Introduction
Atrial fibrillation (AF) is the most common cardiac
arrhythmia and affects millions of individuals worldwide. The
incidence of AF increases with age (1,2).
Although the details are poorly understood, the persistence of AF
is considered a result of atrial remodeling (3). Atrial remodeling includes electrical,
structural and contractile remodeling processes, which are the
primary contributors to the development and maintenance of AF
(3,4). Structural remodeling may be an
adaptive process (dedifferentiation of cardiomyocytes) aimed at
protecting the atrial myocytes or a maladaptive process
(degeneration of cells with fibrotic replacement) (4,5).
Despite the different characteristics, all adaptive processes are
ultimately processes of programmed cell survival (6). Certain myocytes with structural
alterations are able to activate a programmed cell death pathway
(7). Moreover, these dynamic
processes may be modulated by the expression of B cell lymphoma 2
(BCL-2; which protects cells from apoptosis) and BCL-2-associated X
protein (BAX; which opposes the effects of BCL-2, thereby promoting
apoptosis) (8).
Previous studies have indicated that the homogeneous
conduction of atrial activity not only relies on cardiomyocyte
integration but is also affected by the extracellular matrix (ECM)
among the atrial myocytes (9,10).
Atrial fibrosis (abnormal deposition of ECM proteins in the atrium)
may be associated with the substrates of AF by increasing the
heterogeneity of atrial conduction and playing a significant role
in the maintenance of this type of arrhythmia (11,12).
Interstitial fibrosis may facilitate local intra-atrial conduction
block and increase atrial susceptibility to AF, as well as form
stable local sources for atrial micro-reentry and AF induction
(13,14). In addition, matrix
metalloproteinases (MMPs) and their endogenous inhibitors, tissue
inhibitors of MMPs (TIMPs), are regarded as potential etiological
agents in atrial fibrotic remodeling (15).
However, the mechanisms of atrial fibrosis during
ageing and/or in AF remain unclear. The purpose of this study was
to investigate whether abnormal expression of MMP-9/TIMP-1 and
BCL-2/BAX are correlated with histological and ultra-structural
changes, including the characteristic accelerated fibrosis and
apoptosis during ageing and/or in AF induced by chronic rapid
atrial pacing. We also sought to identify potential substrates for
AF.
Materials and methods
Animal preparation
Fourteen adult (1–3 year old) and 14 aged (>8
year old) mongrels weighing 18–26 kg were obtained from the Animal
Center of Xinjiang Medical University (Urumqi, China). The ages of
the dogs were estimated by a veterinarian based on standard
measurements for age, including dentition, coat and eye condition,
as well as musculoskeletal and conformational descriptors. The dogs
were kept in temperature-controlled housing under a 12/12 h
light/dark cycle and fed a standard laboratory diet and water ad
libitum. The Animal Care and Use Committee of the Xinjiang
Medical University approved all experiments in accordance with the
National Institutes of Health Guide for the Care and Use of
Laboratory Animals (Publication No. 85–23, revised 1985).
Experimental design
Six-lead electrocardiogram (ECG) measurements were
made on conscious dogs resting quietly to confirm sinus rhythm
(SR). Echocardiograms were performed to exclude structural heart
disease. The animals were randomly divided into four groups of
seven: the adult SR, aged SR, adult AF and aged AF groups. AF was
induced by chronic rapid atrial pacing and defined as the
persistence of AF for ≥5 days.
Induction of AF
Animals were anesthetized with pentobarbital sodium
(30 mg/kg i.v.) and ventilated with 1.5–2% isoflurane and 2 l/min
O2. In total, 0.15 mg/kg morphine sulfate was injected
into the epidural space for postoperative analgesia. Under sterile
conditions, a right intercostal thoracotomy was performed, the
pericardium was opened and the heart was suspended in a pericardial
cradle. A lead was attached to the epicardium of the left atrial
(LA) appendage. It was tunneled subcutaneously and connected to a
pulse generator (Department of Electronic Engineering, Fudan
University, Shanghai, China). Pulse generators were implanted in
subcutaneous pockets on the left posterior chest wall. Once the
incisions were closed and the dogs had recovered from the
anesthesia, they were monitored for 2–3 days in the recovery room
before being moved for routine care. They were treated
prophylactically with cefazolin (25 mg/kg i.v. BID) for 2 days
after surgery. They were allowed to stabilize for 1 week and were
then subjected to LA appendage pacing at 600 bpm to induce
persistent AF. Dogs that exhibited persistent AF for ≥5 days were
analysed in vitro.
Morphological evaluation
Tissues from the LA wall were immediately fixed in
4% paraformaldehyde at 4°C and embedded in paraffin. Light
microscopy was performed using semi-thin sections (2 μm)
stained with hematoxylin-eosin and Masson’s trichrome stains. At
least two sections per atrial site were examined and ≥200 cells per
section were analyzed to quantify the extent of myolysis in the
cardiomyocytes. The extent of cell change was evaluated only in
cells in which the nucleus was present in the plane of the section.
The myolytic area of the cardiomyocytes was measured with a digital
imaging system (Motic Images Advanced, Richmond, BC, Canada). Cells
were scored as mildly myolytic if myolysis involved 10–25% of the
cytosol and as severely myolytic if >25% of the sarcomeres were
absent.
For electron microscopy, ultrathin sections (50–100
nm) were cut from each sample, counterstained with uranium acetate
and lead citrate and then examined under a transmission electron
microscope at high magnification (×10,000) (Philips 201; Philips,
Sunnyvale, CA, USA) by two professional staff.
Terminal deoxynucleotidyl
transferase-mediated deoxyuridine triphosphate (dUTP) nick end
labeling (TUNEL) assay
The sections were transferred to xylene and
rehydrated in decreasing concentrations of alcohol. Slides were
then incubated (10 min, room temperature) with 10 μg
proteinase K (Sigma-Aldrich, St. Louis, MO, USA) per 1 ml
phosphate-buffered saline (PBS). Endogenous peroxidase (POD) was
inactivated with an ImmunoPure POD suppressor (Pierce Biotechnology
Inc., Rockford, IL, USA) for 30 min. Tissue sections permeabilized
with 1% Triton X-100 (4°C, 2 min) were stained with an in
situ cell detection POD system (Boehringer Mannheim, Mannheim,
Germany). DNA strand breaks were identified by labeling free 3′-OH
termini with dUTP-fluorescein isothiocyanate (FITC) using TUNEL.
