Introduction
A number of studies have demonstrated that
nicotinamide adenine dinucleotide (NAD+) plays an
important role in energy metabolism and mitochondrial function, as
well as gene expression, calcium homeostasis, aging and cell death
(1–3). Multiple studies have also revealed
that NAD+ is a cytoprotective agent for primary cultures
of astrocytes, neurons and myocytes; NAD+ treatment has
been shown to decrease the necrotic cell death in these types of
cells induced by oxidative stress (4), DNA-alkylating agents (5) or oxygen-glucose deprivation (6). A previous study demonstrated that
NAD+ administration significantly reduced ischemic brain
injury in rats (7). One of the key
precursors of NAD+ is nicotinamide mononucleotide (NMN),
which is converted to NAD+ by nicotinamide
mononucleotide adenylyltransferase (2).
Parkinson’s disease (PD) is one of most common types
of movement disorder. A major pathological change of the disease is
the loss of dopaminergic neurons in the substantia nigra pars
compacta (8). Although it has been
suggested that oxidative stress, mitochondrial dysfunction,
inflammation or apoptosis of the dopaminergic neurons in the
substantia nigra may play a key role in the pathogenesis of PD
(9), the precise mechanisms
underlying the pathogenesis of PD are not well understood (10). There has been little effective
therapy for treatment of this debilitating disorder (11). Dysfunction of the mitochondria may
be involved in the pathogenesis of PD and may become a promising
therapeutic target for the disease (12).
In vitro and in vivo studies have
revealed that rotenone, a mitochondrial complex I inhibitor,
induces PD-like behavioral and neuropathological changes, including
the induction of apoptosis and acceleration of α-synuclein
formation, in PD models (13,14).
Since NAD+ treatment is able to attenuate the genotoxic
agent-induced mitochondrial alterations in neurons and astrocytes
(4), it was hypothesized that NMN
treatment may attenuate rotenone-induced cytotoxicity. In the
current study, a cellular model of PD, using rotenone-treated PC12
cells, was established to investigate whether NMN is a protective
agent against rotenone-induced cytotoxicity.
Materials and methods
Cell culture
PC12 cells were purchased from the Cell Resource
Center of the Shanghai Institute of Biological Sciences of the
Chinese Academy of Sciences (Shanghai, China). The cells were
plated at an initial density of 5×104 cells/well onto
24-well culture plates to test cell viability, and at a density of
1×106 cells/well onto 6-well plates in order to prepare
samples for western blot analysis. The culture media used was
Dulbecco’s modified Eagle’s medium containing 4,500 mg/l D-glucose,
584 mg/l L-glutamine and 110 mg/l sodium pyruvate (Thermo
Scientific, Tewksbury, MA, USA), and also containing 1% penicillin
and streptomycin (Invitrogen Life Technologies, Carlsbad, CA, USA)
and 10% fetal bovine serum (PAA Laboratories GmbH, Linz, Austria).
The cells were given 0.5 μM rotenone (Sigma, St. Louis, MO, USA)
with or without co-treatment with different concentrations of NMN
(N3501, Sigma Aldrich, USA). The cells were kept for 24 h in an
incubator with 5% CO2 at 37°C. In total, the following
four concentrations were tested: 0.1mM, 1.0 mM, 5mM and 10mM,
however, the effects of last three appeared to be similar. Thus,
the minimum (0.1 mM) and maximum (1.0 mM) concentrations were
selected in order to indicate the effect of improving the energy
activity and survival rate of rotenone-treated PC12 cells/
Determination of cell survival
Cell survival was measured by a quantitative
colorimetric assay with
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma). Following drug treatment, PC12 cells were incubated for 4 h
with 5 mg/ml MTT. Subsequently the cell cultures were lysed in
dimethyl sulfoxide (DMSO; Sigma) for 15 min. The optical absorption
at 570 nm was determined using a plate reader (Synergy2, Biotek,
Winooski, VT, USA).
Intracellular lactate dehydrogenase (LDH)
assay
Using a previously described method (15), cell survival was quantified by the
measurement of LDH activity in cell lysates. Cells were lysed for
20 min in lysing buffer containing 0.04% Triton X-100, 2 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 0.2 mM
dithiothreitol, 0.01% bovine serum albumin and 0.1% phenol red. A
total of 50 μl cell lysates at pH 7.5 were mixed with 150 μl 500 mM
potassium phosphate buffer (pH 7.5) containing 1.5 mM NADH (Sigma)
and 7.5 mM sodium pyruvate (Sigma). The change in the absorbance at
340 nm (A340 nm) was monitored over 90 sec. Percentage cell
survival was calculated by standardizing the LDH activity of the
sample cell lysates to the LDH activity of the lysates of the
control (wash only) cell cultures.
Extracellular LDH assay
LDH is a cytosolic enzyme that is released into the
cell media upon cell lysis. Extracellular LDH activity is highly
correlated with a major index of cell necrosis: the level of
propidium iodide-positive cells (16). Therefore, extracellular LDH
activity may be used for assessing the number of cells undergoing
cell death (17). In the present
study, the extracellular LDH activity of PC12 cells was assessed
using a previously described method (16). Briefly, 100 μl extracellular media
from the samples was mixed with 150 μl potassium phosphate buffer
(500 mM, pH 7.5) containing 1.5 mM NADH and 7.5 mM sodium pyruvate.
Changes in the A340 nm of the samples was monitored over 90 sec
using a plate reader.
Determination of apoptotic and necrotic
cell death by flow cytometry-based Annexin V/7-aminoactinomycin D
(7-AAD) staining
Following washing twice with phosphate-buffered
saline (PBS), the cells were suspended in cold 1X binding buffer at
a concentration of 1×106 cells/ml. A total of 100 μl
cell suspension was mixed with 10 μl Annexin V R-phycoerythrin (PE)
conjugate (SouthernBiotech, Birmingham, AL, USA). Following
incubation on ice for 15 min, 200 μl cold 1X binding buffer was
added to each tube, followed by the addition of 10 μl 7-AAD
(SouthernBiotech). The number of the cells in early- and late-stage
apoptosis, and necrosis, was assessed by a flow cytometer (BD
FACSAria II; BD Biosciences, Franklin Lanes, NJ, USA).
