Introduction
Neoplastic cell proliferation and inhibition of cell
death is maintained by aberrant gene expression. Histone
deacetylase (HDAC) inhibitors offer a means to modulate these
genetic changes by influencing the status of the epigenome
(1). Valproate (VPA), an
established HDAC inhibitor for the treatment of epilepsy with
advantageous pharmacokinetic properties, causes hyperacetylation of
the N-terminal tails of histones H3 and H4 in vitro and
in vivo (2). Thereby, VPA
directly inhibits HDAC enzymatic activity at concentrations of
approximately 0.5 mM (3).
The associated antitumor effect of VPA is conveyed
by modulating multiple pathways, including cell cycle, apoptosis,
angiogenesis, metastasis, differentiation and senescence. These
effects depend in part on the level of differentiation and the
associated underlying genetic alterations which differ between
tumor entities. This could explain why VPA does not show homogenous
responses in all malignant cell types. The majority of clinical
data on the antitumor effects of VPA have been generated in the
context of malignant hematological diseases (4). Solid tumors have recently been added
to the circle of malignancies whose growth and survival may be
influenced by VPA (5), which is
currently in clinical trials (6).
Information on the activity of VPA and other HDAC inhibitors
against colon cancer is limited and inconsistent among cell lines
(7). For example, the VPA effect
has been shown to partially depend on the expression of wild-type
adenomatous polyposis coli (APC) protein in colon cancer cell
lines, whereas mutant APC-expressing cells showed decreased VPA
sensitivity (8). Additionally, the
response of freshly isolated primary colon cancer cells to VPA
treatment showed inter-individual variations (9).
To clarify the extent and variability of VPA
activity concerning colon cancer cell growth, the in vitro
response of four colon cancer cell lines (Caco-2, SW-480, CX-1 and
WIDR) to VPA exposure was investigated. Furthermore, SW-480 cells
were implanted into NOD/SCID mice to compare the in vitro
findings with the anti-neoplastic effect of VPA in vivo.
Materials and methods
Cell cultures
Human colon cancer cell lines (Caco-2, SW-480, CX-1
and WIDR) were obtained from DSMZ (Braunschweig, Germany). Caco-2,
CX-1 and WIDR cells were cultured in Earles' minimum essential
medium (MEM), and SW-480 cells were cultured in McCoy's 5A medium
(Gibco/Invitrogen, Kalsruhe, Germany), supplemented with 10% fetal
calf serum (FCS), 20 mM HEPES-buffer (pH 7.4), 2% glutamine and 1%
penicillin/streptomycin. Subcultures from passages 7–11 were
selected for experimental use.
Drugs
VPA (a gift from G.L. Pharma GmbH, Lannach, Austria)
was used at a final concentration of 0.25, 0.5, 1 or 2 mM. The
tumor cells were treated with VPA for 3 or 5 days. The controls
remained untreated. To exclude the toxic effects of the compounds,
cell viability was determined by trypan blue
(Gibco/Invitrogen).
Detection of apoptosis
To detect apoptosis, the expression of Annexin
V/propidium iodide (PI) was evaluated using the Annexin V-FITC
Apoptosis Detection kit (BD Pharmingen, Heidelberg, Germany).
Staining was performed according to the manufacturer's
instructions. Briefly, tumor cells were dislodged with accutase
(PAA Laboratories GmbH, Coelbe, Germany), washed once with cold PBS
and resuspended in 1X binding buffer (1×106 cells/ml).
The cell suspension (100 μl; 1×105 cells) was
then incubated with 5 μl of Annexin V-FITC and 5 μl
of PI in the dark for 15 min at room temperature. The cells were
analyzed on a FACScalibur (BD Biosciences, Heidelberg, Germany).
The percentage of apoptotic cells (early and late) in each quadrant
was calculated using CellQuest software (BD Biosciences).
Measurement of tumor cell growth
Cell growth was assessed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
dye reduction assay (Roche Diagnostics, Penzberg, Germany). Treated
vs. non-treated tumor cells (100 μl; 1×104
cells/ml) were seeded into 96-well culture plates. After 24, 48 and
72 h, MTT (0.5 mg/ml) was added to the cells and incubated for 4 h
at 37°C in 5% CO2. Thereafter, solubilization buffer
containing 10% SDS in 0.01 M HCl was added to dissolve the purple
formazan salt crystals by allowing the plate to stand overnight at
37°C in 5% CO2. The absorbance of the solubilized purple
formazan product was spectrophotometrically measured at 570 nm
using Infinite® M 200 microplate reader
(Tecan-Deutschland, Crailsheim, Germany). Each experiment was
carried out in triplicate. After subtracting the background
absorbance, the results were expressed as the mean cell number.
Cell cycle analysis
Tumor cells were grown to 70% confluency and
subsequently treated with VPA (controls remained untreated). After
24 h, tumor cell populations were stained according to the
manufacturer's instruction using a Cycle Test Plus DNA Reagent kit
(Becton Dickinson) and subsequently analyzed by a FACScan flow
cytometer (Becton Dickinson). Events (10,000) were collected from
each sample. Data acquisition was carried out using CellQuest
software, and the cell cycle distribution was calculated using
ModFit software (Becton Dickinson). The number of gated cells in
G1, G2/M or S-phase was presented as a percentage.
Western blot analysis
To explore cell cycle-regulating proteins, tumor
cell lysates were subjected to sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE; 50 μg per
lane). PeqGold pre-stained protein marker IV (Peqlab Biotechnologie
GmbH, Erlangen, Germany) was used as a molecular weight standard.
Following separation, protein was transferred to a polyvinyl
difluoride membrane (PVDF; Hybond P; GE Healthcare, Munich,
Germany). The blots were blocked with 10% low-fat milk for 1 h at
room temperature, followed by incubation overnight at 4°C with
monoclonal antibodies directed against cell cycle proteins: Cdk1
(IgG1, clone 1, 1:2,000), cdk2 (IgG2a, clone 55, 1:2,000), cdk4
(IgG1, clone 97, 1:250), cyclin D1 (IgG1, clone G124-326, 1:500),
cyclin E (IgG1, clone HE12, 1:2,000), p19 (IgG1, clone 52,
1:5,000), p21 (IgG1, clone 2G12, 1:200) and p27 (IgG1, clone 57,
1:200) (all antibodies from BD Biosciences). Apoptosis was analyzed
using bax (IgG1, clone 3, 1:250) and bcl-2 (IgG1, clone 7/bcl-2,
1:500) antibodies from BD Transduction Laboratories (Heidelberg,
Germany). Anti-histone H3 (IgG, clone Y173, 1:5,000),
anti-acetylated H3 (IgG, clone Y28, 1:500), anti-histone H4
(polyclonal IgG, 1:250) and anti-acetylated H4 (Lys8, polyclonal
IgG, 1:500) were all from Biomol GmbH (Hamburg, Germany).