Incorporated fluorescein was detected with an anti-fluorescein
antibody Fab fragment from sheep conjugated with horse-radish POD.
Following the reaction with metal-enhanced diaminobenzidine (DAB)
(Boehringer Mannheim), sections were counterstained with
hematoxylin. Positive controls consisted of fixed and permeabilized
sections incubated with DNase I (1 μg/ml; 10 min, room
temperature). For negative controls, sections were incubated in
labeling solution without terminal deoxynucleotidyl transferase.
The percentage of TUNEL-positive nuclei was calculated by examining
50 randomly selected fields per section corresponding to ∼700 cells
at high magnification (×400).
Detection of gene expression
Total RNA was extracted from samples of the LA wall
using TRIzol (Invitrogen Life Technologies, Carlsbad, CA, USA). The
expression levels of target genes were measured by real-time
quantitative reverse transcription-polymerase chain reaction
(RT-PCR) using SYBR-Green qPCR master mix (Bio-Rad, Hercules, CA,
USA) in a 20 μl reaction volume containing 50 ng cDNA. All
reactions were performed in triplicate and included a negative
control. PCR was carried out using an ABI Prism 7500 Sequence
Detection System (Applied Biosystems, Carlsbad, CA, USA) under the
following conditions: 2 min at 50°C, 10 min at 95°C and 40 cycles
of 15 sec at 95°C and 1 min at 60°C. Relative quantification of
mRNA levels was obtained using the 7500 system, which performs
comparative analysis. Fluorescence signals were normalized to the
housekeeping gene, β-actin. The comparative threshold cycle (CT)
relative quantification method was used (ΔΔCT). For every sample,
each gene was quantified in duplicate in three separate
experiments. The values were averaged and then used to calculate
2−ΔΔCT, which corresponds to the expression relative to
β-actin. The amplicons of expected size were confirmed by gel
electrophoresis. The sequences of the genes studied were obtained
from GenBank and the primers were designed using Primer 5.0
(Applied Biosystems). The amplicon size of the primer sequence and
annealing temperature of the genes are presented in Table I.
 | Table I.Primer sequence and amplicon size of
genes. |
Table I.
Primer sequence and amplicon size of
genes.
| Gene | Primer
sequence | Amplicon size
(bp) | Annealing
temperature (°C) |
|---|
| β-actin | F:
5′-AAGGACCTGTATGCCAACACA-3′ | | |
| R:
5′-ATCCACACAGAATACTTGCGTT-3′ | 152 | 57 |
| MMP-9 | F:
5′-GACGCTATGGGCTATGAGTTAC-3′ | | |
| R:
5′-AGTCCAGGTAGCCCTTTAGGT3′ | 165 | 58 |
| TIMP-1 | F:
5′-TGGATTACAATGAGGCGAAG-3′ | | |
| R:
5′-AGACCCTTCAATTCGGTATCA-3′ | 112 | 60 |
| BCL-2 | F:
5′-CACAAGAGCCAAGGCTACCT-3′ | | |
| R:
5′-CAGGAAAGCAGGAAGTCTCAA-3′ | 158 | 58 |
| BAX | F:
5′-ATTGAGAAACGATTTGCCTACA-3′ | | |
| R:
5′-GGGAAATGGCTTATTCTCCTTTGCTT-3′ | 187 | 59 |
Assessment of protein expression
Protein was extracted from tissue samples of the LA
wall with 5 mmol/l Tris-HCl (pH 7.4), 2 mmol/l ethylenediamine
tetraacetic acid (EDTA), 5 μg/ml leupeptin, 10 μg/ml
benzamidine and 5 μg/ml soybean trypsin inhibitor, followed
by tissue homogenization. All procedures were performed at 4°C.
Equal amounts (100 μg/sample) of LA membrane proteins were
separated on 8% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) gels and transferred to polyvinylidene
difluoride membranes. Membranes were blocked in 5% non-fat dry milk
in TTBS (50 mmol/l Tris-HCl; 500 mmol/l NaCl, pH 7.5; 0.05%
Tween-20) for 2 h (room temperature) and then incubated with the
primary antibody (1:500 dilution) in 5% non-fat dry milk in TTBS
for 4 h at room temperature. Membranes were then incubated with
rabbit polyclonal anti-MMP-9 (Santa Cruz Biotechnology Inc., Santa
Cruz, CA, USA) as well as rabbit polyclonal anti-TIMP-1, BCL-2 and
BAX (Abcam, Cambridge, MA, USA). Membranes were washed three times
in TTBS, re-blocked in 5% non-fat dry milk in TTBS (15 min) and
then incubated with horseradish POD-conjugated goat anti-rabbit
(1:5000) in 5% non-fat dry milk in TTBS (40 min). Immunoreactive
bands were detected by Immun-Star horse-radish peroxidase (HRP)
substrate (Bio-Rad) and quantified by densitometry analysis using
Image Quant 350 and Image Quant TL-1 (GE Healthcare, Fairfield, CT,
USA). Anti-β-actin antibodies (Santa Cruz Biotechnology) were used
to control for equal protein loading and to normalize the target
protein band intensity. All western blotting target bands were
expressed quantitatively by normalization to the control band on
the same lane. Western blotting band intensities were expressed as
optical density (OD) units corresponding to densitometric band
intensity following background subtraction divided by β-actin
signal intensity for the same sample.
Statistical analysis
SPSS 15.0 (SPSS Inc., Chicago, IL, USA) was used in
the statistical analysis. Quantitative data are expressed as mean ±
standard deviation (SD). Comparisons between the quantitative data
were made using the t-test, whereas those for qualitative data were
assessed with χ2 analysis. P<0.05 was considered to
indicate a statistically significant difference.
Results
ECG data
ECG data for the adult and aged groups studied in SR
are shown in Table II. The ECGs of
the aged dogs revealed longer P-wave durations and P-wave
dispersion compared with those of the adult dogs. These groups did
not differ in other variables and the difference between these two
groups in time to onset of persistent AF was not significant, with
the adult dogs having developed persistent AF after 40±5 days and
the aged dogs acquiring it after 52±7 days of atrial pacing
(P>0.05).
| Table II.ECG data of adult and aged canines in
SR. |
Table II.