Hoechst 33258 staining
The nuclear size of cells was assessed by Hoechst
staining (18). Following
treatment, cells were fixed in 4% formaldehyde for 15 min at room
temperature. After washing with PBS, the cells were stained with 20
μg/ml Hoechst 33258 (Sigma) in PBS for 20 min. Images of the
stained nuclei were captured under a fluorescence microscope (Leica
DMI3000B, Leica, Mannheim, Germany).
Western blot analysis of poly
(ADP-ribose) polymerase 1 (PARP-1)
Using a previously described method (19), PC12 cells were lysed in
radioimmunoprecipitation assay (RIPA) cell lysis buffer [50 mM
Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate
and 0.1% sodium dodecyl sulfate (SDS); Sigma] supplemented with 100
μM protease inhibitor cocktail (Roche Diagnostics, Mannheim,
Germany). Membrane fractions were separated by centrifugation at
16,000 × g for 20 min at 4°C. Protein concentrations were
determined using a bicinchoninic acid (BCA) protein assay kit
(Thermo Scientific, Rockford, IL, USA). The samples were normalized
to 30-μg of total protein extract and were fractionated by 10%
SDS-polyacrylamide gel electrophoresis and transferred onto
polyvinylidene difluoride membranes using a standard technique. The
membranes were blocked with 5% skimmed milk at 37°C for 1 h and
incubated with rabbit PARP-1 (p116/p25) antibodies (1:1,000;
Epitomics, Burlingame, CA, USA) at 4°C overnight. Horseradish
peroxidase (HRP)-linked anti-rabbit immunoglobulin G (IgG;
Epitomics) and enhanced chemiluminescence (ECL) substrate (Thermo
Scientific) were added and the bands were visualized. The equal
loading of samples was confirmed by stripping the membranes and
reprobing them with goat polyclonal anti-actin IgG (1:400, Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and HRP-conjugated
rabbit anti-goat IgG (Epitomics). Levels of immunoreactive proteins
were determined by densitometric scanning using a ChemiDoc XRS
system (Bio-Rad, Hercules, CA, USA).
Determination of the levels of ATP
ATP levels were quantified using an ATP
Bioluminescence assay kit HS II (Roche Diagnostics) according to
the manufacturers’ instructions. Cells were lysed with cell lysis
reagent (Roche Diagnostics) and 50 μl lysates were mixed with 150
μl luciferase assay reagent (Roche Diagnostics). The luminescence
was detected using a plate reader (Biotek Synergy 2; BioTek
Instrument, Inc., Winooski, VT, USA). The protein concentrations of
the samples were determined by a BCA assay. The ATP concentrations
of the samples were calculated using an ATP standard and normalized
against the amount of protein in the samples.
NAD+ assay
Following drug treatment, the cells were carefully
washed with PBS. The levels of NAD+ were measured using
a plate reader according to a previously described method (4). Cells were extracted in 0.25 ml 0.5 N
HClO4, scraped, neutralized with 3 M KOH and 100 mM
sodium phosphate buffer (pH 7.0). The levels of NAD+
were assessed based on the reduction of MTT to formazan by NADH,
which was generated by enzymatic cycling with alcohol
dehydrogenase. The rate of optical density (OD) increase at 560 nm
was determined by examining the samples immediately and 20 min
after the addition of the sample extracts.
Statistical analyses
All data are presented as mean + standard error.
Data were assessed by one-way analysis of variance (ANOVA) followed
by the Student-Newman-Keuls post hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Treatment with NMN attenuates
rotenone-induced injury of PC12 cells
The present study determined the effect of NMN
treatment on rotenone-induced changes in the survival rate of PC12
cells using MTT, intracellular LDH and extracellular LDH assays.
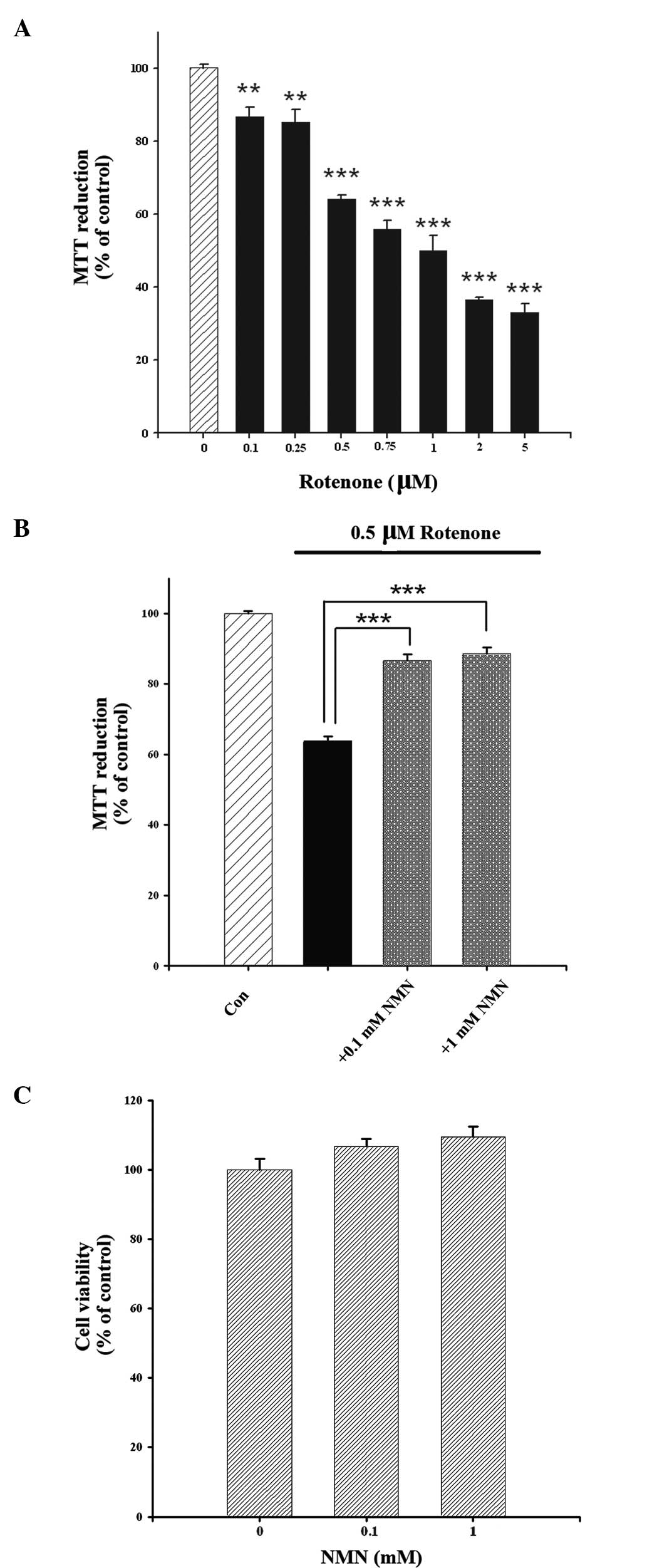
Treatment with rotenone for 24 h decreased the survival rate of
PC12 cells in a concentration-dependent manner, as assessed by MTT
assay (Fig. 1A). Since 0.5 μM
rotenone induced an ~60% decrease in MTT reduction, this
concentration of rotenone was used in subsequent experiments.