HRP-conjugated goat anti-mouse IgG (1:5,000; Millipore GmbH,
Schwalbach/Ts, Germany) served as the secondary antibody. β-actin
(1:1,000; Sigma, Taufenkirchen, Germany) served as a loading
control. The antibodies were diluted in Towbin buffer with 0.5%
Tween-20 and 0.5% bovine serum albumin. The membranes were briefly
incubated with ECL detection reagent (Enhanced
Chemiluminescence-ECL™; GE Healthcare) and exposed to ECL Hyperfilm
(GE Healthcare) developed in Curix 60 (Agfa, Dusseldorf, Germany)
and documented by Gel Doc (BioRad Laboratories GmbH, Munich,
Germany).
Tumor growth in vivo
In vivo experiments were carried out
ethically by EPO-Experimental Pharmacology & Oncology GmbH
(Berlin, Germany). SW-480 cells (1×107) were injected
subcutaneously into female NOD/SCID mice. Treatment was initiated
when the tumors had grown to a palpable size (5–6 mm diameter). VPA
was dissolved in saline, and the mice were treated
intraperitoneally with single doses of 100, 200 and 300 mg/kg bw
VPA once daily (n=10). The control mice received normal saline.
Each amount of VPA was well-tolerated, and the experiment was
terminated on day 25. Tumor size was measured using a vernier
caliper. Body weight and mortality were recorded continuously to
estimate tolerability. The tumor tissue was excised to evaluate
protein expression by Western blot analysis.
Statistics
The experiments were performed 3–6 times.
Statistical significance was calculated by the Wilcoxon
Mann-Whitney U-test. Differences were considered statistically
significant at a p-value of <0.05.
Results
Diminished tumor cell proliferation under
valproate exposure in vitro
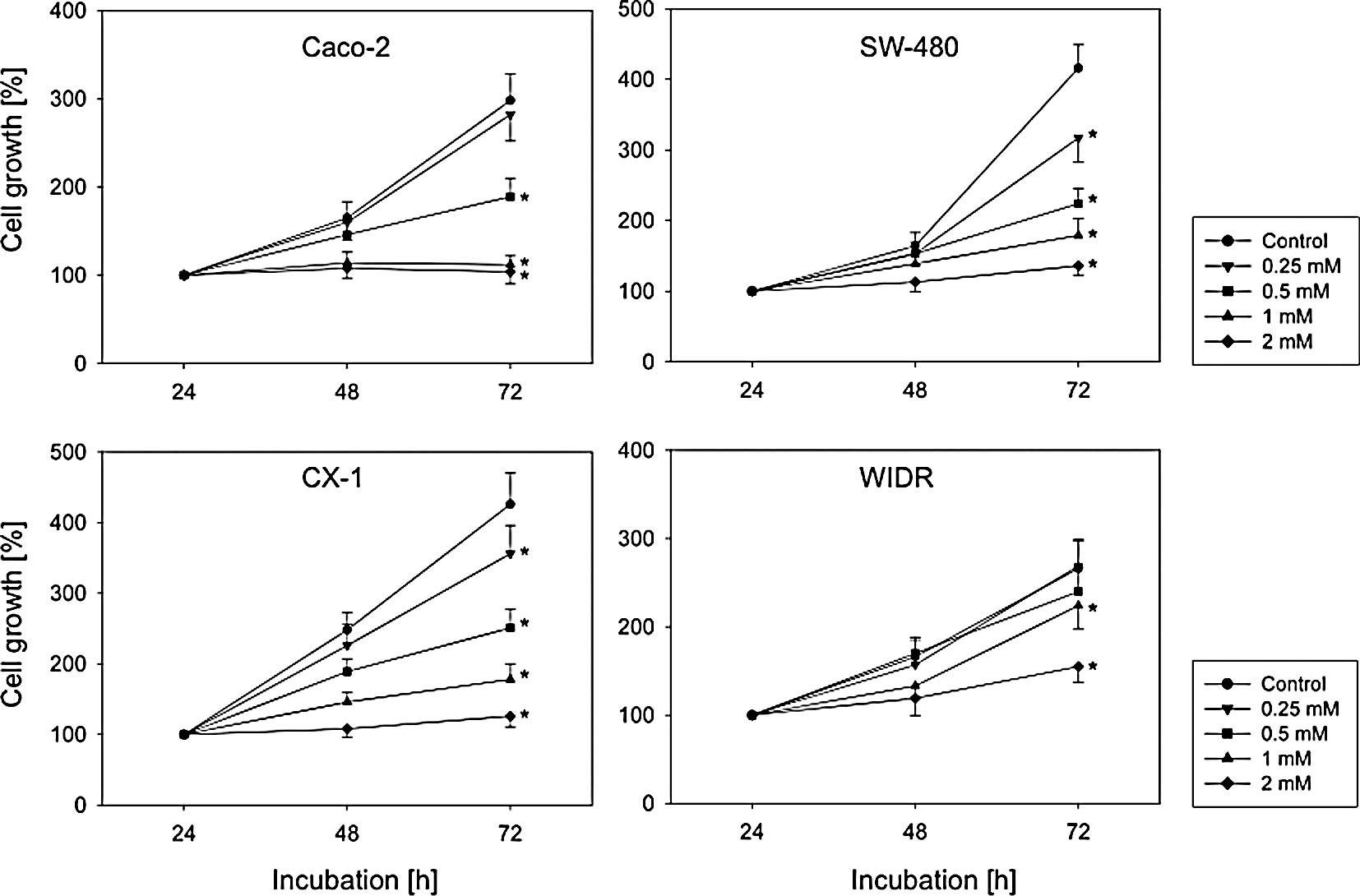
VPA significantly blocked the growth of the cell
lines investigated (Fig. 1). No
differences were noted between 3 and 5 days of pre-treatment.
Therefore, only the results after a 5-day VPA exposure are shown.
Nevertheless, the efficacy of VPA to diminish tumor growth depended
on the cell line used. WIDR cells responded weakly to VPA.
Application of 2 mM VPA was required to distinctly down-regulate
WIDR cell growth. By contrast, 0.5 mM VPA was sufficient to stop
the cell growth of Caco-2 cells, and the growth dynamics of CX-1
and SW-480 were altered even at concentrations of 0.25 mM VPA
(Fig. 1).
