Introduction
Acute-on-chronic liver failure (ACLF) is a recently
introduced term that represents the condition of severe acute
deterioration due to chronic liver disease. According to Asian
Pacific Association for The Study of the Liver (APASL) consensus,
ACLF is the acute and rapid deterioration of liver function
accompanied by subsequent multiple end-organ failure in a patient
with previously well-compensated liver disease due to the effects
of a precipitating event (1). In
the majority of Asian countries, hepatitis B accounts for 70% of
all ACLF cases (1). Although a
number of clinical studies have investigated ACLF, the
pathophysiology of chronic hepatitis B (CHB)-related ACLF remains
poorly understood. Ample evidence suggests that dysregulated
inflammation plays an important role due to an imbalanced host
response to injury, with a self-perpetuating effect of liver
insufficiency (2–6). A previous study suggested that the
transition from a stable cirrhotic condition to a sudden, acute
decompensation leading to liver failure is caused by an acute
systemic inflammatory response that is mainly mediated by cytokines
(7). Inflammation of
cholangiocytes, which are involved in hyperbilirubinemia and
intrahepatic cholestasis, is always observed during ACLF and is
considered a marker of poor prognosis.
High mobility group protein (HMG) is a ubiquitous
nuclear protein that is expressed in a number of eukaryotic cells.
HMG box 1 (HMGB1) belongs to this family, which also includes HMGA,
HMGB and HMGN. There are two binding motifs (the A and B boxes) in
HMGB1, and the active cytokine domain of HMGB1 is localized to the
B box (8,9). HMGB1 is shuttled between the nucleus
and cytoplasm via nuclear pores.
As a nuclear protein, HMGB1 stabilizes nucleosomes
and enables DNA binding to facilitate gene transcription; however,
HMGB1 is also actively released by monocytes and macrophages in
response to inflammatory cytokines and is passively released by
necrotic or damaged cells (10,11).
HMGB1 was recently identified as a late mediator of lethal systemic
inflammation diseases, and this role was also demonstrated in an
animal model of cytokine-mediated disease (11). Elevated levels of HMGB1 have been
observed in patients with acute pancreatitis, acute lung injury,
rheumatoid arthritis, hemorrhagic shock, and ischemia-reperfusion
injury (12–16), suggesting that HMGB1 is closely
associated with these pathogenic processes and may be a central
molecule that triggers and maintains the cascading inflammatory
reactions.
High levels of HMGB1 have also been detected in the
sera of patients with chronic hepatitis (17), acute liver failure (18) and hepatitis B virus-related ACLF
(19), demonstrating that HMGB1
release is associated with liver cell damage. In the present study,
it was investigated whether HMGB1 is released from cholangiocytes
to induce cholangiole inflammation and the exacerbation of
intrahepatic cholestasis in ACLF and if such a release can be
induced by lipopolysaccharide (LPS) or tumor necrosis factor-α
(TNF-α) in vitro.
Materials and methods
Patients and specimens
Between January 2006 and December 2007, 13 patients
with ACLF and 20 patients with CHB were included in this study.
ACLF and chronic hepatitis diagnoses fulfilled the criteria of the
APASL 2008 consensus (1).
Histology was confirmed independently by two pathologists at the
You’an Hospital (Beijing, China). All 13 patients underwent liver
transplantation. Serological data were collected from archived
patient records prior to treatment. Liver function tests included
analysis of alanine aminotransferase (ALT), aspartate
aminotransferase (AST), total bilirubin (TB), direct bilirubin (DB)
and albumin (ALB) levels. The serological marker for hepatitis B
was examined prior to ACLF diagnosis, and HBsAg/HBcAg in the liver
tissues were routinely detected by immunochemistry in the
Department of Pathology. The study conformed to the tenets of the
Declaration of Helsinki, and informed written consent was obtained
from all patients prior to the study.
Immunohistochemical staining of liver
samples
Liver samples from patients with ACLF and CHB were
retrieved from the archives of the Department of Pathology, and
normal liver samples were obtained from donors at the liver
transplantation center at the You’an Hospital. Serial
formalin-fixed, paraffin-embedded samples were deparaffinized in
xylene and rehydrated in a series of graded alcohols and distilled
water. Endogenous peroxidase activity was quenched by incubation in
3% H2O2. Prior to immunostaining, the
sections were incubated for 12 min in citrate buffer (pH 6.0) in a
microwave oven at 99°C to enhance their immunoreactivity.
Subsequent to blocking, the sections were incubated at 4°C
overnight with rabbit polyclonal anti-HMGB1 antibody (ab-18256;
Abcam, Cambridge, MA, USA) or mouse monoclonal anti-CK7 antibody
(Zhongshan Jinqiao Biotechnology Co., Beijing, China). Detection
was performed using the streptavidin-biotin-peroxidase method with
the PV-9000 kit (Zhongshan Jinqiao Biotechnology Co.) according to
the manufacturer’s instructions. Diaminobenzidine was employed as a
chromogen. Normal liver tissues from adult donors served as
negative controls. The slides were counterstained with hematoxylin
and mounted. The immunoreactivity of the cholangiocytes was graded
based on the percentage of immunopositive cells: +++, >67% of
cells stained; ++, 33–67%; +, 5–33%; focal, <5%; and −, no
stained cells. Focal staining was also considered negative
staining.
TFK-1 cell culture
The human cholangiocarcinoma cell line TFK-1
(20) was kindly provided by
Professor Liu Shun Ai (Ditan Hospital, Beijing, China). TFK-1 cells
were maintained in RPMI-1640 medium (HyClone, Logan, UT, USA)
supplemented with 100 IU/ml penicillin, 100 mg/ml streptomycin, and
10% fetal bovine serum (Gibco-BRL, Grand Island, NY, USA). The
cells were cultured at 37°C in a humid atmosphere of 95% air and 5%
CO2. The cells were passaged three times per week.