Co-treatment of the cells with NMN (0.1 or 1 mM) and rotenone led
to a significantly higher survival rate of the PC12 cells when
compared with the survival rate of the PC12 cells treated with
rotenone alone (Fig. 1B). The
effect of treatment with NMN alone is shown in Fig. 1C.
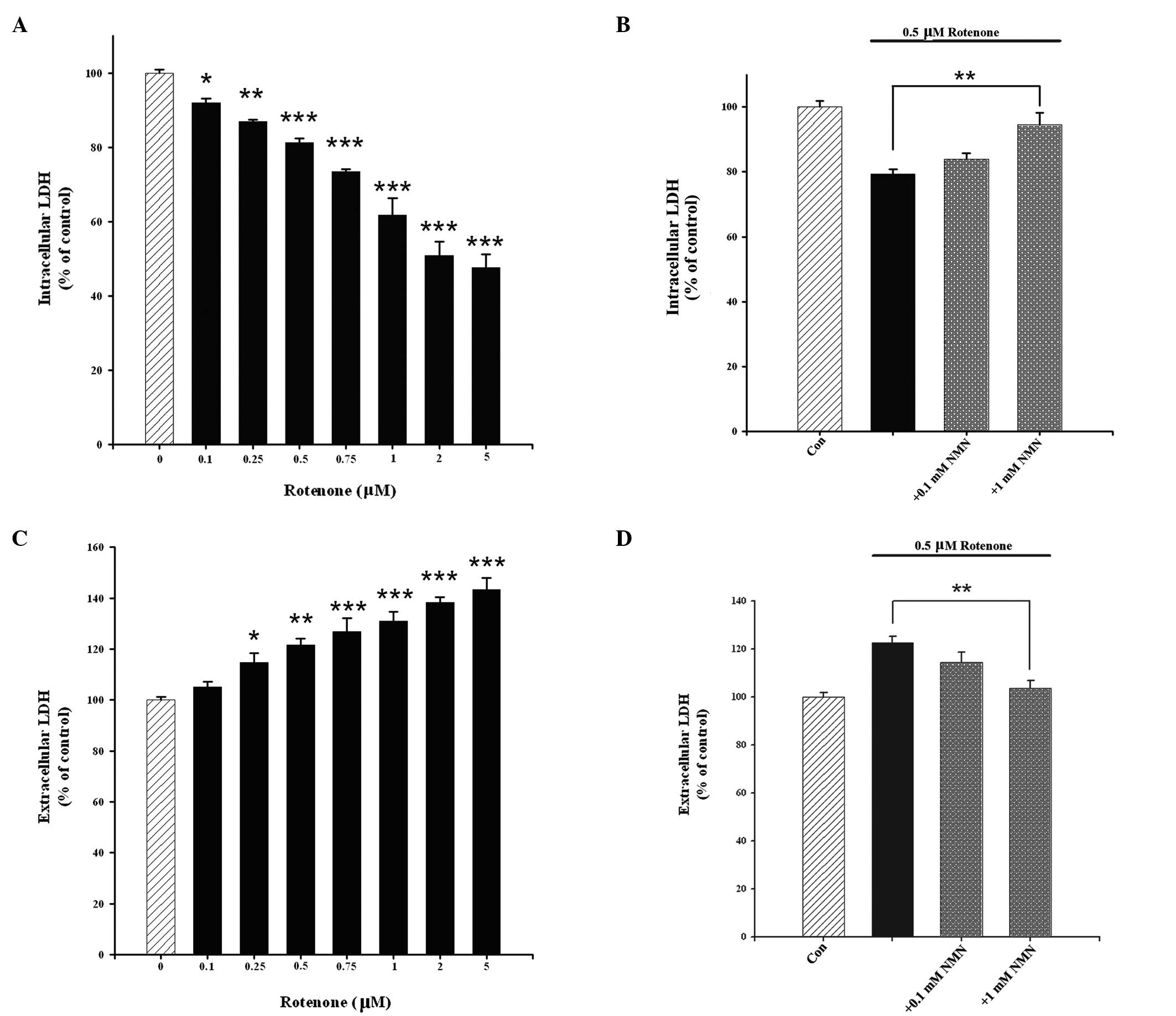
An intracellular LDH assay was also conducted to
determine the effects of NMN and rotenone on cell survival.
Treatment of the cells with rotenone caused a
concentration-dependent reduction in intracellular LDH activity,
which is an index of cell survival (Fig. 2A). This effect was significantly
attenuated by NMN co-treatment (Fig.
2B). Cell death was assessed by extracellular LDH assay. It was
observed that rotenone concentration-dependently induced the death
of the PC12 cells (Fig. 2C), which
was significantly attenuated by NMN co-treatment (Fig. 2D).