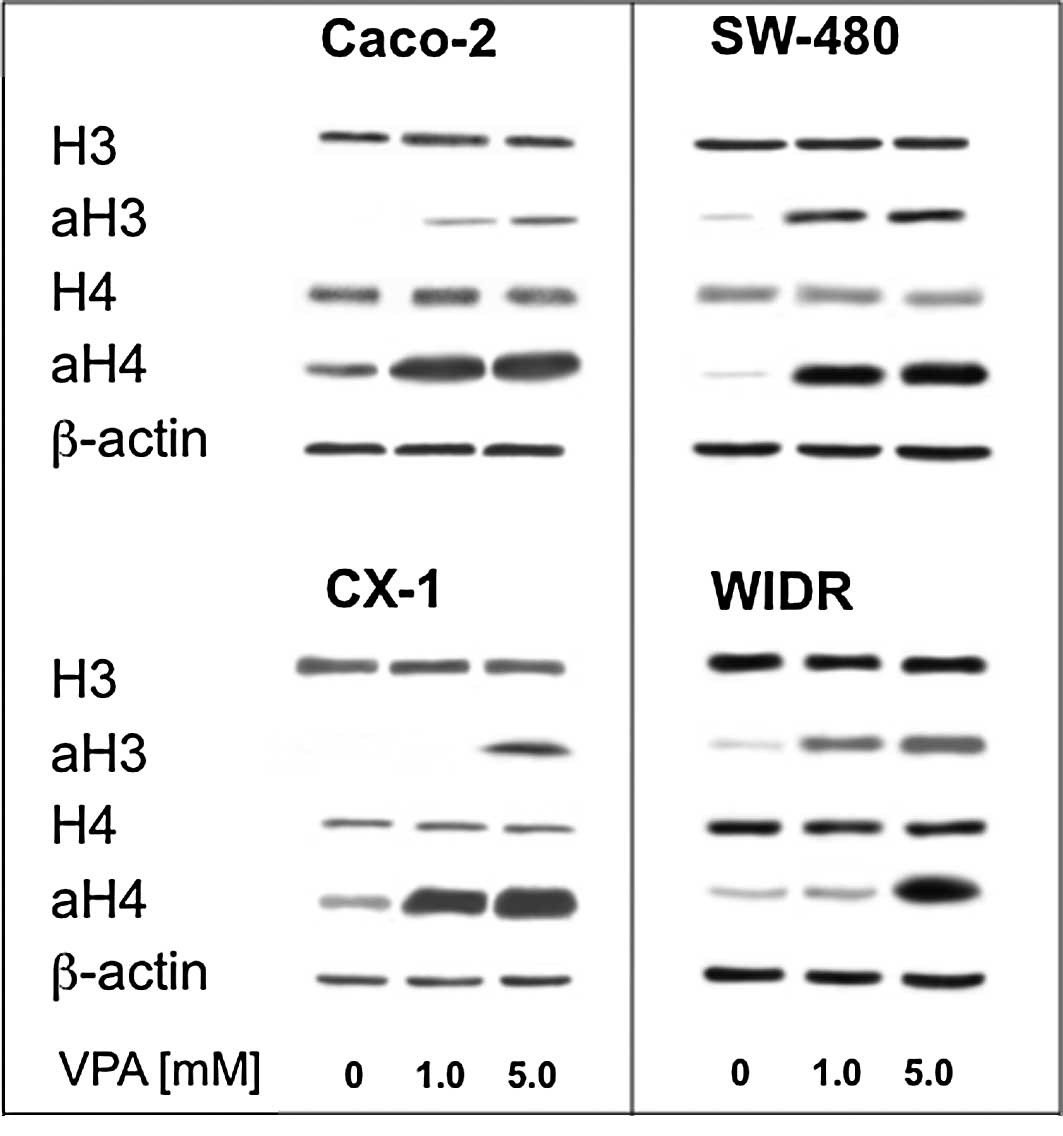
Valproate exposure increases histone
deacetylase-3 and -4 acetylation
As VPA is an inhibitor of histone deacetylase, this
experiment aimed to prove that this property also holds true for
the colorectal cell lines investigated. Therefore, acetylation was
analyzed by Western blotting. The four cell lines showed a
dose-dependent increase in H3 and H4 acetylation under the
influence of VPA, while the amount of histone proteins did not
change (Fig. 2).
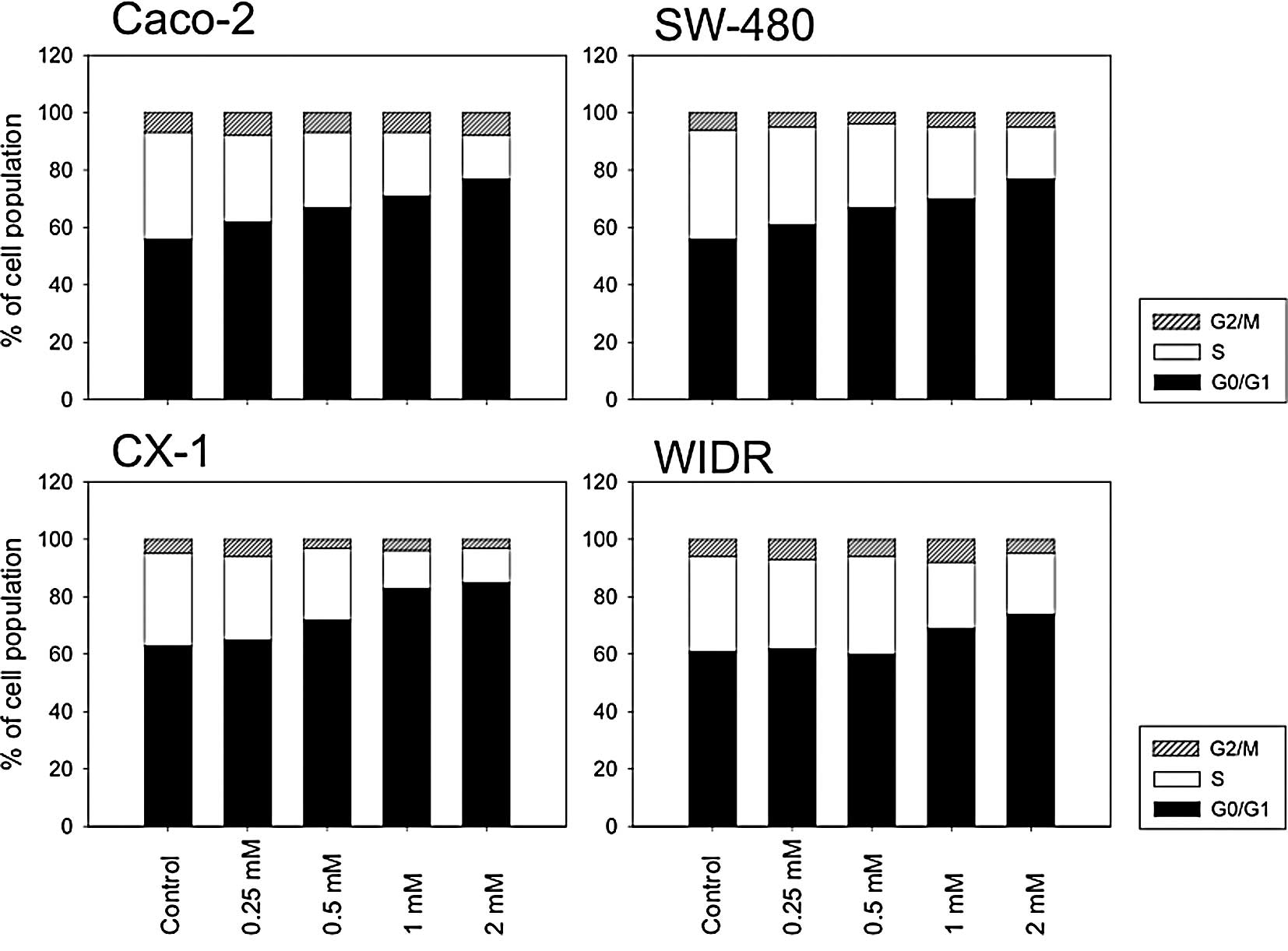
Valproate alters the proportion of cells
in different cell cycle phases
A major finding in cell cultures under
anti-neoplastic treatment is the shift of cell cycle phases. This
