Introduction
Hepatic carcinoma is the sixth most common cancer
worldwide and the third most common cause of mortality from cancer
with 626,000 cases and 598,000 mortalities annually (1). In China, there are 360,000 cases of
hepatic carcinoma and 350,000 associated mortalities a year
(2), and hepatic carcinoma is the
second most common cause of cancer-associated mortalities (1,3).
Hepatitis B virus and aflatoxins are considered the major and
common factors attributed to the etiology of liver cancer, and they
can act individually or synergistically on the liver to cause
cancer (4,5). Other factors, including hepatitis C
virus, genetic susceptibility or genetic polymorphisms, may also
have an important role in the etiology of liver cancer (6).
Previous studies have investigated the mechanism of
hepato-carcinogenesis (7,8). The majority of these studies have
focused on the genetic changes in key tumor suppressor genes and
oncogenes; however, it has been suggested that epigenetic
disruption of gene expression may also have an important role in
the development of cancer (9).
Epigenetic events have been found to be involved in the etiology of
a wide variety of types of human cancer, including hepatic
carcinoma. The current definition of epigenetics is the study of
heritable changes in gene expression that occur independently from
changes in the primary DNA sequence (10). The heritability of gene expression
patterns is primarily mediated by epigenetic modifications, which
include DNA methylation, chromatin remodeling, histone replacement
and alterations to histone tails (8,11,12).
DNA methylation is the most extensively studied epigenetic
modification in mammals, and it provides a stable gene silencing
mechanism that has an important role in the regulation of gene
expression and chromatin architecture (10). Several studies have reported that
there are somatically acquired DNA methylation changes in various
tumor-suppressor genes and other cancer-associated genes (13,14).
Histone deacetylation is a type of histone modification that may
regulate key cellular processes, including transcription, DNA
replication and DNA repair (15).
DNA methylation and histone deacetylation may work independently or
in concert to alter gene expression during tumorigenesis.
Therefore, in the present study, the effect of inhibiting DNA
methylation and histone deacetylation in HepG2 cells was
investigated to determine the potential role of epigenetic
modifications in the development and treatment of hepatic
carcinoma, and to explore a novel therapeutic strategy for the
treatment of hepatic carcinoma.
Materials and methods
Research materials and gene chip
In order to explore the effect of DNA methylation
and histone deacetylation on hepatoma cells, the HepG2 cell line
was used. The cells had been treated with 5-aza-2′-deoxycytidine
(5-aza-dC; aza), trichostatin A (TSA), and a combination of aza and
TSA to inhibit DNA methylation, histone deacetylation and both
methylation and deacetylation, respectively. The gene expression
profiles of the treated cells were compared with those of the
control group to investigate the effects of methylation and
deacetylation on liver cancer cells. GSE5230 sample data from the
Gene Expression Omnibus (GEO) database was used (16), which included 4 gene chips of the
treatment by aza, TSA, combination of aza and TSA and the control
group, respectively.
Acquisition of the differentially
expressed genes
The samples were identified and the microarray data
were analyzed using the R software (v.2.13.0) (17) platform, as well as GEOquery
(18) and the limma package to
further process the data. GEOquery obtains chip expression
profiling data from the GEO database quickly, whilst limma can be
used to statistically analyze the differentially expressed genes
(19,20). The GEOquery package was used to
obtain data of chip expression profiling that had already been
preprocessed, and the chip data as transformed with log2. The
expression profiles of the HepG2 cells treated with aza, TSA, aza
and TSA and the control group were then compared, and the
differentially expressed genes inhibited by methylation and
acetylation were analyzed using the linear regression model package
limma.
Gene Otology (GO) analysis of the
differentially expressed genes
In order to investigate the changes in the
differentially expressed genes at the cellular level and their
functional clustering, classification of gene function and position
was performed using GO (21),
using the GOEAST platform (22).
In the present study, a hyper-geometric algorithm was selected for
the statistical analysis. The entire microarray probe was used as a
background control and the differentially expressed genes from
biological processes were clustered; thus, the effect of these
differentially expressed genes on the cells was determined.
