Introduction
Congenital muscular dystrophies (CMDs) are
genetically and clinically heterogeneous hereditary myopathies that
have a predominantly autosomal recessive mode of inheritance. They
are characterized by congenital hypotonia, delayed motor
development and the early onset of progressive muscle weakness, as
well as a dystrophic pattern on muscle biopsy (1). Merosin-deficient congenital muscular
dystrophy type 1A (MDC1A) is one of the most frequent forms of CMD
in Western countries. It is an autosomal recessive neuromuscular
disorder caused by mutations in the laminin α-2 gene (LAMA2) on
chromosome 6q22–23 that results in a deficiency of the laminin α-2
chain, a component of skeletal muscle extracellular matrix
laminin-2, merosin (2).
In China, MDC1A is an extremely rare condition with
only seven reported cases to date (3). Patients with MDC1A have severe
muscular weakness and atrophy, diffuse contractures, inability to
walk and facial dysmorphism. In addition, they have markedly
increased serum creatine kinase (CK) levels and have characteristic
white matter abnormalities on cranial magnetic resonance imaging
(MRI).
With more widespread use of molecular genetic
testing, these tests are becoming more important for confirming the
diagnosis of CMD subtypes than are muscle biopsies. In the current
study, we report a case of a young female with MDC1A whose
diagnosis was confirmed by clinical presentation, characteristic
white matter abnormalities and molecular genetic testing without
the need for a muscle biopsy.
Case report
Clinical presentation
The 4-year-old patient was the first daughter of
non-consanguineous, healthy parents. The patient first attended the
Department of Pediatrics, Sun Yat-sen Memorial Hospital of Sun
Yat-sen University (Guangdong, China) at the age of 3 years due to
severe hypotonia and marked proximal weakness. Developmental
milestones were delayed at 6 months of age and the patient
exhibited severe axial and peripheral hypotonia with feeding
difficulties. By 2 years of age, the patient was able to hold her
head up, but was unable to roll over or sit alone. At the age of 4
years, the patient was able to sit unsupported, but not stand.
Intellectual and speech development was normal. The individual was
born at 41 weeks of gestation and the birth weight was 3,100 g. The
pregnancy and delivery were uneventful. A family history revealed
no other cases of neuromuscular diseases. The study was approved by
the Ethics Committee of Sun Yat-sen Memorial Hospital of Sun
Yat-sen University. Informed consent was obtained from the
patients’ family.
When the patient was first observed in another
hospital, the serum CK level was 3,008 mU/ml and the
electroencephalogram (EEG) and survival of motor neuron 1 (SMN1)
gene expression were normal. MRI revealed diffuse white matter
dysplasia and the suspected diagnosis was adrenoleukodystrophy.
On physical examination, the patient’s chest and
abdomen were normal, as were the results of cardiac assessment.
However, the patient exhibited severe axial hypotonia with
non-progressive bilateral upper and lower extremity weakness, which
was more proximal than distal and predominantly at the shoulder and
pelvic girdle. The muscle strength of the lower limb muscles was
manually assessed using the standard Medical Research Council (MRC)
scale (4), with a result of grade
3/5. The upper limb proximal muscle strength was determined to be
grade 4/5. The cranial nerves were normal and the patient had no
difficulties on sensory examination and coordination. Deep tendon
reflexes and Babinski’s sign were negative. Tactile, pinprick and
vibration sensations were normal.
Laboratory test results were as follows: leukocyte
count, 8,900 cells/μl; hemoglobin, 11.5 g/dl; hematocrit,
33.5%; and platelet count, 294×103 cells/μl.
Biochemistry test results were as follows: glutamic oxaloacetic
transaminase, 10 IU/l; glutamate alanine aminotransferase, 39 IU/l;
CK, 1,556 IU/l; CK-MB, 98 IU/l; triglycerides, 0.88 mmol/l; and
ammonia, 10.0 μmol/l. Urine and blood screens for hereditary
metabolic diseases were unremarkable.
Neuroimaging
MRI was performed at Sun Yat-sen Memorial Hospital
of Sun Yat-sen University when the patient was 3 years old.