effect has also previously been shown for VPA (5). Therefore, the proportion of the
colorectal cancer cell lines undergoing various cell cycle phases
under VPA exposure was determined. The four cell lines displayed a
concentration-dependent cell cycle shift towards the G0/G1 phase
and a corresponding reduction in S-phase cells (Fig. 3). The strongest alterations were
induced in Caco-2, CX-1 and SW-480 cells, whereas WIDR responded
only poorly to the drug treatment.
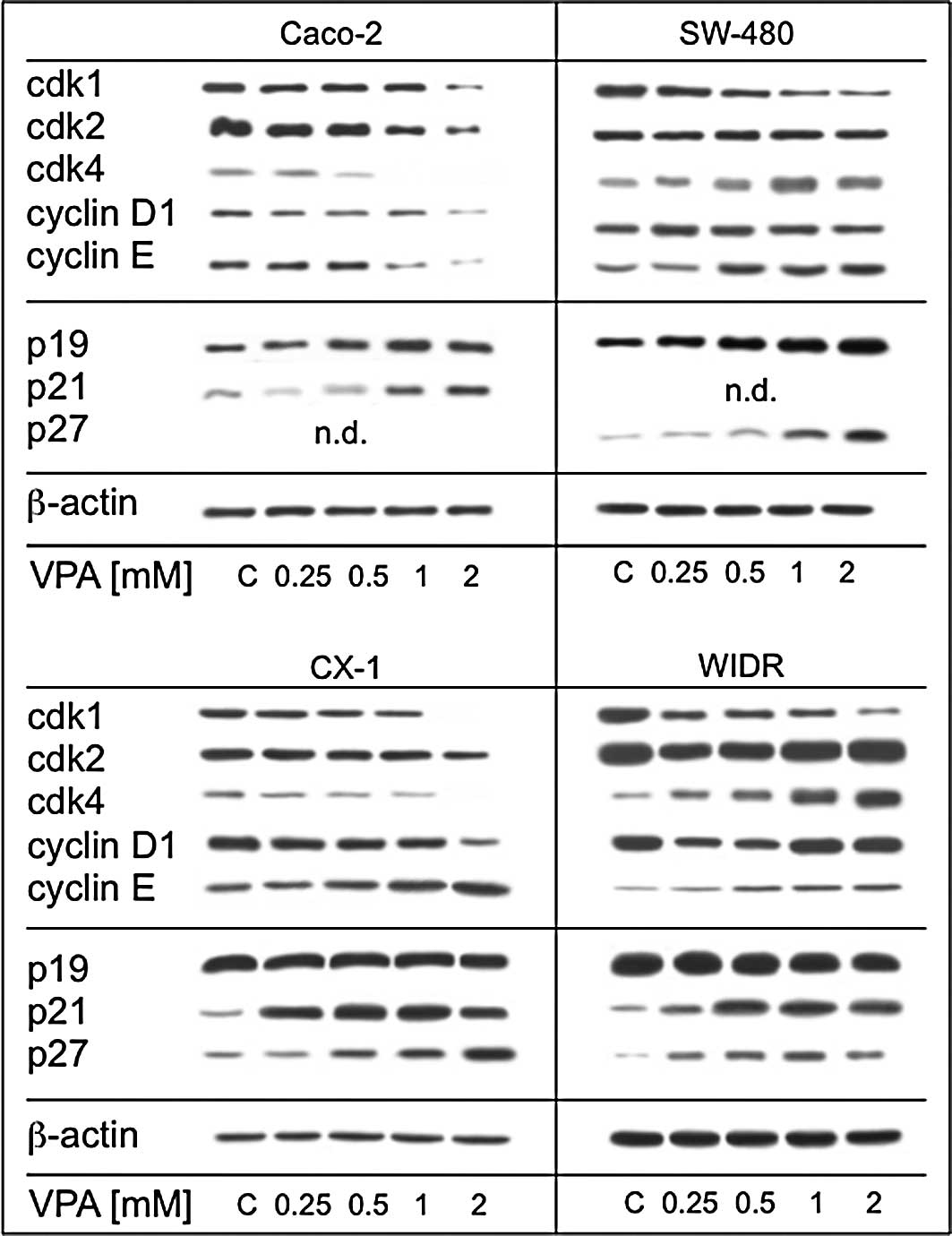
Valproate alters key cell
cycle-regulating proteins
Subsequent experiments concentrated on the
mechanistic background responsible for the cell growth blockade.