Biological pathway data
In order to investigate the changes induced in the
cells as a result of the inhibition of DNA methylation and histone
deacetylation at the molecular level, the effects of these
modifications on biological pathways were examined. All metabolic
and non-metabolic pathways were acquired from the current public
open access database WikiPathways(http://www.wikipathways.org) (23,24),
and the WikiPathways clustering analysis of differentially
expressed genes was achieved through the Gene Set Analysis Toolkit
V2 platform (25,26), in order to determine the changes in
the signal pathways of HepG2 cells.
Identification of potential target sites
for regulatory transcription factors
Based on the gene annotation data arranged by the
MSigDB (http://www.broadinstitute.org/gsea/msigdb/index.jsp)
database, analyzed by gene abundance, with statistical calculations
conducted with a hypergeometric algorithm and calibrated by
Benjamini and Hochberg (BH) procedure, the potential target sites
for regulation by transcription factors were obtained.
Results
Identification of differentially
expressed genes following the inhibition of methylation and
deacetylation
The data were analyzed using a t-test (20) modified by Bayesian model in order
to obtain the differentially expressed genes. P-values were
obtained for all the genes, and they were corrected as the false
discovery rate (FDR). P<0.05 was considered to indicate a
statistically significant difference. The numbers of genes with
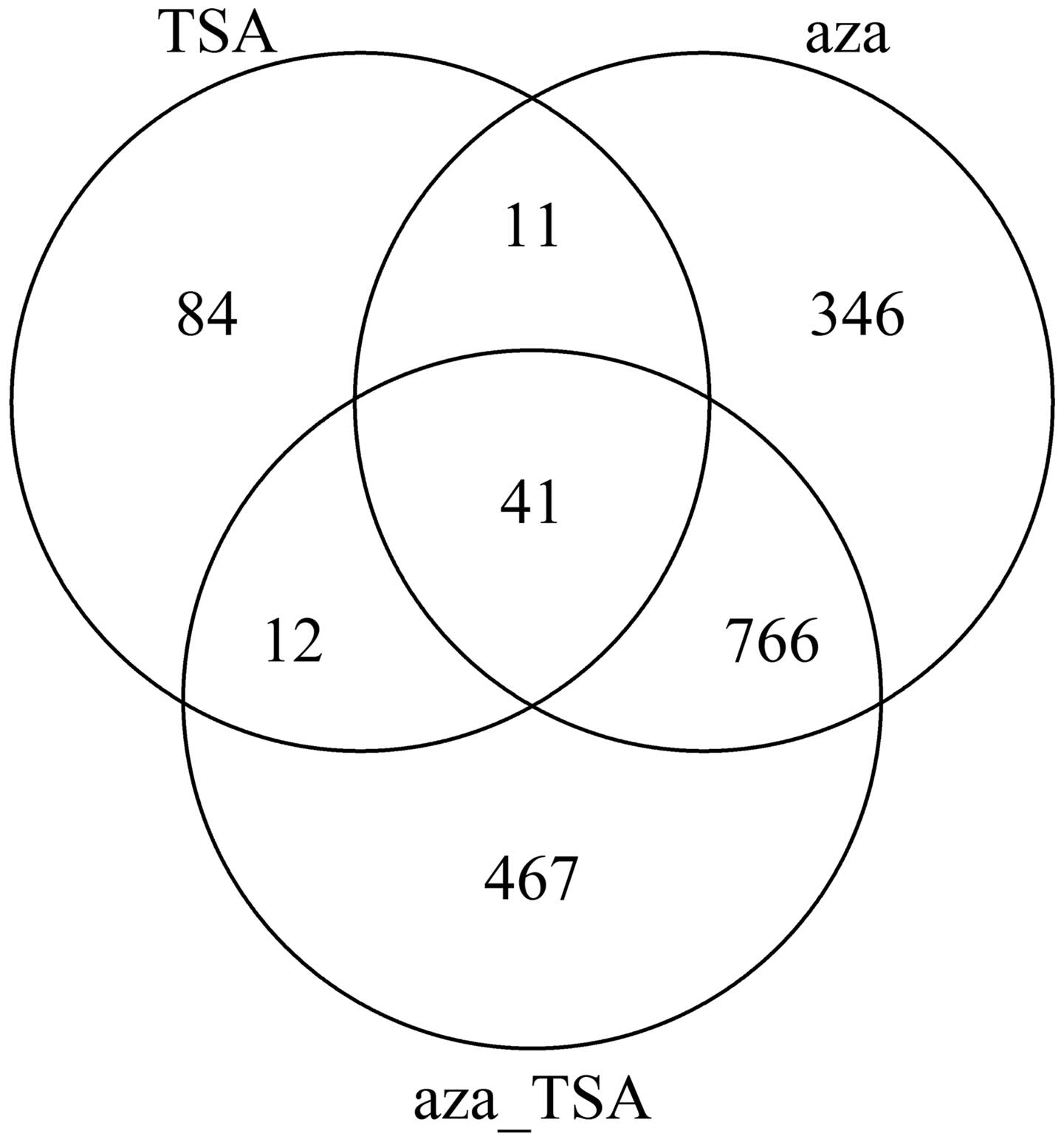
changes in expression levels are shown in Fig. 1.
As seen in Fig. 1,
treatment with aza or TSA induced numerous changes in gene
expression in HepG2 cells. The number of the altered genes
following aza treatment was larger compared with that following TSA
treatment. The results indicate that inhibition of DNA methylation
and histone acetylation affects gene expression of HepG2 cells;
however, methylation has a more significant contribution to the
gene expression and regulation of liver cancer cells.
Biological pathway enrichment regulated
by DNA methylation and histone acetylation
Since the inhibition of DNA methylation and histone
