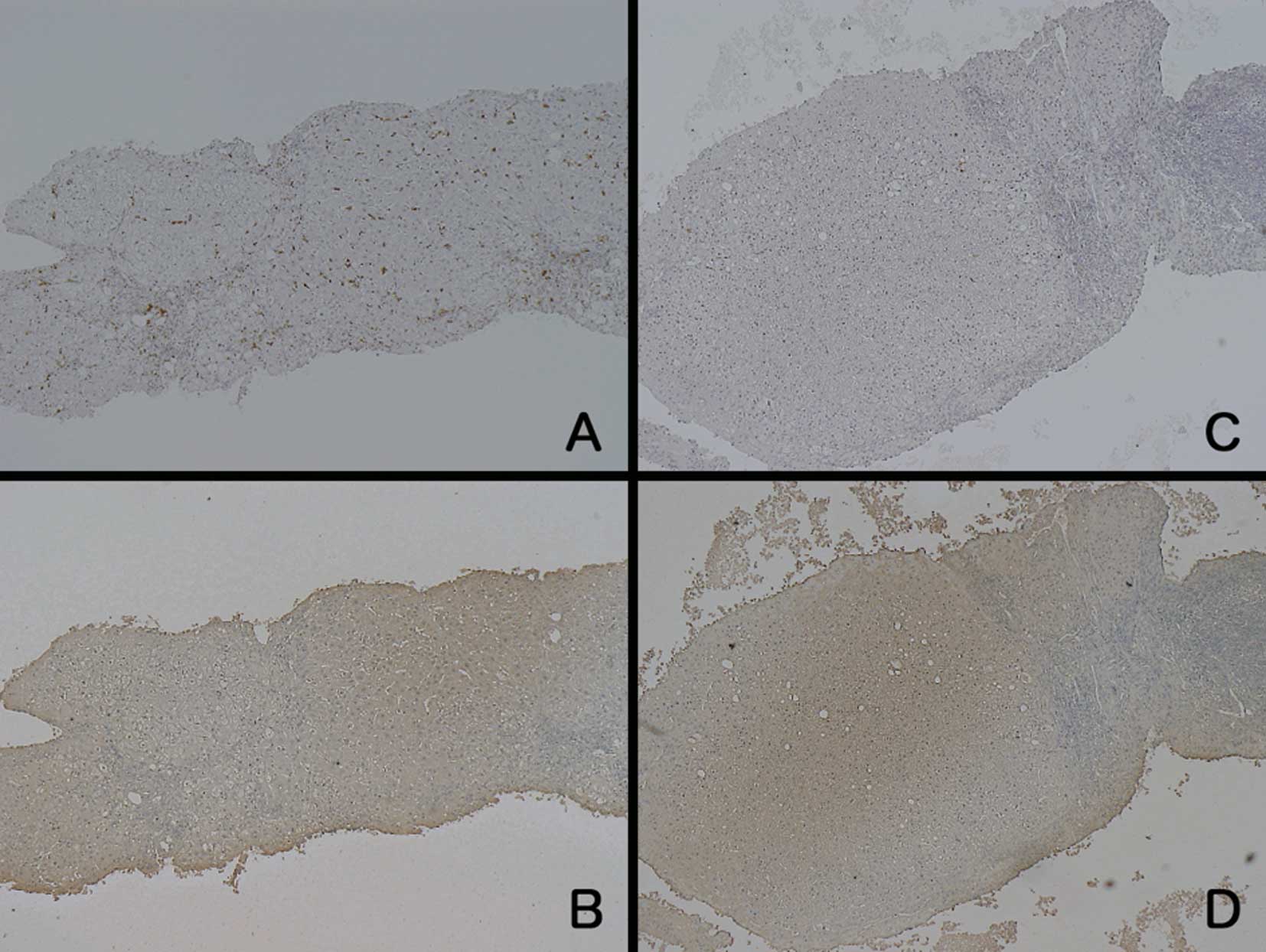
The samples from the LC patients showed different
findings compared to the samples from the patients with the other
diseases (Fig. 4). Regarding LDH
staining, unevenly stained hepatocytes were observed, and the
staining intensity was weaker around the portal area. The
hepatocytes in the zone around the central vein showed relatively
strong staining for LDH, but the intensity was not as strong as
that in the samples from the ALF patients at the acute phase. On
the other hand, the number of CD-68-positive cells was equal or
slightly increased compared to the normal control samples. When the
number of CD-68-positive cells was increased, they were distributed
evenly throughout the liver. The grading results for LDH and CD-68
staining are summarized in Table
II.
| Table II.Grading of staining intensity. |
Table II.
