Introduction
Asthma is a chronic inflammatory disease of the
bronchial airway characterized by chronic airway inflammation,
airway hyperresponsiveness, mucus overproduction and airway
remodeling (1,2). A previous study demonstrated that
airway remodeling is a characteristic feature of asthma and may be
observed from the early stages (3). Structural and functional changes to
the epithelial cells are significant in the process of airway
remodeling (3,4) and airway epithelial cells are able to
promote the process through epithelial-mesenchymal transition (EMT)
(5). During the process of EMT,
the expression levels of type I collagen, fibronectin-1 and
α-smooth muscle actin (α-SMA) have been observed to be upregulated,
while the expression of the epithelial marker, E-cadherin, has been
shown to be reduced (6). The
abnormal process of EMT was observed as a central pathological and
physiological characteristic in the early stages of airway
remodeling (7).
Found in inflammatory zone 1 (FIZZ1) was first
reported in 2000 by Holcomb et al in allergic pulmonary
inflammation (8) and was shown to
play a vital role in pulmonary inflammation and angiogenesis
(9). Our previous study
demonstrated that FIZZ1 was vital in airway remodeling in asthma
and was capable of increasing the expression levels of α-SMA and
type I collagen in the early stages of airway remodeling (10).
However, the mechanism by which FIZZ1 functions in
the process of airway remodeling remains unclear. In the present
study, the hypothesis that FIZZ1 may activate the phosphoinositide
3-kinase (PI3K)/protein kinase B (Akt) signaling pathway through
promoting Akt phosphorylation in vitro was investigated. In
addition, the effect that blocking the PI3K/Akt pathway has on
decreasing inflammatory cell infiltration and alleviating airway
remodeling via regulating the process of EMT was investigated.
Materials and methods
Animals
Specific-pathogen-free, female BALB/c mice (age,
8–10 weeks; weight, 20±2 g; Animal Experiment Center of Shandong
University, Shandong, China) were sensitized on days 1 and 14 by
intraperitoneal injection of 20 μg ovalbumin (OVA; Sigma-Aldrich,
St. Louis, MO, USA) and 4 mg Al(OH)3 (Sigma-Aldrich)
suspended in 0.2 ml saline. On days 21–23 following the initial
sensitization, the mice were challenged for 30 min with an aerosol
of 1% (wt/vol) OVA in saline using an ultrasonic nebulizer (PARI
BOY SX, Starnberg, Germany), while saline alone was used to
challenge the control group. LY294002 (7.5 mg/kg body weight; Cell
Signaling Technology, Inc., Beverly, MA, USA), Akt inhibitor IV (5
mg/kg body weight; Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA) or saline were administered intratracheally 2 h prior to each
OVA aerosol challenge (Table I).
All the animal experiments were approved by the Institutional
Animal Care and Use Committee of Shandong University (Jinan,
China).
 | Table IAnimal model generation. |
Table I
Animal model generation.
| Group | Sensitize on days 1
and 14 | Challenge on days 21,
22 and 23 | Intratracheal
agents |
|---|
| Control | Saline | Saline | Saline |
| OVA | OVA | OVA | Saline |
| LY294002 | OVA | OVA | LY294002 |
| Akt inhibitor IV | OVA | OVA | Akt inhibitor IV |
Analysis of airway responsiveness
At 24 h after the last challenge, the mice were
anesthetized by intraperitoneal injection of chloral hydrate (4
mg/kg body weight). Methacholine was administered at a
concentration of 0, 4, 8, 12 or 16 g/l. Measurements of airway
hyperresponsiveness were conducted using an animal pulmonary
instrument (flexiVent, Hong Kong, China) 1 min after each dose with
2 min between doses. The results were expressed as the maximum
resistance following each dose minus the baseline (saline alone)
resistance.
Histological analysis
Lung tissues were fixed in 10% neutral formalin,
paraffin-embedded, cut into 4-μm sections and stained with
hematoxylin and eosin for examination of inflammatory cell
infiltration.
Immunohistochemistry analysis
Sections were dewaxed, rehydrated and antigen
retrieval was performed with 10 mM sodium citrate (pH 6.1). Next,
the sections were blocked with 5% bovine serum albumin for 20 mins
at 37°C. The sections were incubated with anti-FIZZ1 (1:300),
anti-type I collagen (1:300), anti-E-cadherin (l:300) or
anti-fibronectin-1 (l:300) antibodies (all Santa Cruz
Biotechnology, Inc.) overnight at 4°C. The sections were
subsequently incubated with polyclonal goat anti-rabbit
immunoglobulins/horseradish peroxidase (1:200) for 30 min at 37°C.
The nuclei were counterstained with hematoxylin.
Murine lung epithelial-12 (MLE-12) cell
culture
The MLE-12 cell line was purchased from a cell bank
(American Type Culture Collection, Manassas, VA, USA) and cultured
in Dulbecco’s modified Eagle’s medium/F12 complete medium with 10%
fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin and 100
μg/ml streptomycin at 37°C and 5% CO2. Following a 48-h
culture, the cells were seeded in 6-well culture plates.