deacetylation caused changes in the expression in certain genes,
changes in the biological pathways of hepatoma cells following the
inhibition of DNA methylation and histone deacetylation was further
investigated. The differentially expressed genes were selected and
WikiPathways sub-pathway enrichment analysis was performed. The
genes were clustered by hypergeometric algorithm and then multiplex
detection was proofread using the BH algorithm in order to identify
changes in the signaling pathways of hepatoma cells. Biological
pathways significantly changed under the limiting conditions
(corrected to P<0.1) with at least two genes in the signaling
pathway are shown in Table I.
 | Table IChanges in biological pathways
following inhibition of DNA methylation and histone acetylation by
aza and TSA. |
Table I
Changes in biological pathways
following inhibition of DNA methylation and histone acetylation by
aza and TSA.
| Treatment | Pathway | P-value |
|---|
| Aza | Endochondral
ossification | 0.0816 |
| Integrin-mediated
cell adhesion | 0.0816 |
| Fatty acid
β-oxidation | 0.0816 |
| AMPK signaling | 0.0816 |
| Fluoropyrimidine
activity | 0.0816 |
| Irinotecan
pathway | 0.0816 |
| α6β4 signaling
pathway | 0.0816 |
| Prostaglandin
synthesis and regulation | 0.0816 |
| Prolactin signaling
pathway | 0.0816 |
| Complement and
coagulation cascades | 0.0816 |
| Striated muscle
contraction | 0.0958 |
| TSA | TGF-β signaling
pathway | 0.0015 |
| Striated muscle
contraction | 0.0504 |
| Irinotecan
pathway | 0.0504 |
| Mitochondrial
LC-fatty acid β-oxidation | 0.0588 |
| TSA + aza | Fatty acid
biosynthesis | 0.0655 |
| AMPK signaling | 0.0655 |
| α6β4 signaling
pathway | 0.0877 |
| Fluoropyrimidine
activity | 0.0877 |
| Fatty acid
β-oxidation | 0.0877 |
As the expression of only a few genes changed
following treatment with TSA, clustering of only one signaling
pathway, the transforming growth factor (TGF)-β signaling pathway,
was observed. The TGF-β signaling pathway has very important roles
in the body, including during embryonic development, cell growth,
differentiation and apoptosis, as well as in intracellular
metabolic balance. Therefore, histone deacetylation appears to have
a critical effect on liver cancer cells.