ECG data of adult and aged canines in
SR.
| Group | P (msec) | PWD (msec) | PR (msec) | QRS (msec) | QT (msec) |
|---|
| Adult | 66.1±6.4 | 19.1±4.1 | 123.9±7.2 | 63.1±4.3 | 248.9±11.7 |
| Aged | 75.9±5.3a | 26.7±3.1a | 130.0±7.7 | 64.7±5.4 | 246.5±17.3 |
Fibrotic changes
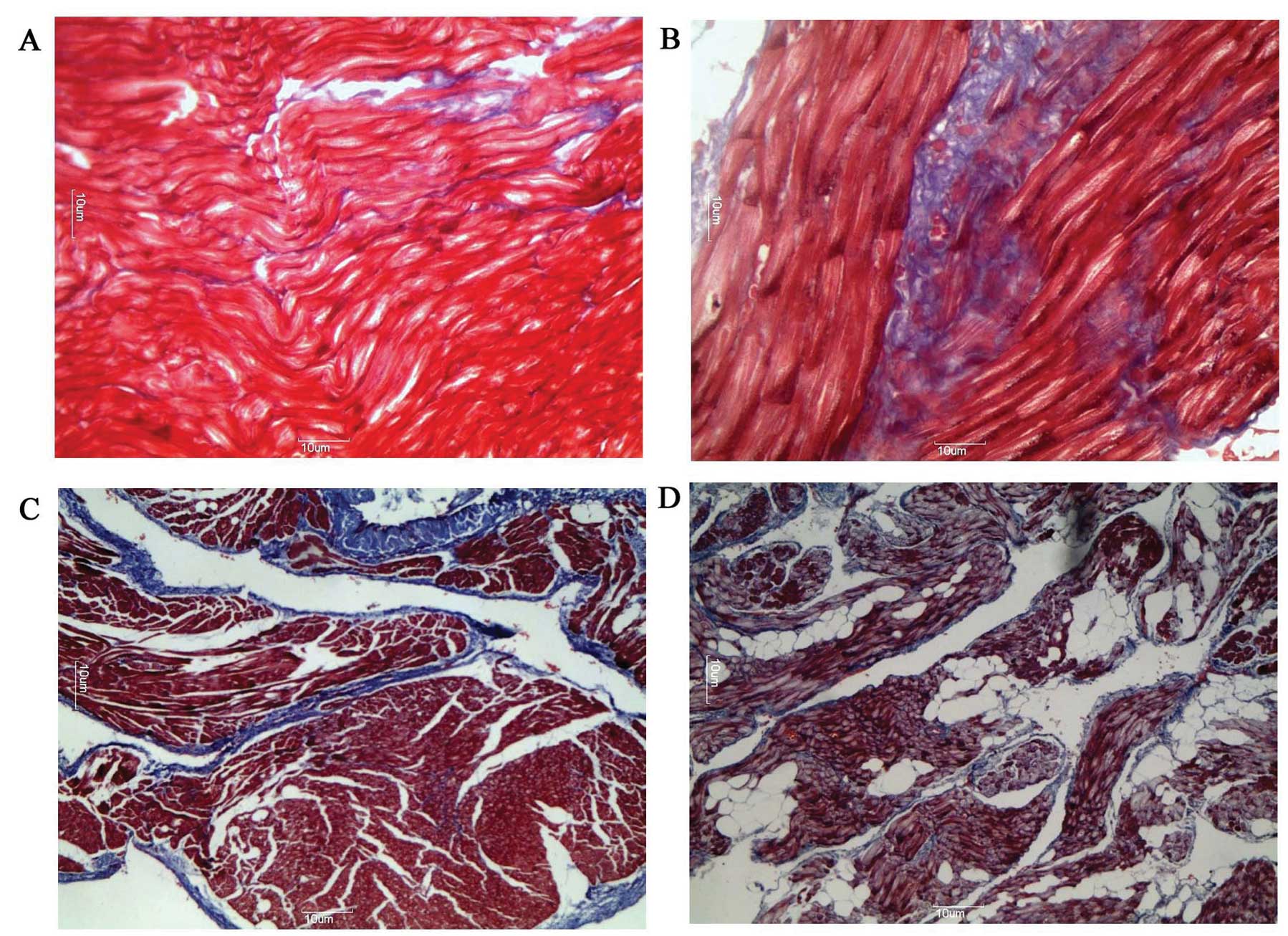
As shown in Fig. 1,
compared with those in the adult group, the myocardial fibres of
tissue from the aged group appeared to be more compact and were
closer together. Muscle bundles were separated by large strands of
connective tissue and a significantly higher deposition of
connective tissue was observed (4.1±0.9 vs. 8.4±1.0%; n=15;
P<0.05). Additionally, the degree of myocardial fiber disarray
was higher in the adult (4.1±0.9 vs. 14.7±2.1%; n=15; P<0.01)
and aged (8.4±1.0 vs. 18.2±2.4%; n=15; P<0.01) groups with AF
than in the corresponding groups in SR. The degree of myocardial
fiber disarray among muscle bundles of tissue was greater in the
aged group with AF than in the adult group with AF (14.7±2.1 vs.
18.2±2.4%; n=15; P<0.05).