Treatment with NMN decreases
rotenone-induced apoptosis of PC12 cells
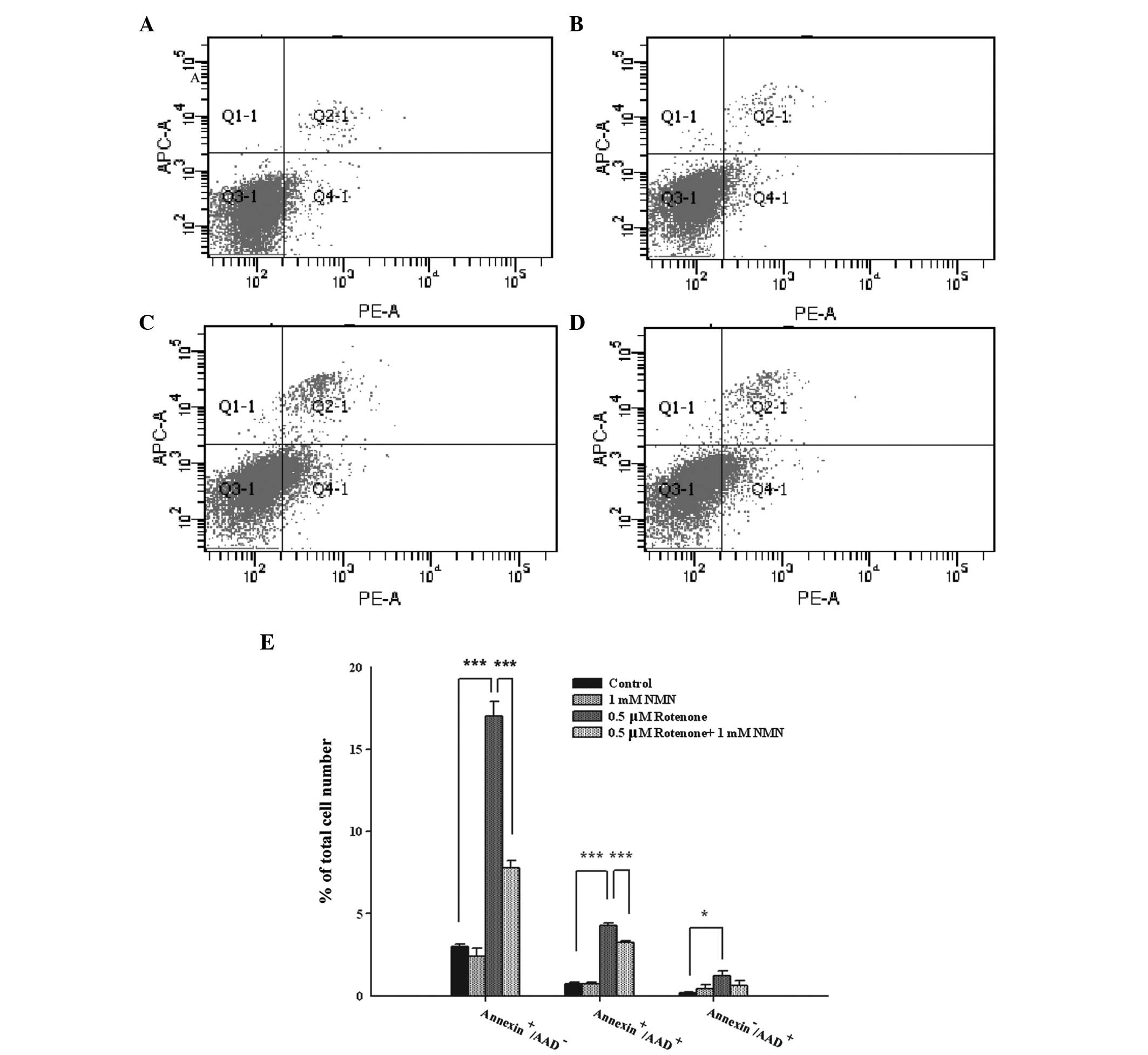
Flow cytometry-based Annexin V/7-AAD staining was
conducted to determine the early and late apoptosis and necrosis of
PC12 cells treated with rotenone and NMN. Phosphatidylserine (PS)
is expressed on the outer leaflet of the plasma membranes of
early-stage apoptotic cells, which may be stained by labeled
Annexin V. Late-stage apoptotic and necrotic cells lose the
integrity of their plasma membranes, which become permeable to the
fluorescent dye 7-AAD (20). Thus,
Annexin V−/7-AAD−, Annexin
V+/7-AAD−, Annexin
V+/7-AAD+ and Annexin
V−/7-AAD+ cells are defined as normal,
early-stage apoptotic, late-stage apoptotic and necrotic cells,
respectively. Treatment of the cells with 1 mM NMN (Fig. 3A) did not significantly affect the
level of cell death compared with that of the control cells
(Fig. 3B). Treatment of the cells
with 0.5 μM rotenone led to a marked increase in the number of
Annexin V+/7-AAD− cells, as well as increases
in the numbers of Annexin V+/7-AAD+ and
Annexin V−/7-AAD+ cells, compared with the
control group (Fig. 3C).
Co-treatment of the cells with rotenone and NMN led to significant
reductions in the numbers of Annexin
V+/7-AAD− and Annexin
V+/7-AAD+ cells when compared with the
rotenone treatment alone (Fig.
3D). Quantification of these results indicates that NMN
significantly decreases the numbers of early- and late-stage
apoptotic cells (Fig. 3E).