DNA methylation was inhibited following treatment
with aza, resulting in a series of genes being expressed
differentially. Multiple biological pathways are associated with
these differentially expressed genes, including signal
transduction-related integrin-mediated cell adhesion, the adenosine
monophosphate-activated protein kinase (AMPK) signaling pathway,
the α6β4 signaling pathway, prostaglandin synthesis and regulation,
the prolactin signaling pathway, metabolism-associated fatty acid
β-oxidation, fluoropyrimidine activity, the drug-related irinotecan
pathway and the cell motility-associated complement and coagulation
cascades pathway.
The signaling pathways altered following treatment
with Aza and TSA were broadly similar to those altered following
treatment with aza alone, which include striated muscle
contraction, the irinotecan pathway, AMPK signaling, the α6β4
signaling pathway, fluoropyrimidine activity and fatty acid
β-oxidation. Furthermore, Aza and TSA co-treatment had a
significant influence on fatty acid metabolism in HepG2 cells;
however, mitochondrial long chain-fatty acid β-oxidation and fatty
acid biosynthesis were indicated to be unaffected, with the
exception of fatty acid β-oxidation.
GO clustering of the differentially
expressed genes
In the organism, various means are required for
regulation of the more important physiological processes.
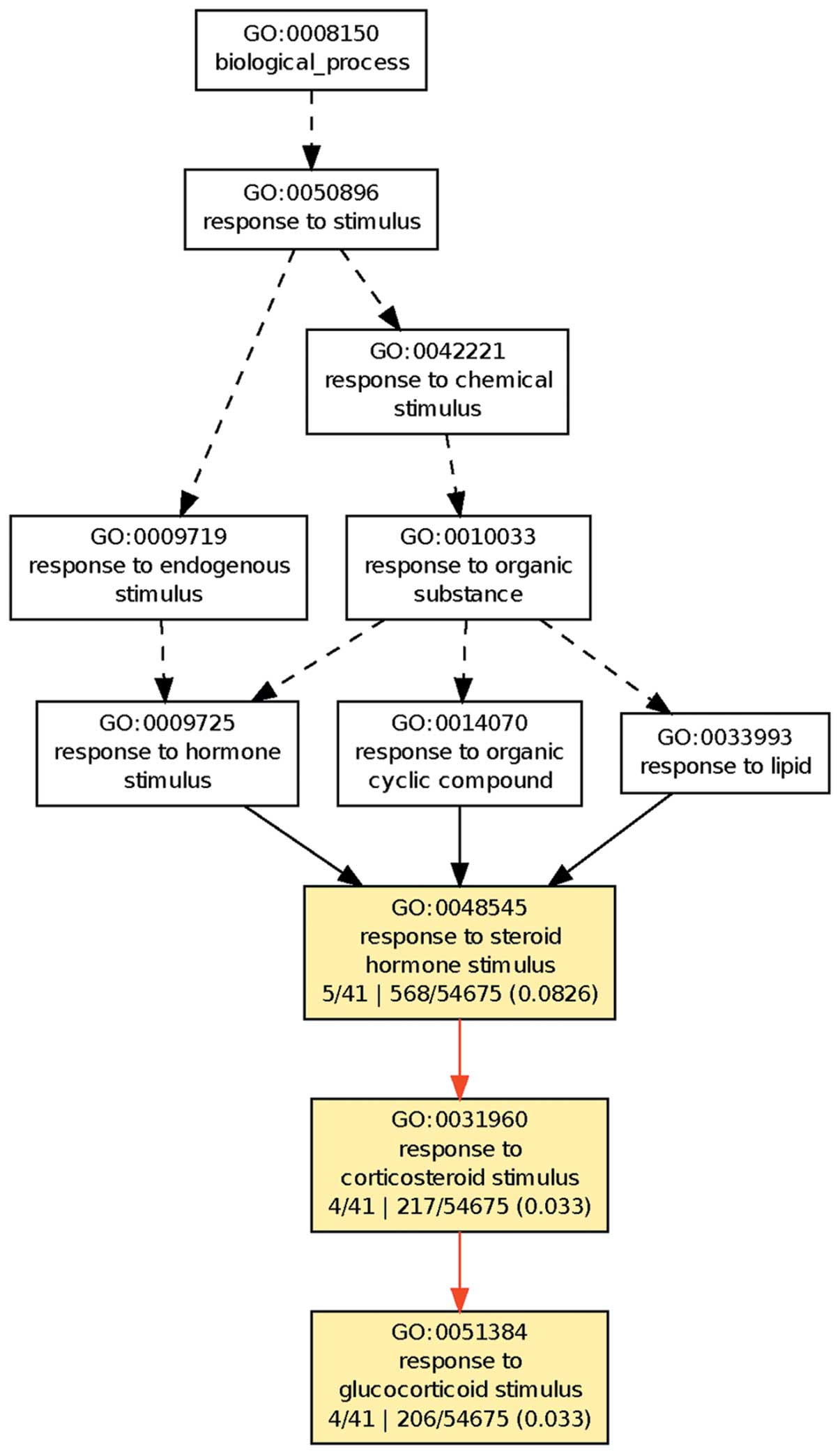
Therefore, the present study focused on the 41 genes expressed
differentially for all three treatments. GO clustering was
performed on their physiological processes using the GOEAST
platform, as shown in Fig. 2.
The results demonstrated that these 41 genes
clustered on the cell response to steroid hormones, in particular
glucocorticoids. This suggests that DNA methylation and histone
deacetylation have important roles in the regulation of the
response to glucocorticoid in hepatoma cells.
Analysis of target sites of the potential
transcription factors
The spatial structure of the chromosome is altered
as a result of the epigenetic modifications of DNA and histones,