TFK-1 cell stimulation with LPS and
TNF-α
TFK-1 cells were seeded at a density of
5×104 cells/ml in 96-well culture dishes (Corning Inc.,
Corning, NY, USA) and stimulated with 10, 100 or 500 ng/ml TNF-α
(Peprotech, Rocky Hill, NJ, USA) or 1, 10 or 40 μg/ml LPS (Sigma,
St. Louis, MO, USA) for 4, 8, 16 or 24 h. Unstimulated cells were
used as a control group.
Cell viability assay
Cell viability was determined using an MTT assay as
previously described (21).
Briefly, TFK-1 cells were cultured in 96-well plates overnight at a
density of 2×103 cells per well. The cells were exposed
to LPS or TNF-α at each concentration in triplicate at a final
volume of 200 μl. Culture medium was used as a control blank. After
4, 8, 16 or 24 h of culture, the absorbance of each well was
measured using a Bio-Rad microplate reader (Bio-Rad, Hercules, CA,
USA). The cell viability (%) was calculated according to the
following formula: Absorbance of experimental well/absorbance of
control well × 100.
HMGB1 concentration in the culture medium
of TFK-1 cells
The concentration of HMGB1 in the medium of the LPS-
or TNF-α-treated and control cells was determined using an ELISA
assay according to the manufacturer’s instructions (HMGB1 ELISA
Kit; R&D systems, Abingdon, UK). The ELISA standards ranged
from 78–5,000 pg/ml. All samples were measured in triplicate.
Cell nuclear-cytoplasmic fractionation
and western blot analysis
Cells were harvested, and nuclear-cytoplasmic
fractionation was conducted using NE-PER® Nuclear and
Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Waltham,
MA, USA) according to the manufacturer’s instructions. Western blot
analysis was performed as previously described (22). Equivalent amounts of protein were
separated by SDS-PAGE and transferred to polyvinylidene difluoride
membranes (Millipore, Bedford, MA, USA). Subsequent to blocking,
the membranes were incubated with the appropriate diluted primary
antibodies, targeting HMGB1, proliferating cell nuclear antigen
(PCNA; Abcam) and β-actin (Sigma). The signal of the target protein
was detected using an enhanced chemiluminescence detection system
(Pierce Biotechnology, Inc., Rockford, IL, USA) and recorded on
film in the linear detection range.
Statistical analysis
All continuous data are expressed as the mean ±
standard deviation. Comparisons between groups were performed using
the Student’s t-test or the χ2 test, including Fisher’s
exact test. Statistical significance was defined as P<0.05. All
statistical analyses were performed using SPSS software, version
16.0 for Windows (SPSS, Inc., Chicago, IL, USA).
Results
Baseline characteristics of the
patients
The baseline characteristics of the 13 patients in
the ACLF group and the 20 patients in the CHB group are shown in
Table I. The TB (428.76±214.40
μmol/l in ACLF vs. 19.49±13.17 μmol/l in CHB) and DB (232.56±115.99
μmol/l in ACLF vs. 6.89±6.65 μmol/l in CHB) levels were highly
elevated and the percentage of prothrombin activity (26.47±11.52%
in ACLF vs. 95.06±11.48% in CHB) was extremely low in the ALCF
group compared with the CHB group. Of the 13 patients with ACLF,
five had encephalopathy (grade II–III), four had ascites, one had
gastrointestinal tract hemorrhage and five had infections. These
complications are consistent with the diagnosis of ACLF. All
patients with CHB were HBsAg/HBcAg positive in liver tissues, and
11 of the 13 patients with ACLF were positive for HBsAg/HBcAg.
 | Table IBaseline characteristics of the
patients in the ACLF and chronic hepatitis B groups. |
Table I
Baseline characteristics of the
patients in the ACLF and chronic hepatitis B groups.
| Characteristics | ACLF (n=13) | Chronic hepatitis B
(n=20) | P-value |
|---|
| Age (years) | 41.84±12.31 | 28.20±7.01 | <0.005 |
| Gender
(female/male) | 3/10 | 8/12 | 0.456 |
| ALT (U/L) | 165.75±221.51 | 162.91±135.73 | 0.968 |
| AST (U/L) | 158.28±158.75 | 91.42±71.59 | 0.157 |
| TB (μmol/l) | 428.76±214.40 | 19.49±13.17 | <0.005 |
| DB (μmol/l) | 232.56±115.99 | 6.89±6.65 | <0.005 |
| ALB (g/l) | 34.16±4.20 | 38.60±5.22 | 0.025 |
| PT (sec) | 28.73±6.74 | 12.51±0.70 | <0.005 |
| PTA (%) | 26.47±11.52 | 95.06±11.48 | <0.005 |
HMGB1 and CK7 expression in patients with
ACLF and CHB
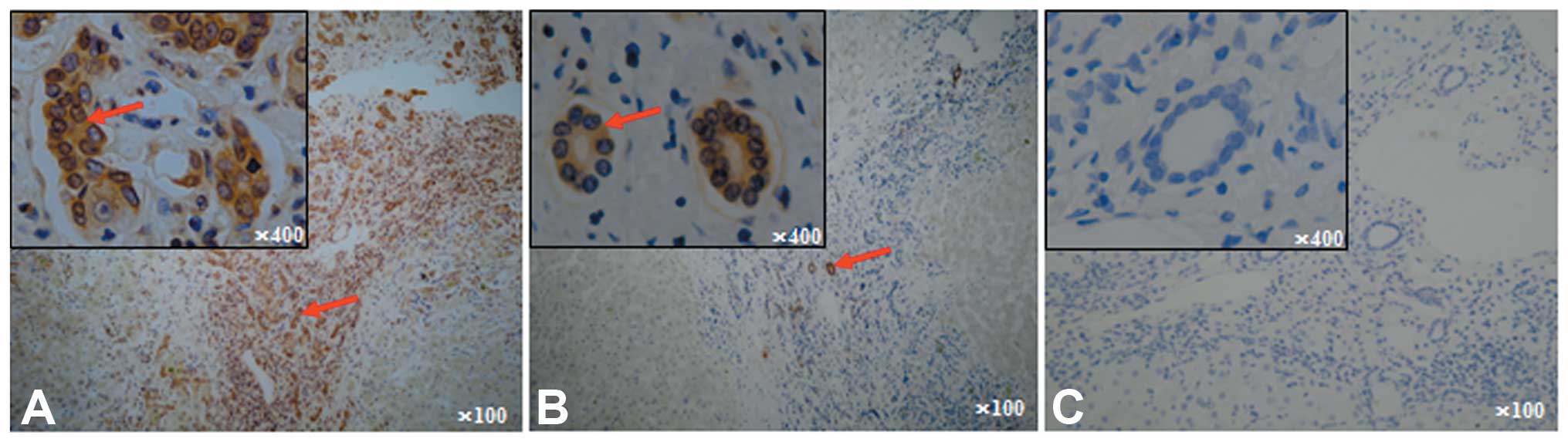
HMGB1-positive staining was predominantly observed
in the cytoplasm and extracellular area of cholangiocytes,
particularly in the newly formed cholangiocytes in most
inflammatory and portal areas of ACLF. Positive staining for HMGB1
was also observed in the nuclei of certain cholangioles (Fig. 1A), suggesting that
nuclear-cytoplasmic HMGB1 translocation occurs in the
cholangiocytes of patients with ACLF. Few cholangioles displayed
positive HMGB1 staining in the CHB liver tissues (Fig. 1B). Negative HMGB1 or focal positive
staining was observed in the normal liver donors (Fig. 1C). In addition, positive HMGB1
staining was also observed in the monocytes, lymphocytes, and
hepatocytes of the inflammatory portal areas in the livers of
patients with ACLF and CHB but not in the livers of normal
donors.