Cell ultrastructural changes
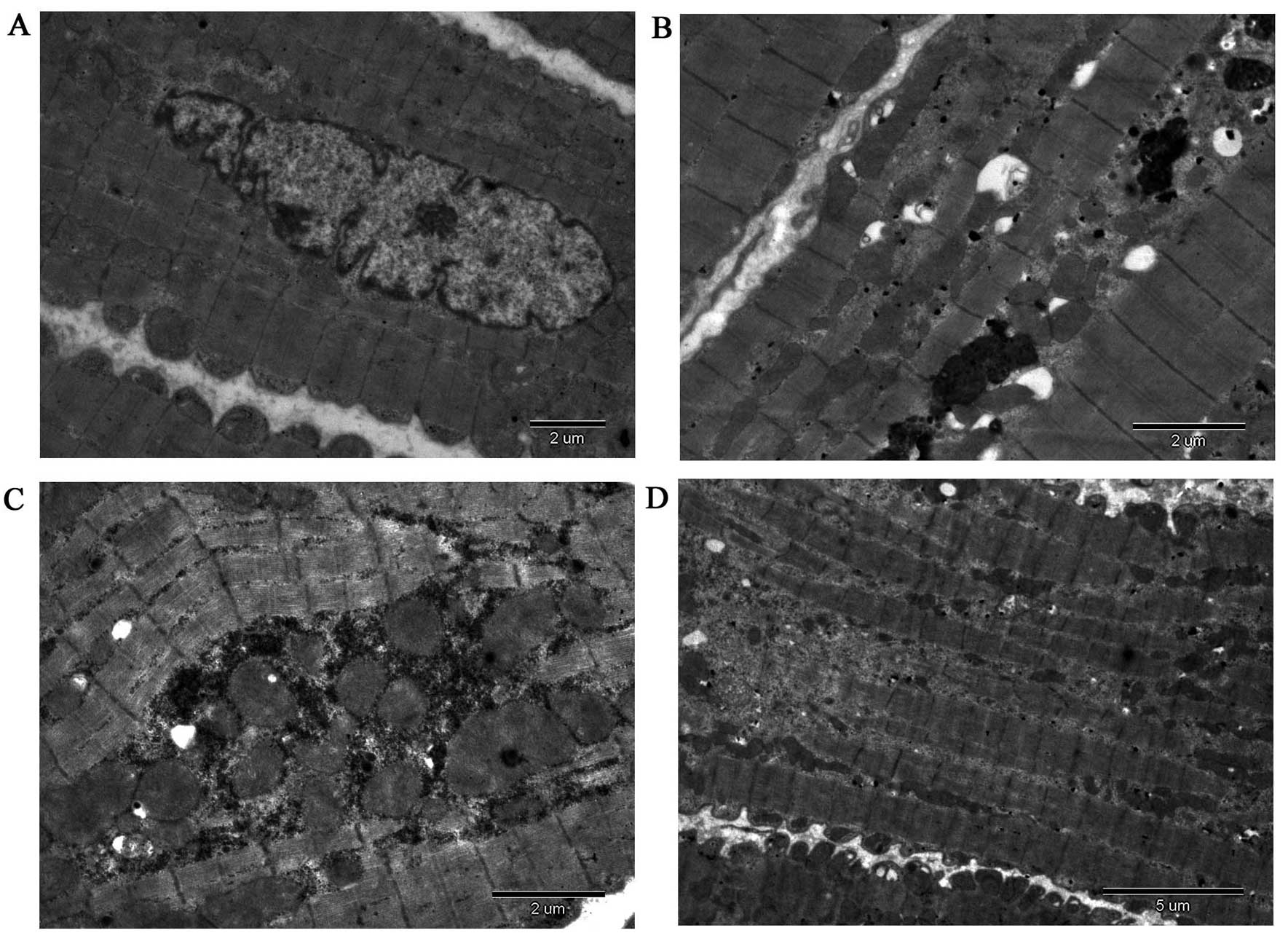
The ultrastructure of the atrial myocardium was
examined by electron microscopy and representative transmission
electron micrographs are shown in Fig.
2. Atrial myocytes of the LA wall obtained from the adult dogs
had a regular sarcomere organization, uniformly sized mitochondria
and nuclei demonstrating normal clumping of chromatin at the
nuclear membrane (n=3, samples from randomly selected adult dogs;
Fig. 2A). Atrial myocytes of the
LA wall obtained from the aged dogs presented an abnormal
ultrastructure, with mild and severe sarcomere degeneration,
mitochondrial swelling with a reduction in the density and
organization of the cristae, karyopyknosis with chromatin
margination to the nuclear membrane, expanding endoplasmic
reticulum, increased glucogen and mildly compact and close
myocardial fibers (n=3, samples from randomly selected aged dogs;
Fig. 2B). Atrial myocytes of the
LA wall obtained from the adult dogs with AF appeared to have a
more abnormal ultrastructure, with severe sarcomere degeneration,
more mitochondrial swelling, karyopyknosis indicating some cell
apoptosis, several secondary lysosomes, expanding endoplasmic
reticulum, decreased glucogen and irregular and disorderly
myocardial fibers (n=3, samples from randomly selected adult dogs
with AF; Fig. 2C). Atrial myocytes
of the LA wall obtained from the aged dogs with AF also exhibited
an abnormal ultrastructure, with more severe sarcomere
degeneration, mitochondria showing vacuoles, more karyopyknosis
indicating cell apoptosis, expanding endoplasmic reticulum and
secondary lysosomes, as well as some disintegration of myofilaments
(n=3, samples from randomly selected aged dogs with AF; Fig. 2D).
Apoptotic indices
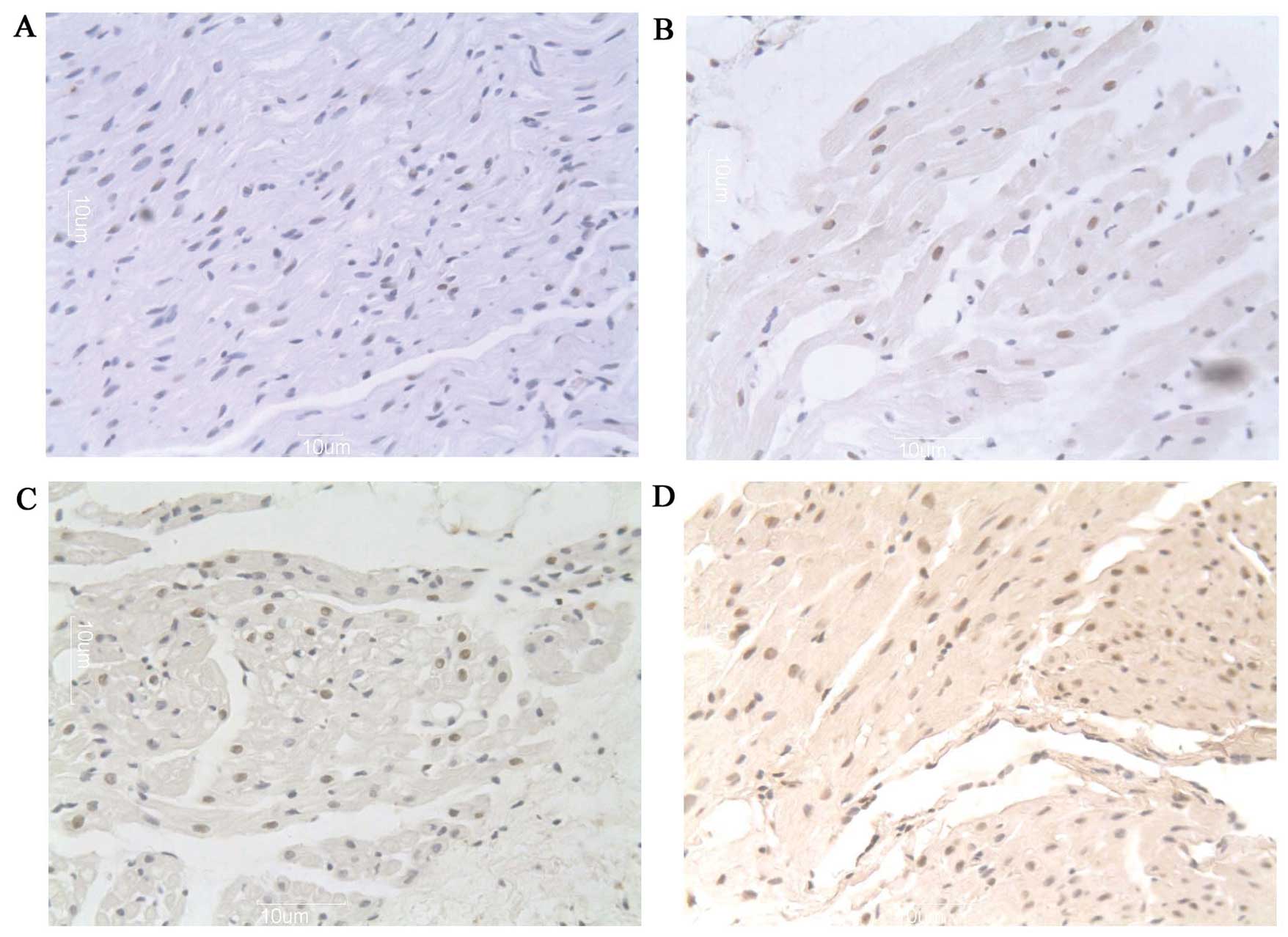
As shown in Fig. 3,
a higher percentage of myocytes with myolysis in aged dogs had
TUNEL-positive nuclei compared with the adult group (22.0±5.45 vs.