and this then alters the binding ability of trans-regulatory
elements. As an important class of trans-regulatory elements,
transcription factors may be a cause of the changes of gene
expression following the inhibition of DNA methylation or histone
deacetylation. Therefore, in the present study common binding sites
for transcription factors shared by the differentially expressed
genes were identified. Upstream sequences of the differentially
expressed genes were used to investigate the potential target sites
for the transcription factors, and the hypergeometric clustering
algorithm was used, with proofreading of the P-value with the BH
algorithm, and 10 target sites for the transcription factors were
then identified (Table II).
| Table IIPotential target sites for
transcription factors. |
Table II
Potential target sites for
transcription factors.
| Drug | Target | P-value |
|---|
| Aza |
hsa_RGAGGAARY_V$PU1_Q6 | 0.3087 |
|
hsa_TATAAA_V$TATA_01 | 0.3087 |
| hsa_V$AP1_Q4 | 0.3544 |
|
hsa_KTGGYRSGAA_UNKNOWN | 0.4030 |
|
hsa_V$E2F1DP1RB_01 | 0.4334 |
|
hsa_CCCNNGGGAR_V$OLF1_01 | 0.4334 |
| hsa_V$E12_Q6 | 0.4334 |
| hsa_V$ER_Q6_02 | 0.4334 |
| hsa_V$AP1_C | 0.4334 |
|
hsa_V$CREBP1_01 | 0.4334 |
| TSA | hsa_V$SMAD_Q6 | 0.0371 |
| hsa_V$IK1_01 | 0.0371 |
|
hsa_V$MYCMAX_02 | 0.0371 |
| hsa_V$ZIC1_01 | 0.0371 |
| hsa_V$PBX1_01 | 0.0621 |
|
hsa_V$FREAC3_01 | 0.0621 |
| hsa_V$USF_01 | 0.0621 |
|
hsa_TGGAAA_V$NFAT_Q4_01 | 0.0621 |
| hsa_V$ARNT_01 | 0.0621 |
| hsa_V$GATA1_02 | 0.0621 |
| Aza + TSA |
hsa_TTTNNANAGCYR_UNKNOWN | 0.0301 |
|
hsa_CTGCAGY_UNKNOWN | 0.7488 |
|
hsa_KRCTCNNNNMANAGC_UNKNOWN | 0.8112 |
| hsa_V$SRF_Q6 | 0.8487 |
| hsa_V$SRF_Q4 | 0.8487 |
| hsa_V$OCT1_03 | 0.8487 |
| hsa_V$ATF_01 | 0.8487 |
| hsa_V$HOXA4_Q2 | 0.8487 |
|
hsa_V$TAL1BETAITF2_01 | 0.8487 |
| hsa_V$E2F_02 | 0.8487 |
Discussion
The results from the WikiPathways clustering
analysis demonstrated that inhibition of histone acetylation in
HepG2 cells by exposure to TSA significantly affected the TGF-β
signaling pathway, which indicates that histone deacetylation has
an important role in the TGF-β signaling pathway in HepG2 cells.