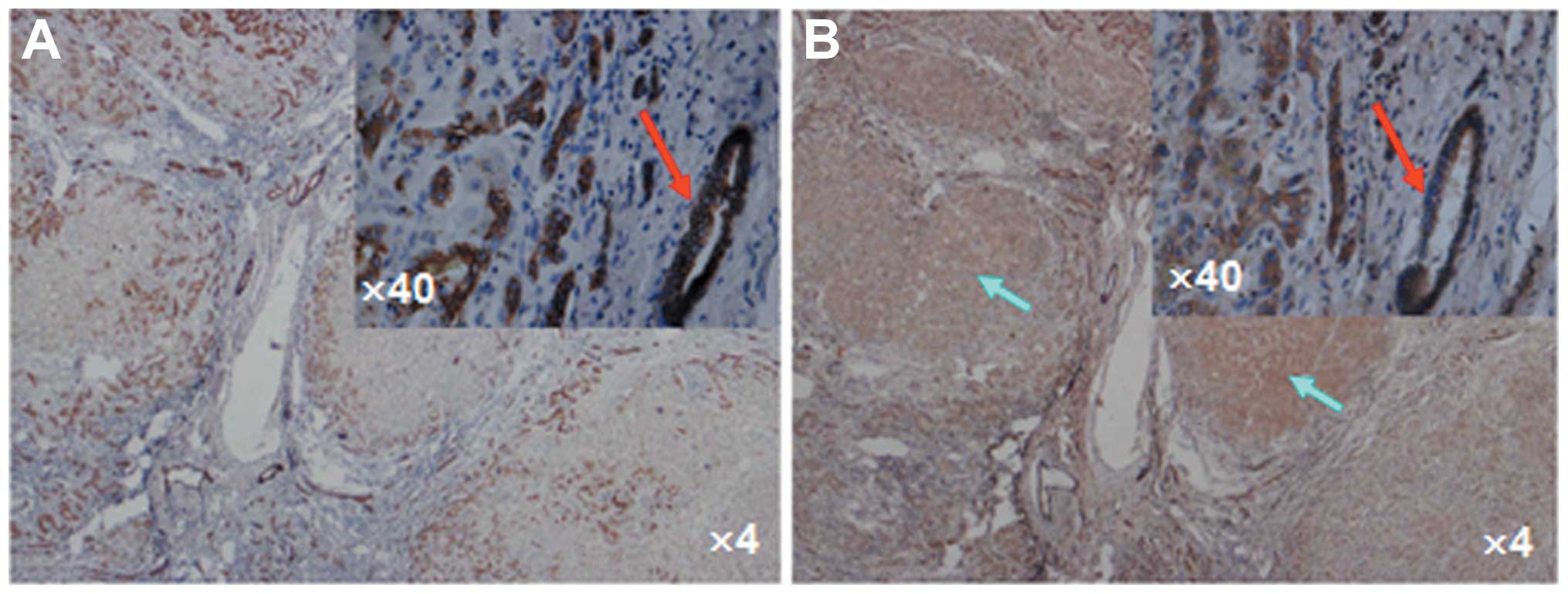
CK7 was used as a biomarker for cholangiocytes to
confirm the cell origin. Positive CK7 staining was observed in the
cytoplasm of the cholangiocytes in the newly formed cholangioles.
The cholangiocytes of the newly formed cholangioles that were
positive for CK7 staining were also positive for HMGB1 staining in
the liver samples of ACLF patients (Fig. 2), indicating that the release of
HMGB1 from cholangiocytes plays a crucial role in the high serum
HMGB1 level in patients with ACLF. The proliferation of large
amounts of newly formed cholangioles accompanied by the release of
the proinflammatory mediator HMGB1 suggests the possibility of
intrahepatic cholestasis caused by inflammation around the
cholangioles and bile duct. In addition, positive HMGB1 staining
was also observed in the hepatocytes of certain pseudolobules
(Fig. 2B), consistent with a
previous study (23).