Grading of staining intensity.
| Anti-LDH5
| Anti-CD-68
|
|---|
| 1+ | 2+ | 3+ | 1+ | 2+ | 3+ |
|---|
| ALF (n=7) | 0 | 1 | 6 | 0 | 1 | 6 |
| AH (n=7) | 3 | 2 (2) | 0 | 1 | 3 | 3 |
| CH (n=8) | 7 | 1 | 0 | 7 | 1 | 0 |
| LC (n=6) | 0 | (5) | (1) | 3 | 3 | 0 |
Discussion
Although various ‘triggers’ have the potential to
elicit ALF, most of them are divided into two major categories,
namely direct hepatotoxic agents, such as acetaminophen, and immune
response-related causes, including hepatitis viruses (2,13).
With the former, the degree of liver damage generally depends on
the ingested dose of the toxin. With the latter, however, it
remains unclear why some patients proceed to ALF, while most
patients with the same etiology suffer no more than self-limiting
AH. The main ‘triggers’ of the latter category are hepatitis
viruses, such as HBV, HAV and hepatitis E virus (HEV), in which
hepatocyte death has been considered to be induced by cytotoxic T
cells that recognize virus-related antigens on the cell surface
(14). However, this cytotoxic
mechanism cannot explain the reason why a limited proportion of
patients develop ALF.
Previous studies using ALF animal models with immune
response processes indicated that hepatic microcirculation
disturbance may play a pivotal role in disease progression, which
may be caused by cytotoxic mediators from activated
sinusoidal-lining cells (6–8).
However, it is difficult to directly prove that the hepatocytes of
patients suffering from ALF exist under hypoxic conditions. Before
beginning the present study, we hypothesized that the amount of
intracellular LDH represents the intracellular oxygen concentration
as it is well known to exhibit increased transcription under
anaerobic conditions (9–12). This phenomenon is widely observed,
regardless of the organ or species. Considering that LDH is only a
crucial enzyme for anaerobic respiration, its increased
transcription appears to be a reasonable response.
In the past, elevated serum levels of LDH in
patients with hepatitis have merely been regarded as a result of
enzyme leakage following destruction of hepatocytes. Furthermore,
the diagnostic value of serum LDH in liver disease has been thought
to be low compared to alanine aminotransferase due to the
widespread distribution of LDH-producing cells in the whole body
(15,16). The only exceptions are congestive
liver and shock liver, which are both associated with decreases in
the hepatic oxygen concentration and distinct increases in the
serum LDH level (17). Taking
these findings for hypoxic liver diseases together with the
observation that LDH transcription increases under hypoxic
conditions, we consider that the degree of LDH production can be
used as a marker of hypoxia in immunohistochemical analysis.
In patients with ALF, we found that LDH production
in the remaining hepatocytes was distinctly increased compared to
that in other liver diseases. Considering that our biopsy samples
were obtained within 1 week of onset, this result indicates that
microcirculation disturbance exists from the early stage of the
disease. Another important point is that the biopsy samples
simultaneously showed marked proliferation of macrophages. In
patients who survived without liver transplantation, decreases in
the number of macrophages and in the amount of LDH production
occurred during the recovery phase. Notably, some of the patients
with AH showed similar macrophage proliferation as the ALF
patients, but their intensity of LDH staining was not strong.
Although it remains unclear what finally drives AH patients with
macrophage overactivation towards ALF, we suggest that a transient
hyper-inflammatory response results in self-limiting AH, while a
persistent and uncontrolled response leads to ALF.
Another important finding of the present study was
the elevated LDH production in patients with LC. Although the
intensity of this staining was not comparable to that in samples
from the ALF patients, it was significantly stronger than that in
samples from CH patients. Although hepatocyte hypoxia in cirrhosis
has been well established in experimental models, it has been
difficult to demonstrate that hepatocytes in cirrhotic patients
exist under hypoxic conditions (18,19).
Our findings suggest that hepatocytes in patients with LC are under
chronic hypoxic conditions, which may be caused by accumulated
collagens that interfere with oxygen diffusion to hepatocytes.
In our preliminary study, we were unable to enroll
any cases of acetaminophen-induced ALF as such cases are scarce in
Japan. Therefore, it remains unclear whether microcirculation
disturbance contributes to the progression of ALF caused by agents
exhibiting direct hepatotoxicity. We presume that the process of
macrophage overactivation and consequent microcirculation
disturbance may have lower contributions to ALF caused by
acetaminophen. Further studies, including animal model experiments,
are required.