FIZZ1 recombinant protein co-culture and
FIZZ1 small hairpin RNA (shRNA) transfection
The MEL-12 cell line was cultured with FIZZ1
recombinant protein (1 μg/ml; Santa Cruz Biotechnology, Inc.),
while the control group used phosphate-buffered saline instead.
Subsequent to 24 h, the protein was extracted from the cell line
for analysis of the Akt phosphorylation levels and the expression
levels of α-SMA and type I collagen.
Escherichia coli JM109 bacteria of the
FIZZ1-shRNA plasmid (sc-39724-SH; Santa Cruz Biotechnology, Inc.)
were amplified in liquid medium. Plasmid DNA was extracted with the
Omega D6950 plasmid extraction kit (Omega Bio-tek, Inc., Norcross,
GA, USA).
Lipofectamine 2000 reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA) was used to transfect the
FIZZ1-shRNA plasmid to the cell line, according to the
manufacturer’s instructions. After 48 h of incubation, the cells
were harvested for analysis of FIZZ1, p-Akt, α-SMA and type I
collagen.
An empty plasmid was added to the control and was
cultured for the same amount of time. The cells were then collected
to analyze the expression of FIZZ1, p-Akt, α-SMA and type I
collagen by western blot analysis.
Quantitative polymerase chain reaction
(PCR)
Total RNA was extracted from the mouse lungs with
TRIzol reagent (Invitrogen Life Technologies), according to the
manufacturer’s instructions, and was reverse transcribed with a
PrimeScript 1st Strand cDNA Synthesis kit (Takara Bio, Inc., Shiga,
Japan). cDNA was amplified by SYBR Premix Ex TaqII (Perfect
Real Time; Takara Bio, Inc.) in Roche Light Cycler 2.0. The primers