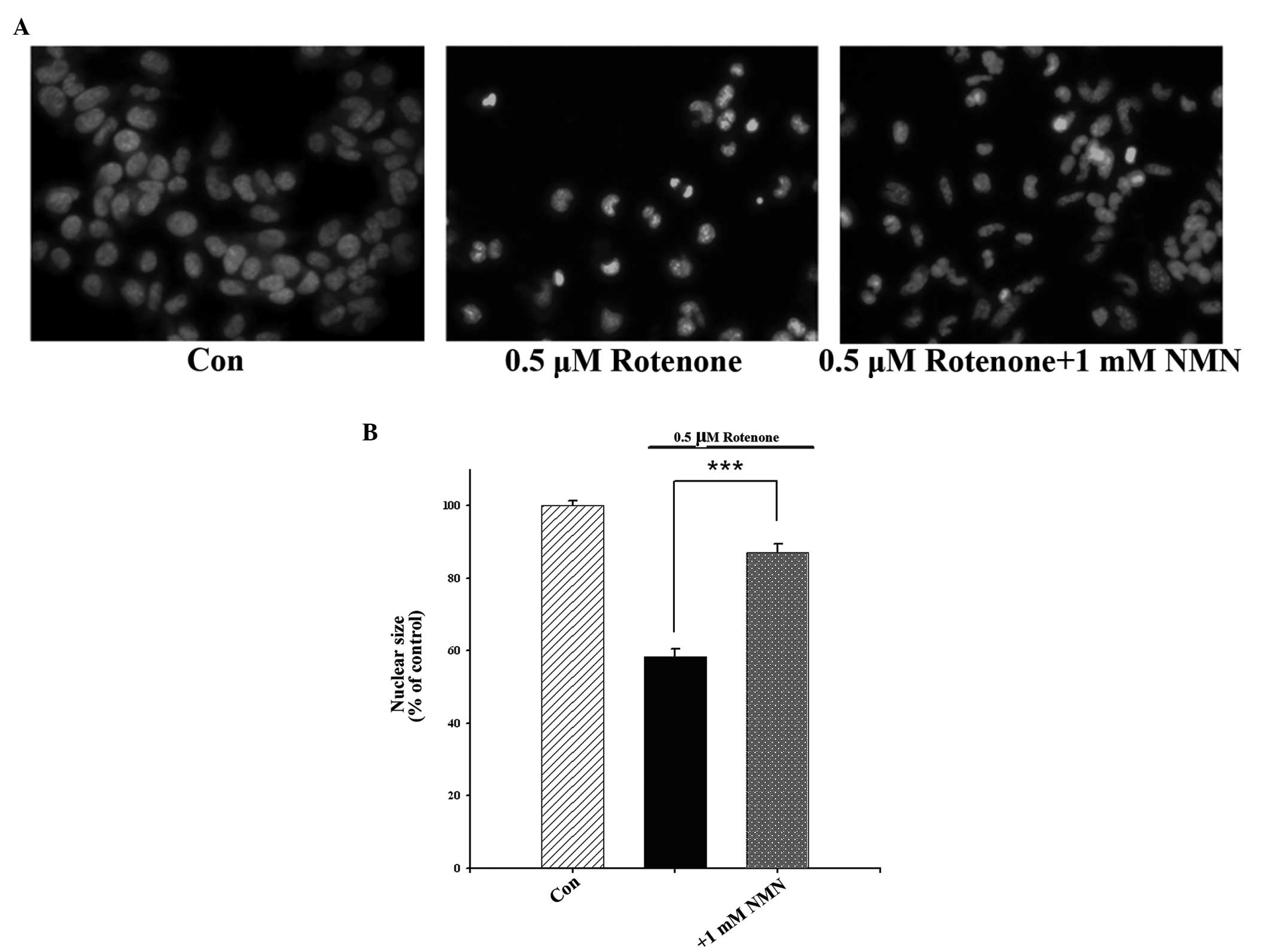
Nuclear condensation is an indication of cellular
apoptosis and may be observed through Hoechst 33258 staining.
Furthermore, PARP-1 is cleaved by active caspase-3 during
caspase-3-dependent apoptotic processes. To further determine
whether NMN is able to decrease rotenone-induced apoptosis, the
levels of nuclear condensation and PARP-1 cleavage in
rotenone-treated PC12 cells were assessed. It was observed that
rotenone concentration-dependently induced nuclear condensation,
and this effect was significantly attenuated by NMN co-treatment
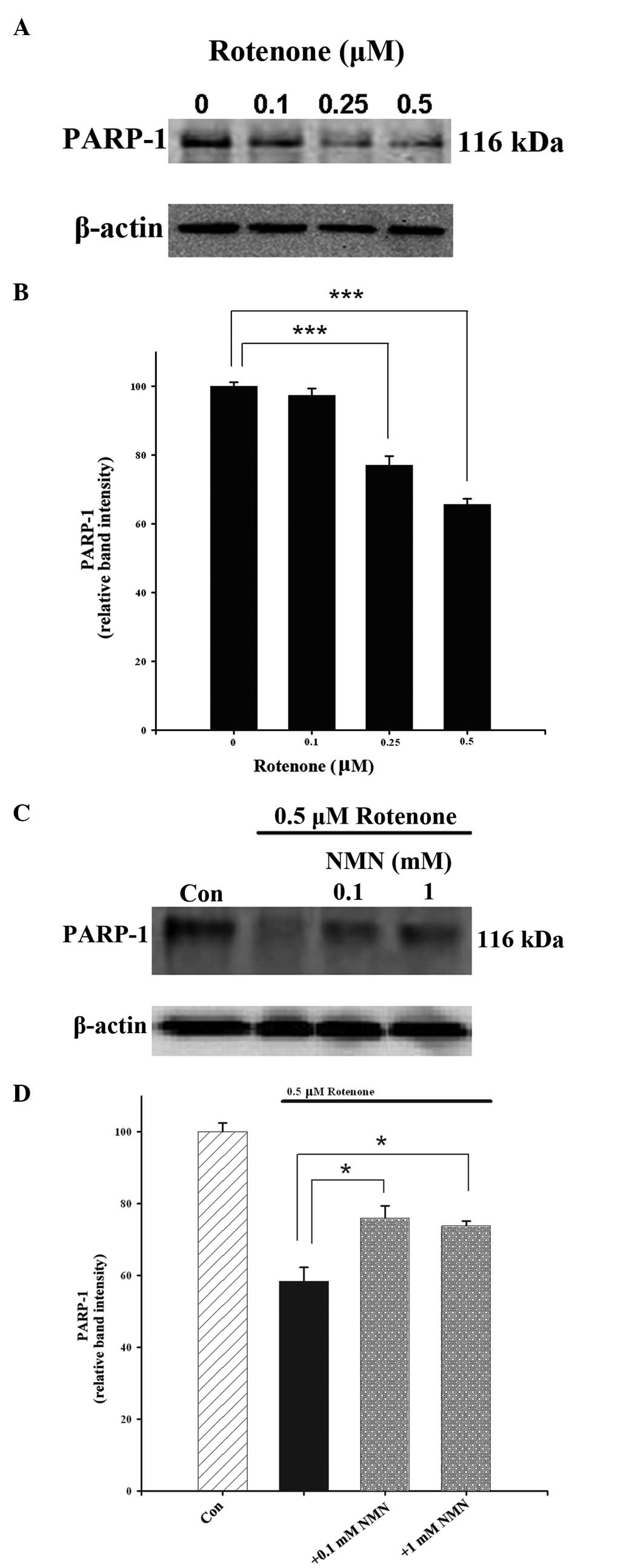
(Fig. 4). Western blot analysis
also revealed that rotenone concentration-dependently reduced the
levels of PARP-1 (Fig. 5A and B).