In conclusion, we demonstrated that LDH production
in hepatocytes varies among ALF, AH, LC and CH patients, and we
consider that this variation reflects the degree of intracellular
oxygen concentration. Elevated macrophage proliferation and
increased LDH production were observed in ALF, suggesting that
overactivation of macrophages and subsequent microcirculation
disturbance in the liver play pivotal roles in the progression of
ALF.
References
|
1.
|
Sass DA and Shakil AO: Fulminant hepatic
failure. Liver Transpl. 11:594–605. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Vaquero J and Blei AT: Etiology and
management of fulminant hepatic failure. Curr Gastroenterol Rep.
5:39–47. 2003. View Article : Google Scholar
|
|
3.
|
Hiraoka A, Horiike N, Akbar SM, Michitaka
K, Matsuyama T and Onji M: Soluble CD163 in patients with liver
diseases: very high levels of soluble CD163 in patients with
fulminant hepatic failure. J Gastroenterol. 40:52–56. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Matsui A, Mochida S, Ohno A, Nagoshi S,
Hirose T and Fujiwara K: Plasma osteopontin levels in patients with
fulminant hepatitis. Hepatol Res. 29:202–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Møller HJ, Gronbaek H, Schiodt FV,
Holland-Fischer P, Schilsky M, Munoz S, Hassanein T and Lee WM; the
US Acute Liver Failure Study Group: Soluble CD163 from activated
macrophages predicts mortality in acute liver failure. J Hepatol.
47:671–676. 2007.PubMed/NCBI
|
|
6.
|
Fujiwara K, Ogata I, Ohta Y, Hirata K, Oka
Y, Yamada S, Sato Y, Masaki N and Oka H: Intravascular coagulation
in acute liver failure in rats and its treatment with antithrombin
III. Gut. 29:1103–1108. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Hirata K, Ogata I, Ohta Y and Fujiwara K:
Hepatic sinusoidal cell destruction in the development of
intravascular coagulation in acute liver failure of rats. J Pathol.
158:157–165. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Takenaka K, Sakaida I, Yasunaga M and
Okita K: Ultrastructural study of development of hepatic necrosis
induced by TNF-alpha and D-galactosamine. Dig Dis Sci. 43:887–892.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Firth JD, Ebert BL, Pugh CW and Ratcliffe
PJ: Oxygen-regulated control elements in the phosphoglycerate
kinase 1 and lactate dehydrogenase A genes: similarities with the
erythropoietin 3′ enhancer. Proc Natl Acad Sci USA. 91:6496–6500.
1994.PubMed/NCBI
|
|
10.
|
Kay HH, Zhu S and Tsoi S: Hypoxia and
lactate production in trophoblast cells. Placenta. 28:854–860.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Shi LB, Huang JH and Han BS: Hypoxia
inducible factor-1alpha mediates protective effects of ischemic
preconditioning on ECV-304 endothelial cells. World J
Gastroenterol. 28:2369–2373. 2007.PubMed/NCBI
|
|
12.
|
Sorensen BS, Alsner J, Overgaard J and
Horsman MR: Hypoxia induced expression of endogenous markers in
vitro is highly influenced by pH. Radiother Oncol. 83:362–366.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Losser MR and Payen D: Mechanisms of liver
damage. Semin Liver Dis. 16:357–367. 1996. View Article : Google Scholar
|
|
14.
|
Guidotti LG and Chisari FV: Immunobiology
and pathogenesis of viral hepatitis. Annu Rev Pathol. 1:23–61.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Rosalki SB and McIntyre N: Biochemical
investigations in the management of liver disease. Oxford Textbook
of Clinical Hepatology. 2nd edition. Bircher J: Oxford University
Press; Oxford: pp. 503–521. 1999
|
|
16.
|
Kaplan MM: Laboratory tests. Diseases of
the Liver. 7th edition. Shiff L: J.B. Lippincott Company;
Philadelphia: pp. 108–144. 1993
|
|
17.
|
Cassidy WM and Reynolds TB: Serum lactic
dehydrogenase in the differential diagnosis of acute hepatocellular
injury. J Clin Gastroenterol. 19:118–121. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
DeLeve LD: Hepatic microvasculature in
liver injury. Semin Liver Dis. 27:390–400. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Chaparro M, Sanz-Cameno P, Trapero-Marugan
M, Garcia-Buey L and Moreno-Otero R: Mechanisms of angiogenesis in
chronic inflammatory liver disease. Ann Hepatol. 6:208–213.
2007.PubMed/NCBI
|