Introduction
Similar to bone resorption by osteoclasts, root
resorption of deciduous teeth progresses with the activation of
odontoclasts of macrophage lineage under physiological conditions
(1). In the late stage of root
resorption, a large number of TRAP-positive odontoclasts are seen
on the resorbing dentin surfaces (1,2). The
activity of such monocyte-derived TRAP-positive cells against
calcified hard tissues is prominent when they show a multinuclear
giant cell-like phenotype. The giant cell-like mature osteoclasts
are differentiated from mononuclear osteoclastic cells that develop
from osteoclast precursor cells (3). It has been shown that mononuclear
osteoclastic cells fuse with each other to form multinuclear cells
(MNCs) during the process of maturation, and that mature
osteoclasts usually contain several or more nuclei (4). The formation of osteoclasts requires
two cellular factors: the macrophage-colony stimulating factor
(M-CSF), also known as colony stimulating factor-1, and the
receptor activator of the nuclear factor-κB ligand (RANKL)
(5). M-CSF is indispensable for
proliferation and differentiation of osteoclast precursor cells
that develop from hemopoietic progenitor cells (6). RANKL is a member of the tumor
necrosis factor (TNF) family of cytokines and can induce the
differentiation of osteoclasts from the precursor cells in the
presence of M-CSF (4). It has been
reported that the formation of multinuclear osteoclasts is
influenced by various cytokines, such as interleukin (IL)-1, IL-6,
IL-11 (7,8) and TNF-α (9).
The macrophage migration inhibitory factor (MIF) has
been characterized as a pro-inflammatory cytokine (10) produced by T-lymphocytes, and has
been shown to be associated with the migration of macrophages
during delayed-type hypersensitivity (11,12).
It has been shown that MIF not only plays a role in the immune
system, but that it is also involved in cell proliferation and
differentiation in a variety of organs (13–15).
Onodera et al (16)
reported that matrix metalloproteinase-13 mRNA expression in
osteoblasts was up-regulated by MIF, suggesting that MIF takes part
in osseous metabolism. As hemopoietic progenitor cells that
differentiate into osteoclast precursor cells are of macrophage
lineage (4,17), it is possible that MIF influences
the differentiation or activity of osteoclasts. Although analyses
of bone marrow cells isolated from both wild-type and MIF
transgenic mice have not provided evidence of the direct effects of
MIF on osteoclast formation (18),
it has been shown that trabecular bone volume in the femurs and
vertebrae of MIF-deficient mice is decreased compared to wild-type
mice, suggesting that MIF-deficiency facilitates osteoclastic bone
resorption (19).
Osteoblasts or stromal cells play a role in
osteoclast differentiation, and the interaction of these cells with
osteoclast precursors is crucial for the formation of mature
osteoclasts (20). Osteoblasts are
known to express RANKL, that promotes the differentiation and
activation of osteoclasts, and they also produce osteoprotegerin
(OPG), a member of the TNF receptor superfamily, that suppresses
bone resorption by inhibiting osteoclast formation (21). When the root resorption of
deciduous teeth occurs, hematopoietic progenitor cells migrate from
blood vessels of the periodontal ligament and alveolar bone toward
the root surface (1). In parallel
with the activation of osteoclasts, periodontal tissues adjacent to
the resorbing root surface degenerate without inflammation
(22). As the periodontal
fibroblasts prevent osteoclast formation in a steady state
condition (23), it is plausible
that RANKL-expressing osteoblasts play a predominant role in the
differentiation and/or maturation of osteoclasts around the tooth
root, and MIF may affect the osteoblast activity and regulate root
resorption during the tooth shedding stage. In the present study,
we examined the role of MIF in the formation and action of
dentin-resorbing multinuclear osteoclasts derived from mouse bone
marrow cells.
Materials and methods
Reagents and culture media
MC3T3-E1 cells and mouse bone marrow cells were
cultured in α-minimal essential medium (α-MEM; Gibco BRL, Grand
Island, NY, USA) containing 10% fetal bovine serum (FBS; Hyclone,
Logan, UT, USA). The recombinant soluble form of human RANKL was
purchased from PeproTech, Inc. (Rocky Hill, NJ, USA). Recombinant
human M-CSF was purchased from Austral Biologicals (San Ramon, CA,
USA). Recombinant MIF was expressed in Escherichia coli
BL21/DE3 (Novagen, Madison, WI, USA) and purified as described
previously (24). It contained
<1 pg of endotoxin/μg protein, as determined by the
chromogenic Limulus amebocyte assay (BioWhittaker, Walkersville,
MD, USA).
Osteoclast formation in a co-culture
system
Mouse bone marrow cells were collected from
4-week-old C57BL/6 mice by flushing the femoral shafts with a
26-gauge sterile needle. The cells (1×106 cells/well)
were seeded on confluent MC3T3-E1 osteoblast-like cells
(2×105 cells/well) cultured in 24-well dishes with α-MEM
containing 10% FBS, 10 nM 1-α, 25-dihydroxyvitamin D3
(vitamin D3) and 100 nM dexamethasone (Dex). MIF was
applied to the cells by dissolving in the medium at concentrations
of 0.01, 0.1 and 1.0 μg/ml, and the medium (1 ml/well) was
changed every 2 days. The experimental work was reviewed and
approved by the Animal Care Committee of Hokkaido University
Graduate School of Dental Medicine. After 6 days of co-culture, the
cells were fixed with 10% formaldehyde in PBS for 10 min at room
temperature. After washing with PBS, the cells were treated with an
acetone/ethanol mixture (50:50, v/v) for 1 min at room temperature.
Then, the cells were dried naturally and stained for TRAP by
incubating in 0.1 M sodium acetate buffer (pH 5.0) containing AS-MX
phosphate (Sigma) and Fast red violet LB salt (Sigma) in the
presence of 50 mM sodium tartrate as described previously (25). Rinsed with distilled water,
TRAP-positive MNCs were counted by using a light microscope
(Eclipse 80i; Nikon, Tokyo, Japan).
Quantitative RT-PCR for RANKL mRNA
Confluent MC3T3-E1 cells (2×105
cells/well) on 24-well dishes were cultured with α-MEM containing
10% FBS, 10 nM vitamin D3, 100 nM Dex and MIF (0.01, 0.1
and 1.0 μg/ml) for 24 or 48 h. Total RNA (tRNA) was prepared
from the cells with ISOGEN (Nippon Gene Co., Tokyo, Japan)
according to the manufacturer’s instructions. The tRNA was treated
with DNase I (Takara Bio Co., Kyoto, Japan) for 1 h at 37°C to
remove genomic DNA.