Fig. 4 shows that the expression
of regulatory proteins was altered differentially by VPA among the
cell lines investigated. A distinct cell cycle protein response was
noted in the Caco-2 cells. Here, the expression of cdk1, 2 and 4 as
well as cyclin D1 and E was reduced in a dose-dependent manner
(Fig. 4A). By contrast, SW-480
(Fig. 4B) displayed a clear
reduction in expression for cdk1. CX-1 cells (Fig. 4C) came closest to the Caco-2
pattern revealed by the loss of expression of cdk1, 2 and 4 as well
as cyclin D1, while the expression of cyclin E increased under VPA.
WIDR cells (Fig. 4D) showed a
reduction in cdk1 with a more pronounced expression of cdk 4 and
cyclin D1. Additionally, Western blot analysis was performed for
three tumor-suppressor proteins. P19 was strongly up-regulated in
the SW-480 cells and moderately enhanced in the Caco-2 cells. No
alterations were induced in the CX-1 and WIDR cells. P21 was not
detectable in the SW-480 cells, but a significant increase over the
control cell cultures was noted in the WIDR, CX-1 and Caco-2 cells.
P27 was profoundly elevated in the SW-480 and CX-1 cells and, to a
lesser extent, in the WIDR cells. P27 was not detectable in the
Caco-2 control cells (Fig. 4).
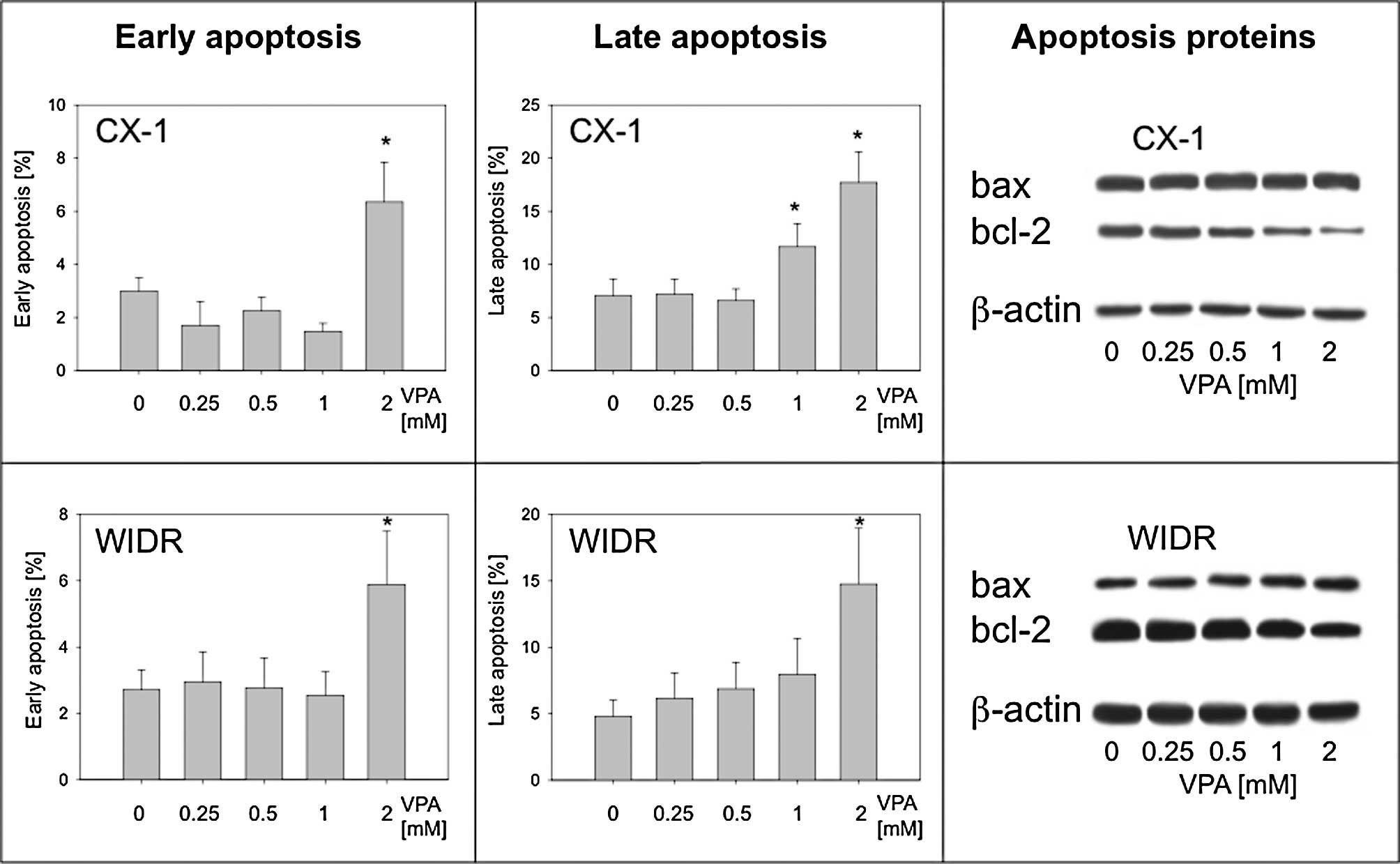
Valproate exposure enhances
apoptosis
The presence of apoptotic events was analyzed and
the alterations of the corresponding key proteins bax and bcl-2 was
evaluated. The proportion of apoptotic Caco-2 cells did not change
in a prominent manner under VPA treatment. However, CX-1 and SW-480
cells showed a significant increase in the cell proportion with
features of early (2 mM VPA) and late apoptosis (1 + 2 mM VPA;
represented by CX-1; Fig. 5A and B,
upper panel). Elevated amounts of apoptotic cells were also
detected with respect to WIDR cells following treatment with 2 mM
VPA (Fig. 5B, lower panel).
Western blot analysis of bax and bcl-2 expression revealed a
dose-dependent decrease in bcl-2, particularly in CX-1 cells,
whereas bax was only slightly enhanced in WIDR cells with 2 mM VPA
exposure (Fig. 5C).