Furthermore, among the significantly upregulated genes in the TGF-β
signaling pathway, lymphoid-enhancing factor 1 (LEF1) was of
particular interest, as it is known to have a functional role in
the Wnt/β-catenin pathway, another important pathway for tumor
growth and invasion (27).
Inhibition of histone acetylation in HepG2 cells may downregulate
LEF1 expression, inhibiting the growth of HepG2 cells. In addition,
bone morphogenetic protein 4 (BMP4) was found to be upregulated
following inhibition of histone deacetylation. BMP4 is a member of
the TGF-β family and is found in the liver. Previous studies have
shown that BMP4 is constitutively expressed in the peribiliary
stroma and endothelial cells in the liver and that its expression
is downregulated following hepatectomy (28,29);
in addition, BMP4 serves as an antiproliferative factor in
hepatocyte proliferation (28).
Altering the TGF-β signaling pathway as a possible
therapeutic treatment for cancer has been previously investigated
in numerous studies (30,31). Therefore, the inhibition of histone
acetylation in HepG2 cells to alter the TGF-β signaling pathway may
provide targeted therapy for hepatic carcinoma; however, further
investigation is required to determine the detailed mechanisms of
TGF-β production and activation.
Treatment with aza and the combination of aza and
TSA inhibited DNA methylation in HepG2 cells, which resulted in
alterations in intracellular biological pathways, including
integrin-mediated cell adhesion. Several of the signaling
transduction pathways have important roles in growth, metabolism
and regulation of differentiation in HepG2 cells. For example,
TNFSF13, a member of the tumor necrosis factor family, was found to
be upregulated following treatment with aza. TNFSF13 has a
pathogenic role in the microenvironments of solid and hematological
tumors (32). Elevated serum
levels of TNFSF13 have been reported in oral cavity cancers
(33), and are correlated with
increased serum TNF levels, angiogenesis and poor prognosis in
multiple myeloma (34). In
addition, the inhibition of DNA methylation following treatment
with aza and TSA resulted in changes in the AMPK signaling pathway.
AMPK is a master regulator of energy homeostasis and is involved in
the regulation of a number of physiological processes, including
the β-oxidation of fatty acids, lipogenesis and protein and
cholesterol synthesis. Previous studies have demonstrated that
changes in these processes occur during cancer due to alterations
in AMPK activity within cancer cells or in their periphery
(35). The results of the present
study demonstrated that tumor suppressor proteins tuberous
sclerosis complex (TSC) 1 and 2, which are substrates of AMPK, were
differentially expressed and tumor suppressor p53 was upregulated.
In addition, DNA methylation has been investigated as a potential
biomarker and therapeutic target in malignant tumors (36). Furthermore, inhibition of DNA
methylation has a similar therapeutic effect as irinotecan, which
is an effective drug for the treatment of certain types of cancer,
including intestinal cancer and small cell carcinoma (37). Therefore, inhibition of DNA
methylation and histone acetylation may provide a novel therapeutic
treatment for hepatic carcinoma.
In addition, the clustering of the GO physiological
processes of the 41 differentially expressed genes for the three
treatment groups in the present study showed that the response of
the HepG2 cells to glucocorticoids changed. Glucocorticoids are a
class of steroid hormones secreted by zona fasciculata in the
adrenal cortex, and have a role in the regulation of glucose and
fat metabolism, and protein biosynthesis and metabolism (38,39).