T1-weighted images revealed symmetrical, low signal intensity in
the white matter of the frontal, parietal, temporal and occipital
lobes; however, the cortex was normal (Fig. 1C). T2-weighted images revealed
lesions with abnormally high signal intensity in the white matter
of the frontal, parietal, temporal and occipital lobes (Fig. 1A and B). The ventricles,
cerebellum, basal ganglia and pons were normal. Semi-quantitative
magnetic resonance spectroscopy (MRS) revealed that the
N-acetylaspartate/creatine (NAA/Cr) and choline/creatine (Cho/Cr)
metabolite ratios were within normal ranges. However, the
myoinositol/creatine (mI/Cr) metabolite ratio was slightly
increased (Fig. 1D).
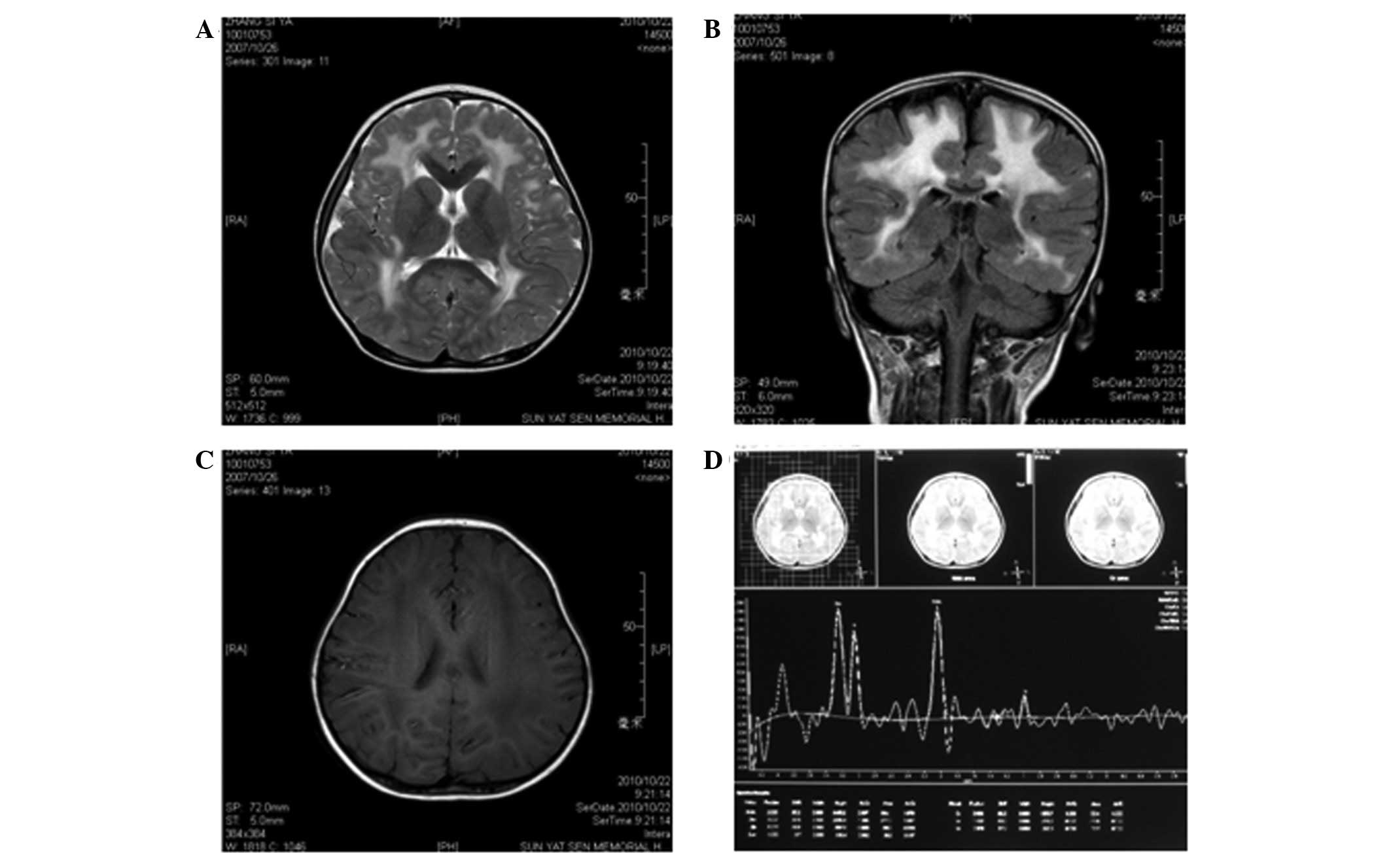 | Figure 1.Magnetic resonance imaging (MRI)
results at 3 years of age. (A) Axial T2-weighted image and (B)
coronal T2-weighted fluid attenuated inversion recovery (FLAIR)
image show diffuse, symmetrical high signal intensities in the
cerebral white matter. (C) Axial T2-weighted image shows diffuse
low signal intensity in the cerebral white matter. (D) MRS [TR/TE =
(2,000 m/sec)/(35 m/sec)] of the parietal white matter at 3 years
of age demonstrated that the NAA/Cr and Cho/Cr ratios were normal,
but the MI/Cr ratio was slightly increased. TR, time of repetition;
TE, time of echo; Cho, choline; Cr, creatine; MI, myo-inositol;
NAA, N-acetylaspartate, MRS, magnetic resonance spectroscopy. |
Electrophysiological studies
Electromyography (EMG) was used to aid in
differentiating whether the patient’s deficits were myogenic or
neurogenic. Needle EMG of the left and right tibialis anterior
muscle suggested a myopathic process with reduced recruitment
potential, decreased amplitude and duration of response, appearance
of variable small amplitudes and short-duration polyphasic myogenic
waves. Nerve conduction studies (NCS) were performed for the left
tibial and left deep peroneal nerves using conventional methods.
These revealed a normal motor nerve conduction velocity (MCV) as
follows: tibial posterior, 52 m/sec and deep peroneal, 50 m/sec
(normal range, 50–58 m/sec in the legs).
Molecular genetic testing
The patient in the present case was suspected of
having MDC1A based on congenital hypotonia, delayed motor
milestones and brain white matter abnormalities on MRI. Thus,
molecular genetic testing was performed without a muscle biopsy.
Genomic DNA from the patient and the patient’s parents was