Real-time reverse transcriptase-polymerase chain
reaction (RT-PCR) was performed for the quantification of RANKL and
GAPDH mRNAs with Smart Cycler (Cepheid, Sunnyvale, CA, USA).
Single-stranded cDNA was synthesized from 2 μg of the
treated RNA as follows: the tRNA was incubated with 0.5 μg
oligo(dT)12–18 primer (Gibco Brl) at 70°C for 10 min,
then chilled on ice and mixed with 1 μl 10 mM dNTP
(Gibco-BRL) for 5 min at 25°C. The mixture was reacted with 1
μl Superscript II RNase H-reverse transcriptase (Gibco Brl)
for 10 min at 25°C, which was followed by incubation at 42°C for 50
min. PCR for the cDNAs was set up in a Smart Cycler reaction tube
(Cepheid). According to the instruction of Takara Ex Taq
R-PCR version kit (Takara Bio), 0.25 μl Ex Taq R-PCR,
2.5 μl 10X R-PCR buffer, 0.3 μl 250 mM
Mg2+ solution for R-PCR, 0.75 μl dNTP mixture and
2.5 μl SYBR-Green I (dilution 1:3,000) were mixed with 2
μl sample cDNA, 15.7 μl dH2O, and 0.5
μl 15 μM forward and reverse primers (RANKL, 5′-TAT
GAT GGA AGG CTC ATG GT-3′ and 5′-TGT CCT GAA CTT TGA AAG CC-3′;
GAPDH, 5′-CGG AGT CAA CGG ATT TGG TCG TAT-3′ and 5′-AGC CTT CTC CAT
GTT GGT GAA GAC-3′).
The PCR reaction was performed at the following
temperature cycle: a denaturation step at 95°C for 30 sec and 50
cycles of denaturation at 95°C for 1 sec, annealing at 55°C for 15
sec and extension at 72°C for 15 sec. The amplification for RANKL
and GAPDH cDNAs was carried out simultaneously and the final
products were quantified by measuring fluorescence from SYBR-Green
I bound to the double-stranded cDNA in the reaction tubes. The
fluorescence intensity from the SYBR-Green I-labeled RANKL cDNA was
normalized to that from the labeled GAPDH cDNA.
Measurement of OPG in the culture
media
Confluent MC3T3-E1 cells (2×105
cells/well) on 24-well dishes were cultured with α-MEM containing
10% FBS, 10 nM vitamin D3, 100 nM Dex and MIF (0.01, 0.1
and 1.0 μg/ml) for 24 or 48 h. The concentration of OPG in
the conditioned medium was measured using a mouse OPG immunoassay
kit (ANALYZA Immunoassay System; Techne Co., Minneapolis, MN, USA)
that was based on the sandwich enzyme immunoassay technique. In
brief, standard solutions and the collected samples were placed in
a 96-well microplate pre-coated with monoclonal antibody specific
to mouse OPG and incubated for 2 h at room temperature. After
washing, a peroxidase-labeled polyclonal antibody specific to OPG
was added to each well and incubated for 2 h. Then, unbound
antibody-enzyme reagent was removed by washing and a substrate
reagent was added to the wells. Following incubation for 30 min at
room temperature, absorbance at 450 nm was measured with a
microplate reader.
Osteoclast formation in a bone marrow
cell culture
Bone marrow cells were collected from 4-week-old
C57BL/6 mice as described above. They were applied to a Sephadex
G-10 column (Pharmacia, Uppsala, Sweden) and incubated for 45 min
at 37°C. Then, the cell suspension was passed through the column
with the addition of α-MEM containing 10% FBS and collected. By
this process, adhesive cells mixed in the extract of bone marrow
were trapped in the column (26),
and we used the passed cells as osteoclast precursors for the
subsequent experiment. The cells (2×104 cells/well) were
plated in 24-well plates with 1 ml of α-MEM containing 10% FBS, 5
ng/ml M-CSF, 50 ng/ml soluble RANKL and 0.1 μg/ml MIF. After
6 days of culture, the cells were fixed and stained for TRAP as
described earlier.
Scanning electron microscope (SEM)
observation of resorption pits
The activity of osteoclasts in terms of degrading
calcified tissue was assessed by a comparison of the size of the
resorption pits which appeared on mammoth tusk-derived dentin
slices (Hokudo Co., Sapporo, Japan). Osteoclast precursor cells
(2×104 cells/well) were plated on a dentin slice (14 mm
in diameter, 300 μm in thickness) placed in the well of
culture plates containing 1 ml of α-MEM supplemented with 10% FBS,
5 ng/ml M-CSF, 50 ng/ml soluble RANKL and 0.1 μg/ml MIF.
After 6 days of culture, the cells were removed and the dentin
slices were dehydrated through a graded series of ethanol and
critical-point-dried with liquid CO2. The dentin slices
were coated with platinum and palladium in an ion coater (E-1030;
Hitachi, Tokyo, Japan) and observed with (SEM).
Statistical analysis
The results were expressed as the means ± SD.
Overall comparisons were made using ANOVA and statistical
differences between the groups were determined by Bonferroni’s
test. Data with a P-value <0.05 were considered significant.