32.9±3.4%; n=12; P<0.05). The majority of the nuclei with weak
TUNEL staining were large with a uniform distribution of
heterochromatin (Fig. 3A and B).
Additionally, the frequency of apoptosis was significantly
increased in the adult (22.0±5.45 vs. 33.4±3.9%; n=12; P<0.01)
and aged (32.9±3.4 vs. 51.2±3.4%; n=12; P<0.001) groups with AF
compared with their counterpart groups in SR, with the frequency in
the aged group being higher. In addition, several nuclei decreased
in size and were stained strongly by TUNEL, indicating that more
extensive DNA cleavage was associated with these nuclear
alterations (Fig. 3C and D).
LA mRNA and protein expression of
MMP-9/TIMP-1 and BCL-2/BAX
As shown in Tables
III and IV, and Figs. 4 and 5, the mRNA and protein expression levels
of MMP-9 and BAX significantly increased (P<0.05), whereas the
mRNA and protein expression of TIMP-1 and BCL-2 were down-regulated
in the aged group compared with the adult group (P<0.05).
Compared with the control groups, the adult and aged groups with AF
exhibited significantly increased mRNA and protein expression
levels of MMP-9 and BAX (P<0.05), with the expression of BAX
being higher in the two AF groups (P<0.05). By contrast, the
mRNA and protein expression levels of TIMP-1 and BCL-2 were
down-regulated in the adult and aged groups with AF (P<0.05),
with the expression of BCL-2 being lower in the two AF groups
(P<0.05).
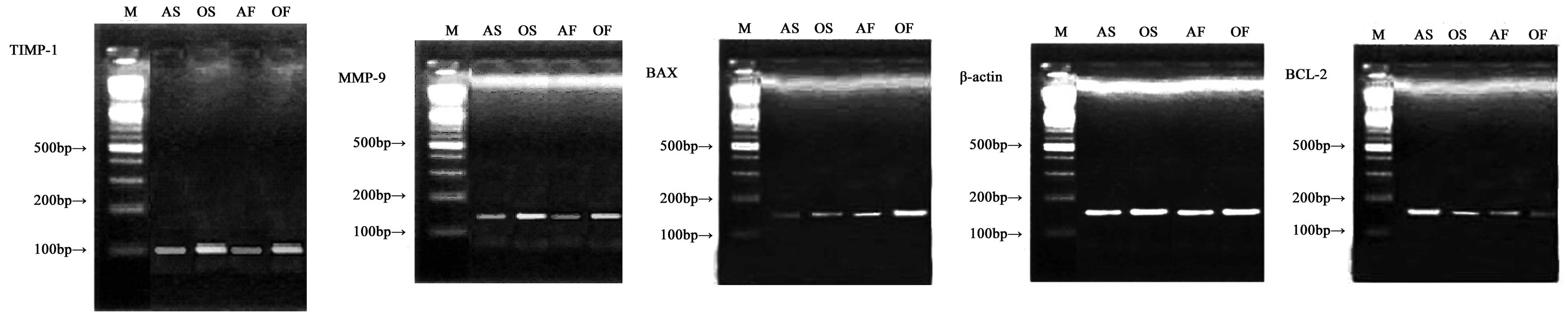 | Figure 4.Representative gels of MMP-9/TIMP-1
and BCL-2/BAX mRNA expression in the LA myocardium. M, DL-2000
marker; AS, adult SR group; OS, aged SR group; AF, adult AF group;
OF, aged AF group. MMP, matrix metalloproteinase; TIMP, tissue
inhibitors of MMPs; BCL-2, B cell lymphoma 2; BAX, BCL-2-associated
X protein; LA, left atrium; SR, sinus rhythm; AF, atrial
fibrillation. |
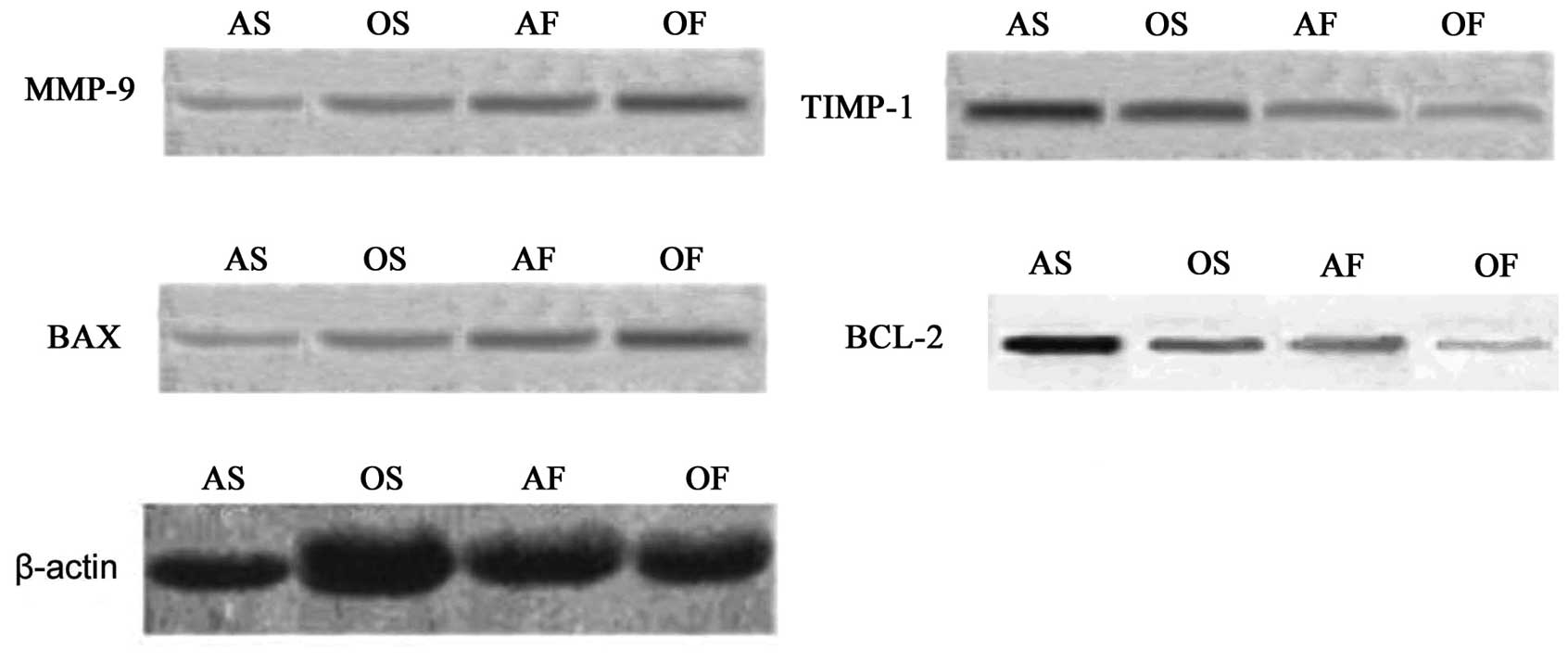 | Figure 5.Representative immunoblots (western
blotting) showing MMP-9/TIMP-1 and BCL-2/BAX protein expression in
the LA myocardium. AS, adult SR group; OS, aged SR group; AF, adult
AF group; OF, aged AF group; MMP, matrix metalloproteinase; TIMP,
tissue inhibitors of MMPs; BCL, B cell lymphoma; BAX,
BCL-2-associated X protein; LA, left atrium; SR, sinus rhythm; AF,
atrial fibrillation. |
| Table III.Comparison of target gene mRNA levels
between the atrial fibrillation groups and the sinus rhythm
groups. |
Table III.
Comparison of target gene mRNA levels
between the atrial fibrillation groups and the sinus rhythm
groups.
| Group | n | MMP-9 | TIMP-1 | BAX | BCL-2 |
|---|
| SR adult | 7 | 1.1483±0.2371 | 3.1602±0.3029 | 1.2520±0.2831 | 2.9571±0.3745 |
| SR aged | 7 |
1.8520±0.3029a |
2.1139±0.2273a |
1.9520±0.1824a |