Of the 13 patients with ACLF, 11 showed >67%
positive (+++) staining for HMGB1 in the cholangiocytes, and two
showed 33–67% positive staining (++) for HMGB1. However, none of
the CHB patients showed >33% positive HMGB1 staining (+++~++) in
cholangioles; 2 of the 20 CHB patients showed weakly positive
(<5%) HMGB1 (+) staining, and 18 of the 20 patients showed
negative HMGB1 staining. Almost all the normal donors exhibited
negative HMGB1 staining, except for one or two focal positive
cholangioles. Thus, HMGB1 release was significantly higher in the
patients with ACLF than in the patients with CHB (P<0.001;
Table II).
| Table IIHMGB1 expression in cholangiocytes in
the liver tissues of patients with ACLF and CHB. |
Table II
HMGB1 expression in cholangiocytes in
the liver tissues of patients with ACLF and CHB.
| HMGB1
expression |
|---|
|
|
|---|
| Patient groups | − | + | ++ | +++ |
|---|
| ACLF (n=13) | 0 | 0 | 2 | 11 |
| CHB (n=20)a | 18 | 2 | 0 | 0 |
| P-value | | | <0.001 | |
Active and passive secretion of HMGB1
from TFK-1 cells stimulated with LPS and TNF-α
To examine the active secretion of HMGB1 by TFK-1
cells, it was investigated whether LPS and TNF-α induced
HMGB1 release from TFK-1 cells. Cultured TFK-1 cells were subjected
to increasing concentrations of LPS or TNF-α for 4, 8, 16 and 24 h,
as described in Materials and methods. The HMGB1 concentration in
the medium of the cultured cells was determined at different time
points using an ELISA assay simultaneously with an examination of
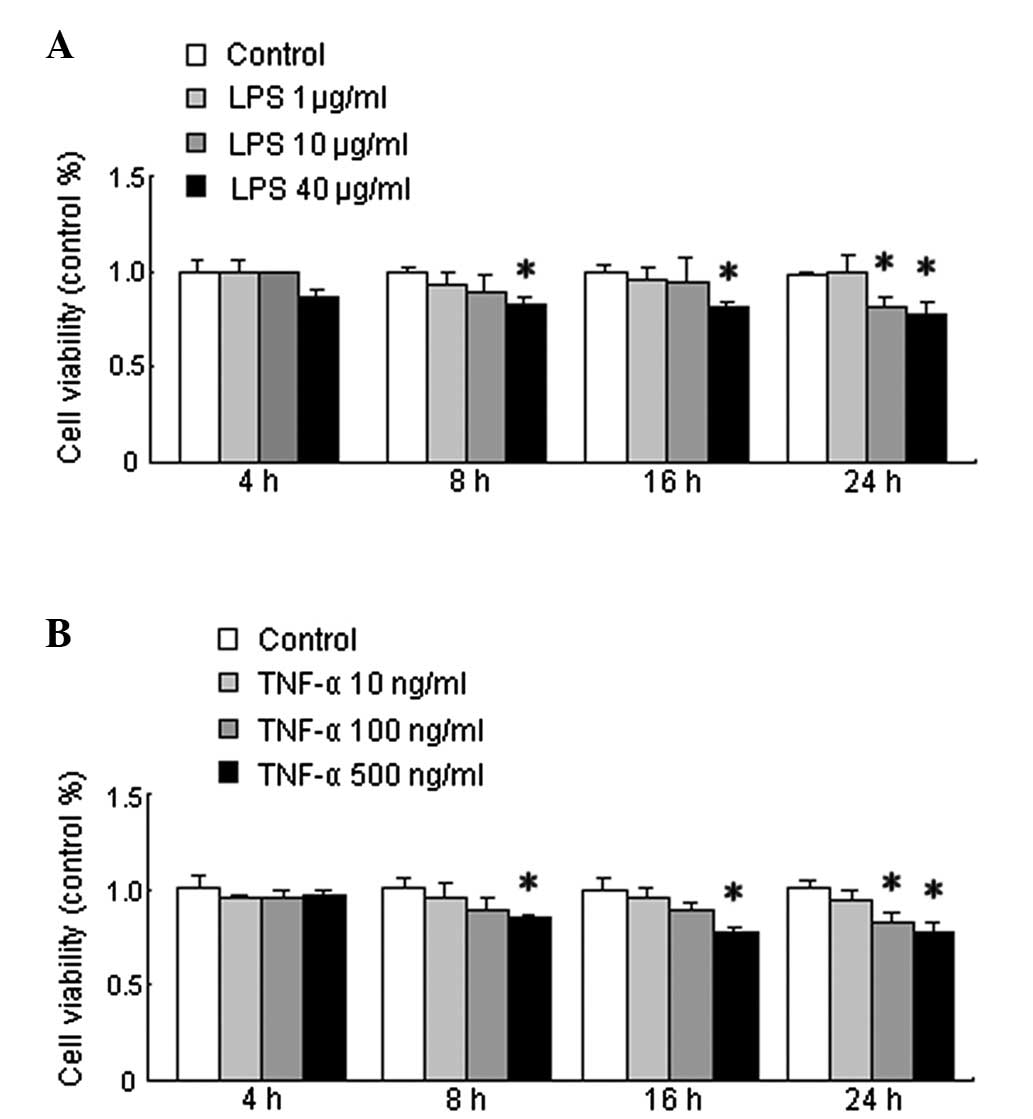
cell viability. As shown in Fig.