Results
Effects of MIF on osteoclast formation in
the co-culture
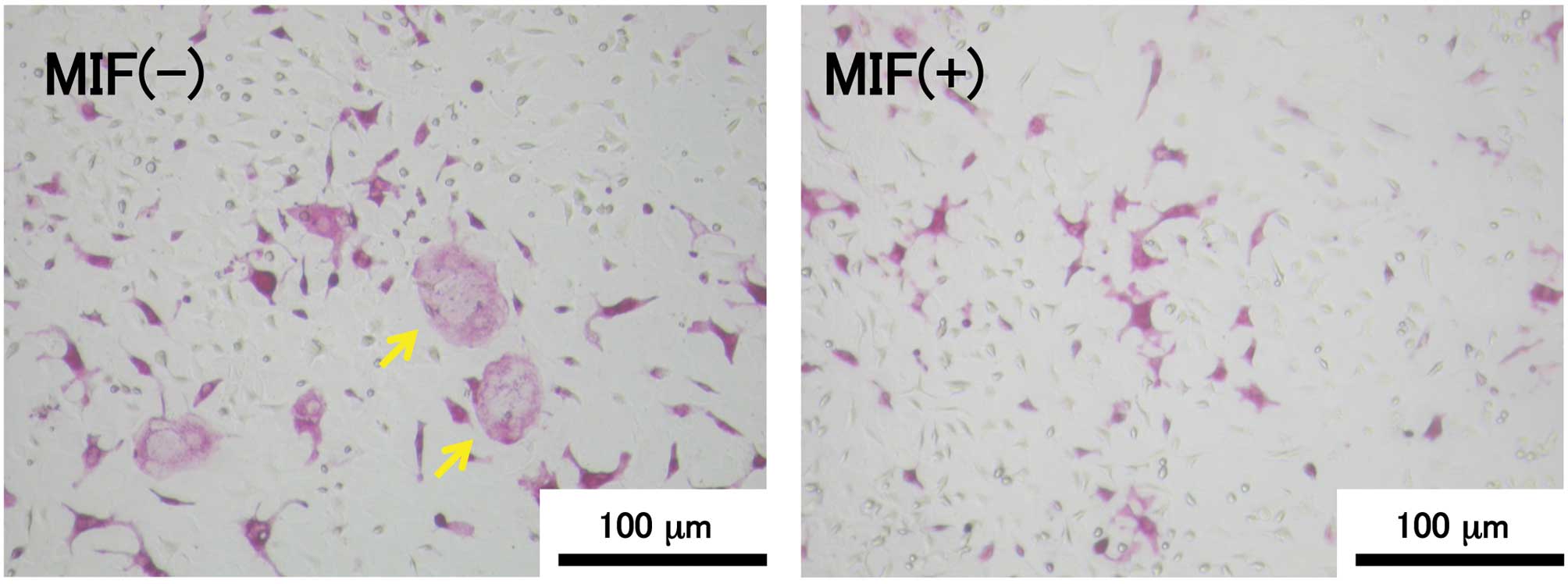
TRAP-positive MNCs were observed in the 6-day
co-culture of MC3T3-E1 cells and bone marrow cells in the presence
of vitamin D3 and Dex (Fig.
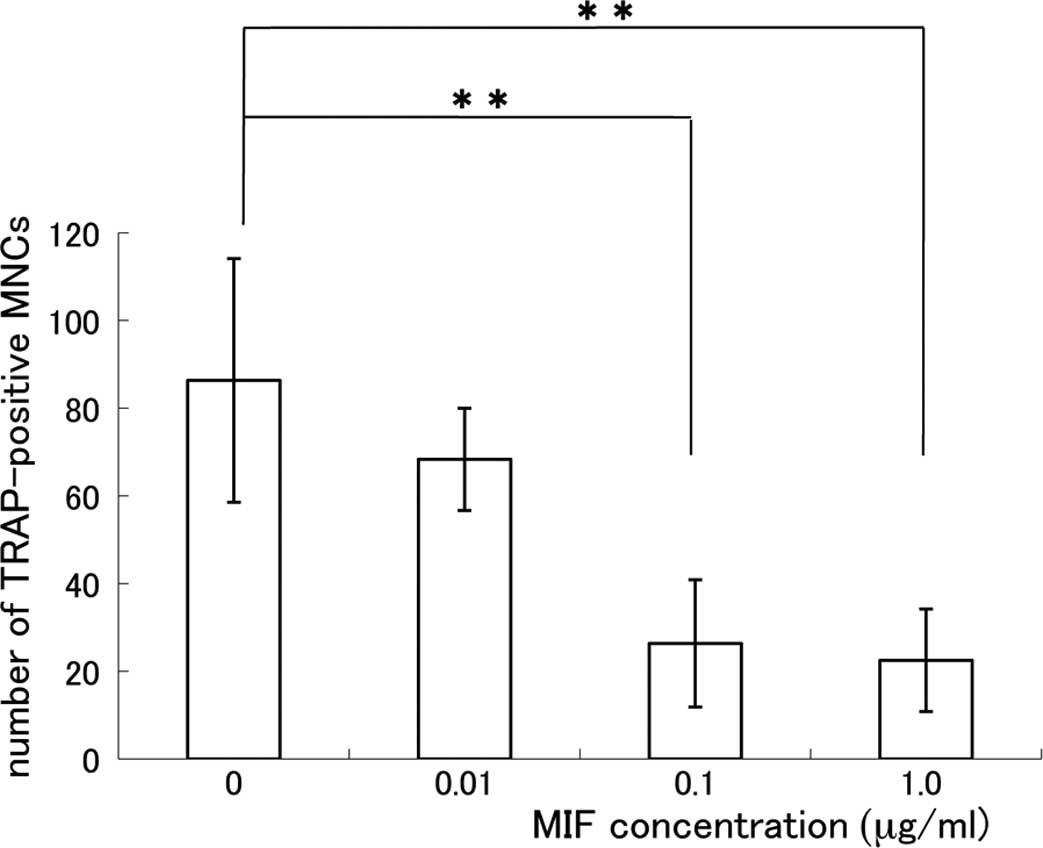
1). The relationship between the MIF concentration and the
number of TRAP-positive MNCs is shown in Fig. 2. The formation of TRAP-positive
MNCs was significantly suppressed in the MIF-treated co-culture and
the effect of MIF was dose-dependent. The suppressive effect of MIF
on the osteoclast formation was remarkable at the concentrations of
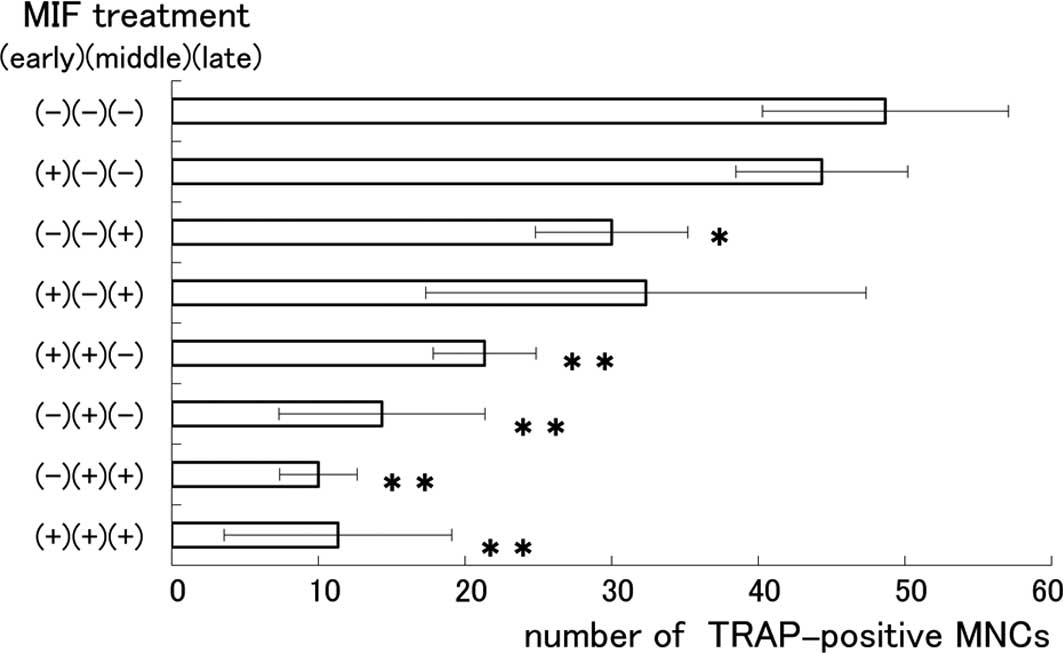
0.1 and 1.0 μg/ml. To determine the exact stage at which MIF
influenced the formation of MNCs, we divided the 6-day cultivation
period into 3 stages: the early stage (days 1–2), the middle stage
(days 3–4) and the late stage (days 5–6). Eight co-culture groups
were prepared and MIF (0.1 μg/ml) was applied at each of the
stages of a 2-day period, as indicated in Fig. 3. The number of MNCs was apparently
reduced in the groups where MIF was applied at the middle and late
stages in the cultivation period. The suppressive effect of MIF on
the formation of MNCs was most remarkable when applied at the
middle stage. The present results are consistent with a recent
report demonstrating that the formation of MNCs in a culture of RAW
264.7 cells, a mouse macrophage cell line, was suppressed more
effectively in the presence of RANKL when MIF was applied at the
last 2 days in a 4-day culture (19). It is noteworthy that the middle
stage in our experiment coincides with the time when multinuclear
osteoclasts were first confirmed in the same co-culture system
(27).