1.8623±0.3382a |
| AF adult | 7 |
2.1372±0.2981a |
1.9126±0.3812a |
2.7361±0.2937a |
1.5620±0.2490a |
| AF aged | 7 |
2.8072±0.3369b |
1.1683±0.1927b |
3.2532±0.3271b |
0.8524±0.2176b |
| Table IV.Comparison of target protein levels
between the atrial fibrillation groups and the sinus rhythm
groups. |
Table IV.
Comparison of target protein levels
between the atrial fibrillation groups and the sinus rhythm
groups.
| Group | n | MMP-9 | TIMP-1 | BAX | BCL-2 |
|---|
| SR adult | 7 | 0.1738±0.0329 | 0.7620±0.0572 | 0.2165±0.0263 | 0.6850±0.0562 |
| SR aged | 7 |
0.3093±0.0462a |
0.5833±0.0429a |
0.4281±0.0390a |
0.5192±0.0509a |
| AF adult | 7 |
0.4473±0.0528a |
0.3982±0.0320a |
0.5018±0.0472a |
0.3278±0.0297a |
| AF aged | 7 |
0.6172±0.0627b |
0.2207±0.0313b |
0.7163±0.0572b |
0.1629±0.0162b |
The protein expression levels of MMP-9 (r=0.348;
P=0.019) and TIMP-1 (r=−0.331; P=0.027) were correlated with the
degree of myocardial fiber disarray, whereas the protein expression
levels of BAX (r= 0.451; P= 0.002) and BCL-2 were correlated with
the frequency of apoptosis (r=−0.309; P= 0.038).
Discussion
Myocardial fibrosis is a dynamic process during
which normal collagen chains are degraded and replaced by fibrous
interstitial deposits (9,10,13).
MMPs are involved in matrix degradation and collagen synthesis.
They play a crucial role in ECM homeostasis in a number of
physiological and pathological situations, including during ageing
and/or in AF (3,12,13,16).
These proteolytic enzymes are regulated at transcriptional and
translational levels, as well as by endogenous physiological
inhibitors, including TIMPs (11,15).
In the majority of disease processes, a dynamic interplay between
these MMPs and the TIMPs (mainly MMP-9/TIMP-1) determines the
phenotypic changes (14,17). Additionally, MMP-9 and TIMP-1
interact with tissue necrosis factors, angiotensin and other
cytokines in the LA remodeling process in AF (17,18).
Profibrotic signals act on the balance between MMPs and TIMPs
(9,11,18).
Thus, TIMP-1 deficiency has been shown to result in LA and left
ventricular dilatation, cardiomyocyte hypertrophy and contractile
dysfunction (10,15,17).
The samples from our dog models of ageing and/or AF demonstrated
impaired balance between MMP-9 and TIMP-1, with increased levels of
MMP-9 and lack of TIMP-1 upregulation. In addition, our study
demonstrated that the protein expression levels of MMP-9 and TIMP-1
were correlated with the levels of myocardial fibrosis in the LA
with ageing and/or in AF. These results suggest that the altered
MMP-9/TIMP-1 stoichiometry may result in loss of control of MMP-9
activity and contribute to the development of interstitial fibrosis
in atrial remodeling.