In addition, they may inhibit the immune response and have
anti-inflammatory effects (38,40).
Therefore, inhibition of DNA methylation and histone acetylation
not only affects the metabolism of HepG2 cells, but also the immune
activity.
Furthermore, a large number of the genes that were
found to be differentially expressed following inhibition of DNA
methylation and histone deacetylation may have the same target
sites for transcription factors, and these sites may have an
important role in the regulation of gene expression. For example,
the differentially expressed gene BMP4 mentioned previously may be
regulated by SMAD1. BMP4 signal transduction is dependent on SMAD
phosphorylation via alk3 and SMAD signaling is associated with
decreased hepatocyte proliferation following hepatectomy (41).
In conclusion, the present study identified a range
of differentially expressed genes associated with DNA methylation
and histone deacetylation blockage in HepG2 cells. Further studies
of these genes and their regulation may aid in elucidating the
underlying mechanism of the development of hepatocellular
carcinoma.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Chen JG, Zhang SW and Chen WQ: Analysis of
liver cancer mortality in the national retrospective sampling
survey of death causes in China, 2004–2005. Zhonghua Yu Fang Yi Xue
Za Zhi. 44:383–389. 2010.(In Chinese).
|
|
3
|
Chen JG and Zhang SW: Liver cancer
epidemic in China: past, present and future. Semin Cancer Biol.
21:59–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luo RH, Zhao ZX, Zhou XY, Gao ZL and Yao
JL: Risk factors for primary liver carcinoma in Chinese population.
World J Gastroenterol. 11:4431–4434. 2005.PubMed/NCBI
|
|
5
|
Groopman JD, Johnson D and Kensler TW:
Aflatoxin and hepatitis B virus biomarkers: a paradigm for complex
environmental exposures and cancer risk. Cancer Biomark. 1:5–14.
2005.PubMed/NCBI
|
|
6
|
Chen TH, Chen CJ, Yen MF, et al:
Ultrasound screening and risk factors for death from hepatocellular
carcinoma in a high risk group in Taiwan. Int J Cancer. 98:257–261.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aguirre-Ghiso JA: Models, mechanisms and
clinical evidence for cancer dormancy. Nat Rev Cancer. 7:834–846.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ducasse M and Brown MA: Epigenetic
aberrations and cancer. Mol Cancer. 5:602006. View Article : Google Scholar
|
|
9
|
Dolinoy DC, Weidman JR and Jirtle RL:
Epigenetic gene regulation: linking early developmental environment
to adult disease. Reprod Toxicol. 23:297–307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar
|
|
11
|
Jenuwein T: The epigenetic magic of
histone lysine methylation. FEBS J. 273:3121–3135. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barkess G: Chromatin remodeling and genome
stability. Genome Biol. 7:3192006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Esteller M: Cancer epigenomics: DNA
methylomes and histone-modification maps. Nat Rev Genet. 8:286–298.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vucic EA, Brown CJ and Lam WL: Epigenetics
of cancer progression. Pharmacogenomics. 9:215–234. 2008.
View Article : Google Scholar
|
|
15
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dannenberg LO and Edenberg HJ: Epigenetics
of gene expression in human hepatoma cells: expression profiling
the response to inhibition of DNA methylation and histone
deacetylation. BMC Genomics. 7:1812006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
R Development Core Team. R: A language and
environment for statistical computing. R Foundation for Statistical
Computing; Vienna, Austria: 2008
|
|
18
|
Davis S and Meltzer PS: GEOquery: a bridge
between the Gene Expression Omnibus (GEO) and BioConductor.