Effects of MIF on the expression of RANKL
and OPG in MC3T3-E1 cells
To investigate whether the suppressive effect of MIF
on the formation of MNCs is dependent on the amounts of RANKL
and/or OPG that are produced by MC3T3-E1 cells (28), we measured RANKL mRNA and OPG
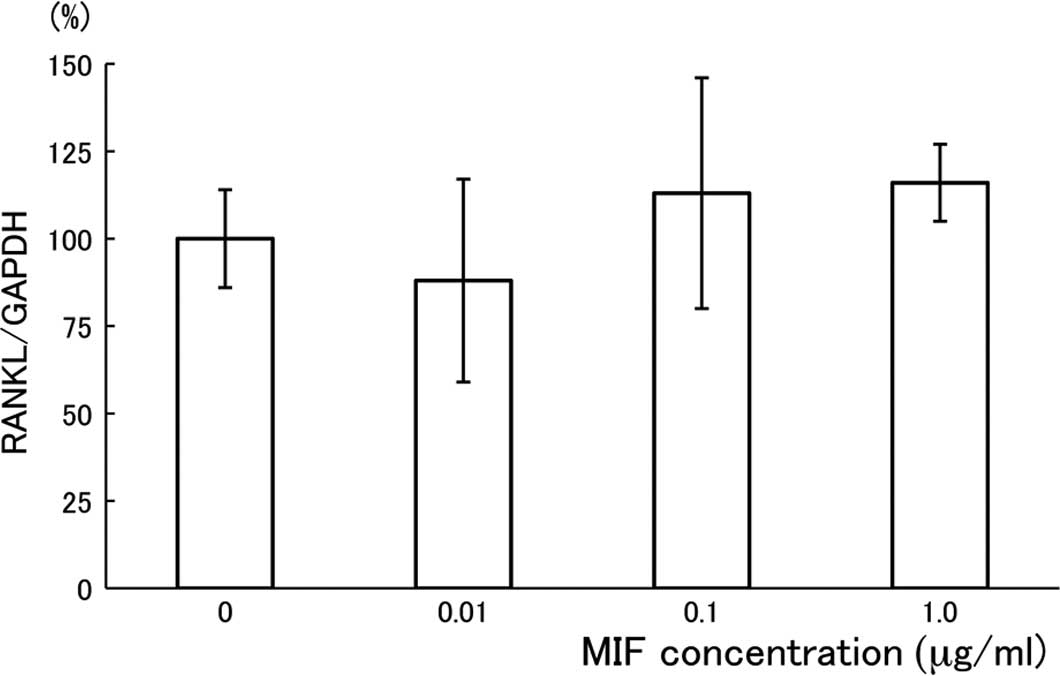
production in the MC3T3-E1 cell culture. The expression of RANKL
mRNA was evaluated using quantitative RT-PCR, and the result is
shown in Fig. 4 as a relative
expression ratio of RANKL to GAPDH. After the 24-h cultivation
period with MIF, there was no substantial effect of the treatment
on the RANKL mRNA expression in MC3T3-E1 cells. The treatment of
MC3T3-E1 cells with MIF for 48 h under the same conditions also had
no effect (data not shown). For the assay of OPG production by
MC3T3-E1 cells, conditioned media were collected from MC3T3-E1 cell
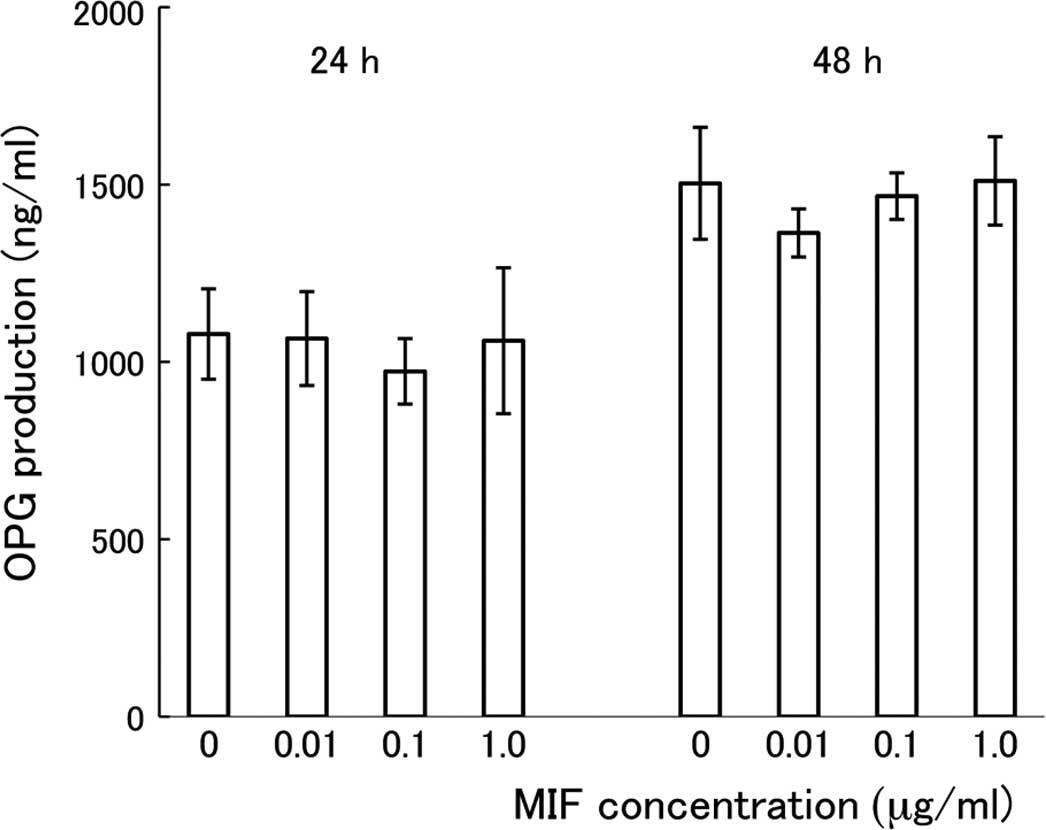
cultures treated with MIF for 24 or 48 h. As shown in Fig. 5, there was no significant
difference in the concentration of OPG among the cultures treated
with different doses of MIF. Cultivation of the cells for a longer
period with MIF (24 vs. 48 h) resulted in a further accumulation of
OPG in the media, but there was no difference in the OPG
concentration among the cultures treated with different doses of
MIF. We did not find a difference in the number of MC3T3-E1 cells
per well among the cultures after the treatment with MIF (data not
shown).