extracted from peripheral blood leukocytes using standard
procedures. PCR and DNA direct sequencing were used to analyze all
65 exons of LAMA2 to determine if there were any gene
mutations.
DNA analysis revealed that the patient had a
homozygous nonsense mutation in the LAMA2 gene in exon 50; a C>T
exchange in nucleotide 7147 causing a stop codon (Arg2383X stop).
The patient’s parents were heterozygotes for this mutation. This
finding confirmed the diagnosis of MDC1A.
Discussion
MDC1A is caused by mutations in the LAMA2 gene and
was first described by Tomé et al in 1994 (2). The estimated prevalence of CMDs is ~1
in 7 million (5). In Europe, MDC1A
accounts for ~40% of CMD cases (6). MDC1A is characterized by congenital
muscle hypotonia, delayed or arrested motor milestones and feeding
difficulties. Muscle weakness is absent or slowly progressive and
is accompanied by contractures that mostly affect the elbows, hips,
knees and ankles. The majority of patients may achieve unsupported
sitting; however, <10% achieve ambulation (7). The common life-threatening
complications of MDC1A include respiratory failure and feeding
difficulties.
The patient in the present study was only 4 years
old and had not suffered from any severe respiratory infections.
However, with this disease, pulmonary infection is the most common
cause of mortality, which may occur during the first decade or
anytime thereafter. Treatment with non-invasive ventilation and
tracheostomy may greatly improve health.
In CMDs, the serum levels of CK are mildly to
markedly elevated. In general, CMD subtypes with primary or
secondary merosin deficiency, including dystroglycanopathies, show
high serum CK concentrations, while those with no merosin
deficiency show normal or mildly increased serum CK concentrations
(8). In the present case, the
serum CK level was elevated to 1,556 IU/l, which indicated primary
or secondary merosin deficiency.
EMG and NCS are recommended for all patients with
suspected CMDs to confirm myopathy and to exclude other diseases.
In the present case, EMG confirmed a myopathic process with early
recruitment and decreased amplitude and duration of response, while
the results of NCS were normal. In a number of cases of MDC1A, mild
neuropathic changes may be observed since laminin α-2 is absent in
the basement membranes surrounding Schwann cells and myelin sheaths
(9).