used were as follows: α-SMA forward, 5′-CCACCGCAAATGCTTCTAAGT-3′
and reverse, 3′-GGCAGGAATGATTTGGAAAGG-5′; type I collagen forward,
5′-CGCCATCAAGGTCTACTGC-3′ and reverse, 3′-GAATCCATCGGTCATGCTCT-5′;
and GADPH forward, 5′-GTGGCAAAGTGGAGATTGTT-3′ and reverse,
3′-CTCGCTCCTGGAAGATGG-5′. The PCR conditions were 95°C for 10 sec,
then 40 cycles at 95°C for 5 sec, 57°C for 30 sec and 72°C for 30
sec. The expression levels of α-SMA and type I collagen were
assessed using the comparative Ct method.
Western blot analysis
In total, 30 μg protein extracted from the lung was
separated by 10% SDS-PAGE, transferred onto a polyvinylidene
fluoride membrane and subjected to immunoblotting with anti-α-SMA,
anti-type I collagen or anti-β-actin antibodies (Santa Cruz
Biotechnology, Inc.).
Statistical analysis
Data are presented as the mean ± SEM. Continuous
variables were analyzed using the Student’s t-test between the
groups studied. P<0.05 was considered to indicate a
statistically significant difference.
Results
Asthma model in mice
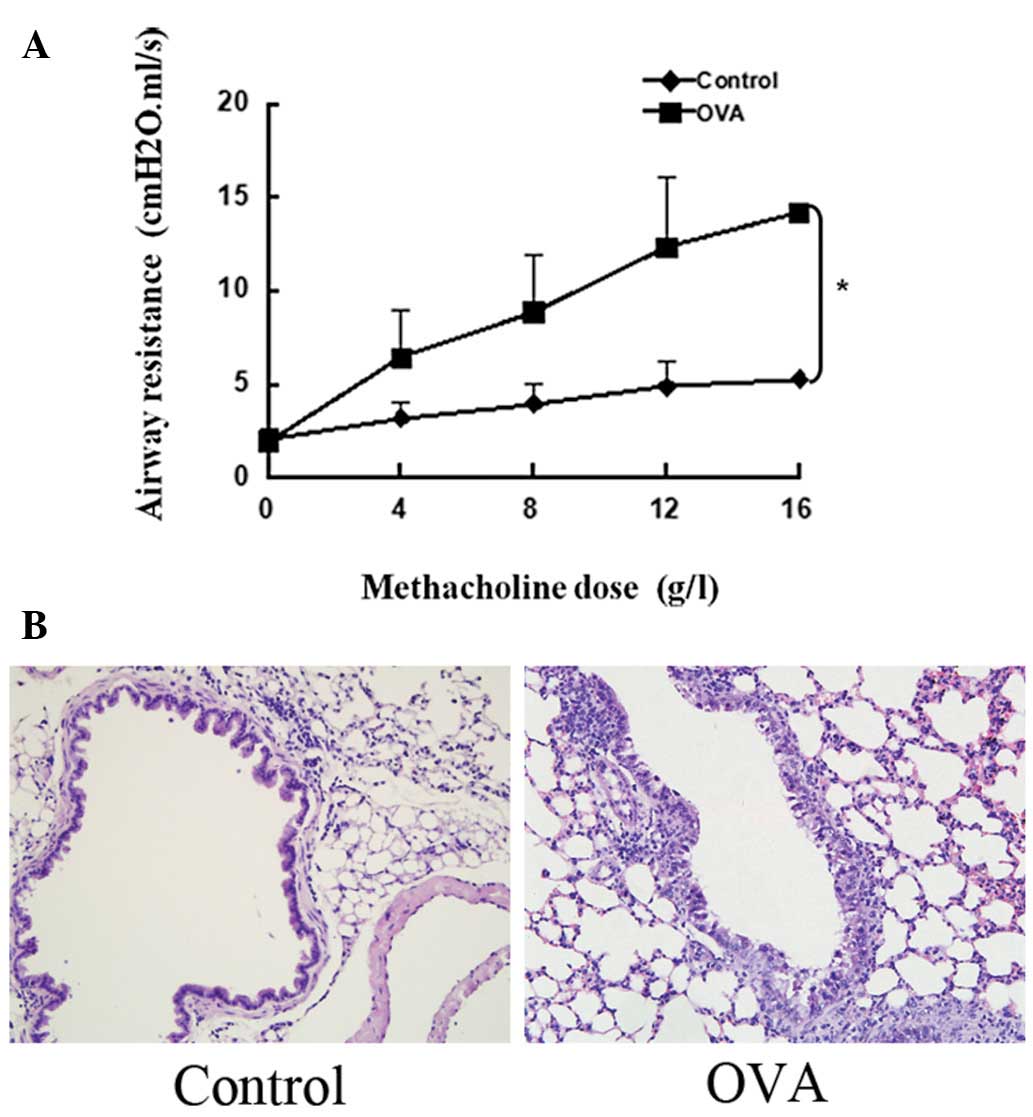
The murine asthma model was confirmed by detecting
airway hyperresponsiveness and histopathology. As shown in Fig. 1A, the OVA-treated mice developed
airway hyperresponsiveness to inhaled methacholine compared with
the saline-challenged group. Furthermore, the OVA aerosol challenge
induced the infiltration of inflammatory cells around the airway
(Fig. 1B).
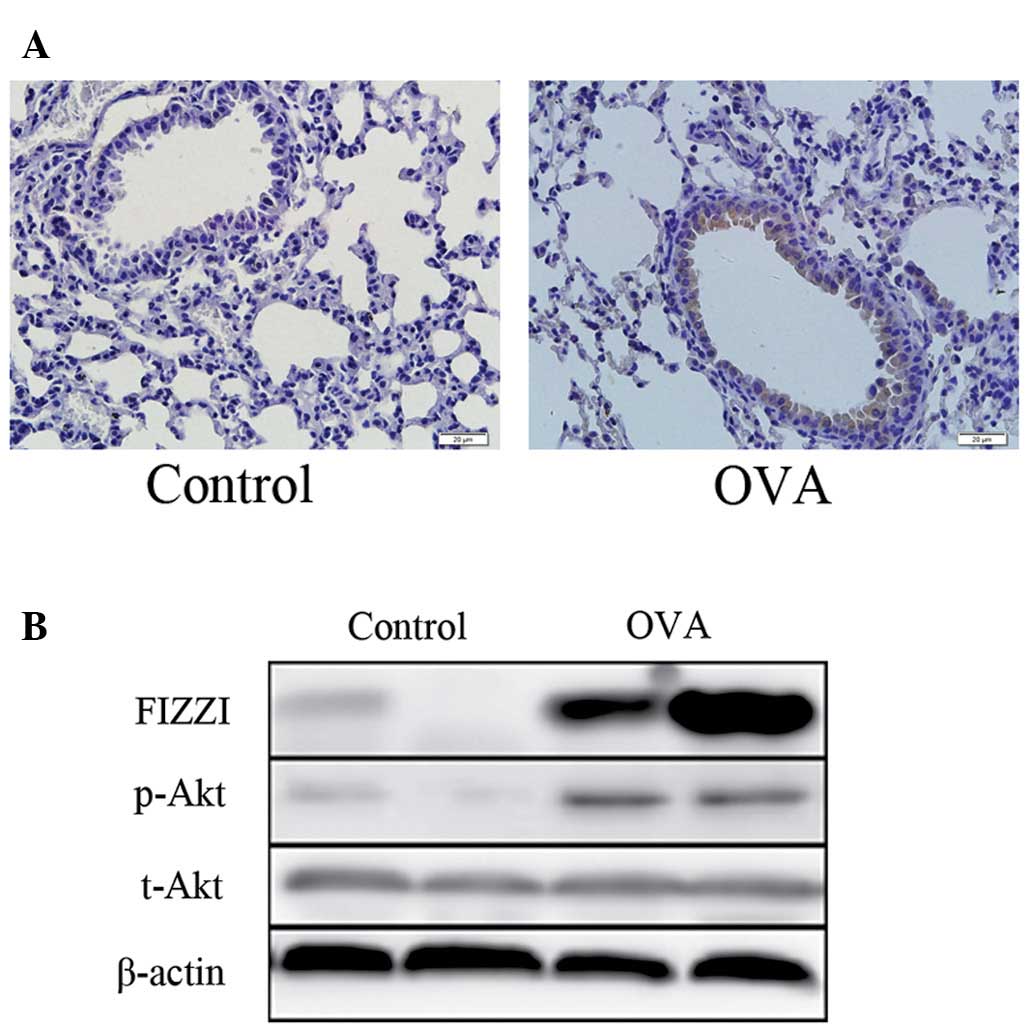
FIZZ1 and p-Akt expression in mice
The expression of FIZZ1 in mouse airway epithelium
cells was upregulated in the OVA-induced asthmatic mice compared
with the saline control group (Fig.