Effects of MIF on the osteoclast
formation in the bone marrow cell culture
Osteoclast formation was observed in the mouse bone
marrow cells cultured with M-CSF and soluble RANKL for 6 days.
Without applications of these factors, TRAP-positive cells were not
induced, and treatment of the bone marrow cells with either M-CSF
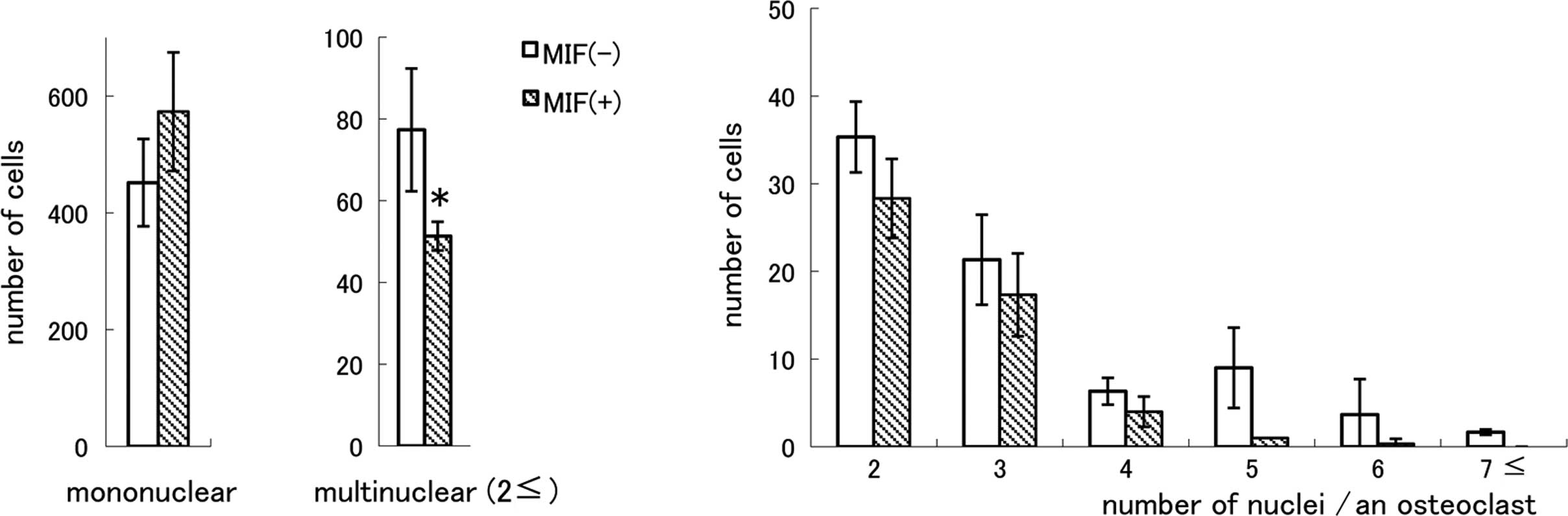
or RANKL had no effect (data not shown). As shown in Fig. 6A, TRAP-positive mononuclear cells
were present in MIF-treated cultures and the cell number was not
smaller than that in the control cultures. However, the number of
MNCs was significantly smaller in the MIF-treated cultures compared
to the control cultures (P<0.05). When MNCs were classified into
subgroups by the number of nuclei (Fig. 6B), it became apparent that there
were very few TRAP-positive MNCs having five or more nuclei in the
MIF-treated cultures. The ratio of MNC formation in the absence of
MIF was 14.6% of the total TRAP-positive cells and in the presence
of MIF it was 9.4%. There was no difference in the total number of
the nuclei of TRAP-positive cells between the control and
MIF-treated cultures, and the numbers were 696.1±81.5 and
706±117.5/well, respectively. Fig.