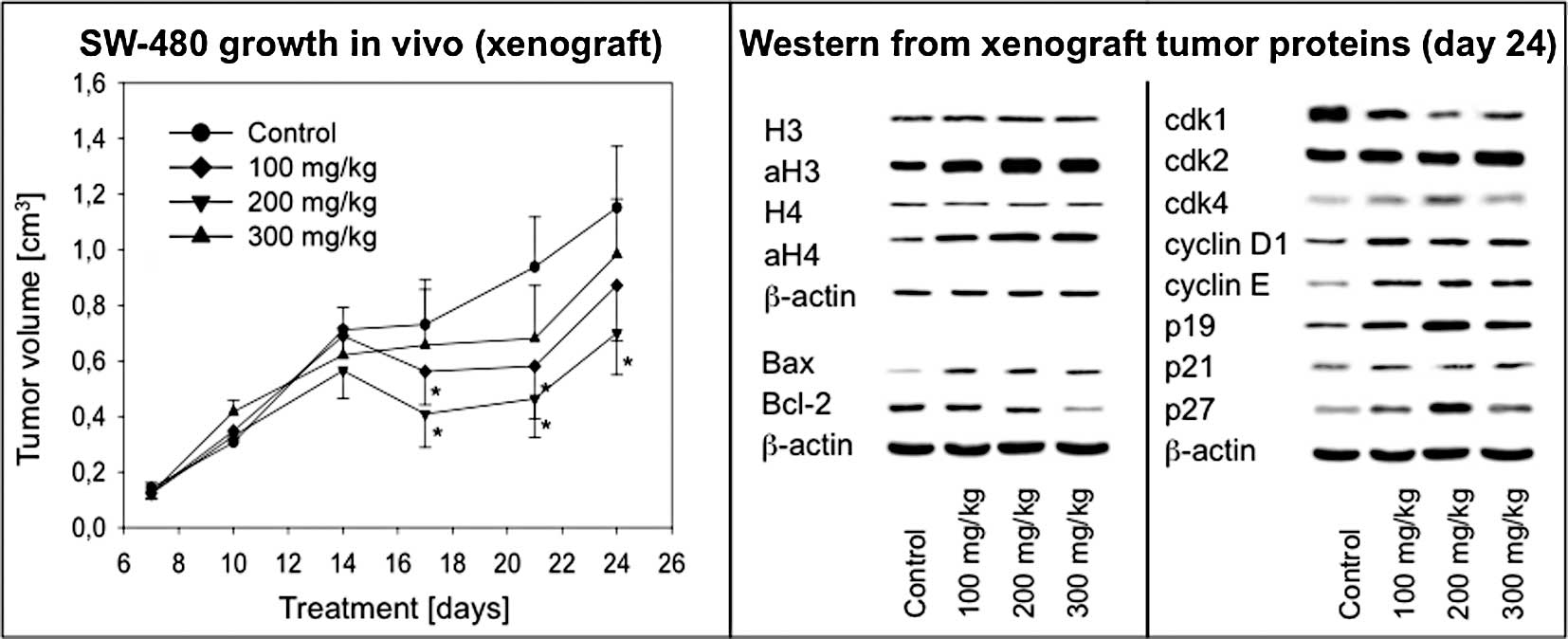
Valproate treatment suppresses colon
cancer growth in vivo
To confirm the in vitro findings in an in
vivo model, tumor xenografts were established in NOD/SCID mice
using the SW-480 cell line. VPA applied at a 200 mg/kg dosage
significantly inhibited tumor growth as measured by tumor volume
(Fig. 6A). At day 21, the tumor
volume was calculated to be 0.51±0.17 cm3 (VPA) vs.
0.89±0.25 cm3 (control). Notably, a VPA dose of 300 mg/kg was less
effective compared to the 200 mg/kg schedule. Tumors showed a
growth pattern recovery at day 25 in the presence of VPA, but
remained significantly smaller than tumors taken from untreated
animals.
Valproate treatment changes histone
acetylation, cell cycle- and apoptosis-related proteins in
vivo
In analogy to the in vitro experiments,
histone acetylation, cell cycle-regulating and apoptosis-related
proteins were analyzed. VPA treatment resulted in an increase in
acetylated H3 and H4 in a dose-dependent manner with a plateau
phase at 200 mg/kg (Fig. 6B). Cdk1
expression was reduced, whereas cdk4 increased with maximum effects
noted at 200 mg/kg VPA (Fig. 6C).
Cyclin D1 and E expression was more prominent under VPA treatment
when compared to the control, although no dose-dependent effects
were observed. Notably, p19 and p27 were considerably elevated with
a maximum response at 200 mg/kg VPA. Finally, VPA treatment
resulted in an increase in the pro-apoptotic protein bax and a
decrease in the anti-apoptotic protein bcl-2 (Fig. 6B).
Discussion
Our findings point to distinct growth blocking
properties of VPA on colon tumor cells in vitro and in
vivo, which are in accordance with previous reports that HDAC
inhibition triggers growth inhibition of colon cancer cell lines
alone or in combination with other drugs (10,11).
Of note, the antiproliferative effect of VPA in vivo peaked
between treatment days 17 and 21. Thereafter, the treatment effect
subsided slightly as tumor growth increased again at day 24 of the
administration period. The anti-neoplastic properties of VPA were
found to be dependent on its exposure time in a kidney cancer cell
model (5). Moreover, the results
of clinical studies on VPA in the context of anti-neoplastic
therapy suggest that chronic application of the drug is required
(12). Nevertheless, in light of
the finding that our in vivo model partially regained its
growth capability under VPA treatment, the potential of VPA to
induce resistance in colon cancer cells must be taken into
consideration (13).
Since VPA is a known HDAC inhibitor, it was of
interest to monitor the alterations of histone acetylation in our
models. High expression of HDAC class I has previously been
described as a possible treatment target and a prognostic factor in
colorectal cancer cells (14).
Additionally, it has been shown that the HDAC inhibitory properties
of VPA are essential for its anti-neoplastic activity (15). Therefore, it is of relevance to
note that histone acetylation increased significantly in our in
vitro and in vivo models. Among the cell cultures,
SW-480 exhibited the strongest acetylation response of H3 and H4 in
comparison to the other cell lines. This finding, together with the
previously described strong expression of HDAC in these cells
(16), contributed to the choice
of SW-480 for the in vivo experiments. High levels of HDAC3
expression in SW-480 cells correlated with a strong response to
HDAC inhibition through VPA treatment (17). A substantial increase in
acetylation under VPA treatment was observed in our SW-480
xenografts, although a certain degree of acetylation was already
prominent in the control animals. Taken together, the findings
suggest that VPA exhibits its anti-neoplastic effect in colorectal
cancer cell lines through alteration of histone acetylation.