2A). In terms of protein levels, an enhancement of FIZZ1
expression and Akt phosphorylation was detected in the OVA group by
western blot analysis (Fig.
2B).
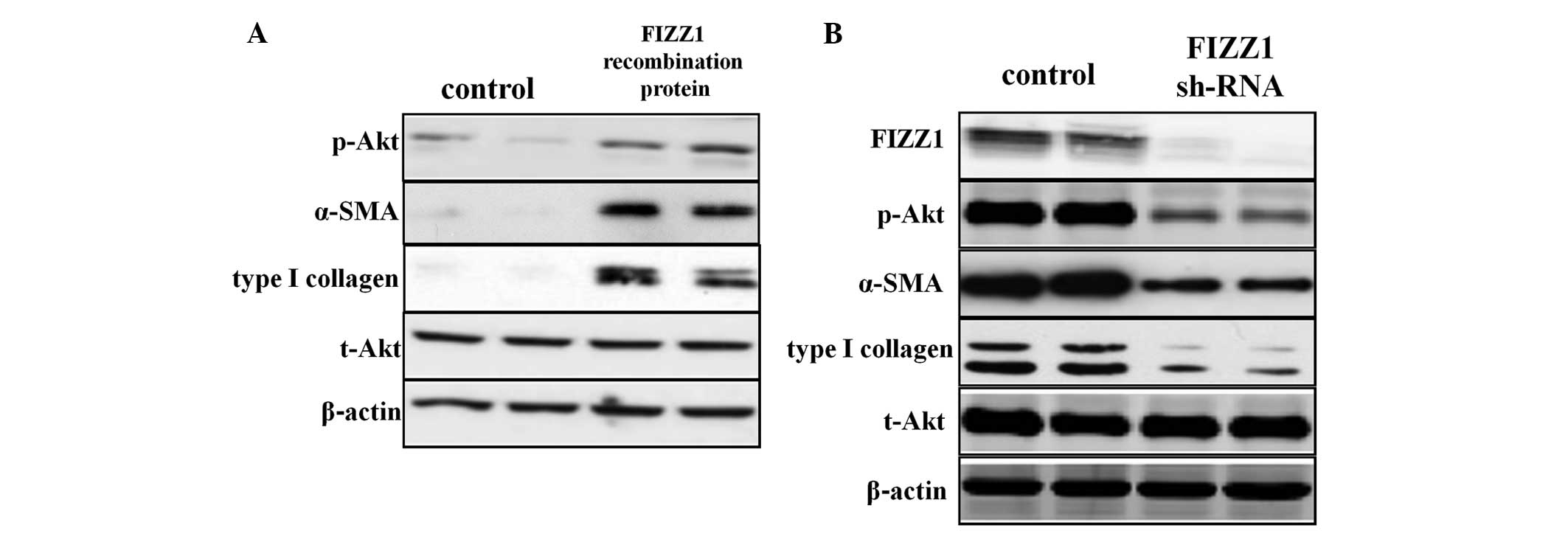
Expression of FIZZ1 and Akt
phosphorylation in MLE-12 cells following FIZZ1 shRNA transfection
by western blot analysis
To verify whether FIZZ1 was able to promote Akt
phosphorylation, the MLE-12 cell line was cultured with FIZZ1
recombinant protein following FIZZ1 shRNA transfection and the
phosphorylation levels of Akt were detected. Following FIZZ1
recombinant protein co-culture, Akt phosphorylation and α-SMA and
type I collagen expression were upregulated (Fig. 3A). The level of FIZZ1 expression in
the MLE-12 cells following FIZZ1 shRNA transfection was
downregulated, as were the levels of Akt phosphorylation and α-SMA
and type I collagen expression (Fig.
3B).