The rotenone-induced reductions in the levels of PARP-1 were
significantly attenuated by NMN co-treatment (Fig. 5C and D).
Treatment with NMN restores the
intracellular levels of NAD+ in rotenone-treated PC12
cells
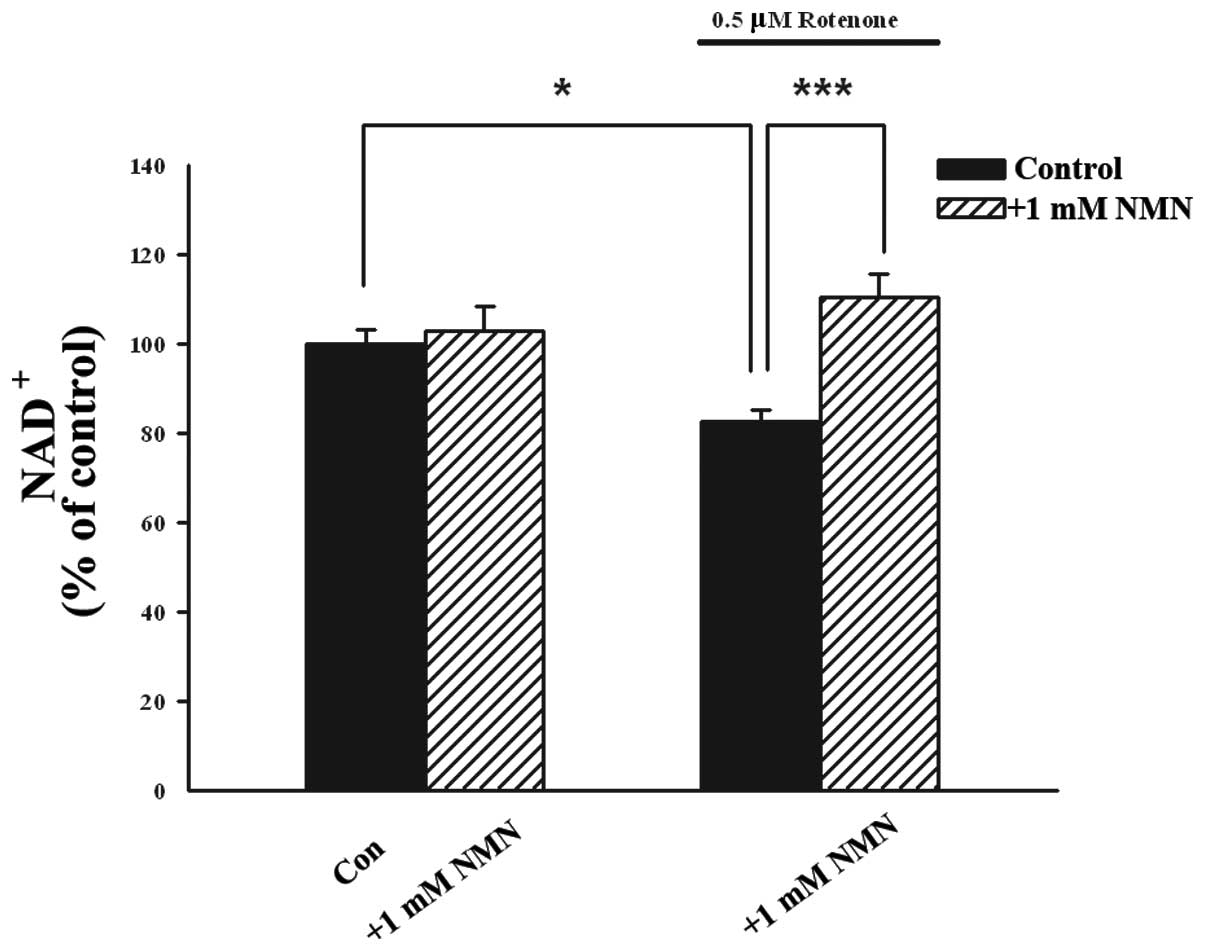
Since NMN is a precursor of NAD+, the
present study aimed to determine whether NMN treatment enhances the
intracellular levels of NAD+ in PC12 cells. It was
observed that treatment of PC12 cells with 0.5 mM rotenone led to a
significant reduction in the intracellular levels of
NAD+, which was prevented by co-treatment with 1 mM NMN
for 24 h (Fig. 6).