Structurally, the most important change in aged
atria is an enhancement of the fibrous tissue that is interspersed
between myocytes. Additionally, these alterations predominate in
fibrillating and ageing atria. Cardiac fibrosis is characterized by
the excessive accumulation of fibrillar collagen in the
extracellular space. Fibrosis is ubiquitous in the atria of the
ageing heart (18). Interstitial
fibrosis reduces electrical coupling in the heart (19) and significantly increases the
complexity of the myocardial architecture by electrically
insulating cardiac cells and/or muscle bundles (20,21),
a direct consequence of which is that the typical uniform
anisotropic conduction in the atrial myocardium is replaced by
non-uniform anisotropic conduction (22,23).
Studies have shown that such fibrosis preferentially affects
lateral (or transverse) connections over longitudinal cell-cell
connections (24,25), resulting in a much slower and
zigzag transverse propagation (26,27).
Thus, a premature response occurring in the aged atrial myocardium
has a higher probability of undergoing a unidirectional block from
an imbalance between source/sink currents and initiating reentry
due to the underlying arrhythmogenic substrate created as a result
of the electrical and/or structural remodeling (14,27,28).
The results of the current study indicate that
ageing and/or fibrillating atria contain a number of apoptotic
myocytes and that myocytes with myolysis may exhibit several
features of dedifferentiation. We identified an impaired balance
between BAX and BCL-2, with increased levels of BAX and lower
levels of BCL-2 with ageing and/or in AF and determined that the
protein expression of BAX and BCL-2 were correlated with the
frequency of apoptosis in LA organisation with ageing and/or in AF.
A balance therefore exists between protective anti-apoptotic
signals (which likely take part in the molecular basis of the ‘cell
survival syndromes’ described above) and pro-apoptotic pathways.
Chronic or repetitive stress with ageing and/or in AF may lead to
the progressive downregulation of protective mechanisms and
sustained activation of pathways that eventually induce
apoptosis.
Several observations relate the onset of apoptosis
to metabolic dysfunction (6,7,8,29).
To begin with, a major determinant of the apoptosis cascade is
mitochondrial destabilization, characterized by loss of membrane
potential, generation of reactive oxygen species and liberation of
cytochrome c(6,7,8,30).
As mitochondria are the main site of cardiac adenosine triphosphate
(ATP) production, it is not surprising that apoptosis is related to
ATP depletion (6,7,8,31).
Decreased ATP content promotes the transfer of the pro-apoptotic
protein BAX to the mitochondria (32). Insertion of BAX into the
mitochondrial membrane creates pores through which cytochrome
c is extruded to the cytosol (33). In other cell types, activation of
the apoptotic pathway is accompanied by an impairment of glucose
uptake and downregulation of the anerobic production of ATP
(31,32,34).
Interestingly, overexpression of BCL-2 counteracts the onset of
apoptosis under conditions of ATP depletion (30,33,35).
This protective effect is multifactorial, including an inhibition
of BAX-induced release of cytochrome c and inhibition of
APAF-1 (30,31,35).
BCL-2 is now well established as an anti-apoptotic protector in
cardiac cells. Studies have suggested that BCL-2 induces a state of
‘metabolic hibernation’ that improves cell resistance in stress
conditions (35,36). As apoptosis requires energy, severe
depletion of ATP is followed by necrosis rather than apoptosis
(37,38). Cardiac apoptosis therefore results
from ‘mild’ but repetitive or prolonged episodes of stress
(ischemia, stretch or overload with ageing and/or in AF), which
progressively downregulates protective mechanisms (downregulation
of BCL-2 expression) and activates pro-apoptotic pathways
(overexpression of BAX). When adaptive mechanisms of cardiac
survival are no longer able to sustain cellular homeostasis, the
disturbance of energy metabolism, contractile function and gene
expression trigger cardiac apoptosis.
We did not subject the animals in our models of
chronic AF to atrioventricular nodal ablation, rendering
excessively rapid ventricular rates accompanied by rapid atrial
pacing inevitable. In fact, the majority of the dogs with chronic
AF in this study had a certain degree of atrial dilation,
suggesting chronic hemodynamic overload of their atria besides AF,
which may be an important factor in triggering a programmed cell
death pathway and atrial fibrosis. For instance, abnormal levels of
resting tension have been shown to induce apoptosis in ventricular
myocytes (29–31). This mechanism may be particularly
important for myocytes of thin atrial walls, in which even moderate
hemodynamic overload may induce substantial overstretching. This
may also explain why atrial samples from the aged dogs with AF,
which often had a greater degree of hemodynamic overload of the
atria, also had marked histological abnormalities and apoptotic
cells.
Acknowledgements
This study was supported by the
Program of National Natural Science Foundation of China (No.
308660299), the Program of the Natural Science Foundation of the
Xinjiang Uygur Autonomous Region (No. 200821143 and 2011211A074)
and the Program of the Doctoral Fund of the Ministry of Education
(200807600004). The funders had no role in study design, data
collection and analysis, decision to publish or preparation of the
manuscript.
References
|
1.
|
Nattel S: New ideas about atrial
fibrillation 50 years on. Nature. 415:219–226. 2002.PubMed/NCBI
|
|
2.
|
Chen LY and Shen WK: Epidemiology of
atrial fibrillation: A current perspective. Heart Rhythm. 4:S1–S6.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Allessie M, Ausma J and Schotten U:
Electrical, contractile and structural remodeling during atrial
fibrillation. Cardiovasc Res. 54:230–246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Kostin S, Klein G, Szalay Z, Hein S, Bauer
EP and Schaper J: Structural correlate of atrial fibrillation in
human patients. Cardiovasc Res. 54:361–379. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Everett TH, Wilson EE, Verheule S, Guerra
JM, Foreman S and Olgin J: Structural atrial remodeling alters the
substrate and spatiotemporal organization of AF: a comparison in
canine models of structural and electrical atrial remodeling. Am J
Physiol Heart Circ Physiol. 291:H2911–H2923. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Williams GT: Programmed cell death:
apoptosis and oncogenesis. Cell. 65:1097–1098. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Lockshin RA, Facey CO and Zakeri Z: Cell
death in the heart. Cardiol Clin. 19:1–11. 2001. View Article : Google Scholar
|
|
8.
|
Yang E, Zha J, Jockel J, Boise LH,
Thompson CB and Korsmeyer SJ: Bad, a heterodimeric partner for
bcl-XL and bcl-2, displaces bax and promotes cell death. Cell.