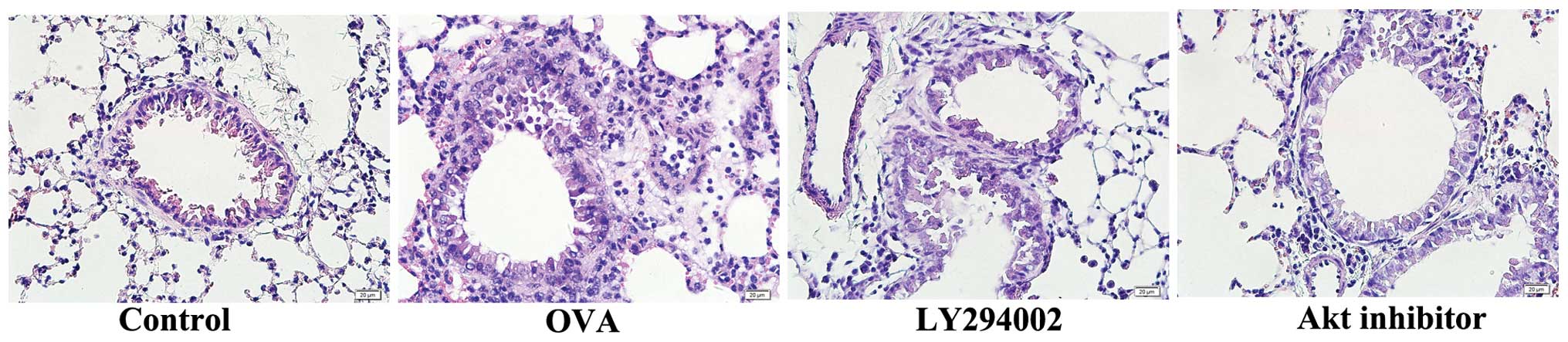
Effects of LY294002 and Akt inhibitor IV
on OVA-induced inflammatory cell infiltration
To determine the role of the PI3K/Akt signaling
pathway in inflammatory cell infiltration, LY294002 and Akt
inhibitor IV were administered intratracheally to the asthmatic
mouse model. Histological analysis, as presented in Fig. 4, demonstrated that LY294002 and Akt
inhibitor IV may attenuate inflammatory cell infiltration as
compared with the OVA-induced asthmatic group.
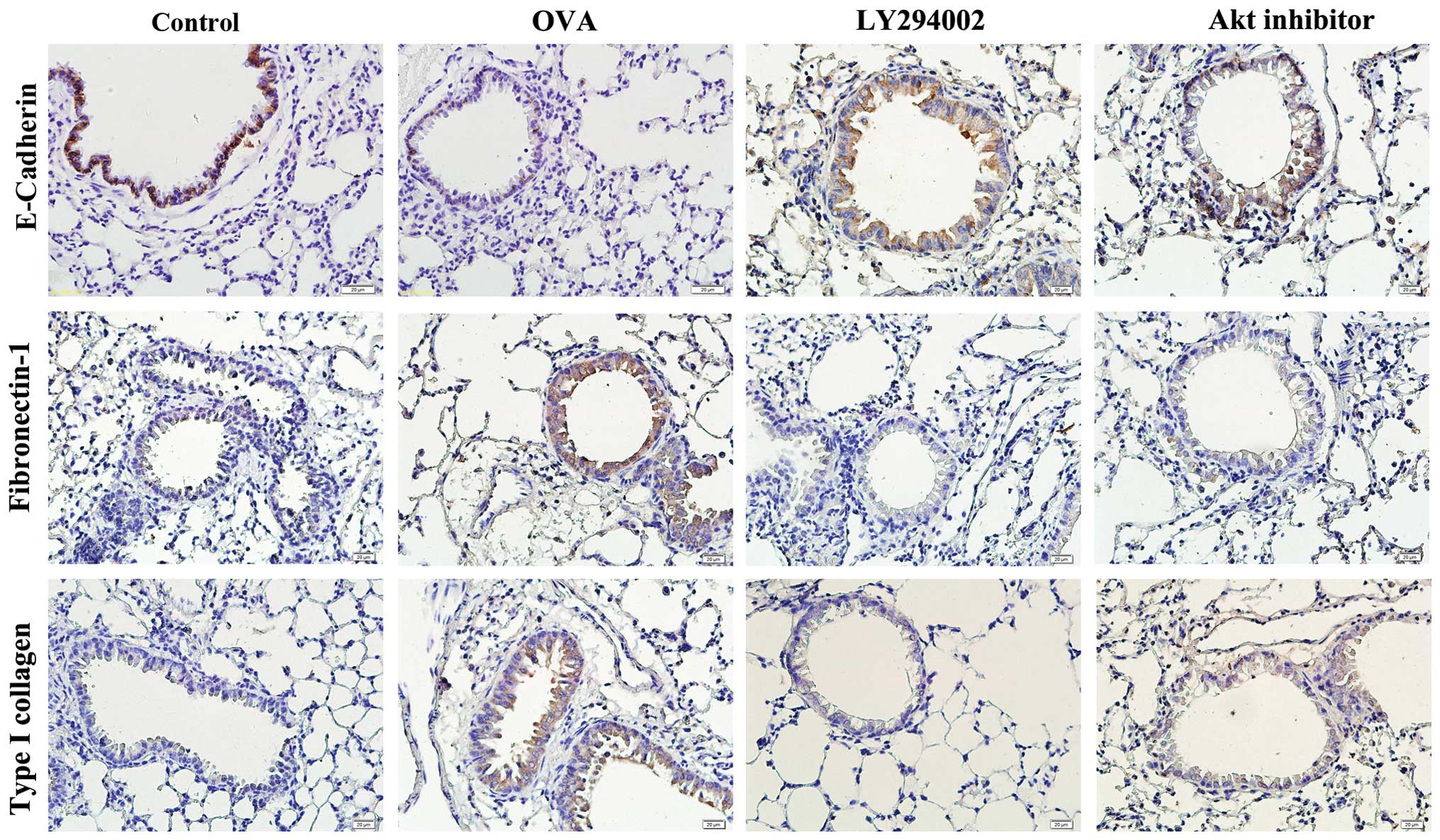
LY294002 and Akt inhibitor IV may
overcome airway remodeling
To verify the effects of the PI3K/Akt pathway on
OVA-induced airway remodeling, the expression levels of E-cadherin,
fibronectin-1 and type I collagen in the airway epithelium cells
were detected by immunohistochemistry following LY294002 and Akt
inhibitor IV intratracheal administration. High expression levels
of fibronectin-1 and type I collagen were identified in the
epithelial cells of the airway in the asthmatic mice; LY294002 and
Akt inhibitor IV reduced this expression compared with the OVA
group. By contrast, the expression level of E-cadherin decreased in
the OVA-induced mice, and LY294002 and Akt inhibitor IV were shown
to upregulate the expression (Fig.