Treatment with NMN attenuates the
rotenone-induced reduction in intracellular levels of ATP in PC12
cells
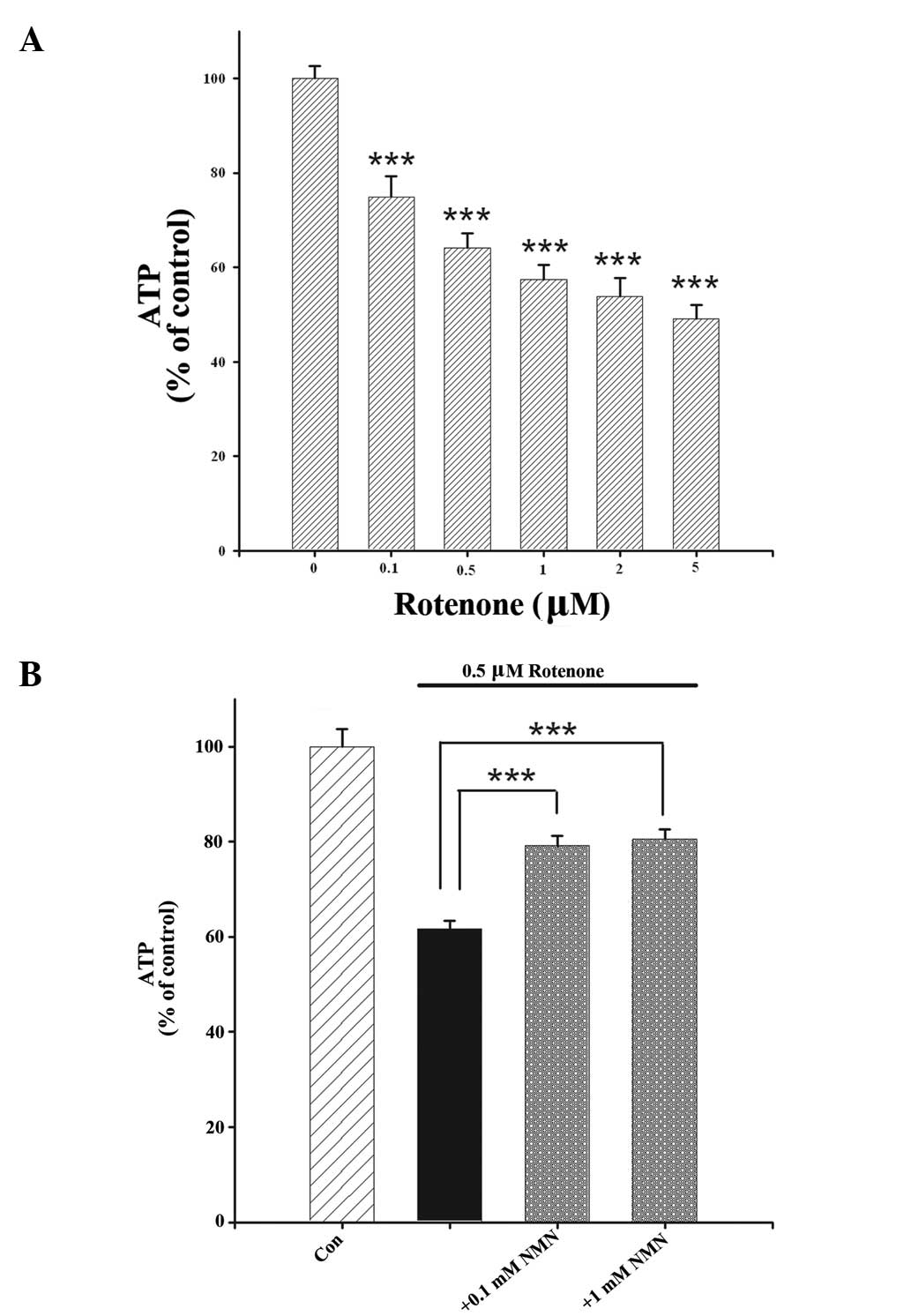
To investigate the mechanisms underlying the
protective effect of NMN on rotenone-induced cell death, the effect
of NMN treatment on the intracellular levels of ATP in
rotenone-treated cells was determined. Rotenone
concentration-dependently reduced the intracellular levels of ATP
in the cells (Fig. 7A); this
effect was significantly attenuated when the cells were co-treated
with NMN (Fig. 7B).
Discussion
The major results of the present study include: i)
NMN attenuated the rotenone-induced reduction in the survival rate
of PC12 cells; ii) NMN reduced the early- and late-stage apoptosis
of rotenone-treated PC12 cells; iii) NMN restored the intracellular
levels of NAD+ in rotenone-treated PC12 cells; and iv)
the protective effects of NMN against rotenone-induced cell death
were demonstrated through the prevention of rotenone-induced ATP
depletion.
The results of the present study suggest that NMN
attenuates cell apoptosis and decreases the intracellular levels of
ATP in rotenone-treated PC12 cells. Cumulative evidence has
suggested that NAD+ plays significant roles in a variety
of biological processes, including energy metabolism, mitochondrial
functions, calcium homeostasis, antioxidation/generation of
oxidative stress, gene expression, immunological functions, aging
and cell death (2).
NAD+ treatment has also been found to decrease the rate
of apoptosis of primary cultures of neurons, astrocytes and
myocytes, induced by various insults (4). NAD+ acts as a
neuroprotective agent via several mechanisms, including the
prevention of mitochondrial impairment (4,21),
prevention of ATP depletion and glycolytic inhibition (4,5,21),
and the enhancement of DNA repair (6).
NMN is a major precursor of NAD+ in the
salvage pathway of NAD+ synthesis, where it is converted
to NAD+ in cells by nicotinamide mononucleotide
adenylyltransferase (2). The
current study demonstrated that NMN treatment was highly protective
against the rotenone-induced cytotoxicity of PC12 cells in a
cellular model of PD. This was revealed through various cell
apoptosis assays, including LDH and MTT assays, and flow
cytometry-based Annexin V/7-AAD and Hoechst staining. Furthermore,
the present study demonstrated that NMN treatment was able to
significantly decrease the rotenone-induced apoptosis of cells, as
indicated by experiments that applied flow cytometry-based Annexin
V and Hoechst staining, and Western blot analysis of PARP-1.
The mechanisms underlying the protective effect of
NMN on the neurotoxicity of rotenone were investigated. The results
of the present study demonstrated that treatment with NMN restored
the intracellular levels of NAD+ and attenuated the
reduction in the levels of ATP in rotenone-treated PC12 cells.
Since intracellular ATP (22) and
NAD+ (2) are mediators
of cell survival, the beneficial effects of NMN on the levels of
ATP and NAD+ may at least partially account for the
protective effects of NMN against rotenone-induced cell death. As
NAD+ restoration may lead to a reduction in the ATP
consumption used for NAD+ synthesis (2), the NMN-induced restoration of
intracellular levels of NAD+ may account for the
beneficial effects of NMN on the intracellular levels of ATP.