3A, there was no difference between the cell viability of the
untreated cells and that of the cells treated with LPS for 4 h at
each concentration. The viability of the cells treated with LPS for
8, 16, and 24 h at a concentration of 40 μg/ml and for 24 h at 10
μg/ml was significantly decreased compared with that of the
untreated cells (P<0.05). The HMGB1 concentration in the medium
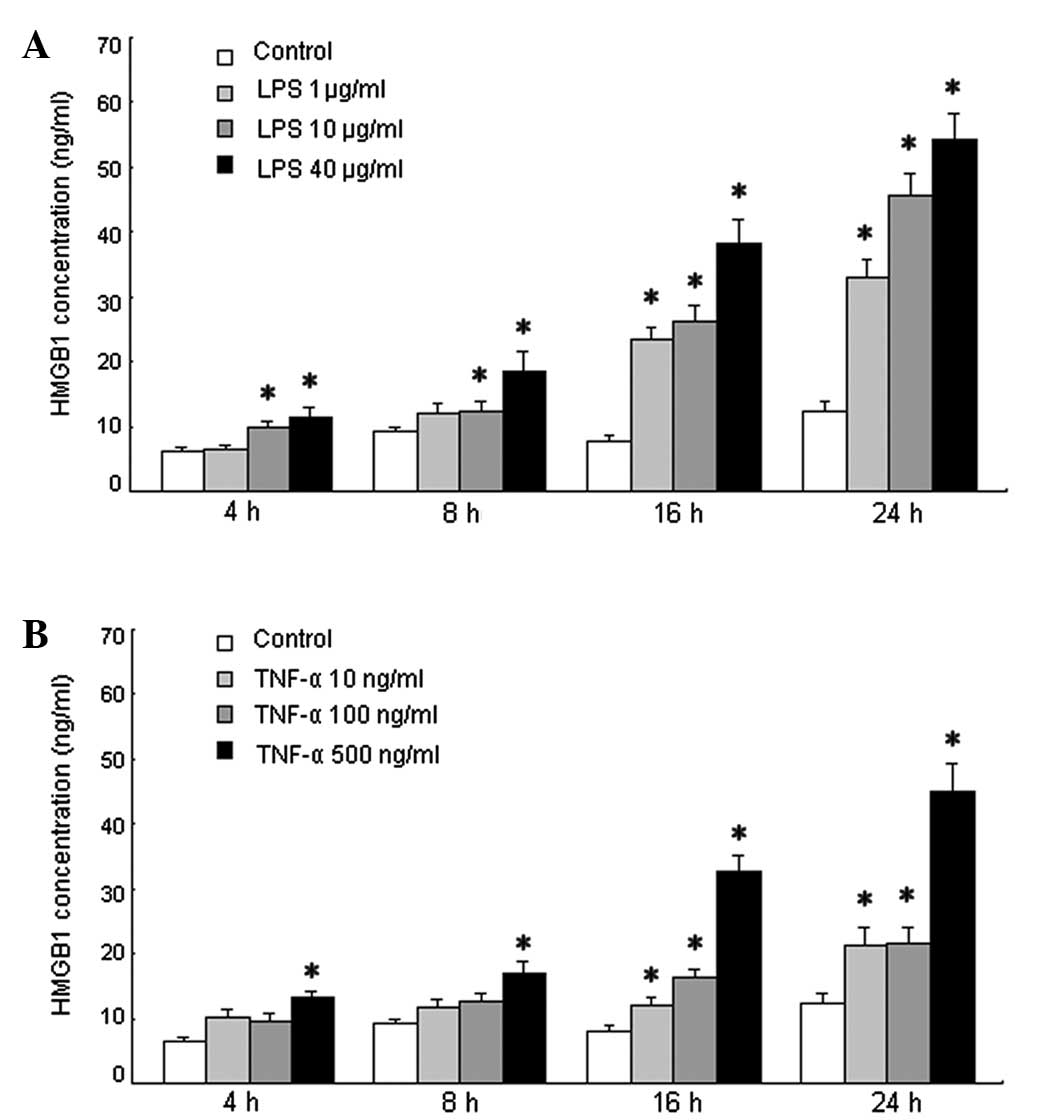
of the LPS-treated TFK-1 cells at each LPS concentration at each
time point is shown in Fig. 4A.
The HMGB1 concentration increased after stimulation with LPS at
each time point compared with that for the untreated cells
(P<0.05).
There was no difference between the cell viability
of the untreated cells and the cells treated with TNF-α for 4 h at
each concentration. As shown in Fig.
3B, cell viability was significantly decreased in the cells
treated with TNF-α for 8, 16 and 24 h at a concentration of 500
ng/ml and for 24 h at 100 ng/ml compared with that of the untreated
cells (P<0.05). The HMGB1 concentration in the culture medium of
the TNF-α-treated TFK-1 cells at each TNF-α concentration at each
time point is shown in Fig. 4B.
The HMGB1 concentration gradually increased after stimulation with
TNF-α compared with that for the untreated cells at each time
point. The HMGB1 concentration in the medium of the cells treated
with 500 ng/ml TNF-α was increased significantly at each time point
compared with that for the untreated cells (P<0.05). In
addition, the HMGB1 concentration in the medium of the cells
treated with 10 or 100 ng/ml TNF-α for 16 and 24 h was also
significantly increased compared with that for the untreated cells
(P<0.05). Therefore, exogenous (LPS) and endogenous (TNF-α)
inflammatory stimuli induced HMGB1 release in TFK-1 cell cultures,
starting at 4 h post-LPS or -TNF-α stimulation. The release of
HMGB1 was not completely dependent on cell death since the cell
viability was not significantly altered by 1 or 10 μg/ml LPS or by
10 or 100 ng/ml TNF-α, even at 16 h post treatment (Fig. 3). Furthermore, LPS- or
TNF-α-stimulated TFK-1 cells released HMGB1 in a
concentration-dependent manner (Fig.
4), starting at concentrations as low as 10 μg/ml LPS (Fig. 4A) or 500 ng/ml TNF-α (Fig. 4B). The increase in HMGB1
concentration was mainly due to active release of HMGB1 by the
TFK-1 cells. At slightly cytotoxic dosages, LPS (40 μg/ml) or TNF-α
(500 ng/ml) may also cause passive HMGB1 leakage from necrotic
TFK-1 cells.