In concordance with previously published results,
cell cycle analysis revealed a shift towards the G0/G1 phase under
the influence of VPA, with gradual differences in the investigated
tumor cells. The corresponding Western blot analysis of crucial
cell cycle regulating proteins revealed that only cdk1 expression
was uniformly decreased by VPA in all of the cell lines. We
therefore postulate that this mechanism is (at least partially)
responsible for the cell cycle delay and growth blockade. In this
context, VPA at a high dose of 300 mg/kg applied in vivo
triggered a slight increase in cdk1 expression compared to the 200
mg/kg dose. The reversal of cdk1 response with high VPA doses may
contribute to the corresponding impairment of VPA-induced growth
reduction. Based on the SW-480 results, cdk2 does not appear to
play a role in the context of VPA-induced cell cycle changes. This
can be valued in light of the fact that cdk2 is dispensable for S
phase initiation as shown in cdk2 knockout studies (18). Regarding cdk4, it is of note that
Caco-2 and CX-1 cells responded with down-regulation, and SW-480
and WIDR with the opposite phenomenon. Since the deregulation of
the cyclin D/cdk4 pathway has been identified as a feature of
multiple tumor types contributing to their growth advantage
(19), this finding would not have
been expected.
Furthermore, it is unclear why cyclin E showed
decreased expression solely in Caco-2 cells, while the other cell
lines and the in vivo tumors displayed an increase in its
expression. The fact that cyclin E-deficient mice develop normally
(20) may aid in the assessment of
the role of cyclin E in the anti-neoplastic effect of VPA. Cyclin
D1 is of interest regarding the mitogenic potential of the analyzed
cell lines. Cyclin D1 is overexpressed in a number of cancers
(21) and additionally its
repression promotes cellular differentiation (22). Consequently, the suppression of
cyclin D1 may be relevant for maintaining cellular quiescence and
preventing cell proliferation (23).
In this context, HDAC1 has been described to
participate in a complex that inhibits the cyclin D1 promotor
(24). Therefore, cyclin D1 may be
an ideal target of VPA activity, and its subsequent suppression
under VPA treatment would be expected. However, this held true only
for Caco-2 and CX-1 cells. This again underlines the variability of
cellular events initiated by VPA. Although growth repression and
reduced cell cycle activity were present in all four cell lines,
this cannot be stringently connected with uniform alterations of
cell cycle-regulating proteins. However, it is possible that the
observed increases in cell cycle proteins are initiated to
counteract the antiproliferative effect of VPA, thereby maintaining
mitogen requirements to uphold cellular proliferation (25). Since these changes were also
observed in the in vivo model, we hypothesize that the
up-regulation of cyclins contributed to the recovery of tumor
growth after VPA treatment at day 21 as the tumor becomes adjusted
to the anti-neoplastic effect of VPA (13).
The second group of proteins that impact cell
proliferation are tumor suppressors or cyclin-dependent kinase
inhibitors. We investigated p19, p21 and p27. While p19 was not
altered, p21 and p27 were increased in all cell lines. The in
vivo situation differed somewhat with constant expression of
p21 and increased expression of p19 and p27 in the SW-480 xenograft
tumors under VPA treatment. In light of these observations it is
evident that all three tumor-suppressor proteins were involved in
the anti-neoplastic effect of VPA in the colon cancer cell lines.
p19 has been shown to promote cell cycle arrest or apoptosis via
p53 degradation (26). As a member
of the INK4 protein group, it inhibits the catalytic subunits of
cdk4 and cdk6 (25). Examples for
P19 involvement in HDAC inhibitor activity are scarce. Trichostatin
A and sodium butyrate up-regulated p19 in murine fibroblasts, while
fibrosarcomas in ARF knockout mice were more resistant to HDAC
inhibitor treatment (27).
Notably, the in vivo model exhibited a slightly reduced p19
and p27 induction when treated with 300 mg/kg VPA. This finding may
have contributed to the reduced effectiveness of the higher dose
against tumor growth when compared to 200 mg/kg of VPA.
p21, which was mostly induced by VPA application in
our in vitro experiments, has already been established as a
protagonist in the mode of VPA or HDAC inhibitor activity,
respectively, and recently was confirmed in the treatment of
rhabdomyosarcoma cells with VPA resulting in cell cycle arrest
(28). A similar effect of VPA
treatment with G2/M phase blockade caused by p21 activation has
been shown for human mesenchymal stem cells (29). Moreover, HDAC4-induced growth of
the colon cancer cell line HCT 116 appeared to be dependent on p21,
since its absence abrogated the growth response (30). p27 belongs to a more broadly acting
Cip/Kip protein family which is able to inhibit cyclin D-, E- and
A-dependent kinases (31).
Thereby, it carries the capability to inhibit the progression from
G1 to S phase (cyclin E-dependent) and S phase progression (cyclin
A-dependent) (32). Our results of
up-regulation of p27 in both the in vivo and in vitro
model are in agreement with a number of reports, which have
documented p27 involvement with VPA treatment and G0/G1 phase
arrest (33).
Significant induction of apoptosis in vitro
was recorded only at the highest VPA concentrations. Although
apoptosis is an established process involved in the anti-neoplastic
mechanisms of VPA, we assume that it does not play a predominant
role in our experimental setup and the investigated cell lines.
Similar results have been described for cultured thoracic cancer
cells, where VPA induced apoptosis in less than 20% of the cells in
culture (34), which corroborates
our results.
Taken together, our findings contribute to the
understanding of VPA-associated anti-neoplastic effects. VPA
exhibited a robust growth inhibition of colorectal cancer cell
lines in the in vitro and in vivo models mainly by
modification of the cell cycle and corresponding regulating
proteins. We suggest that VPA may become an innovative option for
the treatment of colorectal malignancies, since VPA is a clinically
established drug with few side effects (12). Nevertheless, a number of questions
have yet to be answered. The regain of tumor growth after a longer
VPA treatment period challenges the concepts of long-term VPA
application to inhibit tumor progression. Perhaps combination
therapy with other cytotoxic agents could come into play.
References
|
1.
|
Taunton J, Hassig CA and Schreiber SL: A
mammalian histone deacetylase related to the yeast transcriptional
regulator Rpd3p. Science. 272:408–411. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Gurvich N, Tsygankova OM, Meinkoth JL and
Klein PS: Histone deacetylase is a target of valproic acid-mediated
cellular differentiation. Cancer Res. 64:1079–1086. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Gottlicher M, Minucci S, Zhu P, et al:
Valproic acid defines a novel class of HDAC inhibitors inducing
differentiation of transformed cells. EMBO J. 20:6969–6978. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Kuendgen A and Gattermann N: Valproic acid
for the treatment of myeloid malignancies. Cancer. 110:943–954.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Jones J, Juengel E, Mickuckyte A, Hudak L,
Wedel S, Jonas D and Blaheta RA: The histone deacetylase inhibitor
valproic acid alters growth properties of renal cell carcinoma in
vitro and in vivo. J Cell Mol Med. 13:2376–2385. 2009. View Article : Google Scholar
|
|
6.
|
Tan J, Cang S, Ma Y, Petrillo RL and Liu
D: Novel histone deacetylase inhibitors in clinical trials as
anti-cancer agents. J Hematol Oncol. 3:52010. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Lea MA, Ibeh C, Shah N and Moyer MP:
Induction of differentiation of colon cancer cells by combined
inhibition of kinases and histone deacetylase. Anticancer Res.