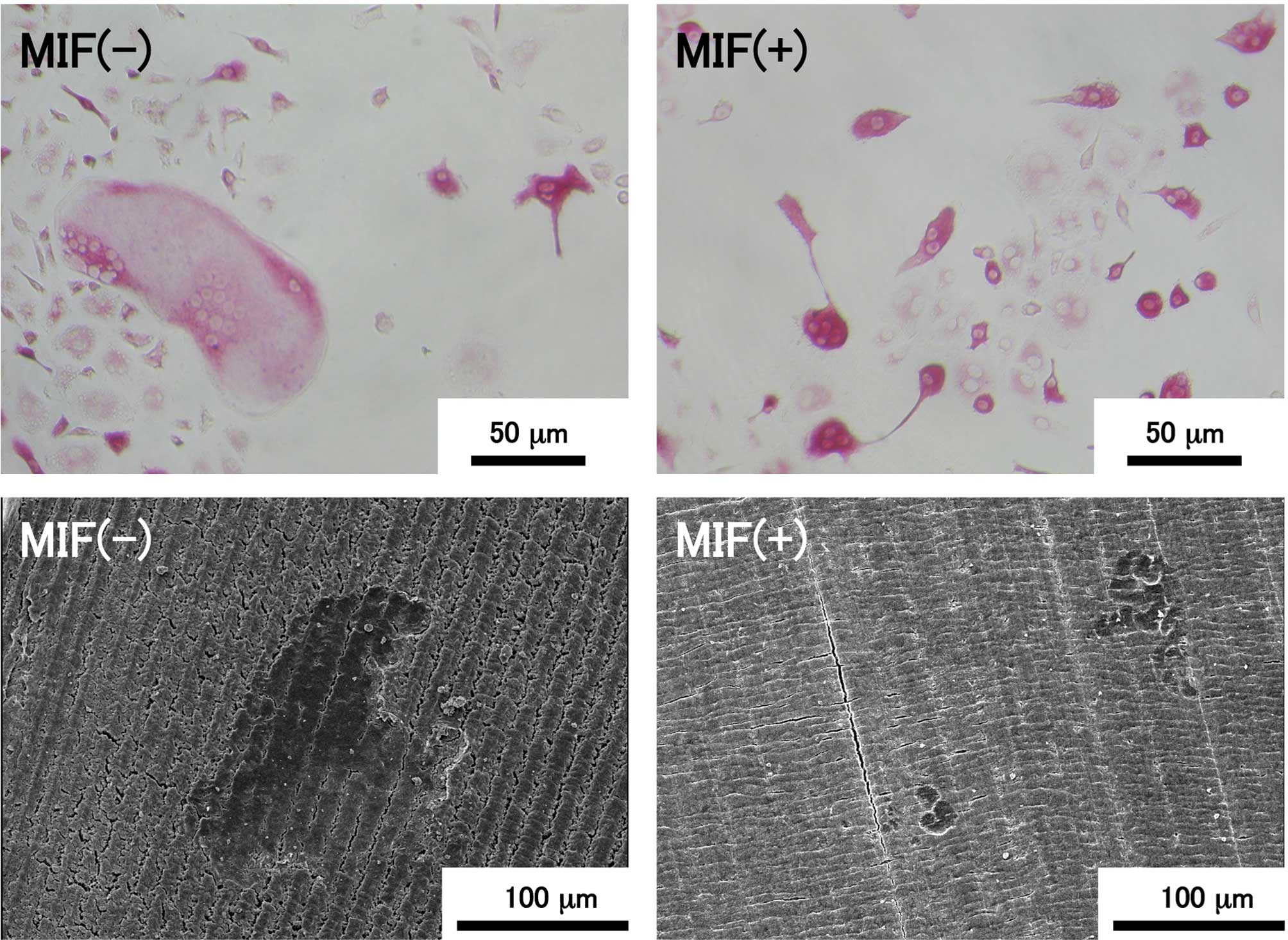
7 shows representative images of TRAP-positive cells in the
bone marrow cell culture and resorption pits formed by osteoclasts
on the dentin slices. TRAP-positive multinuclear giant cells with
more than ten nuclei were present in the control culture, as shown
in the upper left image, although such a large cell was not found
in the MIF-treated culture (upper right image). The maximum number
of nuclei in individual MNC in the control culture was 17, but that
in the MIF-treated culture was 6. TRAP-positive cells containing a
few nuclei were present in both the control and MIF-treated
cultures, and the majority of the TRAP-positive cells in both
cultures were mononuclear cells. Resorption pits formed on the
dentin slices were visualized by SEM (Fig. 7; lower images). A large resorption
pit of >100 μm in diameter was formed on the control
slice, but such a large pit was never found on the slices exposed
to MIF-treated bone marrow cells.
Discussion
In the present study, we examined the role of MIF in
the formation of dentin-resorbing MNCs from mouse bone marrow
cells, and showed that MIF inhibited the formation of MNCs by
preventing cell fusion at the middle stage and, to a lesser extent,
the late stage of the 6-day cultivation period in co-culture with
MC3T3-E1 cells. Osteoblast/stromal cells play a role in the
differentiation of osteoclasts by producing RANKL, an essential
factor for the osteoclast formation. They also produce OPG that
suppresses osteoclast formation by acting as a decoy receptor for
RANKL (4). It can be speculated
that the inhibitory effect of MIF on the formation of MNCs may be
mediated by the suppression of RANKL or by the up-regulation of
OPG, as the amount of these factors is closely related to the
formation of MNCs, as shown in our previous study (23). However, as is evident from the
present experiments using MC3T3-E1 cell culture, neither RANKL mRNA
expression nor OPG production was influenced by MIF. In these
experiments, 24- or 48-h exposure of MC3T3-E1 cells to 1.0
μg/ml of MIF, which is 10-fold higher than the dose
sufficient to inhibit formation of MNCs in the co-culture, had no
effect. Exposure of murine calvarial osteoblasts to MIF for 12 or
24 h also had no substantial effects on cellular RANKL and OPG mRNA
levels (18). Although these
results do not exclude the possibility that MIF has some effects on
the production of other cytokines or factors in MC3T3-E1 cells and
influences the maturation of osteoclasts, it is apparent that MIF
does not affect the RANKL-dependent intercellular processes of
osteoclast differentiation from the precursors.
MNCs can be induced from bone marrow cells in a
culturing condition that does not contain osteoblastic cells
(29). Instead of the cooperation
with osteoblastic cells, TRAP-positive cells were induced
abundantly in the culture of bone marrow cells stimulated with
M-CSF and RANKL. We did not find a significant difference in the
number of mononuclear osteoclastic cells between the control and
MIF-treated cultures, although a slightly larger number of
mononuclear osteoclastic cells was observed in the MIF-treated
cultures. By contrast, a significant reduction in the number of
MNCs was observed in the MIF-treated cultures and the suppressive
effect of MIF on the fusion of osteoclastic cells was evident when
the number of mature osteoclasts was counted; we found very few
MNCs containing five or more nuclei in the MIF-treated
cultures.
For the isolation of bone marrow cells, we removed
osteoblastic and fibroblastic cells effectively from the
heterogenous cell mixture extracted from mouse femoral shafts by
purifying it using a Sephadex G-10 column in which adhesive cells,
such as osteoblasts, were trapped. This treatment excluded the
possible methodological flaw such that MIF activated contaminated
cells and affected the maturation of osteoclasts. Our histological
studies further support the direct action of MIF on differentiated
osteoclasts. In contrast to the formation of extremely large
osteoclasts containing more than ten nuclei in the MIF-free
culture, mature MNCs were rarely found in the bone marrow cell
culture treated with MIF. The assay of pit formation on dentin
slices suggests a close relationship between the sizes of MNCs and
their dentin-resorbing potential. The large resorption pits formed
on the dentin surface mean strong phagocytic action of giant
osteoclasts. Taken together with the results reported by another
group (19), it can be concluded
that MIF inhibits the formation of MNCs by preventing the fusion of
osteoclastic cells.