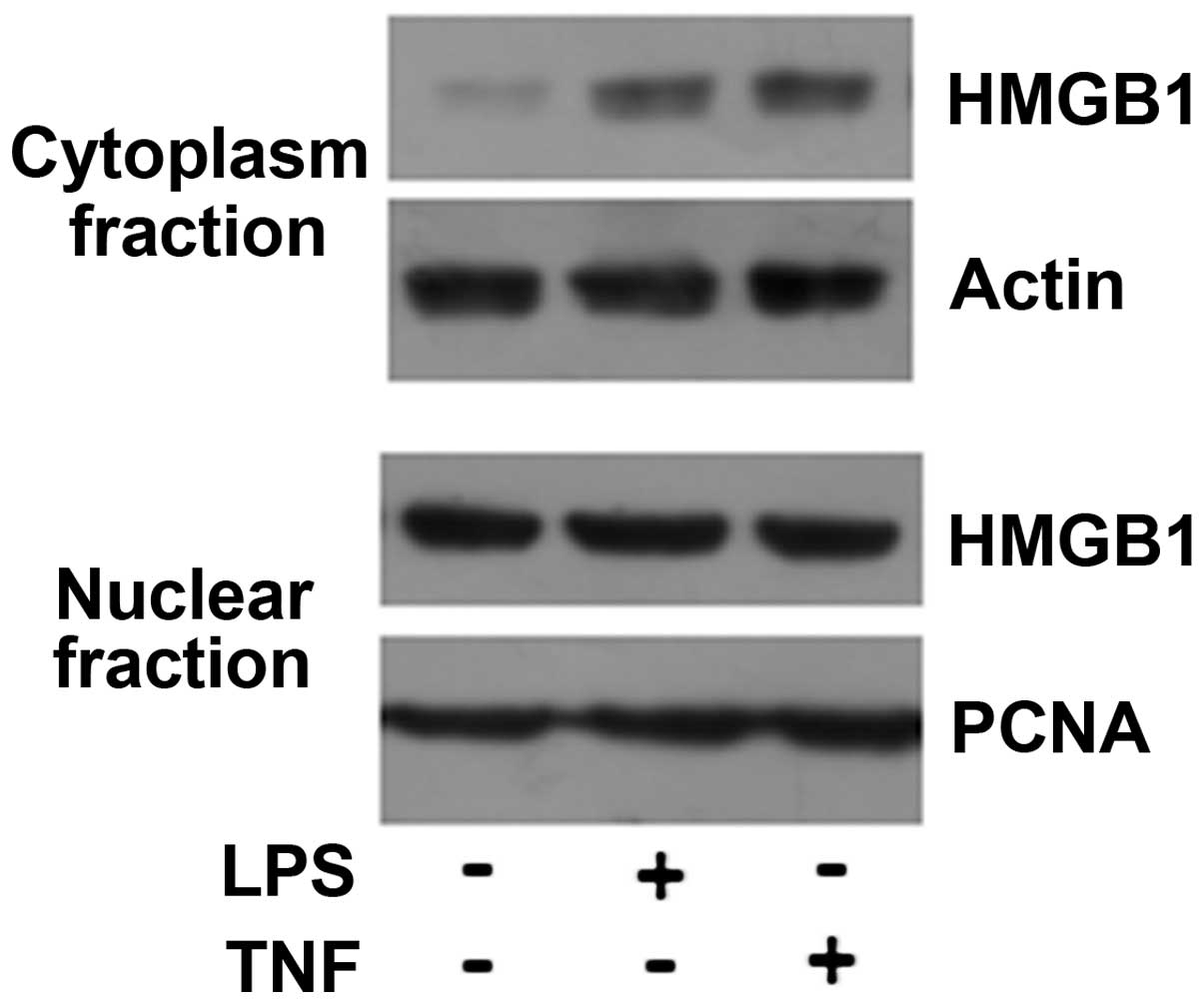
Immunohistochemical analysis of the liver tissues of
the ACLF patients indicated cytoplasmic translocation of HMGB1 in
the cholangiocytes. TFK-1 cells were stimulated with 10 μg/ml LPS
or 100 ng/ml TNF-α for 8 h, and the cells were then collected and
prepared for cytoplasmic and nuclear fractionation. The results of
the western blot analysis are shown in Fig. 5; the HMGB1 level in the cytoplasmic
fraction increased following stimulation with LPS or TNF-α. Taken
together, these results suggest that endogenous and exogenous
inflammatory stimuli can induce HMGB1 nuclear-cytoplasmic
translocation in cholangiocytes and active HMGB1 release.
Discussion
The most common clinical manifestation of liver
failure in Asia, particularly in China, is ACLF, which is defined
as an acute episode of liver function decompensation in a liver
that is already impaired by chronic hepatitis B (24). Although the etiology of ACLF varies
between Western and Eastern countries, the clinical manifestations
are remarkably similar, suggesting that the various pathogenic
factors induce common patterns of innate immune responses (25). In addition, many other
proinflammatory cytokines, including TNF-α and IL-6, may play a
common role in the pathophysiology of ACLF (26). It has been reported that
monocytes/macrophages actively release HMGB1 in response to
exogenous stimuli, such as LPS, or endogenous inflammatory stimuli,
including TNF-α and IL-1β (11).
Active HMGB1 release has been demonstrated in non-immune cells
including pituicytes and enterocytes (27,28).
HMGB1 release has been observed in hepatocytes from patients
suffering from various liver diseases (23,29),
and the cytoplasmic translocation of HMGB1 has been observed in
patients with acute liver failure (23). Inflammation of the cholangioles is
also important in ACLF; therefore, whether HMGB1 is released from
cholangiocytes in patients suffering from ACLF was
investigated.
In this study, cytoplasmic translocation of HMGB1
was observed in cholangiocytes from patients with ACLF caused by
HBV infection. Furthermore, the experiments revealed that active
nuclear-cytoplasmic HMGB1 translocation and release occurred in
TFK-1 cells upon stimulation with exogenous (LPS) and endogenous
(TNF-α) inflammatory stimuli.
In China, a large proportion of ACLF cases are
caused by HBV infection (1); thus,
the present study evaluated the cytoplasmic translocation of HMGB1
in patients with ACLF caused by hepatitis B. Consistent with
previous reports, histopathological changes were observed,
including massive, sub-massive, or bridging necrosis with immune
cell infiltration in the cholangiocytes. Notably, HMGB1 was clearly
observed in the cholangiocytes, particularly in newly formed
cholangioles, in samples from patients with ACLF caused by HBV
infection but not in samples from patients with chronic HBV
infection or normal donors. One limitation of this clinical study
is that the liver tissue samples were obtained during liver
transplantation surgery and thus did not represent the early
clinical manifestation immediately following the onset of ACLF.