The majority of patients with MDC1A have normal
intellectual and speech development, although cases of learning
disabilities and mental retardation have been reported (10). Epilepsy has been estimated to occur
in ~6–8% of these cases; seizures are partial and complex, with no
consistent pattern (10). In the
present case, an EEG was normal; however, it is essential that a
standard EEG is performed periodically for MDC1A patients.
Despite a minority that has clinical central nervous
system findings, a consistent finding common to all patients >6
months of age is the presence of cerebral white matter
abnormalities on neuroimaging. In the present case, cranial MRI
revealed signal abnormalities in the white matter of the frontal,
parietal, temporal and occipital lobes, whereas the cortex was
normal. Children may initially be misdiagnosed as having a
leukodystrophy. White matter changes do not regress with time.
Although the pathophysiology of the white matter changes has not
been completely elucidated, the majority of investigators postulate
that disruption of the blood-brain barrier associated with laminin
α-2 leads to increased water content, which results in abnormal
white matter signal intensity (11,12).
The pattern of white matter abnormalities associated with MDC1A is
characteristic as compared with other CMD subtypes. A small number
of patients have structural changes with mild ventricular
enlargement, focal cortical dysplasia, occipital polymicrogyria and
hypoplasia of the pons and cerebellum (13).
Previously, a diagnosis of MDC1A was based on the
clinical findings of severe congenital hypotonia, weakness
associated with high CK blood levels, white matter abnormalities
and dystrophy associated with negative immunostaining of biopsied
muscle for merosin (14). A muscle
biopsy appears to be an essential factor in the diagnosis of MDC1A.
However, with the more widespread use of molecular genetic testing
for confirming the diagnosis of a CMD subtype, the recent trend has
been to perform molecular genetic testing without a muscle biopsy
when the medical history, physical examination and neurological
examination support the diagnosis of a CMD.
In the present case, due to the congenital
hypotonia, delayed motor milestones, markedly elevated CK
concentration and brain white matter abnormalities on MRI, the
patient was suspected of having MDC1A. Thus, we directly proceeded
to molecular genetic testing without performing a muscle biopsy.
Ultimately, a nonsense mutation in the LAMA2 gene confirmed our
diagnosis of MDC1A.
LAMA2 is located on chromosome 6q22–23 in humans and
on chromosome 10 in mice (15).
This gene comprises 65 exons that encode for the α2 chain subunit
of laminin-2. Laminin-2 is a heterotrimer consisting of laminin
α-2, β-1 and γ-1 subunits (16).
Mutations in LAMA2 include nonsense, missense, deletion and
splice-site mutations, which all result in a primary deficiency in
the laminin α-2 chain of merosin (15,17).
Thus, for MDC1A, an evaluation may proceed directly to molecular
genetic testing without a biopsy, depending on a typical
presentation and following exclusion of other more common
diagnoses. By contrast, if multiple genes need to be tested, such
as for confirming a diagnosis of a dystroglycanopathy, the
immunohistochemical analysis of a muscle biopsy may identify the
subtype prior to molecular genetic testing.
MDC1A is the most common form of CMD. MDC1A is
caused by a mutation of LAMA2 located on human chromosome 6q22–23.
The typical presentations of MDC1A are severe congenital hypotonia,
muscle weakness, elevated serum levels of CK and white matter
abnormalities. To confirm a diagnosis of MDC1A, the evaluation may
proceed directly to LAMA2 molecular genetic testing without the
need for a muscle biopsy.
Abbreviations:
|
CMDs
|
congenital muscular dystrophies;
|
|
MDC1A
|
merosin-deficient congenital muscular
dystrophy type 1A;
|
|
MRI
|
magnetic resonance imaging;
|
|
EEG
|
electroencephalogram;
|
|
SMN1
|
survival of motor neuron 1;
|
|
MRC
|
Medical Research Council;
|
|
CK
|
creatine kinase;
|
|
CK-MB
|
creatine kinase-MB;
|
|
MRS
|
magnetic resonance spectroscopy;
|
|
NAA/Cr
|
N-acetylaspartate/creatine;
|
|
Cho/Cr
|
choline/creatine;
|
|
MI/Cr
|
myoinositol/creatine;
|
|
EMG
|
electromyography;
|
|
NCS
|
nerve conduction studies;
|
|
MCV
|
motor nerve conduction velocity;
|
|
LAMA2
|
laminin α-2 gene
|
Acknowledgements
This study was supported by a grant
from the Medical Scientific Research Foundation of Guangdong
Province (B2011091).