Apoptotic changes are major pathological
transformations in PD and numerous other neurological diseases
(23,24). A compromise in energy metabolism
may also play a significant role in the pathology of
neurodegenerative disorders (9).
The present study indicates that NMN treatment may be highly
protective against not only apoptosis, but also energy compromise,
in rotenone-treated PC12 cells.
These results suggest that NMN may become a
promising drug for the treatment of PD and multiple other diseases
in which apoptosis and energy compromise play significant
pathological roles. Further in vivo studies of the effect of
NMN on PD are required.
Acknowledgements
The authors would like to thank all participants of
this study. This study was supported by the ‘973’ National Program
Grants for Basic Research No. 2007CB947900 (to SC), No.
2010CB945200 (to WY) and No. 2011CB504104 (to SC), the Key
Discipline Program Grant of Shanghai Municipality No. S30202 (to
SC), the Chinese National Science Foundation Grant No. 81171098 (to
WY), the Pujiang Scholar Program Grant No. 09PJ1405900 (to WY) and
the Research Grant of Shanghai Jiao Tong University for
Interdisciplinary Research on Engineering and Physical Sciences (to
WY).
References
|
1
|
Ying W: NAD+ and NADH in
cellular functions and cell death. Front Biosci. 11:3129–3148.
2006.
|
|
2
|
Ying W: NAD+/NADH and
NADP+/NADPH in cellular functions and cell death:
regulation and biological consequences. Antioxid Redox Signal.
10:179–206. 2008.
|
|
3
|
Xia W, Wang Z, Wang Q, et al: Roles of
NAD(+)/NADH and NADP(+)/NADPH in cell death. Curr Pharm Des.
15:12–19. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Alano CC, Ying W and Swanson RA:
Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes
requires NAD+ depletion and mitochondrial permeability
transition. J Biol Chem. 279:18895–18902. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ying W, Garnier P and Swanson RA:
NAD+ repletion prevents PARP-1-induced glycolytic
blockade and cell death in cultured mouse astrocytes. Biochem
Biophys Res Commun. 308:809–813. 2003.
|
|
6
|
Wang S, Xing Z, Vosler PS, et al: Cellular
NAD replenishment confers marked neuroprotection against ischemic
cell death: role of enhanced DNA repair. Stroke. 39:2587–2595.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ying W, Wei G, Wang D, et al: Intranasal
administration with NAD+ profoundly decreases brain
injury in a rat model of transient focal ischemia. Front Biosci.
12:2728–2734. 2007.
|
|
8
|
Fearnley JM and Lees AJ: Ageing and
Parkinson’s disease: substantia nigra regional selectivity. Brain.
114:2283–2301. 1991.
|
|
9
|
Beal MF: Mitochondria, oxidative damage,
and inflammation in Parkinson’s disease. Ann NY Acad Sci.
991:120–131. 2003.
|
|
10
|
Bredesen DE, Rao RV and Mehlen P: Cell
death in the nervous system. Nature. 443:796–802. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Simola N, Pinna A and Fenu S:
Pharmacological therapy of Parkinson’s disease: current options and
new avenues. Recent Pat CNS Drug Discov. 5:221–238. 2010.
|
|
12
|
Beal MF: Therapeutic approaches to
mitochondrial dysfunction in Parkinson’s disease. Parkinsonism
Relat Disord. 15(Suppl 3): S189–S194. 2009.
|
|
13
|
Olanow CW: The pathogenesis of cell death
in Parkinson’s disease - 2007. Mov Disord. 22(Suppl 17): S335–S342.
2007.
|
|
14
|
Bové J, Prou D, Perier C and Przedborski
S: Toxin-induced models of Parkinson’s disease. NeuroRx. 2:484–494.
2005.
|
|
15
|
Ying W and Swanson RA: The
poly(ADP-ribose) glycohydrolase inhibitor gallotannin blocks
oxidative astrocyte death. Neuroreport. 11:1385–1388. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ying W, Han SK, Miller JW and Swanson RA:
Acidosis potentiates oxidative neuronal death by multiple
mechanisms. J Neurochem. 73:1549–1556. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Roy MK, Takenaka M, Kobori M, Nakahara K,
Isobe S and Tsushida T: Apoptosis, necrosis and cell
proliferation-inhibition by cyclosporine A in U937 cells (a human
monocytic cell line). Pharmacol Res. 53:293–302. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ma Y, Chen H, Xia W and Ying W: Oxidative
stress and PARP activation mediate the NADH-induced decrease in
glioma cell survival. Int J Physiol Pathophysiol Pharmacol.
3:21–28. 2011.PubMed/NCBI
|
|
19
|
Nie H, Chen H, Han J, et al: Silencing of
SIRT2 induces cell death and a decrease in the intracellular ATP
level of PC12 cells. Int J Physiol Pathophysiol Pharmacol. 3:65–70.
2011.PubMed/NCBI
|
|
20
|
Zimmermann M and Meyer N: Annexin V/7-AAD
staining in keratinocytes. Methods Mol Biol. 740:57–63. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alano CC, Garnier P, Ying W, Higashi Y,
Kauppinen TM and Swanson RA: NAD+ depletion is necessary
and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal
death. J Neurosci. 30:2967–2978. 2010.
|
|
22
|
Huang F, Vemuri MC and Schneider JS:
Modulation of ATP levels alters the mode of hydrogen
peroxide-induced cell death in primary cortical cultures: effects
of putative neuroprotective agents. Brain Res. 997:79–88. 2004.
View Article : Google Scholar
|
|
23
|
Schulz JB: Anti-apoptotic gene therapy in
Parkinson’s disease. J Neural Transm Suppl. 467–476. 2006.
|
|
24
|
Liou AK, Clark RS, Henshall DC, Yin XM and
Chen J: To die or not to die for neurons in ischemia, traumatic
brain injury and epilepsy: a review on the stress-activated
signaling pathways and apoptotic pathways. Prog Neurobiol.
69:103–142. 2003. View Article : Google Scholar : PubMed/NCBI
|