HepG2 cells (human hepatocellular carcinoma cell
line) can be induced to release HMGB1 by treatment with LPS or
TNF-α in a time- and dose-dependent fashion (23). HMGB1 may also be released passively
following necrosis (10,30) and apoptosis, depending on the cell
type (31,32). It has also been reported that human
gingival fibroblasts release HMGB1 protein through both active and
passive pathways (32). In the
present study, positive HMGB1 staining was predominantly observed
in the cytoplasm and extracellular areas of cholangiocytes in the
most inflamed and portal area in the samples from the patients with
ACLF, indicating cytoplasmic translocation of HMGB1 in these cells.
Furthermore, HMGB1 translocation from the nucleus to the cytoplasm
was confirmed by western blot analysis in TFK-1 cells incubated
with LPS and TNF-α. It has been widely suggested that
re-localization and accumulation of HMGB1 in the cytoplasm is a
necessary step for its extracellular release, raising the
possibility that activated cholangiocytes, particularly newly
formed ones, could be a source of extracellular HMGB1, which might
contribute to the inflammatory response during ACLF. During liver
injury/failure, HMGB1 may be released as a danger signal from the
activated or damaged cholangiocytes as well as from immune cells
and necrotic cells.
Clinical signs and symptoms may not indicate whether
the stimulating event was active or passive because complications,
particularly infections, usually accompany ACLF. Of the 13 ACLF
patients investigated, all were positive for serum HBsAg, and 11
were positive for HBsAg in liver tissue. In addition, five patients
suffered from bacterial infections, four from ascites, and one from
a gastrointestinal tract hemorrhage. Thus, HMGB1 may be
synergistically released by the above two pathways as a result of
infection and cell damage, leading to exacerbated inflammation and
injury of the bile duct, particularly the newly formed
cholangioles, intrahepatic cholestasis, and hyperbilirubinemia
during the process of ACLF. HMGB1 release was observed in the
cholangiocytes, particularly in the newly formed cholangioles, in
the liver tissues of ACLF patients. Active and passive release of
HMGB1 was observed in TFK-1 cells induced with LPS or TNF-α in
vitro. Therefore, HMGB1 may act as a late inflammatory mediator
triggered by LPS and TNF-α in the inflammatory process of ACLF.
Cellular and ductular cholestasis indicate acute injury of the
liver. Features of cholestasis and bile duct proliferation are very
common in patients with acute liver injury, particularly in
patients with ACLF. The release of HMGB1 from the newly formed
cholangiocytes was confirmed by CK7 staining in the inflamed portal
area. It may be hypothesized that the increased inflammation
triggered by HMGB1 and/or TNF-α could be at least partly
responsible for intrahepatic cholestasis and could further lead to
hyperbilirubinemia in patients with ACLF.
To the best of our knowledge, this is the first
study to demonstrate that HMGB1 is actively released from
cholangiocytes, particular from newly formed cholangioles, in ACLF.
As an effective inflammatory mediator of TNF-α or endotoxin (LPS),
HMGB1 may progressively exacerbate the inflammation and damage of
the bile duct, intrahepatic cholestasis, and hyperbilirubinemia
during the early stages of ACLF. Infection is the most common cause
of exacerbated hyperbilirubinemia in the clinic. The prevalence of
ductular bilirubinostasis in biopsy specimens from patients with
developed infections is significantly higher than in those without
developed infections (33). Since
HMGB1 is elevated during infection, strategies to reduce HMGB1
activity using antibodies may be therapeutically effective
(34,35). Duan et al (36) indicated that an enhanced serum
level of HMGB1 was associated with the development of HBV-related
ACLF in patients with CHB. The strong correlation between HMGB1 and
AST levels suggest that HMGB1 may be a useful prognostic marker for
the development of ACLF.
The present results demonstrate the
nuclear-cytoplasmic translocation of HMGB1 in the cholangiocytes of
patients with ACLF caused by HBV infection as well as in TFK-1
cells upon stimulation with either exogenous (LPS) or endogenous
(TNF-α) inflammatory stimuli. These observations raise important
questions regarding the potential pathogenic roles of HMGB1 in
cholangiocytes in ACLF and suggest that HMGB1 may be a therapeutic
target.
Acknowledgements
The authors would like to thank their pathologist,
Lijie Zhang (Department of Pathology, Beijing You’an Hospital), for
assistance with collecting and reviewing the samples. This study
was supported by the ‘215’ high-level health technology project
(2011) and the ‘12th Five-Year’ plan of major science and
technology projects for major infectious disease prevention and
control (2014ZX10002002-001-002).
References
|
1
|
Sarin SK, Kumar A, Almeida JA, et al:
Acute-on-chronic liver failure: consensus recommendations of the
Asian Pacific Association for the study of the liver (APASL).
Hepatol Int. 3:269–282. 2009. View Article : Google Scholar
|
|
2
|
Shawcross DL, Wright GA, Stadlbauer V, et
al: Ammonia impairs neutrophil phagocytic function in liver
disease. Hepatology. 48:1202–1212. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tandon P and Garcia-Tsao G: Bacterial
infections, sepsis, and multiorgan failure in cirrhosis. Semin
Liver Dis. 28:26–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mookerjee RP, Stadlbauer V, Lidder S, et
al: Neutrophil dysfunction in alcoholic hepatitis superimposed on
cirrhosis is reversible and predicts the outcome. Hepatology.
46:831–840. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bone RC, Grodzin CJ and Balk RA: Sepsis: a
new hypothesis for pathogenesis of the disease process. Chest.
112:235–243. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cazzaniga M, Dionigi E, Gobbo G, et al:
The systemic inflammatory response syndrome in cirrhotic patients:
relationship with their in-hospital outcome. J Hepatol. 51:475–482.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rolando N, Wade J, Davalos M, et al: The
systemic inflammatory response syndrome in acute liver failure.