Osteoclast formation occurs through a cell
differentiation pathway that implies the formation of osteoclast
precursors from hemopoietic cells, subsequent differentiation of
the precursors to mononuclear osteoclastic cells, and a process of
cell fusion to form multinuclear osteoclasts (30). The processes of the osteoclast
differentiation require several days in vitro; in fact, it
took 4 days to form MNCs in the co-culture of mouse bone marrow
cells and MC3T3-E1 cells (27). To
specify the time when MIF affected the formation of MNCs, we
divided the 6-day cultivation period into 3 stages, and bone marrow
cells and MC3T3-E1 cells were co-cultured either with or without
MIF by changing the medium every 2 days. We paid particular
attention to adjusting the number of both bone marrow cells and
MC3T3-E1 cells in the culture dishes at the beginning of
co-culture. When MIF was applied to the co-culture at the middle
stage, the formation of MNCs was markedly inhibited. The
application of MIF at the late stage had a substantial, although
lesser effect on the formation of MNCs compared to the MIF
application at the middle stage. By contrast, the application of
MIF at the early stage had no effect, or rather slightly promoted
MNC formation. The middle stage corresponds to the period when
mononuclear osteoclastic cells are formed nearly completely and
they start to fuse with each other in the experimental condition
(27,30). The present findings strongly
indicate that MIF exerts an inhibitory effect on the formation of
MNCs by preventing the multinucleation of osteoclasts, and that MIF
has little effect on the differentiation of osteoclast precursors
to mononuclear osteoclastic cells.
The mechanism of inhibition of the formation of
mature osteoclasts by MIF has not yet been elucidated. In order to
further investigate the action of MIF, the process of cell fusion
must be clarified. Kukita et al (31) showed that the dendritic
cell-specific transmembrane protein (DC-STAMP), a putative
seven-transmembrane-spanning receptor, was induced in osteoclastic
cells by RANKL, and that target inhibition of this molecule by
small interfering RNAs suppressed the formation of MNCs. Yagi et
al (32) also demonstrated
that DC-STAMP is essential for the fusion of osteoclast precursor
cells and macrophages. Vignery (33) proposed a possible mechanism of the
cell fusion; the DC-STAMP-expressing osteoclastic cell acts as the
master fusing cell and fuses with a DC-STAMP-negative follower
cell. Although the ligand for DC-STAMP has not yet been identified,
the possibility that MIF influences the resorption of calcified
tissues by exerting some effects on DC-STAMP itself or its ligand
is a matter of concern.
There is accumulating evidence that MIF may be
associated with juvenile idiopathic arthritis (JIA). A novel
5′-flanking region polymorphism of MIF has been shown to be
associated with systemic-onset JIA (34). Systemic-onset JIA patients carrying
a -173 single-nucleotide G-to-C polymorphism of the MIF gene
(MIF-173*C) had serum and synovial fluid levels of MIF
significantly higher than those in patients carrying a
MIF-173*G allele, and the duration of clinical response
to intra-articular injection of triamcinolone hexacetonide was
significantly shorter in patients carrying the MIF-173*C
allele (35). Notably, a clinical
survey of panoramic radiographs of school children revealed that
dental maturity was significantly advanced in JIA patients compared
to healthy children (36).
Although the effect of corticosteroids used for the treatment of
JIA should also be taken into account, MIF may be involved in the
mechanism of altered dental development in JIA children.
Acknowledgements
This study was supported in part by
KAKENHI (Grant-in-Aid for Scientific Research from the Japan
Society for the Promotion of Science: 18390551, 19659544, 22592274
and 21592584); a grant from the ‘Strategic Research Base
Development’ Program for Private Universities from the Ministry of
Education, Culture, Sports, Science, and technology, Japan (MEXT,
S1001024); a grant from the Dental Research Center, Nihon
University School of Dentistry, and the Sato and Uemura Funds from
the Nihon University School of Dentistry.
References
|
1.
|
N SaharaA ToyokiY AshizawaT DeguchiK
SuzukiCytodifferentiation of the odontoclast prior to the shedding
of human deciduous teeth: an ultrastructural and cytochemical
studyAnat
Rec2443349199610.1002/(SICI)1097-0185(199601)244:1%3C33::AID-AR4%3E3.0.CO;2-G8838422
|
|
2.
|
T DomonM YasudaM OsanaiIncrease in
odontoclast nuclei number by cell fusion: a three-dimensional
reconstruction of cell fusion of human odontoclastsAnat
Rec252462471199810.1002/(SICI)1097-0185(199811)252:3%3C462::AID-AR14%3E3.0.CO;2-29811224
|
|
3.
|
N UdagawaN TakahashiT AkatsuOrigin of
osteoclasts: mature monocytes and macrophages are capable of
differentiating into osteoclasts under a suitable microenvironment
prepared by bone marrow-derived stromal cellsProc Natl Acad Sci
USA8772607264199010.1073/pnas.87.18.7260
|
|
4.
|
T SudaN TakahashiN UdagawaE JimiMT
GillespieTJ MartinModulation of osteoclast differentiation and
function by the new members of the tumor necrosis factor receptor
and ligand familiesEndocr
Rev20345357199910.1210/edrv.20.3.036710368775
|
|
5.
|
DL LaceyE TimmsHL TanOsteoprotegerin
ligand is a cytokine that regulates osteoclast differentiation and
activationCell93165176199810.1016/S0092-8674(00)81569-X9568710
|
|
6.
|
DM BiskobingX FanJ RubinCharacterization
of MCSF-induced proliferation and subsequent osteoclast formation
in murine marrow cultureJ Bone Miner
Res1010251032199510.1002/jbmr.56501007067484277
|
|
7.
|
SK LeeJ KalinowskiS JastrzebskiJA
Lorenzo1,25(OH)2 vitamin D3-stimulated osteoclast formation in
spleen-osteoblast cocultures is mediated in part by enhanced IL-1α
and receptor activator of NF-κB ligand production in osteoblastsJ
Immunol16923742380200212193704
|
|
8.
|
O KudoA SabokbarA PocockI ItonagaY
FujikawaNA AthanasouInterleukin-6 and interleukin-11 support human
osteoclast formation by a RANKL-independent
mechanismBone3217200310.1016/S8756-3282(02)00915-812584029
|
|
9.