80:285–291. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Verheule S, Wilson E, Everett T IV,
Shanbhag S, Golden C and Olgin J: Alterations in atrial
electrophysiology and tissue structure in a canine model of chronic
atrial dilatation due to mitral regurgitation. Circulation.
107:2615–2622. 2003.PubMed/NCBI
|
|
10.
|
Spinale FG: Myocardial matrix remodeling
and the matrix metalloproteinases: influence on cardiac form and
function. Physiol Rev. 87:1285–1342. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Cardin S, Libby E, Pelletier P, et al:
Contrasting gene expression profiles in two canine models of atrial
fibrillation. Circ Res. 100:425–433. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Bouzeghrane F, Reinhardt DP, Reudelhuber
TL and Thibault G: Enhanced expression of fibrillin-1, a
constituent of the myocardial extracellular matrix in fibrosis. Am
J Physiol Heart Circ Physiol. 289:H982–H991. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Mihm MJ, Yu F, Carnes CA, Reiser PJ,
McCarthy PM, Van Wagoner DR and Bauer JA: Impaired myofibrillar
energetics and oxidative injury during human atrial fibrillation.
Circulation. 104:174–180. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Disertori M, Latini R, Barlera S, et al:
Valsartan for prevention of recurrent atrial fibrillation. N Engl J
Med. 360:1606–1617. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Mukherjee R, Herron AR, Lowry AS, et al:
Selective induction of matrix metalloproteinases and tissue
inhibitor of metalloproteinases in atrial and ventricular
myocardium in patients with atrial fibrillation. Am J Cardiol.
97:532–537. 2006. View Article : Google Scholar
|
|
16.
|
Anyukhovsky EP, Sosunov EA, Plotnikov A,
Gainullin RZ, Jhang JS, Marboe CC and Rosen MR: Cellular
electrophysiologic properties of old canine atria provide a
substrate for arrhythmogenesis. Cardiovasc Res. 54:462–469. 2002.
View Article : Google Scholar
|
|
17.
|
Lin CS, Lai LP, Lin JL, et al: Increased
expression of extracellular matrix proteins in rapid atrial
pacing-induced atrial fibrillation. Heart Rhythm. 4:938–949. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Moe GW, Laurent G, Doumanovskaia L, Konig
A, Hu X and Dorian P: Matrix metalloproteinase inhibition
attenuates atrial remodeling and vulnerability to atrial
fibrillation in a canine model of heart failure. J Card Fail.
14:768–776. 2008. View Article : Google Scholar
|
|
19.
|
Tanaka K, Zlochiver S, Vikstrom KL, et al:
The spatial distribution of fibrosis governs fibrillation wave
dynamics in the posterior left atrium during heart failure. Circ
Res. 101:839–847. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Wit AL and Boyden PA: Triggered activity
and atrial fibrillation. Heart Rhythm. 4:S17–S23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Weiss JN, Qu Z, Chen PS, et al: The
dynamics of cardiac fibrillation. Circulation. 112:1232–1240. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Everett TH IV and Olgin JE: Atrial
fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm.
4:S24–S27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Qu Z, Garfinkel A, Chen PS and Weiss JN:
Mechanisms of discordant alternans and induction of reentry in
simulated cardiac tissue. Circulation. 102:1664–1670. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Miragoli M, Gaudesius G and Rohr S:
Electrotonic modulation of cardiac impulse conduction by
myofibroblasts. Circ Res. 98:801–810. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Koura T, Hara M, Takeuchi S, et al:
Anisotropic conduction properties in canine atria analyzed by high
resolution optical mapping: preferential direction of conduction
block changes from longitudinal to transverse with increasing age.
Circulation. 105:2092–2098. 2002. View Article : Google Scholar
|
|
26.
|
Thijssen VL, Ausma J, Liu GS, Allessie MA,
van Eys GJ and Borgers M: Structural changes of atrial myocardium
during chronic atrial fibrillation. Cardiovasc Pathol. 9:17–28.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Mandapati R, Skanes A, Chen J, Berenfeld O
and Jalife J: Stable microreentrant sources as a mechanism of
atrial fibrillation in the isolated sheep heart. Circulation.
101:194–199. 2000.PubMed/NCBI
|
|
28.
|
Ausma J, Wijffels M, Thone F, Wouters L,
Allessie M and Borgers M: Structural changes of atrial myocardium
due to sustained atrial fibrillation in the goat. Circulation.
96:3157–3163. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Olivetti G, Abbi R, Quaini F, et al:
Apoptosis in the failing human heart. N Engl J Med. 336:1131–1141.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Hockenbery D, Nunez G, Milliman C,
Schreiber RD and Korsmeyer SJ: Bcl-2 is an inner mitochondrial
membrane protein that blocks programmed cell death. Nature.
348:334–336. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Goodwin G, Taylor C and Taegtmeyer H:
Regulation of energy metabolism of the heart during acute increase
in heart work. J Biol Chem. 273:29530–29539. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, bax, that
accelerates programed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Yin XM, Oltvai ZN and Korsmeyer SJ: BH1
and BH2 domains of bcl-2 are required for inhibition of apoptosis
and heterodimerization with Bax. Nature. 369:321–333. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
van Bilsen M, van der Vusse G and Reneman
R: Transcriptional regulation of metabolic processes: implications
for cardiac metabolism. Pflügers Arch. 437:2–14. 1998.PubMed/NCBI
|
|
35.
|
Cheng EH, Kirsch DG, Clem RJ, et al:
Conversion of Bcl-2 to a Bax-like death effector by caspases.
Science. 278:1966–1968. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Marton A, Mihalik R, Bratincsák A, et al:
Apoptotic cell death induced by inhibitors of energy conservation.
Eur J Biochem. 250:467–475. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Tanaka M, Ito H, Adachi S, et al: Hypoxia
induces apoptosis with enhanced expression of Fas antigen messenger
RNA in cultured neonatal rat cardiomyocytes. Circ Res. 75:426–433.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Depre C, Rider M and Hue L: Mechanisms of
control of heart glycolysis. Eur J Biochem. 258:277–290. 1998.
View Article : Google Scholar : PubMed/NCBI
|