Hepatology. 32:734–739. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li J, Kokkola R, Tabibzadeh S, et al:
Structural basis for the proinflammatory cytokine activity of high
mobility group box 1. Mol Med. 9:37–45. 2003.PubMed/NCBI
|
|
9
|
Wang H, Yang H and Tracey KJ:
Extracellular role of HMGB1 in inflammation and sepsis. J Intern
Med. 255:320–331. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scaffidi P, Misteli T and Bianchi ME:
Release of chromatin protein HMGB1 by necrotic cells triggers
inflammation. Nature. 418:191–195. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang H, Bloom O, Zhang M, et al: HMG-1 as
a late mediator of endotoxin lethality in mice. Science.
285:248–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yasuda T, Ueda T, Shinzeki M, et al:
Increase of high-mobility group box chromosomal protein 1 in blood
and injured organs in experimental severe acute pancreatitis.
Pancreas. 34:487–488. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ueno H, Matsuda T, Hashimoto S, et al:
Contributions of high mobility group box protein in experimental
and clinical acute lung injury. Am J Respir Crit Care Med.
170:1310–1316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hamada T, Torikai M, Kuwazuru A, et al:
Extracellular high mobility group box chromosomal protein 1 is a
coupling factor for hypoxia and inflammation in arthritis.
Arthritis Rheum. 58:2675–2685. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fan J, Li Y, Levy RM, et al: Hemorrhagic
shock induces NAD(P)H oxidase activation in neutrophils: role of
HMGB1-TLR4 signaling. J Immunol. 178:6573–6580. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tsung A, Sahai R, Tanaka H, et al: The
nuclear factor HMGB1 mediates hepatic injury after murine liver
ischemia-reperfusion. J Exp Med. 201:1135–1143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu HB, Fan XG, Huang JJ, et al: Serum
level of HMGB1 in patients with hepatitis B and its clinical
significance. Zhonghua Gan Zang Bing Za Zhi. 15:812–815. 2007.(In
Chinese).
|
|
18
|
Oshima G, Shinoda M, Tanabe M, et al:
Increased plasma levels of high mobility group box 1 in patients
with acute liver failure. Eur Surg Res. 48:154–162. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Duan XZ, Hu JH, Li C, et al: Relation
between serum levels of high mobility group box 1 and hepatitis B
virus-related acute-on-chronic liver failure. Zhonghua Gan Zang
Bing Za Zhi. 21:434–437. 2013.(In Chinese).
|
|
20
|
Saijyo S, Kudo T, Suzuki M, et al:
Establishment of a new extrahepatic bile duct carcinoma cell line,
TFK-1. Tohoku J Exp Med. 177:61–71. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang D, Gao Q, Guo L, et al: Isolation
and identification of cancer stem-like cells in esophageal
carcinoma cell lines. Stem Cells Dev. 18:465–473. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li LW, Yu XY, Yang Y, et al: Expression of
esophageal cancer related gene 4 (ECRG4), a novel tumor suppressor
gene, in esophageal cancer and its inhibitory effect on the tumor
growth in vitro and in vivo. Int J Cancer. 125:1505–1513. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou RR, Zhao SS, Zou MX, et al: HMGB1
cytoplasmic translocation in patients with acute liver failure. BMC
Gastroenterol. 11:212011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liver Failure and Artificial Liver Group,
Chinese Society of Infectious Diseases, Chinese Medical
Association; Severe Liver Diseases and Artificial Liver Group,
Chinese Society of Hepatology, Chinese Medical Association.
Diagnostic and treatment guidelines for liver failure. Zhonghua Gan
Zang Bing Za Zhi. 14:643–646. 2006.(In Chinese).
|
|
25
|
Leifeld L, Dumoulin FL, Purr I, et al:
Early up-regulation of chemokine expression in fulminant hepatic
failure. J Pathol. 199:335–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ambrosino G, Naso A, Feltracco P, et al:
Cytokines and liver failure: modification of TNF- and IL-6 in
patients with acute on chronic liver decompensation treated with
Molecular Adsorbent Recycling System (MARS). Acta Biomed. 74(Suppl
2): 7–9. 2003.PubMed/NCBI
|
|
27
|
Liu S, Stolz DB, Sappington PL, et al:
HMGB1 is secreted by immunostimulated enterocytes and contributes
to cytomix-induced hyperpermeability of Caco-2 monolayers. Am J
Physiol Cell Physiol. 290:C990–C999. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang H, Vishnubhakat JM, Bloom O, et al:
Proinflammatory cytokines (tumor necrosis factor and interleukin 1)
stimulate release of high mobility group protein-1 by pituicytes.
Surgery. 126:389–392. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sitia G, Iannacone M, Muller S, Bianchi ME
and Guidotti LG: Treatment with HMGB1 inhibitors diminishes
CTL-induced liver disease in HBV transgenic mice. J Leukoc Biol.
81:100–107. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rovere-Querini P, Capobianco A, Scaffidi
P, et al: HMGB1 is an endogenous immune adjuvant released by
necrotic cells. EMBO Rep. 5:825–830. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bell CW, Jiang W, Reich CF III and
Pisetsky DS: The extracellular release of HMGB1 during apoptotic
cell death. Am J Physiol Cell Physiol. 291:C1318–C1325. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Feghali K, Iwasaki K, Tanaka K, et al:
Human gingival fibroblasts release high-mobility group box-1
protein through active and passive pathways. Oral Microbiol
Immunol. 24:292–298. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Katoonizadeh A, Laleman W, Verslype C, et
al: Early features of acute-on-chronic alcoholic liver failure: a
prospective cohort study. Gut. 59:1561–1569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pisetsky DS, Erlandsson-Harris H and
Andersson U: High-mobility group box protein 1 (HMGB1): an alarmin
mediating the pathogenesis of rheumatic disease. Arthritis Res
Ther. 10:2092008. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Andersson U and Tracey KJ: HMGB1 is a
therapeutic target for sterile inflammation and infection. Annu Rev
Immunol. 29:139–162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Duan XZ, Hu JH, Li C, et al: Relation
between serum levels of high mobility group box 1 and hepatitis B
virus-related acute-on-chronic liver failure. Zhonghua Gan Zang
Bing Za Zhi. 21:434–437. 2013.(In Chinese).
|