References
|
1.
|
Banker BQ and Engel AG: Basic reactions of
muscle. Myology: Basic and Clinical. Engel AG and
Franzini-Armstrong C: 3rd edition. McGraw-Hill; New York, NY: pp.
691–748. 2004
|
|
2.
|
Tomé FM, Evangelista T, Leclerc A, et al:
Congenital muscular dystrophy with merosin deficiency. C R Acad Sci
III. 317:351–377. 1994.
|
|
3.
|
Xiong H, Yao S, Yuan Y, et al: Diagnosis
of congenital muscular dystrophy and clinical significance of
merosin expression. Zhonghua Er Ke Za Zhi. 44:918–923. 2006.(In
Chinese).
|
|
4.
|
Paternostro-Sluga T, Grim-Stieger M, Posch
M, Schuhfried O, Vacariu G, Mittermaier C, Bittner C and
Fialka-Moser V: Reliability and validity of the Medical Research
Council (MRC) scale and a modified scale for testing muscle
strength in patients with radial palsy. J Rehabil Med. 40:665–671.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Voit T and Tomé FS: The congenital
muscular dystrophies. Myology: Basic and Clinical. Engel AG and
Franzini-Armstrong C: 3rd edition. McGraw-Hill; New York, NY: pp.
1203–1238. 2004
|
|
6.
|
Mostacciuolo ML, Miorin M, Martinello F,
et al: Genetic epidemiology of congenital muscular dystrophy in a
sample from north-east Italy. Hum Genet. 97:277–279. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Jones KJ, Morgan G, Johnston H, et al: The
expanding phenotype of laminin alpha2 chain (merosin)
abnormalities: case series and review. J Med Genet. 38:649–657.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Sparks S, Quijano-Roy S, Harper A, et al:
Congenital Muscular Dystrophy Overview. GeneReviews™ [Internet].
Pagon RA, Adam MP, Bird TD, et al: University of Seattle; Seattle,
WA: 2001, http://www.ncbi.nlm.nih.gov/books/NBK1291/uri.
|
|
9.
|
Di Muzio A, De Angelis MV, Di Fulvio P, et
al: Dysmyelinating sensory-motor neuropathy in merosin-deficient
congenital muscular dystrophy. Muscle Nerve. 27:500–506.
2003.PubMed/NCBI
|
|
10.
|
Philpot J, Sewry C, Pennock J and Dubowitz
V: Clinical phenotype in congenital muscular dystrophy: correlation
with expression of merosin in skeletal muscle. Neuromusc Disord.
5:301–305. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Sijens PE, Fock JM, Meiners LC, et al: MR
spectroscopy and diffusion tensor imaging of the brain in
congenital muscular dystrophy with merosin deficiency: metabolite
level decreases, fractional anisotropy decreases, and apparent
diffusion coefficient increases in the white matter. Brain Dev.
29:317–321. 2007. View Article : Google Scholar
|
|
12.
|
Brockmann K, Dechent P, Bönnemann C, et
al: Quantitative proton MRS of cerebral metabolites in laminin
alpha2 chain deficiency. Brain Dev. 29:357–364. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Sunada Y, Edgar TS, Lotz BP, et al:
Merosin-negative congenital muscular dystrophy associated with
extensive brain abnormalities. Neurology. 45:2084–2089. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Fardeau M, Tomé FM, Helbling-Leclerc A, et
al: Congenital muscular dystrophy with merosin deficiency:
clinical, histopathological, immunocytochemical and genetic
analysis. Rev Neurol (Paris). 52:11–19. 1996.(In French).
|
|
15.
|
Helbling-Leclerc A, Zhang X, Topaloglu H,
et al: Mutations in the laminin alpha 2-chain gene (LAMA2) cause
merosin-deficient congenital muscular dystrophy. Nat Genet.
11:216–218. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Burgeson RE, Chiquet M, Deutzmann R, et
al: A new nomenclature for the laminins. Matrix Biol. 14:209–211.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Di Blasi C, Piga D, Brioschi P, et al:
LAMA2 gene analysis in congenital muscular dystrophy: new
mutations, prenatal diagnosis, and founder effect. Arch Neurol.
62:1582–1586. 2005.PubMed/NCBI
|