27:741–748. 2007.PubMed/NCBI
|
|
8.
|
Huang X and Guo B: Adenomatous polyposis
coli determines sensitivity to histone deacetylase
inhibitor-induced apoptosis in colon cancer cells. Cancer Res.
66:9245–9251. 2006. View Article : Google Scholar
|
|
9.
|
Friedmann I, Atmaca A, Chow KU, Jager E
and Weidmann E: Synergistic effects of valproic acid and mitomycin
C in adenocarcinoma cell lines and fresh tumor cells of patients
with colon cancer. J Chemother. 18:415–420. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Biran A, Brownstein M, Haklai R and Kloog
Y: Downregulation of survivin and aurora A by histone deacetylase
and RAS inhibitors: a new drug combination for cancer therapy. Int
J Cancer. 128:691–701. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Koyama M, Izutani Y, Goda AE, et al:
Histone deacetylase inhibitors and
15-deoxy-Delta12,14-prostaglandin J2 synergistically induce
apoptosis. Clin Cancer Res. 16:2320–2332. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Muller S and Kramer OH: Inhibitors of
HDACs – effective drugs against cancer? Curr Cancer Drug Targets.
10:210–228. 2010.
|
|
13.
|
Fedier A, Dedes KJ, Imesch P, von Bueren
AO and Fink D: The histone deacetylase inhibitors suberoylanilide
hydroxamic (Vorinostat) and valproic acid induce irreversible and
MDR1-independent resistance in human colon cancer cells. Int J
Oncol. 31:633–641. 2007.
|
|
14.
|
Weichert W, Roske A, Niesporek S, et al:
Class I histone deacetylase expression has independent prognostic
impact in human colorectal cancer: specific role of class I histone
deacetylases in vitro and in vivo. Clin Cancer Res. 14:1669–1677.
2008. View Article : Google Scholar
|
|
15.
|
Venkataramani V, Rossner C, Iffland L, et
al: Histone deacetylase inhibitor valproic acid inhibits cancer
cell proliferation via down-regulation of the Alzheimer amyloid
precursor protein. J Biol Chem. 285:10678–10689. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Spurling CC, Godman CA, Noonan EJ,
Rasmussen TP, Rosenberg DW and Giardina C: HDAC3 overexpression and
colon cancer cell proliferation and differentiation. Mol Carcinog.
47:137–147. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Chavez-Blanco A, Perez-Plasencia C,
Perez-Cardenas E, et al: Antineoplastic effects of the DNA
methylation inhibitor hydralazine and the histone deacetylase
inhibitor valproic acid in cancer cell lines. Cancer Cell Int.
6:22006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Satyanarayana A and Kaldis P: Mammalian
cell-cycle regulation: several Cdks, numerous cyclins and diverse
compensatory mechanisms. Oncogene. 28:2925–2939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Liu N, Fang H, Li Y and Xu W: Recent
research in selective cyclin-dependent kinase 4 inhibitors for
anti-cancer treatment. Curr Med Chem. 16:4869–4888. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Hanashiro K, Kanai M, Geng Y, Sicinski P
and Fukasawa K: Roles of cyclins A and E in induction of centrosome
amplification in p53-compromised cells. Oncogene. 27:5288–5302.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Yamamoto M, Tamakawa S, Yoshie M, Yaginuma
Y and Ogawa K: Neoplastic hepatocyte growth associated with cyclin
D1 redistribution from the cytoplasm to the nucleus in mouse
hepatocarcinogenesis. Mol Carcinog. 45:901–913. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Mejlvang J, Kriajevska M, Vandewalle C, et
al: Direct repression of cyclin D1 by SIP1 attenuates cell cycle
progression in cells undergoing an epithelial mesenchymal
transition. Mol Biol Cell. 18:4615–4624. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Klein EA and Assoian RK: Transcriptional
regulation of the cyclin D1 gene at a glance. J Cell Sci.
121:3853–3857. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Rampalli S, Pavithra L, Bhatt A, Kundu TK
and Chattopadhyay S: Tumor suppressor SMAR1 mediates cyclin D1
repression by recruitment of the SIN3/histone deacetylase 1
complex. Mol Cell Biol. 25:8415–8429. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Sherr CJ and Roberts JM: CDK inhibitors:
positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Weber HO, Samuel T, Rauch P and Funk JO:
Human p14 (ARF)-mediated cell cycle arrest strictly depends on
intact p53 signaling pathways. Oncogene. 21:3207–3212. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Matheu A, Klatt P and Serrano M:
Regulation of the INK4a/ARF locus by histone deacetylase
inhibitors. J Biol Chem. 280:42433–42441. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Hecker RM, Amstutz RA, Wachtel M, Walter
D, Niggli FK and Schafer BW: p21 Downregulation is an important
component of PAX3/FKHR oncogenicity and its reactivation by HDAC
inhibitors enhances combination treatment. Oncogene. 29:3942–3952.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Lee S, Park JR, Seo MS, et al: Histone
deacetylase inhibitors decrease proliferation potential and
multilineage differentiation capability of human mesenchymal stem
cells. Cell Prolif. 42:711–720. 2009. View Article : Google Scholar
|
|
30.
|
Wilson AJ, Byun DS, Nasser S, et al: HDAC4
promotes growth of colon cancer cells via repression of p21. Mol
Biol Cell. 19:4062–4075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Toyoshima H and Hunter T: p27, a novel
inhibitor of G1 cyclin-Cdk protein kinase activity, is related to
p21. Cell. 78:67–74. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Edgar BA and Orr-Weaver TL:
Endoreplication cell cycles: more for less. Cell. 105:297–306.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Freidkin I, Herman M, Tobar A, Chagnac A,
Ori Y, Korzets A and Gafter U: Effects of histone deacetylase
inhibitors on rat mesangial cells. Am J Physiol Renal Physiol.
298:F426–F434. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Ziauddin MF, Yeow WS, Maxhimer JB, et al:
Valproic acid, an antiepileptic drug with histone deacetylase
inhibitory activity, potentiates the cytotoxic effect of
Apo2L/TRAIL on cultured thoracic cancer cells through
mitochondria-dependent caspase activation. Neoplasia. 8:446–457.
2006. View Article : Google Scholar
|