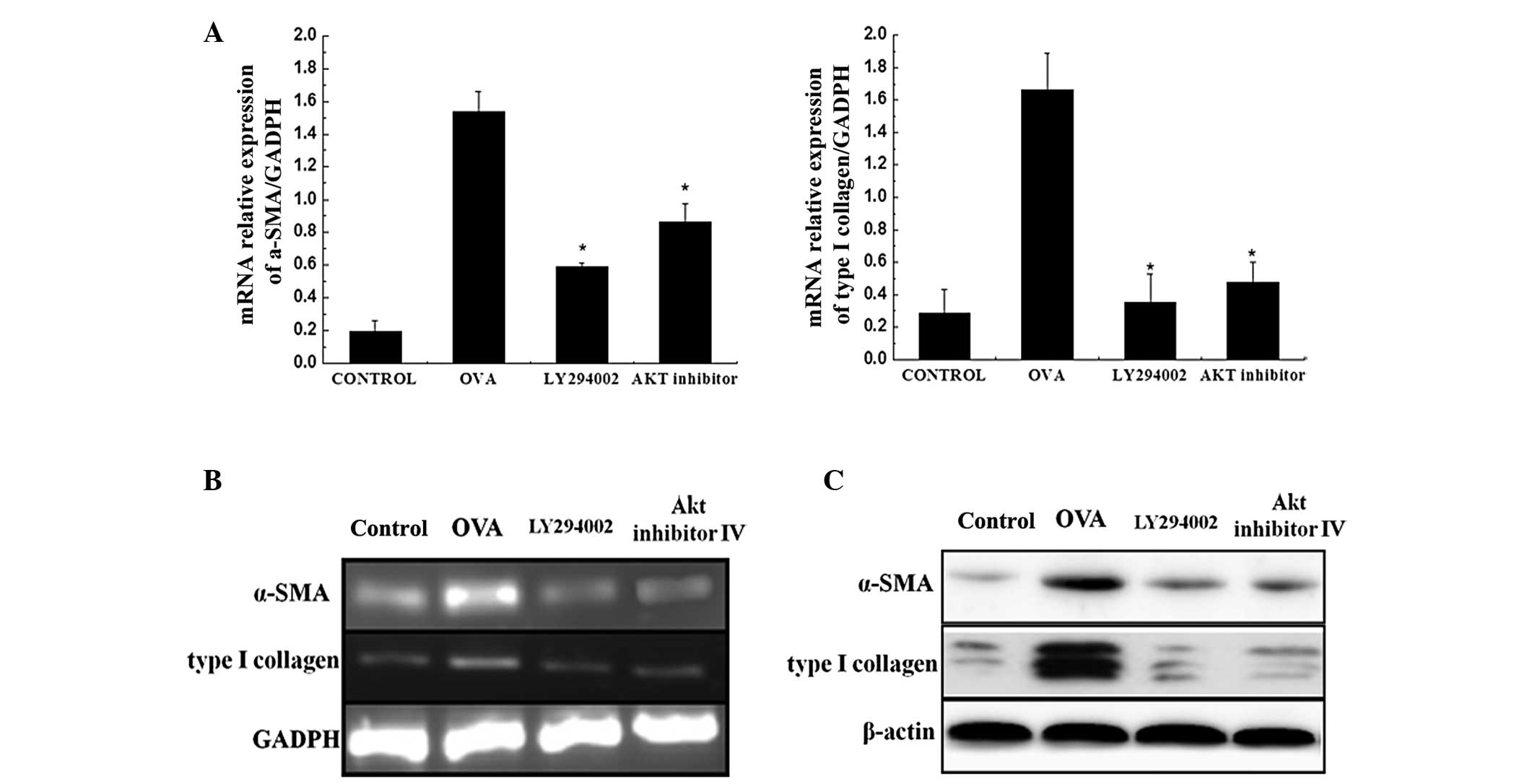
5). In addition, higher expression levels of α-SMA and type I
collagen were identified at the mRNA and protein levels in the
OVA-induced mice. Therefore, blocking the PI3K/Akt pathway may
inhibit the production of α-SMA and type I collagen (Fig. 6).