Bioinformatics. 23:1846–1847. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Diboun I, Wernisch L, Orengo C and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7:2522006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Smyth GK: Linear models and empirical
Bayes methods for assessing differential expression in microarray
experiments. Stat Appl Genet Mol Biol. 3:2004.PubMed/NCBI
|
|
21
|
Ashburner M, Ball CA, Blake JA, et al:
Gene Ontology: tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng Q and Wang XJ: GOEAST: a web-based
software toolkit for Gene Ontology enrichment analysis. Nucleic
Acids Res. 36:W358–W363. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kelder T, van Iersel MP, Hanspers K, et
al: WikiPathways: building research communities on biological
pathways. Nucleic Acids Res. 40:D1301–D1307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pico AR, Kelder T, van Iersel MP, Hanspers
K, Conklin BR and Evelo C: WikiPathways: pathway editing for the
people. PLoS Biol. 6:e1842008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang B, Kirov S and Snoddy J: WebGestalt:
an integrated system for exploring gene sets in various biological
contexts. Nucleic Acids Res. 33:W741–W748. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Duncan D, Prodduturi N and Zhang B:
WebGestalt2: an updated and expanded version of the Web-based Gene
Set Analysis Toolkit. BMC Bioinformatics. 11(Suppl 4): P102010.
View Article : Google Scholar
|
|
27
|
Jeanes A, Gottardi CJ and Yap AS:
Cadherins and cancer: how does cadherin dysfunction promote tumor
progression? Oncogene. 27:6920–6929. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Do N, Zhao R, Ray K, et al: BMP4 is a
novel paracrine inhibitor of liver regeneration. Am J Physiol
Gastrointest Liver Physiol. 303:G1220–G1227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xia Y, Babitt JL, Sidis Y, Chung RT and
Lin HY: Hemojuvelin regulates hepcidin expression via a selective
subset of BMP ligands and receptors independently of neogenin.
Blood. 111:5195–5204. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giannelli G, Mazzocca A, Fransvea E, Lahn
M and Antonaci S: Inhibiting TGF-β signaling in hepatocellular
carcinoma. Biochim Biophys Acta. 1815:214–223. 2011.
|
|
31
|
Ikushima H and Miyazono K: TGFβ
signalling: a complex web in cancer progression. Nat Rev Cancer.
10:415–424. 2010.
|
|
32
|
Mackay F, Sierro F, Grey ST and Gordon TP:
The BAFF/APRIL system: an important player in systemic rheumatic
diseases. Curr Dir Autoimmun. 8:243–265. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jablonska E, Slodczyk B,
Wawrusiewicz-Kurylonek N, et al: Overexpression of B
cell-activating factor (BAFF) in neutrophils of oral cavity cancer
patients-preliminary study. Neoplasma. 58:211–216. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fragioudaki M, Tsirakis G, Pappa CA, et
al: Serum BAFF levels are related to angiogenesis and prognosis in
patients with multiple myeloma. Leuk Res. 36:1004–1008. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brown KA, Samarajeewa NU and Simpson ER:
Endocrine-related cancers and the role of AMPK. Mol Cell
Endocrinol. 366:170–179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Akhurst RJ and Derynck R: TGF-beta
signaling in cancer - a double-edged sword. Trends Cell Biol.
11:S44–S51. 2001.PubMed/NCBI
|
|
37
|
de Jong FA, de Jonge MJ, Verweij J and
Mathijssen RH: Role of pharmacogenetics in irinotecan therapy.
Cancer Lett. 234:90–106. 2006.PubMed/NCBI
|
|
38
|
Taves MD, Gomez-Sanchez CE and Soma KK:
Extra-adrenal glucocorticoids and mineralocorticoids: evidence for
local synthesis, regulation, and function. Am J Physiol Endocrinol
Metab. 301:E11–E24. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rose AJ, Vegiopoulos A and Herzig S: Role
of glucocorticoids and the glucocorticoid receptor in metabolism:
insights from genetic manipulations. J Steroid Biochem Mol Biol.
122:10–20. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Amsterdam A, Tajima K and Sasson R:
Cell-specific regulation of apoptosis by glucocorticoids:
implication to their anti-inflammatory action. Biochem Pharmacol.
64:843–850. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Goldman DC, Bailey AS, Pfaffle DL, Al
Masri A, Christian JL and Fleming WH: BMP4 regulates the
hematopoietic stem cell niche. Blood. 114:4393–4401. 2009.
View Article : Google Scholar : PubMed/NCBI
|