|
J LamS TakeshitaJE BarkerO KanagawaFP
RossSL TeitelbaumTNF-α induces osteoclastogenesis by direct
stimulation of macrophages exposed to permissive levels of RANK
ligandJ Clin Invest160148114882000
|
|
10.
|
T CalandraT RogerMacrophage migration
inhibitory factor: a regulator of innate immunityNat Rev
Immunol3791800200310.1038/nri120014502271
|
|
11.
|
JR DavidDelayed hypersensitivity in vitro:
its mediation by cell-free substances formed by lymphoid
cell-antigen interactionProc Natl Acad Sci
USA567277199610.1073/pnas.56.1.725229858
|
|
12.
|
BR BloomB BennettMechanism of a reaction
in vitro associated with delayed-type
hypersensitivityScience1538082196610.1126/science.153.3731.805938421
|
|
13.
|
A LanahanJB WilliamsLK SandersD
NathansGrowth factor-induced delayed early response genesMol Cell
Biol123919392919921508193
|
|
14.
|
GJ WistowMP ShaughnessyDC LeeJ HodinPS
ZelenkaA macrophage migration inhibitory factor is expressed in the
differentiating cells of the eye lensProc Natl Acad Sci
USA9012721275199310.1073/pnas.90.4.12727679497
|
|
15.
|
RA MitchellCN MetzT PengR BucalaSustained
mitogen-activated protein kinase (MAPK) and cytoplasmic
phospholipase A2 activation by macrophage migration inhibitory
factor (MIF). Regulatory role in cell proliferation and
glucocorticoid actionJ Biol
Chem2741810018106199910.1074/jbc.274.25.18100
|
|
16.
|
S OnoderaJ NishihiraK IwabuchiMacrophage
migration inhibitory factor up-regulates matrix metalloproteinase-9
and -13 in rat osteoblastsJ Biol
Chem27778657874200210.1074/jbc.M10602020011751895
|
|
17.
|
L DanksA SabokbarR GundleNA
AthanasouSynovial macrophage-osteoclast differentiation in
inflammatory arthritisAnn Rheum
Dis61916921200210.1136/ard.61.10.91612228163
|
|
18.
|
S OnoderaS SasakiS OhshimaTransgenic mice
overexpressing macrophage migration inhibitory factor (MIF) exhibit
high-turnover osteoporosisJ Bone Miner
Res21876885200610.1359/jbmr.06031016753018
|
|
19.
|
C JacquinB Koczon-JaremkoHL
AguilaMacrophage migration inhibitory factor inhibits
osteoclastogenesisBone45640649200910.1016/j.bone.2009.06.02819591967
|
|
20.
|
T KatagiriN TakahashiRegulatory mechanisms
of osteoblast and osteoclast differentiationOral
Dis8147159200210.1034/j.1601-0825.2002.01829.x12108759
|
|
21.
|
H YasudaN ShimaN NakagawaOsteoclast
differentiation factor is a ligand for
osteoprotegerin/osteoclastogenesis-inhibitory factor and is
identical to TRANCE/RANKLProc Natl Acad Sci
USA9535973602199810.1073/pnas.95.7.3597
|
|
22.
|
AR Ten CateOral Histology: Development,
Structure, and FunctionMosby-Year BookSt. Louis3253291994
|
|
23.
|
T HasegawaY YoshimuraT KikuiriExpression
of receptor activator of NF-kappa B ligand and osteoprotegerin in
culture of human periodontal ligament cellsJ Periodontal
Res37405411200210.1034/j.1600-0765.2002.01603.x12472833
|
|
24.
|
J NishihiraT KuriyamaM SakaiThe structure
and physiochemical properties of rat liver macrophage migration
inhibitory factorBiochim Biophys
Acta1247159162199510.1016/0167-4838(94)00215-37873586
|
|
25.
|
K ShibataY YoshimuraT KikuiriEffect of the
release from mechanical stress on osteoclastogenesis in RAW264.7
cellInt J Mol Med287379201121491081
|
|
26.
|
H AmanoS YamadaR FelixColony-stimulating
factor-1 stimulates the fusion process in osteoclastsJ Bone Miner
Res13846853199810.1359/jbmr.1998.13.5.8469610749
|
|
27.
|
Y ShiraiY YoshimuraY YawakaEffect of
extracellular calcium concentrations on osteoclast differentiation
in vitroBiochem Biophys Res
Commun265484488199910.1006/bbrc.1999.166410558894
|
|
28.
|
Y DeyamaS TakeyamaKoshikawaOsteoblast
maturation suppressed osteoclastogenesis in coculture with bone
marrow cellsBiochem Biophys Res
Commun274249254200010.1006/bbrc.2000.312710903926
|
|
29.
|
H YasudaN ShimaN NakagawaA novel molecular
mechanism modulating osteoclast differentiation and
functionBone25109113199010.1016/S8756-3282(99)00121-010423033
|
|
30.
|
T TsurukaiN UdagawaK MatsuzakiN TakahashiT
SudaRoles of macrophage-colony stimulating factor and osteoclast
differentiation factor in osteoclastogenesisJ Bone Miner
Metab18177184200010.1007/s00774007001810874596
|
|
31.
|
T KukitaN WadaA KukitaRANKL-induced
DC-STAMP is essential for osteoclastogenesisJ Exp
Med200941946200410.1084/jem.2004051815452179
|
|
32.
|
M YagiT MiyamotoY SawataniDC-STAMP is
essential for cell-cell fusion in osteoclasts and foreign body
giant cellsJ Exp Med202345351200510.1084/jem.2005064516061724
|
|
33.
|
A VigneryMacrophage fusion: the making of
osteoclasts and giant cellsJ Exp
Med202337340200510.1084/jem.2005112316061722
|
|
34.
|
RP DonnE ShelleyWE OllierW ThomsonA novel
5′-flanking region polymorphism of macrophage migration inhibitory
factor is associated with systemic-onset juvenile idiopathic
arthritisArthritis Rheum44178217852001
|
|
35.
|
F De BenedettiC MeazzaM
VivarelliFunctional and prognostic relevance of the -173
polymorphism of the macrophage migration inhibitory factor gene in
systemic-onset juvenile idiopathic arthritisArthritis
Rheum4813981407200312746913
|
|
36.
|
A LehtinenT OksaH HeleniusO
RönningAdvanced dental maturity in children with juvenile
rheumatoid arthritisEur J Oral
Sci108184188200010.1034/j.1600-0722.2000.108003184.x10872987
|