Discussion
In the present study, FIZZ1 was demonstrated to be
overexpressed in the OVA-induced asthmatic mouse model and was
shown to promote Akt phosphorylation in vitro. By blocking
the PI3K/Akt pathway, inflammatory cell infiltration in the OVA
group was alleviated. In the OVA group, the expression levels of
α-SMA, type I collagen and fibronectin-1 were upregulated in the
epithelial cells, while the expression of E-cadherin was decreased,
indicating that EMT is induced in the airways of asthmatic mice.
LY294002 and Akt inhibitor IV can intervene with this EMT process
by increasing E-cadherin expression and downregulating the
expression of α-SMA, type I collagen and fibronectin-1. Thus, we
hypothesize that FIZZ1 may promote airway remodeling in asthmatic
mice via the PI3K/Akt signaling pathway, and that blocking the
PI3K/Akt pathway may relieve airway remodeling by regulating the
abnormal process of EMT in OVA-induced mouse models.
Bronchial asthma is a chronic inflammatory disease
of the airways. Airway remodeling, first reported by Huber in 1922
(11), is the main pathological
feature of asthma and is the result of sustained inflammation and
epithelial cell damage in response to airway injuries (12). The main characteristics of airway
remodeling include epithelial detachment, subepithelial fibrosis,
airway smooth muscle cell and goblet cell hypertrophy and
hyperplasia. All the pathological changes are the major cause of
the clinical symptoms and loss of lung function (3,13).
Therefore, further studies of airway remodeling may provide a novel
treatment strategy for asthma.
Previous studies have demonstrated that EMT promotes
airway remodeling (7,14). In the present study, the expression
levels of α-SMA, type I collagen and fibronectin-1 were observed to
be significantly increased and E-cadherin expression was reduced
compared with the saline control, indicating the occurrence of the
EMT process.
Preliminary study has revealed that the expression
of FIZZ1 was enhanced in the epithelium of the asthmatic rats,
which stimulated the expression of α-SMA and type I collagen in
fibroblasts (10). However, the
mechanism by which FIZZ1 functions in this process remains unknown.
In the present study, FIZZ1 was demonstrated to be capable of
regulating this process via the PI3K/Akt signaling pathway.
PI3K has a wide range of biological effects in
numerous types of immune cells. A previous study revealed that PI3K
enzyme activity was increased in OVA-induced murine models of
asthma, along with p-Akt, one of the downstream signaling molecules
of PI3K (15). LY294002, a
specific PI3K inhibitor, is able to significantly downregulate Akt
phosphorylation and suppress inflammatory cell infiltration, mucus
production and airway hyperresponsiveness in a murine asthmatic
model (16). Inflammation and
airway remodeling is reduced in PI3Kγ-deficient mice (17). The PI3K/Akt signaling pathway is
important in asthma and can promote airway inflammation and
hyperresponsiveness, upregulate T-helper 2 cytokine levels and
increase mucus production (18).
In the present study, following intratracheal administration of
LY294002 and Akt inhibitor IV, the infiltration of inflammatory
cells was relieved and the markers of EMT, α-SMA, type I collagen
and fibronectin-1, expression levels were downregulated with
E-cadherin expression increased when compared with the asthmatic
mouse model. This result indicates that the PI3K/Akt pathway is
involved in the EMT process. Thus, we hypothesize that by blocking
the signaling pathway, the process of EMT may be interrupted, which
in turn relieves airway remodeling in the mouse asthmatic
model.
The PI3K/Akt signaling pathway may be blocked by
phosphatase and tensin homolog deleted on chromosome 10 (PTEN)
through dephosphorylating the signaling lipid, phosphatidylinositol
3,4,5-triphosphate (19). PTEN
protein expression and activity are decreased in allergen-induced
asthma (20). Intratracheal
administration of adenoviruses carrying PTEN cDNA can reduce the
levels of interleukin-4 and −5 in bronchoalveolar lavage fluid,
bronchial inflammation and airway hyperresponsiveness (21). A previous study demonstrated that
PTEN inhibited the proliferation and migration of human airway
smooth muscle mass cells by downregulating the activity of the Akt
signaling pathway (22). Numerous
studies indicate that PTEN plays a significant role in asthma.
However, whether FIZZ1 is an upstream regulator of PTEN, that
affects the process of EMT by regulating the PI3K/Akt pathway,
remains unknown. Thus, this is the direction of research for future
studies.
In summary, the results of the present study
demonstrate that FIZZ1 promotes airway remodeling via the PI3K/Akt
signaling pathway. In addition, blocking the PI3K/Akt signaling
pathway may alleviate airway remodeling in the early stages by
intervening with the EMT process. Antagonism of the PI3K/Akt
signaling pathway may be a potentially useful strategy in the
therapeutic intervention for asthma.
Acknowledgements
Our study was supported by National Natural Science
Foundation of China (no. 81070016 and no. 81270072) and Natural
Science Funding committee of Shandong province (no.
ZR2011HM020).
References
|
1
|
Nakagome K and Nagata M: Pathogenesis of
airway inflammation in bronchial asthma. Auris Nasus Larynx.
38:555–563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Holtage ST: Pathogenesis of asthma. Clin
Exp Allergy. 38:872–897. 2008. View Article : Google Scholar
|
|
3
|
Sumi Y and Hamid Q: Airway remodeling in
asthma. Allergol Int. 56:341–348. 2007. View Article : Google Scholar
|
|
4
|
Holgate ST, Davies DE, Lackie PM, Wilson
SJ, Puddicombe SM and Lordan JL: Epithelial-mesenchymal
interactions in the pathogenesis of asthma. J Allergy Clin Immunol.
105:193–204. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hackett TL: Epithelial-mesemchymal
transition in the pathophysiology of airway remodelling in asthma.
Curr Opin Allergy Clin Immunol. 12:53–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hackett TL, Warner SM, Stefanowicz D, et
al: Induction of epithelial-mesenchymal transition in primary
airway epithelial cells from patients with asthma by transforming
growth factor-beta1. Am J Respir Crit Care Med. 180:122–133. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Folli C, Descalzi D, Scordamaglia F,
Riccio AM, Galamero C and Canonica GW: New insights into airway
remodelling in asthma and its possible modulation. Curr Opin
Allergy Clin Immunol. 8:367–375. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Holcomb IN, Kabakoff RC, Chan B, et al:
FIZZ1, a novel cysteine-rich secreted protein associated with
pulmonary inflammation, defines a new gene family. EMBO J.
19:4046–4055. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamaji-Kegan K, Su Q, Angelini DJ,
Champion HC and Johns RA: Hypoxia-induced mitogenic factor has
proangiogenic and proinflammatory effects in the lung via VEGF and
VEGF receptor-2. Am J Physiol Lung Cell Mol Physiol.
291:L1159–L1168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dong L, Wang SJ, Camoretti-Mercado B, Li
HJ, Chen M and Bi WX: FIZZ1 plays a crucial role in early stage
airway remodeling of OVA-induced asthma. J Asthma. 45:648–653.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huber P, Menif K and Jouvet P: Acute
severe asthma in pediatric resuscitation. The French Study Group on
Pediatric Resuscitation. Arch Pediatr. 1:346–349. 1994.(In
French).
|
|
12
|
Cho JY: Recent advances in mechanisms and
treatments of airway remodeling in asthma: a message from the bench
side to the clinic. Korean J Intern Med. 26:367–383. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Halwani R, Al-Muhsen S and Hamid Q: Airway
remodeling in asthma. Curr Opin Pharmacol. 10:236–245. 2010.
View Article : Google Scholar
|
|
14
|
Yadav UC, Naura AS, Aguilera-Aguirre L, et
al: Aldose reductase inhibition prevents allergic airway remodeling
through PI3K/AKT/GSK3β pathway in mice. PLoS One.
8:e574422013.PubMed/NCBI
|
|
15
|
Lee KS, Lee HK, Hayflick JS, Lee YC and
Puri KD: Inhibition of phosphoinositide 3-kinase delta attenuates
allergic airway inflammation and hyperresponsiveness in murine
asthma model. FASEB J. 20:455–465. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Duan W, Aguinaldo Datiles AM, Leung BP,
Vlahos CJ and Wong WS: An anti-inflammatory role for a
phophoinositide 3-kinase inhibitor LY294002 in a mouse asthma
model. Int Immunopharmacol. 5:495–502. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lim DH, Cho JY, Song DJ, Lee SY, Miller M
and Broide DH: PI3K gamma-deficient mice have reduced levels of
allergen-induced eosinophilic inflammation and airway remodeling.
Am J Physiol Lung Cell Mol Physiol. 296:L210–L219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Medina-Tato DA, Ward SG and Watson ML:
Phosphoinositide 3-kinase signalling in lung disease: leucocytes
and beyond. Immunology. 121:448–461. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee KS, Kim SR, Park SJ, et al:
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN)
reduces vascular endothelial growth factor expression in
allergen-induced airway inflammation. Mol Pharmacol. 69:1829–1839.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee YC: The role of PTEN in allergic
inflammation. Arch Immunol Ther Exp (Warsz). 52:250–254.
2004.PubMed/NCBI
|
|
21
|
Kwark YG, Song CH, Yi HK, Hwang PH, Kim
JS, Lee KS and Lee YC: Involvement of PTEN in airway
hyperresponsiveness and inflammation in bronchial asthma. J Clin
Invest. 111:1083–1092. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lan H, Zhong H, Gao Y, et al: The PTEN
tumor suppressor inhibits human airway smooth muscle cell
migration. Int J Mol Med. 26:893–899. 2010.PubMed/NCBI
|